
Biocompatible and biodegradable fluorescent microfibers physiologically secreted by live cells upon spontaneous uptake of thiophene fluorophore†
Ilaria E.
Palamà
*a,
Francesca
Di Maria
bc,
Stefania
D'Amone
a,
Giovanna
Barbarella
cd and
Giuseppe
Gigli
abe
aInstitute Nanoscience CNR (NNL, CNR-NANO), via Arnesano, Lecce, Italy. E-mail: ilariaelena.palama@nano.cnr.it
bDept. Matematica e Fisica ‘Ennio De Giorgi’, University of Salento, via Monteroni, Italy
cConsiglio Nazionale Ricerche (CNR-ISOF), Via P. Gobetti 101, Bologna, Italy
dMediteknology srl, Via P. Gobetti 101, Bologna, Italy
eItalian Institute of Technology (IIT) – Center for Biomolecular Nanotechnologies, via Barsanti, Arnesano, Italy
First published on 22nd October 2014
Abstract
Live cells can form multifunctional and environmentally responsive multiscale assemblies of living and non-living components. We recently reported the results of a unique approach to introduce supplementary properties, fluorescence in particular, into fibrillar proteins produced by live fibroblasts and extruded into the ECM. In this work, we demonstrate that the physiological secretion of fluorescent nanostructured microfibers upon the spontaneous uptake of the appropriate fluorophore extends to living cells derived by different tissue contexts. We also show that live cells seeded on fluorescent microfibers have a different fate in terms of the cellular morphology, cytoskeleton rearrangement and viability. These results suggest that the microfibers, which are biocompatible and biodegradable, can be used as multiscale biomaterials to direct the cell behaviour.
Introduction
A living cell contains complex substructures that are spatially and functionally self-organizing1 and present unique characteristics useful for materials production and patterning. Our understanding of these systems and their dynamics is largely dependent on the possibility to visualize their presence inside the cells, particularly by fluorescence imaging. To this aim, a variety of organic and inorganic tools have been developed, ranging from fluorescent proteins2 to nanoparticles,3,4 quantum dots5 and small organic molecules.6 A great advantage of the latter is the possibility to employ the methodologies of synthetic chemistry to tailor their optical properties and direct the position of the fluorophore inside live cells through appropriate functionalization.7An emerging class of probes for biochemical imaging is that of thiophene oligomers and polymers. K. P. R. Nilsson et al. reported that a charged quinquethiophene can mark amyloid fibers8in vitro and in vivo and can also discriminate between various types of cells.9 Recently, we reported that some cell-permeant and biocompatible thiophene fluorophores have the capability to spontaneously cross the cell membrane without the need for any type of vector and selectively recognize specific proteins inside living cells.10–12 In particular, the green fluorescent thiophene fluorophore DTTO – namely 3,5-dimethyl-2,3′-bis(phenyl) dithieno[3,2-b;2′,3′-d]thiophene-4,4-dioxide (Fig. 1) – can spontaneously cross the membrane of live embryonic fibroblasts (3T3), be specifically recognized by the hydroxyproline component of protocollagen polypeptide chains and be incorporated via nonbonding interactions during the self-assembly of protocollagen into triple helices. The process leads to the secretion of helical fluorescent microfibres subsequently extruded into the extracellular matrix.11 DTTO is a semiconducting molecule, and collagen–DTTO microfibers are also electroactive.12
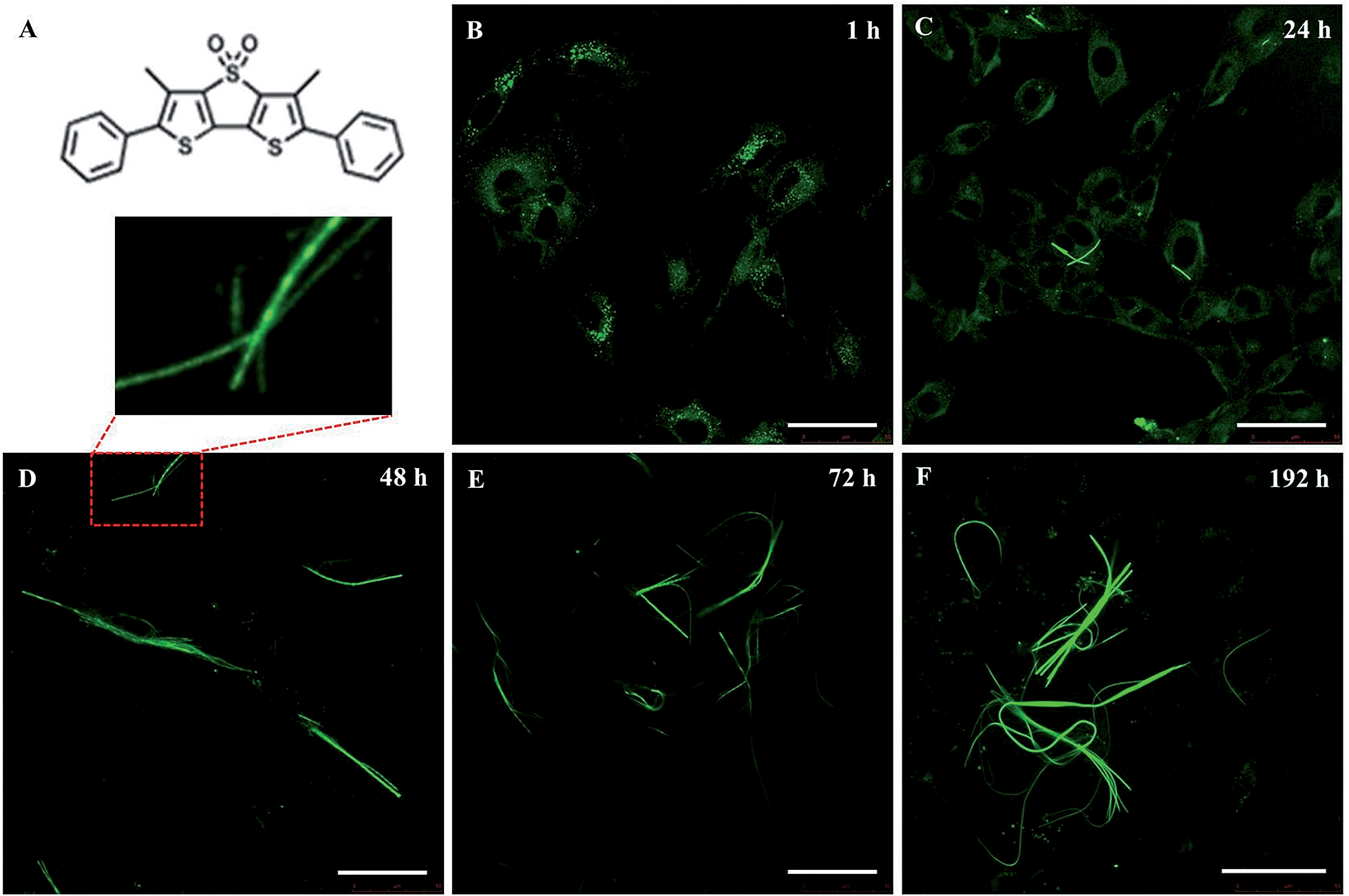 | ||
| Fig. 1 LSCM images of the intracellular formation of fluorescent microfibers in mouse neuroblastoma cells (B104 cell line). (A) Molecular structure of DTTO. (B–F) Time course of the cells incubated with DTTO at a concentration of 0.05 mg mL−1 in DMEM medium. Scale bars: 50 μm. | ||
Here, we report the capability of DTTO to spontaneously cross the membrane of live cells, be recognized by specific proteins and physiologically form fluorescent protein(s)–DTTO microfibers extending to mouse neuroblastoma cells (B104). The interest in testing the B104 cells line is that these cells do not produce collagen, thus the protein(s) targeted by DTTO must be of a different nature. We show that QTOF analysis of the fluorescent microfibers isolated from the cell context indicates that the fluorescent microfibers are mainly made of vimentin,13 a fibrillar protein that is overexpressed in various types of cancers,14 as well as in the Alzheimer's disease,15 and that it is likely to play an important role in cancer cell invasion.16 In view of their potential use as innovative biomaterials, we tested the fluorescent collagen–DTTO microfibers produced by fibroblasts and vimentin–DTTO microfibers produced by neuroblastoma cells as substrates, on which live 3T3 fibroblasts, C2Cl2 myoblasts and B104 neuroblastoma cells, were seeded. We show that the protein–DTTO microfibers have significantly different effects on the cells in terms of the cellular morphology, cytoskeleton rearrangement and viability. These effects span from cell death to microfibers fragmentation into pieces by the cells and subsequent internalization without adverse effects on the cell viability. These results suggest that the physiologically produced protein(s)–DTTO microfibers can be used to direct cell behaviour and be active delivery systems for various bioagents and be used in tissue engineering applications.
Materials and methods
DTTO was synthesized and characterized according to already described modalities.11Cell culture
Mouse neuroblastoma cell cultures (B104) and mouse embryonic fibroblasts (3T3) were maintained in DMEM supplemented with 10% FBS, 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, 5% L-glutamine and 5% sodium pyruvate. Mouse myoblast cells (C2C12) were maintained in growth medium: DMEM medium supplemented with 15% FBS, 50 U mL−1 penicillin, 0.05 mg mL−1 streptomycin, 4 mM glutamine and 10 mM HEPES solution. The cells were grown in a humidified incubator at 37 °C, 5% CO2 and 95% relative humidity. Upon reaching confluence, the cells were passaged using 0.25% trypsin–EDTA. The cells at passages 3–10 were used in the following experimental assays.Live neuroblastoma cell labelling and LSCM monitoring
Mouse neuroblastoma cells (B104) were seeded at a density of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells on a tissue culture plate in 1 mL of complete culture medium. DTTO dye was dissolved in the minimum amount of DMSO to obtain a stock solution and was then administered to the cells by adding the appropriate dilution in serum free DMEM to obtain the final concentration of 0.05 mg mL−1 and incubated at 37 °C in 5% CO2, 95% relative humidity for 1 h. At the end of the incubation period, the unbound dye was removed, and the cell cultures were washed with serum free DMEM medium. The samples were examined after 1, 24, 48, 72 hours and 7 days upon treatment with DTTO by laser scanning confocal microscopy (LSCM). Confocal micrographs were taken using a Leica confocal scanning system mounted into a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), equipped with a 63× oil immersion objective and spatial resolution of approximately 200 nm in x–y and 100 nm in z.
000 cells on a tissue culture plate in 1 mL of complete culture medium. DTTO dye was dissolved in the minimum amount of DMSO to obtain a stock solution and was then administered to the cells by adding the appropriate dilution in serum free DMEM to obtain the final concentration of 0.05 mg mL−1 and incubated at 37 °C in 5% CO2, 95% relative humidity for 1 h. At the end of the incubation period, the unbound dye was removed, and the cell cultures were washed with serum free DMEM medium. The samples were examined after 1, 24, 48, 72 hours and 7 days upon treatment with DTTO by laser scanning confocal microscopy (LSCM). Confocal micrographs were taken using a Leica confocal scanning system mounted into a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), equipped with a 63× oil immersion objective and spatial resolution of approximately 200 nm in x–y and 100 nm in z.
Isolation of fluorescent microfibers from cell lysates
Fluorescent microfibers inside the cells were purified by whole cell lysates in 50 mM Tris HCl, pH 7.4, 1% Triton X-100, 5 mM EDTA, 150 mM NaCl, 1 mM Na3VO4, 1 mM NaF, and 1 mM phenylmethylsulfonyl fluoride (PMSF), in the presence of a protease inhibitor cocktail (10 μM benzamidine-HCl and 10 μg each of aprotinin, leupeptin and pepstatin A per mL) followed by incubation at 4 °C. The DTTO-conjugates were left to decant, harvested into fresh reaction tubes, washed three times with fresh lysis buffer by centrifugation, and stored at 4 °C or −80 °C until being examined by AFM.SDS-PAGE
Sample dilutions of the DTTO-microfibers conjugates in the SDS-loading buffer (1:1) were separated on SDS-polyacrylamide gels without prior heating. The resolved protein bands were visualized by Coomassie staining (Sigma Chemical Co., St. Louis, MO, USA), according to the manufacturer's instructions.
Tryptic digestion and mass spectrometry analysis
Fiber pellets isolated by mouse neuroblastoma cells (B104) were resuspended with urea 8 M and treated with 5 μl of ammonium bicarbonate (AMBIC) 100 mM, reduced with dithiothreitol (DTT 10 mM, 1 μl in AMBIC 100 mM) at 56 °C for 30 min and alkylated with iodoacetamide (55 mM, 1 μl in AMBIC 100 mM) at room temperature in the dark for 1 h. The resulting protein mixture was digested with TPCK-modified sequencing grade trypsin (final ratio of enzyme to substrate was 1:50 w/w) at 37 °C overnight. The samples were then acidified with 1 μl 5% formic acid (FA) solution and dried in a vacuum evaporator. Trypsinized microfibers were resuspended in 30 μl of a 1% FA/acetonitrile 98:2 solution. Chromatographic separation was performed on a high capacity loading chip, with a 150 mm, 300 Å, C18 column prior to a desalting step through a 160 nL trap column. The injected sample (2 μl, 0.8 μg) was loaded on the trap column with a 4 μl min−1 0.1% FA:ACN 98:2 phase; after 3 min, the pre-column was switched in-line with the nanoflow (400 nL min−1, phase A: water:ACN:FA 97:3:0.1, phase B: ACN:water:FA 97:3:0.1), equilibrated in 3% B. The peptides were eluted from the RP column through the following gradient: 3–45% B over a period of 35 minutes, 45–90% in 7 min, 90% B hold for 5 min, and back to 3% B in 3 min – a total of 60 min of runtime, including a 10 min post-run reconditioning step.
Mass spectrometry was performed by ESI-Q-TOF Accurate-Mass G6520AA (Agilent Technologies), controlled by MassHunter (v. B.02.00) and interfaced by a CHIP-cube to an Agilent 1200 nanopump. Centroided MS scan spectra were acquired in positive mode in the range of 300–1700 Da with a 6 Hz sampling rate; top 5 ions, preferring +2 and +3 species, were selected for MS/MS analysis, setting an active exclusion of the same precursor after 1 spectrum over 0.15 minutes. Tandem mass spectra (N2 CID cell) were recorded in the mass range 50–1700 Da with a sampling rate of 3 Hz. Collision energy was calculated using the following expression: [3.6(m/z/100) − 3] V. Automatic QToF calibration was performed before each run. MassHunter (B02.00, from Agilent Technologies) produced mzData.xml raw data that was searched against the SwissProt (v. 57.15) database using an in-house Mascot Server (v. 2.3, Matrix Science, UK) with the following settings: 30 ppm parent ion tolerance, 0.15 Da fragment ion tolerance, semi-tryptic cleavage with one allowed missed cleavage, carbamidomethyl-cysteine as fixed modification and oxidised methionine, lysine and N-terminus carbamylation as the variable modifications, narrowing the search to a rodent proteome. A forward-decoy concatenated protein sequence database was used to evaluate the search false discovery rate.
Immuno co-localization of vimentin
The fluorescent microfibers isolated by mouse neuroblastoma cells (B104) were incubated with the anti-vimentin monoclonal antibody (1:150, Sigma-Aldrich, USA) in PBS buffer at 37 °C for 1 hour. The primary antibody was revealed using the TRITC conjugated anti-mouse antibody (1:1000, Millipore, USA) as a secondary antibody and mounting with fluoroshield (Sigma-Aldrich, USA). Confocal micrographs were taken with a Leica confocal scanning system mounted into a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), equipped with a 63× oil immersion objective and spatial resolution of approximately 200 nm in x–y and 100 nm in z. 3D confocal scanning was performed by reconstructing the photoluminescence emitted from different focalized slices with a sequential image acquisition. The optical sections were collected in transverse x–z and y–z planes.
Morphological characterization by atomic force microscopy (AFM)
The morphological characterization of the isolated fluorescent microfibers by neuroblastoma cells was performed by tapping mode AFM using a Solver PRO Scanning Probe Microscopy (NT-MDT Europe BV, Eindhoven, The Netherlands) in air at room temperature. We used TESPA (Veeco, Santa Barbara, CA) silicon cantilevers with a 20–80 N m−1 spring constant and resonance frequency around 300 kHz and a scan size of 5 μm × 5 μm.Cellular adhesion
Prior to microfiber deposition, cover slip glass was cleaned with a piranha solution consisting of a 3:1 mixture of H2SO4 and H2O2, rinsed sequentially with deionized water, ethanol and acetone, and finally dried with nitrogen. 1 mg mL−1 of isolated fluorescent microfibers (Collagen I hybrid microfibers11,12 and vimentin hybrid microfibers) were deposited on the cover slip glass and dried overnight. All the substrates were sterilized for 15 min using UV light before seeding the cells.
Mouse myoblasts (C2C12), mouse neuroblastoma cells (B104) and mouse embryonic fibroblasts (3T3) were seeded on microfibers at 100000 cell per mL (approximately 1000 cells permm2 of substrate). After 48 hours, each specimen was rinsed twice with PBS 1× to remove any unattached cells. Cytoskeleton morphology was investigated by phalloidin–TRITC at a final concentration of 1 mg mL−1 (Sigma-Aldrich) labeling. The cells were fixed in situ for 5 min in 3.7% formaldehyde, permeabilized with 1% Triton X-100 in PBS 1×, and washed again in PBS 1×.
Thereafter, filamentous actin was stained with phalloidin–TRITC for 40 min at room temperature. Subsequently, the cells were washed several times with PBS to remove the unbound phalloidin conjugate. The samples were examined using a Leica confocal scanning system mounted on a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany) and equipped with a 63× oil immersion objective.
Proliferation assays
Mouse myoblasts (C2C12), mouse neuroblastoma cells (B104) and mouse embryonic fibroblasts (3T3) seeded on Collagen I hybrid microfibers and vimentin hybrid microfibers were analyzed using the cytotoxicity test, MTT, measuring the activity of living cells via the mitochondrial dehydrogenase activity whose key component is 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide.17 The controls were realized by a cell culture on glass cover slip surfaces. The mitochondrial dehydrogenases of the viable cells cleave the tetrazolium ring, yielding purple MTT formazan crystals, which are insoluble in aqueous solutions. The crystals were then dissolved in acidified isopropanol and the resulting purple solution was spectrophotometrically measured. An increase in cell number results in an increase in the amount of MTT formazan formed and an increase in absorbance. The B104 cells (100000 cells per mL) were added to the well culture plates at 1 mL per well and incubated at 37 °C in 5% CO2, 95% relative humidity for 24–48–72 and 192 hours with the DTTO dye suspension. The control was entirely culture medium. After the appropriate incubation period, the cultures were removed from the incubator and a MTT solution was added in amounts equalling 10% of the culture volume. The cultures were then returned to the incubator and incubated for 3 hours. After the incubation period, the cultures were removed from the incubator and the resulting MTT formazan crystals were dissolved in an acidified isopropanol solution to an equal culture volume. The plates were read within 1 hour after adding an acidified isopropanol solution. The absorbance was spectrophotometrically measured at a wavelength of 570 nm and the background absorbance measured at 690 nm was subtracted. The percentage viability is expressed as the relative growth rate (RGR) by following equation:| RGR = (Dsample/Dcontrol) × 100% |
Results and discussion
Vimentin microfibers formation and characterization
Live mouse neuroblastoma cells were incubated with micromolar DTTO in a buffered solution (0.05 mg mL−1). Following the removal of unbound fluorophore and extensive washing, the cells were cultured and monitored continuously for 7 days. Fig. 1 shows the LSCM images of the evolution with time of the neuroblastoma cells after incubation with DTTO. Because of the correct hydrophilicity/hydrophobicity balance and the amphiphilicity due to the presence of the oxygen atoms, DTTO spontaneously crosses the cell membrane and diffuses into the cells. Fig. 1B shows that 1 h after washing, the fluorophore was gathered inside the cells forming globular aggregates around the perinuclear region, as already observed for other thiophene fluorescent dyes.10–12 After 24 h (Fig. 1C), the cytoplasm of the cells appears intensely green fluorescent, whereas the nuclei remain unstained and dark. Moreover, within the cytoplasm of several cells the formation of fluorescent microfibers of a micrometer size is apparent. The microfibers progressively accumulate inside the cells (Fig. 1E and F), whereas the fluorescent cells are visible in the background, indicating that fluorescence is transmitted from the mother to daughter cells during the replication process. It is noteworthy that many microfibers display details where helical green fluorescent structures are present, for example, the details shown in Fig. 1D or S1.†The fluorescent microfibers formed after 7 days from the treatment of the cells with DTTO were separated from the cell lysate, picked up by LSCM and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The results are shown in Fig. 2. SDS-PAGE unambiguously indicated the proteic nature of the fluorescent fibrils. Only one single signal was observed in the region around a 60 kDa molecular weight.
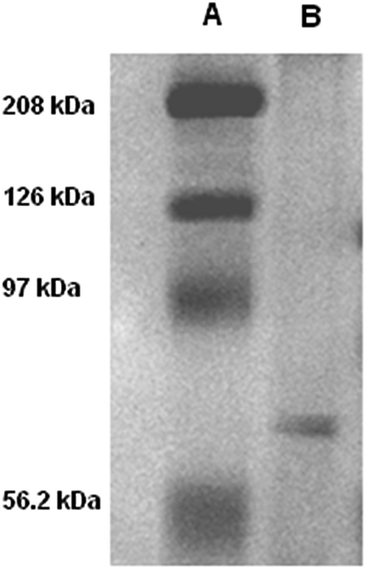 | ||
| Fig. 2 SDS-PAGE of the fluorescent microfibers isolated by the live mouse neuroblastoma cells after 192 hours of treatment with DTTO (A = high molecular weight marker, B = sample). | ||
The fluorescent microfibers isolated by neuroblastoma cells are mainly made of vimentin
The fluorescent microfibers formed 7 days after the treatment with DTTO and separated from the cell lysate were analyzed, after the appropriate treatment (Material & Methods section), by HPLC-ESI-QToF using the Mascot software18 for protein identification. It was found that 73.4% (supplementary attached file denominated ‘Fibrille_Rattus’) of the fluorescent microfibers was made of vimentin, a protein of the intermediate filaments of the cytoskeleton with a molecular weight of 57 kDa.19 A few percent of nestin, plectin and lamin A were also detected (‘Fibrille_Rattus’). The presence of the latter proteins is in agreement with the fact that vimentin presents a subcellular distribution and interaction with many proteins, such as lamin, plectin, etc. In particular, at the nuclear envelope, the 6.6 kDa tail region of vimentin interacts with lamin B,20 whereas another study showed that vimentin interacts with β3 integrins and plectin, which together regulate the organization and distribution of vimentin in several cell types.21In agreement with the mass spectrometry data, co-localization experiments of isolated fluorescent microfibers with the monoclonal vimentin antibody showed that there was in fact co-localization between the antibody and the microfibers. Fig. 3 shows LSCM images of isolated green fluorescent microfibers (a), isolated red fluorescent anti-vimentin antibody (b), merged images (c) and corresponding z-stack sections (d) with a z-resolution of 200 nm. Fig. 3D shows that there is spatial co-localization with the red fluorescent anti-vimentin antibody inside the green fluorescent microfibers (supplementary attached file denominated ‘LCSM_zeta stack sections of vimentin co-localization’).
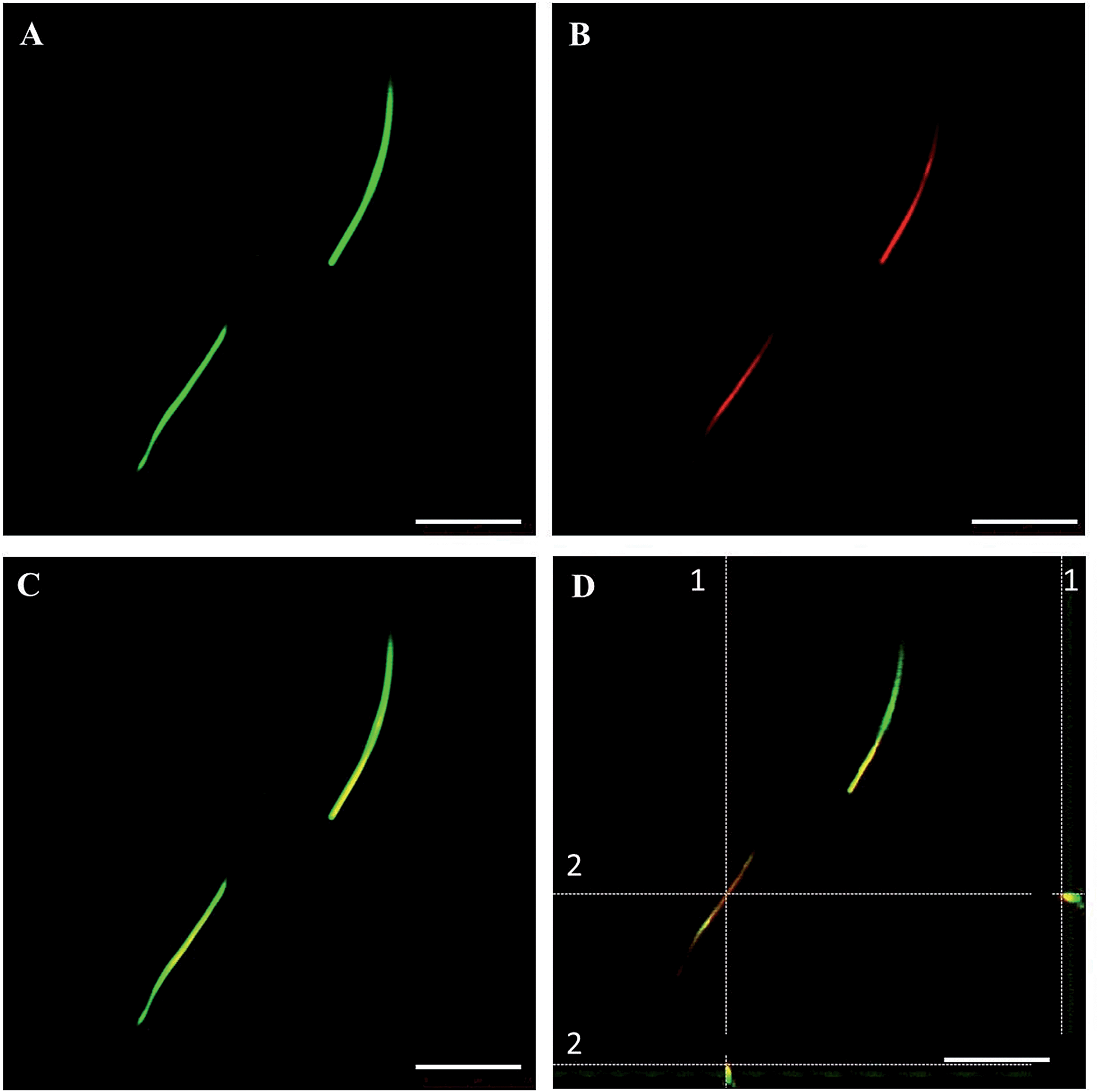 | ||
| Fig. 3 LSCM images of the co-localization experiment between the isolated fluorescent microfibers secreted by the B104 mouse neuroblastoma cells and anti-vimentin antibody. (A) Isolated green fluorescent microfibers. (B) Isolated red fluorescent anti-vimentin antibody. (C) Merge image. (D) Corresponding z-stack sections with a z-resolution of 200 nm, showing the spatial co-localization of DTTO (green) with the anti-vimentin antibody (red) inside the microfibers. Scale bar: 7.5 μm. | ||
Fig. 4 shows AFM images of the fluorescent microfibers isolated from the cell lysate. The morphology of the largest fibers recalls the structure of the dimer of vimentin representing the first level of vimentin self-assembly.22–26 The figure also displays the coiled coil arrangement of two smaller microfibers. Fig. S2† shows another fluorescent microfiber isolated from the cell lysate and provides evidence of the lateral growth of the microfibers.
 | ||
| Fig. 4 (A) AFM image of the fluorescent microfibers isolated from the cellular milieu. The morphology of the largest fiber recalls the structure of the dimer of vimentin representing the first level of the vimentin self-assembly.22 (B) Magnification of Figure A. The arrows indicate the coiled coil arrangement of two smaller microfibers. | ||
Vimentin is a polypeptide comprising 466 amino acids with a highly conserved α-helical ‘rod’ domain that is flanked by non-α-helical N-and C-terminal end domains, the ‘head’ domain and ‘tail’ domain, respectively.13 The self-organization of these molecules leads to the formation of coiled coils, which are the basic structural building blocks for the entire IF family of proteins. Vimentin is also known to form homopolymers and heteropolymers (i.e., it associates with other type III and type IV IFs), which is a common feature among the members of the IF family ascribed to the presence of a coiled-coil α-helical domain. Highly stable polymers are formed, the stability of which is controlled by the phosphorylation status of the integral proteins. The astonishing similarity between the sophisticated shape of the fluorescent microfiber analyzed by AFM in Fig. 4 and the shape of human vimentin dimer, as determined based on X-ray diffraction at the atomic scale (Fig. 4C of ref. 26), is unlikely to be the outcome of mere coincidence. The fact that the cell viability remains unaltered upon DTTO uptake (Fig. S3†) suggests that the fluorescent dye is progressively embedded during protein growth without substantially altering the self-assembly process. DTTO is a small neutral molecule that can be accommodated easily within the alpha helical collagen strand of fibroblasts of different origin via hydrogen bonds, leading to the stabilization of the helical structure.11,12 All IF proteins, including vimentin, although varying greatly in their primary structure, are characterized by an alpha-helical central rod domain having the intrinsic capability to form coiled-coil filaments and are terminated by the non-helical N- and C-domains of variable size, which play a role in the protein self-assembly process. Therefore, the α-helical structure present in vimentin and its ability to form non-covalent bonds with DTTO via non-bonding interactions cannot alone justify the fact that it is mainly vimentin that is concerned by DTTO in live neuroblastoma cells. It is more probable that the choice is determined by some co-factors related to the general ‘economy’ of the cell and its metabolism.
Cell interaction with collagen–DTTO and vimentin–DTTO microfibers
The possibility of separating the physiologically formed microfibers, by LSCM, from the cell lysate owing to their fluorescence offers an unprecedented way of testing their potential as innovative biomaterials. Here, we describe the initial experiments to test their effect on various live cell lines.As shown in Fig. 5, the cells seeded on collagen–DTTO or vimentin–DTTO microfibers assumed different morphologies and configurations depending on the stimuli experimented.
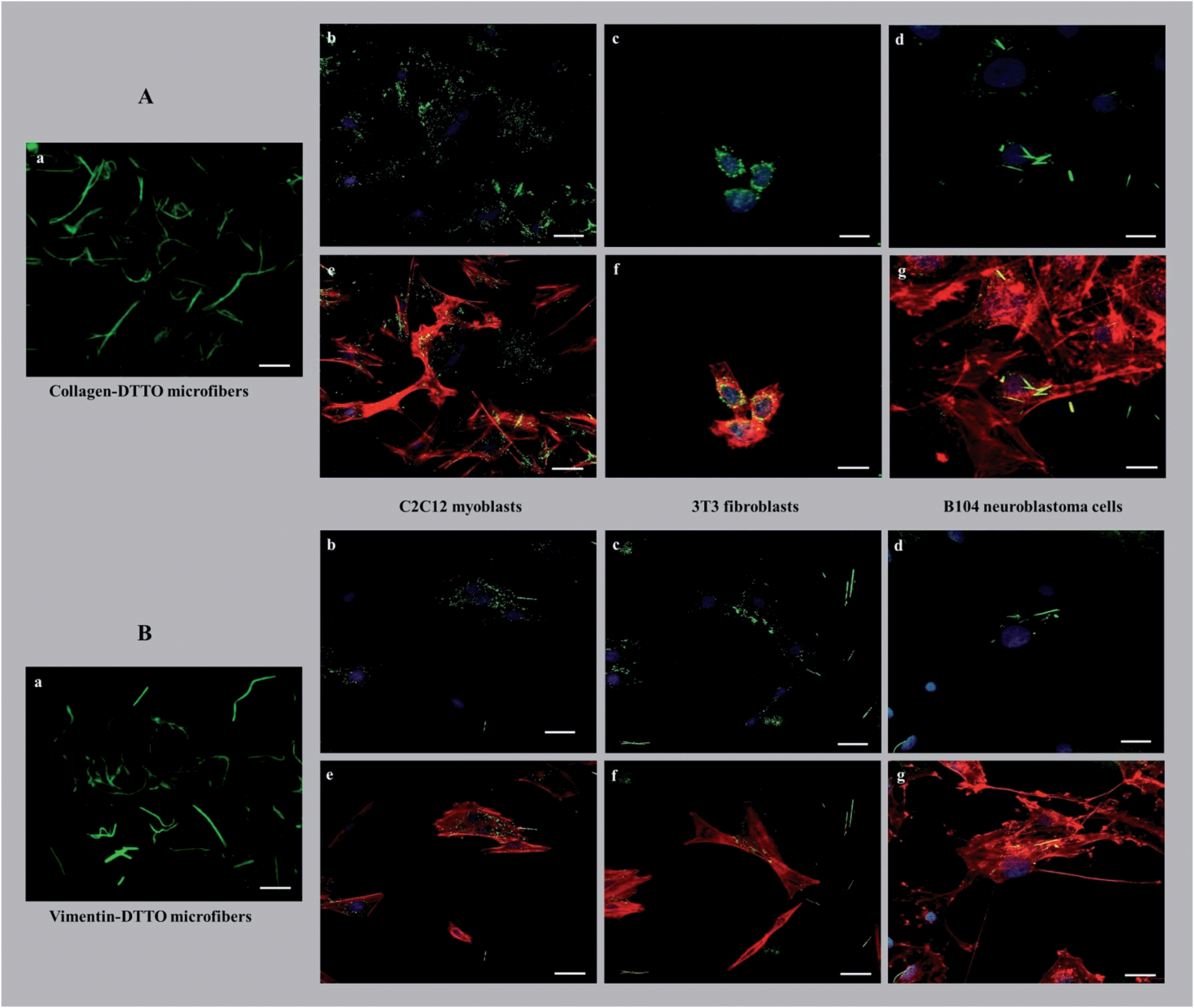 | ||
| Fig. 5 (Aa and Ba) LSCM images of the pristine collagen–DTTO and vimentin–DTTO microfibers used as substrates for the deposition of live C2C12 myoblasts, 3T3 fibroblasts and B104 neuroblastoma cells. (A) LSCM images of fixed C2C12 myoblasts (b and e), 3T3 fibroblasts (c and f) and B104 neuroblastoma cells (d and g) seeded on collagen–DTTO microfibers for 48 hours, stained with DAPI (blue, b–d) and actin (red, e–g). (B) The same for vimentin–collagen microfibers. Scale bar: 10 μm. | ||
After seeding on collagen–DTTO microfibers for 48 hours, C2C12 myoblasts and B104 neuroblastoma cells (Fig. 5Ae and Ag) showed cytoskeleton remodeling, with a remarkable organization of F-actin into the intricate branching networks and polarized filopodia protrusions associated to a more elongated shape, whereas 3T3 fibroblasts (Fig. 5Af) assumed a spindle-shaped appearance displaying a round and contracted cell body. Conversely, when seeded for 48 hours on vimentin–DTTO microfibers, 3T3 fibroblasts (Fig. 5Bf) and B104 neuroblastoma cells (Fig. 5Bg) maintained their epithelial morphology of a polygonal shape with many actin cytoplasmic protrusions (filopodia-like processes), whereas C2C12 myoblasts showed a contracted cell body. As shown in the figure, after 48 h of cell culture, the collagen–DTTO and vimentin–DTTO microfibers appeared as green fluorescent dots, with a uniform distribution inside the cytoplasm of C2C12 myoblasts (Fig. 5Ab and Bb) and 3T3 fibroblasts (Fig. 5Ac and Bc). In this case, it can be suggested that both types of microfibers were degraded by the cell,27,28 conceivably by action of the matrix metalloproteases modifying in this way the microenvironment and resulting in an alteration of the cellular morphology and cytoskeleton rearrangement. On the contrary, in B104 neuroblastoma cells, collagen–DTTO microfibers were partially degraded, as evident in Fig. 5Ad, whereas the vimentin–DTTO microfibers showed their native morphology (Fig. 5Bd). The degradation and possible recycling of the microfibers can be a process used to create the protein fragments required to build new cellular structures.
We also evaluated the viability of C2C12 myoblasts, 3T3 fibroblasts and B104 neuroblastoma cells cultured on glass cover slips coated with collagen–DTTO or vimentin–DTTO microfibers by performing a MTT proliferation assay at 24, 48, 72, and 192 h. As shown in Fig. 6, the substrates coated with vimentin microfibers did not affect the cell viability or proliferation, as judged by the increases in the total cell numbers over time. The same was true for the myoblasts and neuroblastoma cells coated on the collagen–DTTO microfibers. On the contrary, the viability of 3T3 fibroblasts was compromised when the cells were seeded on collagen–DTTO microfibers. As evident in Fig. 5, all microfibers (1 mg mL−1) present on the substrate were degraded and internalized by fibroblasts after 48 h. These results indicate that the massive storage of collagen–DTTO fragments inside fibroblasts leads to apoptosis. This does not exclude the fact that collagen–DTTO microfibers having concentrations below 1 mg mL−1 might not induce cytotoxic effects on the fibroblasts.
 | ||
| Fig. 6 Proliferation assays of C2C12 myoblasts (A), 3T3 fibroblasts (B) and B104 neuroblastoma cells (C) cultured on a uncoated glass cover slip (NT), glass cover slip coated with collagen I or vimentin hybrid microfibers for 24, 48, 72, and 192 h. The results are expressed as mean ± standard deviations and are representative measurements of three distinct sets of data. No significant difference between the values at different time points is observed at P < 0.05 using-Student's t-test. | ||
Further experiments are in progress to elucidate the biochemical mechanisms involved in the degradation of the fluorescent microfibers and to obtain information on how their mechanical and electrical characteristics direct the cellular behaviour.
Conclusions
The results showed that different types of cells employ the organic dithienothiophene fluorophore DTTO for the physiological production of fluorescent fibrillar proteins organized in nanostructured microfibres, namely vimentin with neuroblastoma cells (this study) and type-I collagen with fibroblasts of different origins (ref. 11). Moreover, various types of live cells seeded on these microfibers can internalize and degrade them, experiencing in turn different effects on their morphology and viability.The composition and distribution of the specific extracellular matrix components vary with the type of tissue, and can also be altered by the tissue's development stages and/or pathological state. The cells sourced from different tissues typically yield matrices that mimic the relative composition of the natural tissue matrix. In this work, we demonstrate that the cell source is the primary determinant of the resulting multiscale hybrid microfibers. In particular, we used live neuroblastoma cells that produce hybrid vimentin microfibers upon the uptake of DTTO. The different compositions induce dissimilar cellular behaviour when these multiscale microfibers are used as substrates for cell cultures.
The requirements for materials used in biomedical applications are biocompatibility and biodegradability. The fluorescent microfibers physiologically secreted by the live cells described here present both characteristics. These microfibers represent innovative multiscale biomaterials useful not only for the imaging of cellular processes but also potentially for drug delivery and tissue engineering as well as to confer bioactivity to synthetic scaffolds by the loading of bioactive molecules as growth factors.
Acknowledgements
This study was supported by MAAT-molecular nanotechnology to human health and the environment (PON R & C 2007-2013 project's code PON02_00563_3316357). The authors acknowledge the 'Fondazione Cassa di Risparmio di Modena' funding for the HPLC-ESI-QTOF system at the Interdepartmental Centre Great Tools (CIGS) of the University of Modena and Reggio Emilia. No writing assistance was utilized in the production of this manuscript. The authors have no competing interests to disclose.Notes and references
- F. M. Harold, Microbiol. Mol. Biol. Rev., 2005, 69, 544–564 CrossRef CAS PubMed.
- P. Dedecker, F. C. De Schryver and J. Hofkens, J. Am. Chem. Soc., 2013, 135, 2387–2402 CrossRef CAS PubMed.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- Z. Li, Q. Sun, Y. Zhu, B. Tan, Z. P. Xu and S. Dou, J. Mater. Chem. B, 2014, 2, 2793–2818 RSC.
- F. Song, P. S. Tang, H. Durst, D. T. Cramb and W. C. W. Chan, Angew. Chem., Int. Ed., 2012, 51, 8773–8777 CrossRef CAS PubMed.
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- S. Gandor, S. Reisewitz, M. Venkatachalapathy, G. Arrabito, M. Reibner, H. Schröder, K. Ruf, C. M. Niemeyer, P. I. H. Bastiaens and L. Dehmelt, Angew. Chem., Int. Ed., 2013, 52, 4790–4794 CrossRef CAS PubMed.
- A. Aslund, C. J. Sigurdson, T. Klingstedt, S. Grathwohl, T. Bolmont, D. L. Dickstein, E. Glimsdal, S. Prokop, M. Lindgren, P. Konradsson, D. M. Holtzman, P. R. Hof, F. L. Heppner, S. Gandy, M. Jucker, A. Aguzzi, P. Hammarström and K. P. R. Nilsson, ACS Chem. Biol., 2009, 4, 673–684 CrossRef PubMed.
- A. Ciéslar-Pobuda, M. Bäck, K. Magnusson, M. V. Jain, M. Rafat, S. Ghavami, K. P. R. Nilsson and M. J. Łos, Cytometry, Part A, 2014, 85A, 628–635 CrossRef PubMed.
- F. Di Maria, I. E. Palamà, M. Baroncini, A. Barbieri, A. Bongini, R. Bizzarri, G. Gigli and G. Barbarella, Org. Biomol. Chem., 2014, 12, 1603–1610 CAS.
- I. E. Palamà, F. Di Maria, I. Viola, E. Fabiano, G. Gigli, C. Bettini and G. Barbarella, J. Am. Chem. Soc., 2011, 133, 17777–17785 CrossRef PubMed.
- I. Viola, I. E. Palamà, A. M. L. Coluccia, M. Biasiucci, B. Dozza, E. Lucarelli, F. Di Maria, G. Barbarella and G. Gigli, Integr. Biol., 2013, 5, 1057–1066 RSC.
- K. N. Goldie, T. Wedig, A. K. Mitra, U. Aebi, H. Herrmann and A. Hoenger, J. Struct. Biol., 2007, 158, 378–385 CrossRef CAS PubMed.
- A. Satelli and S. Li, Cell. Mol. Life Sci., 2011, 68, 3033–3046 CrossRef CAS PubMed.
- E. C. Levin, N. K. Acharya, J. C. Sedeyn, V. Venkataraman, M. R. D'Andrea, H. Y. Wang and R. G. Nagele, Brain Res., 2009, 1298, 194–207 CrossRef CAS PubMed.
- M. S. Yoneyama, S. Hatakeyama, T. Habuchi, T. Inoue, T. Nakamura, T. Funyu, G. Wiche, C. Ohyama and S. Tsuboi, Eur. J. Cell Biol., 2014, 93, 157–169 CrossRef PubMed.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS.
- http://www.matrixscience.com/search_intro.html .
- D. Dahl, D. C. Rueger, A. Bignami, K. Weber and M. Osborn, Eur. J. Cell Biol., 1981, 24, 191–196 CAS.
- D. Georgatos and G. Blobel, J. Cell Biol., 1987, 105, 117–125 CrossRef.
- R. Bhattacharya, A. M. Gonzalez, P. J. Debiase, H. E. Trejo, R. D. Goldman, F. W. Flitney and J. C. Jones, J. Cell Sci., 2009, 122, 1390–1400 CrossRef CAS PubMed.
- H. Herrmann and U. Aebi, Annu. Rev. Biochem., 2004, 73, 749–789 CrossRef CAS PubMed.
- M. Schaffeld, H. Herrmann, J. Schultess and J. Markl, Eur. J. Cell Biol., 2001, 80, 692–702 CrossRef CAS PubMed.
- N. Pinto, F. C. Yang, A. Negishi, M. Rheinstadter, T. E. Gillis and D. S. Fudge, Biomacromolecules, 2014, 15, 574–581 CrossRef CAS PubMed.
- H. Herrmann, M. Häner, M. Brettel, S. A. Müller, K. N. Goldie, B. Fedtke, A. Lustig, W. W. Franke and U. Aebi, J. Mol. Biol., 1996, 264, 933–953 CrossRef CAS PubMed.
- A. A. Chernyatina, S. Nicolet, U. Aebi, H. Herrmann and S. V. Strelkov, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13620–13625 CrossRef CAS PubMed.
- L. Monica, B. Andersson and M. J. Warburton, Biochim. Biophys. Acta, Mol. Cell Res., 1995, 1268, 27–34 CrossRef.
- I. Virtanen, O. Närvänen and V. Lehto, Int. J. Cancer, 1988, 42, 256–260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01562b |
| This journal is © The Royal Society of Chemistry 2015 |