
Diethylamino functionalized tetraphenylethenes: structural and electronic modulation of photophysical properties, implication for the CIE mechanism and application to cell imaging†
Yiliu
Lin‡
a,
Gan
Chen‡
a,
Lifang
Zhao
a,
Wang Zhang
Yuan
*a,
Yongming
Zhang
*a and
Ben Zhong
Tang
*bc
aSchool of Chemistry and Chemical Engineering, Shanghai Key Lab of Electrical Insulation and Thermal Aging, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: wzhyuan@sjtu.edu; ymzsjtu@gmail.com
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
cHKUST-Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China
First published on 23rd October 2014
Abstract
Deciphering structural and electronic effects on photophysical properties of aggregation- or crystallization-induced emission (AIE or CIE) luminogens is essential to the related mechanistic understanding, molecular design and applications. Herein, based on AIE-active tetraphenylethene (TPE), a group of diethylamino (DEA) functionalized analogues, i.e. DEATPE, DFDEATPE and CNDEATPE were designed and prepared. The introduction of DEA groups makes DEATPE CIE-active rather than typically AIE-active. Further incorporation of fluorine atoms renders DFDEATPE with AIE and crystallization-induced emission enhancement (CIEE) characteristics. Furthermore, unlike the nonmechanochromic TPE, these luminogens exhibit high contrast mechanochromism. Specifically, DEATPE and DFDEATPE demonstrate rapid self-recovery within a few minutes or even several seconds. Such swift restoration of solid emission clearly indicates active intramolecular motions even in the solid states, which is indicative of the CIE mechanism. Additionally, luminogenic nanoparticles were successfully utilized in cell imaging, thanks to their high efficiency and biocompatibility.
Introduction
Fluorescent materials have drawn considerable attention due to their wide applications in organic light emitting diodes (OLEDs),1 chemo- and bio-sensors,2,3 bioimaging,4etc. Most traditional luminogens, however, suffer from the notorious aggregation-caused quenching (ACQ) problem.5 They are highly emissive in dilute solutions while being weakly fluorescent or even nonemissive in concentrated solutions or aggregated solids. Such a thorny ACQ problem has greatly restricted their practical applications because they are normally used in aggregated or condensed solid states. It is thus essential to develop high efficiency solid emitters. To attain efficient solid-state emission, several physical and chemical approaches including doping into transparent polymeric films,6 J-aggregation,7 dendritic or bulky substituent protection,8 cross-dipole stacking,9 aggregation-induced emission (AIE)10–13 and lateral boryl substitution of π-conjugated frameworks14 were developed. Among these methods, the construction of AIE-active compounds enjoys its unique advantage: while the other approaches try to prevent the natural aggregation process, it encourages aggregation.1d,10–13 As opposed to ACQ dyes, AIE fluorogens are practically nonluminescent in solutions but highly emissive in aggregates, thus perfectly resolve the ACQ problem.10–15The discovery of the AIE phenomenon opens a new way to design and synthesize novel luminogenic molecules.10–12 Experimental and theoretical investigations suggest restriction of intramolecular rotations (RIR) as the main cause for the AIE effect.10,16 Thanks to their high solid-state efficiencies, AIE molecules have found a variety of applications in optoelectronic devices1c,17 and as bioprobes.2a,3b In addition, their twisting conformations easily undergo conformation planarization under external pressure or shear stress, which would result in a remarkable emission change, thus making them highly promising as mechanochromic luminogens.18 The switching and tuning of solid-state emission of these smart materials is of great importance to the fundamental research and such potential applications as sensors, memory chips, optical information storage media, display devices and security inks.18–21 It is also noteworthy that structural and electronic modulation exerts great influence on their emission behaviors.22 For example, propeller-shaped tetraphenylethene (TPE) is a typical AIE compound,23 whereas triphenylacrylonitrile (TPAN)11d and diphenyldibenzofluorene (DPDBF)24 (Chart 1) are almost nonemissive in solutions and amorphous states, but highly emissive in the crystalline states, exhibiting featured crystallization-induced emission (CIE) behaviors; as another example, HPS is AIE-active,25 whereas isopropyl substituted HPS2,5 and HPS3,4 are emissive in solutions, particularly, HPS3,4 shows a high quantum efficiency of 69% in tetrahydrofuran (THF).22a Therefore, the design and synthesis of diverse luminogens with different structures and varying electronic communication are not only important to explore their practical applications, but also fundamentally significant to reveal the underlying mechanism.
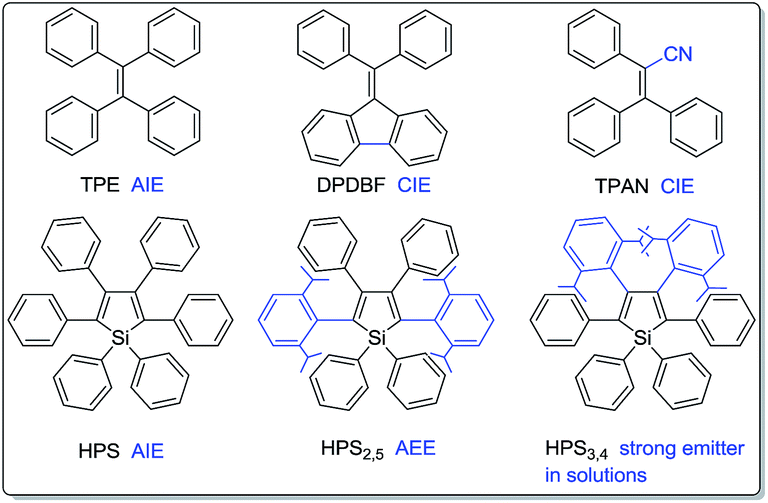 | ||
| Chart 1 Structures of TPE, DPDBF, TPAN, HPS, HPS2,5 and HPS3,4. | ||
In this study, to create high efficiency, mechanochromic and biocompatible AIE-active luminogens suitable for optoelectronic and bioimaging applications, and moreover, to gain further insights into the structural and electronic effects on the AIE system, a group of luminogens consisting of TPE and DEA groups, namely DEATPE, DFDEATPE and CNDEATPE (Chart 2) were designed and synthesized. The incorporation of DEA is believed to yield the following effects: (1) endow the resulting luminogens with good biocompatibility;26 (2) tune the electronic states and thus photophysical properties of the molecules owing to its strong electron-donating ability. Herein, photophysical properties including mechanochromic behaviors and their implication for the CIE mechanism, as well as cell imaging application of the luminogens are investigated.
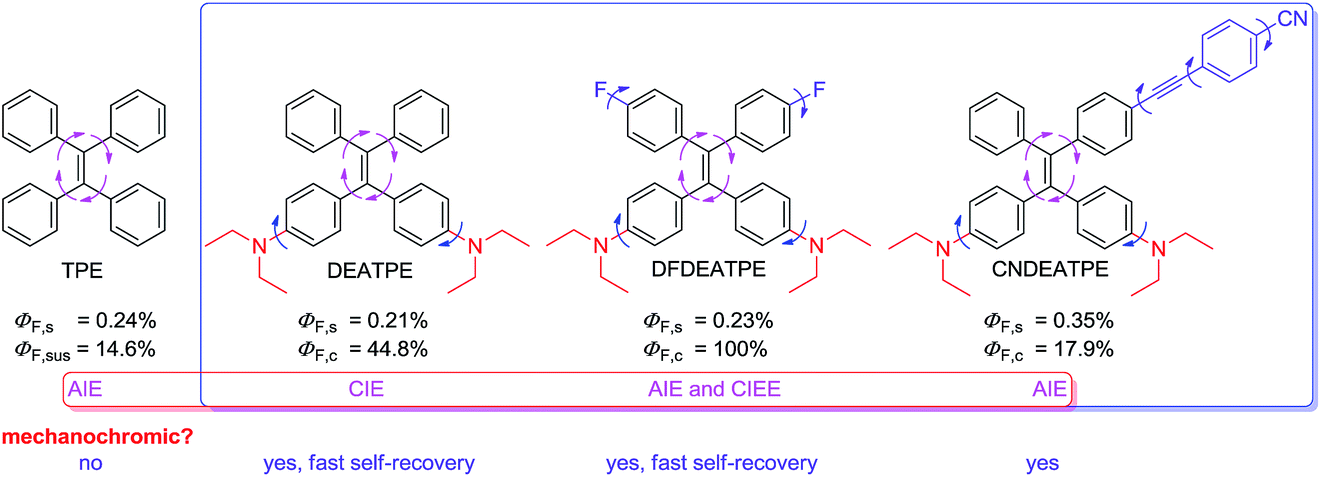 | ||
| Chart 2 Structures and general photophysical properties of TPE, DEATPE, DFDEATPE and CNDEATPE. s = solution; sus = suspension; c = crystal. | ||
Results and discussion
Synthesis and absorption
DEATPE, DFDEATPE and CNDEATPE were prepared according to the synthetic routes shown in Scheme 1. Briefly, DEATPE and DFDEATPE were synthesized by cross McMurry coupling between 4,4′-diethylaminobenzophenone (1) and benzophenone (2a) or 4,4′-difluorobenzophenone (2b) in moderate yields (67%, 70%) by utilizing TiCl4–Zn as catalysts.27 And CNDEATPE was obtained by Sonogashira coupling between 1,1-bis[4-(diethylamino)phenyl]-2-(4-ethynylphenyl)-2-phenylethylene (3)28 and 4-bromobenzonitrile (4) in the presence of Pd(PPh3)2Cl2 catalyst. These target products were spectroscopically characterized by NMR and high resolution mass spectra (HRMS), together with single crystal structures, and satisfactory data were obtained (Experimental section, Fig. S1–S5, Table S1 and S2, ESI†).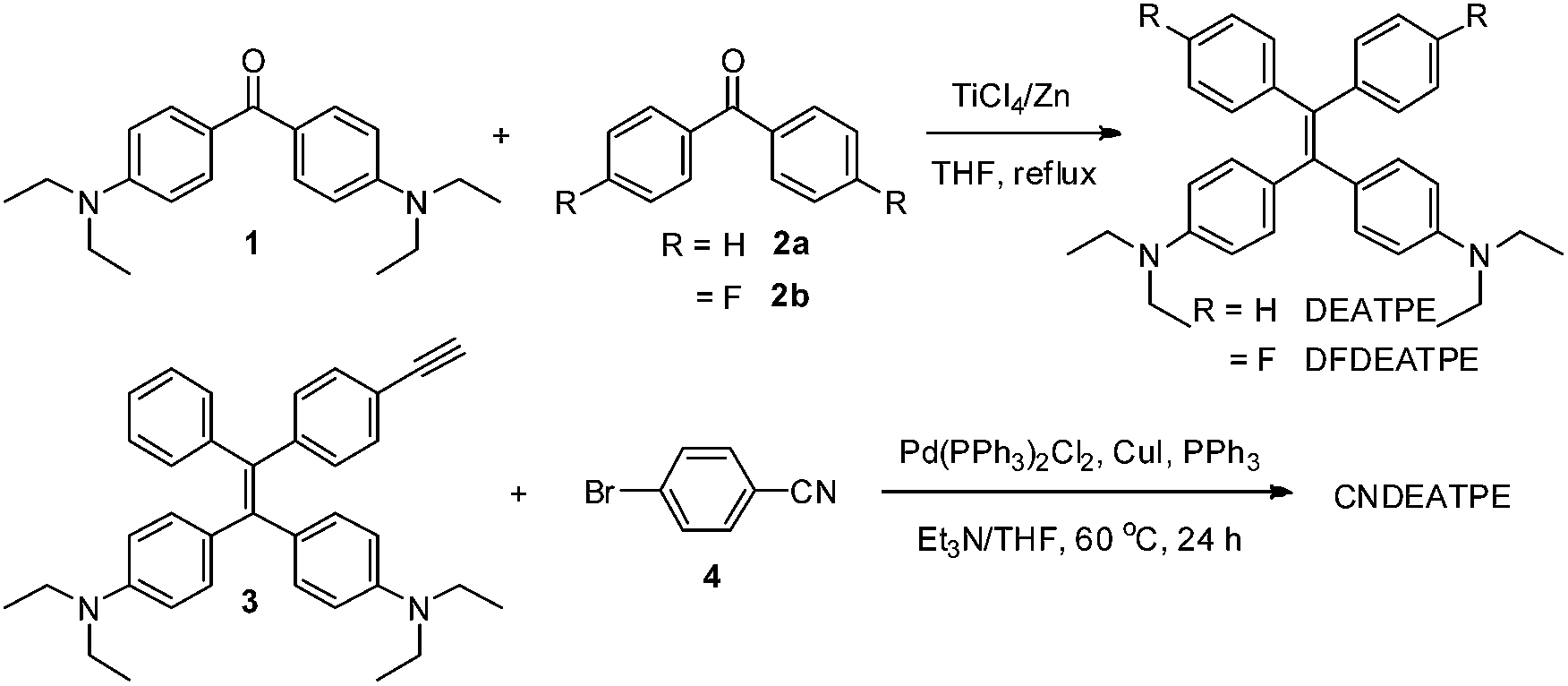 | ||
| Scheme 1 Synthetic routes to DEATPE, DFDEATPE and CNDEATPE. | ||
DEATPE and DFDEATPE show absorption profiles with a peak and a shoulder at 318/364 and 322/364 nm (Fig. 1), respectively. While the shorter bands are attributed to the π–π* transitions, which are slightly red-shifted compared to that of TPE (309 nm), the redder shoulders might be attributed to ICT transitions, owing to the strong electron-donating capability of DEA groups and the reasonable electron-accepting ability of aromatic phenyls. For CNDEATPE, besides the π–π* transitions at 300 and 320 nm, an even redder ICT transition at 416 nm is observed, which should be ascribed to the incorporation of the strong electron-accepting cyano group.
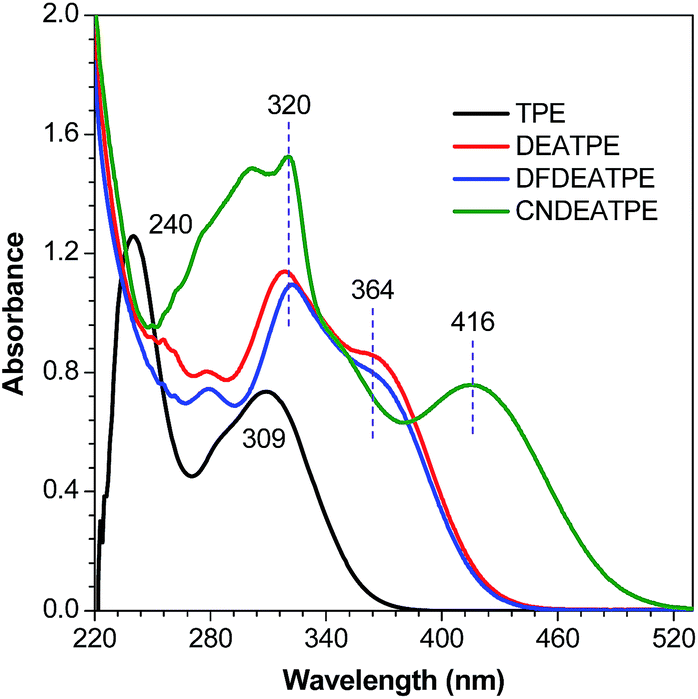 | ||
| Fig. 1 Absorption spectra of TPE, DEATPE, DFDEATPE and CNDEATPE in THF. Concentration = 40 μM. | ||
Notably, the decoration of TPE with DEA groups generates a new ICT band, whereas further incorporation of fluorine atoms or cyano group impacts differently: the former exerts a negligible impact, whereas the latter significantly promotes the ICT process with a remarkably red-shifted (by 42 nm) transition. These results demonstrate distinct structural and electronic effects on the optical absorption. In an effort to understand the electron transition processes, electron densities of the compounds were calculated.29 The electron clouds of both HOMO and LUMO levels for TPE are homogeneously distributed (Fig. 2A and B), confirming the absence of the ICT process. The electron densities of the HOMO levels of DEATPE and DFDEATPE are mainly localized at the phenyls connecting with DEA and the central double bond (Fig. 2C and E), whereas those of the LUMO levels are predominantly distributed at the double bond and the other two phenyls (Fig. 2D and F), showing the explicit ICT feature and the ignorable effect of fluorine atoms. CNDEATPE depicts a similar HOMO density distribution to those of DEATPE and DFDEATPE (Fig. 2G), but rather different LUMO levels, wherein the electron density is essentially immobilized onto the cyano connecting the tolane group (Fig. 2H), suggesting a typical ICT process. These calculation results are highly consistent with the absorption data, further testifying the structural and electronic effects.
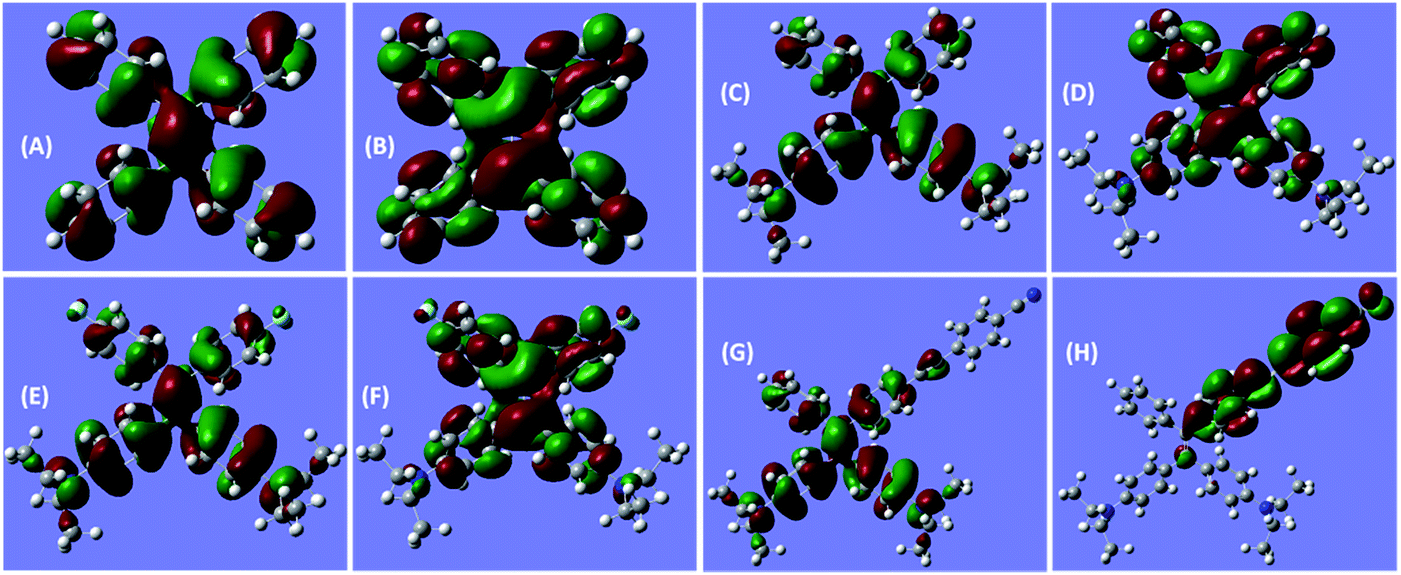 | ||
| Fig. 2 B3LYP/6-31G(d) calculated molecular orbital amplitude plots of (A, C, E, G) HOMO and (B, D, F, H) LUMO levels for (A, B) TPE, (C, D) DEATPE, (E, F) DFDEATPE and (G, H) CNDEATPE, respectively. | ||
Emission property of solutions and nanosuspensions
Since TPE is an archetype AIE luminogen, it is expected that its DEA-containing derivatives are also AIE-active. To check this feature, emission behaviors of the compounds in THF and THF–water mixtures are monitored. Water is used because it is a typical nonsolvent to induce aggregation of the luminogens. As can be observed in Fig. 3A and B, DEATPE shows no light emission in THF, with rather low photoluminescence (PL) intensity being recorded. Such a situation does not change until the water fraction (fw) reaches 70%, at which the luminogenic molecules are aggregated into floating solids and emit bright cyan light with maximum at ∼477 nm. However, when fw is increased to 80%, several emission peaks at 427, 485 and 527 nm are detected with decreased intensity. Further increase in fw (90%) promotes the PL intensity with the retained emission profile. The additional experiment reveals the CIE feature of DEATPE: while sluggish yellowish-green light is observed for the dotted point on a thin-layer chromatography (TLC) plate, strong cyan emission can be observed for its crystalline powders (Fig. 3A).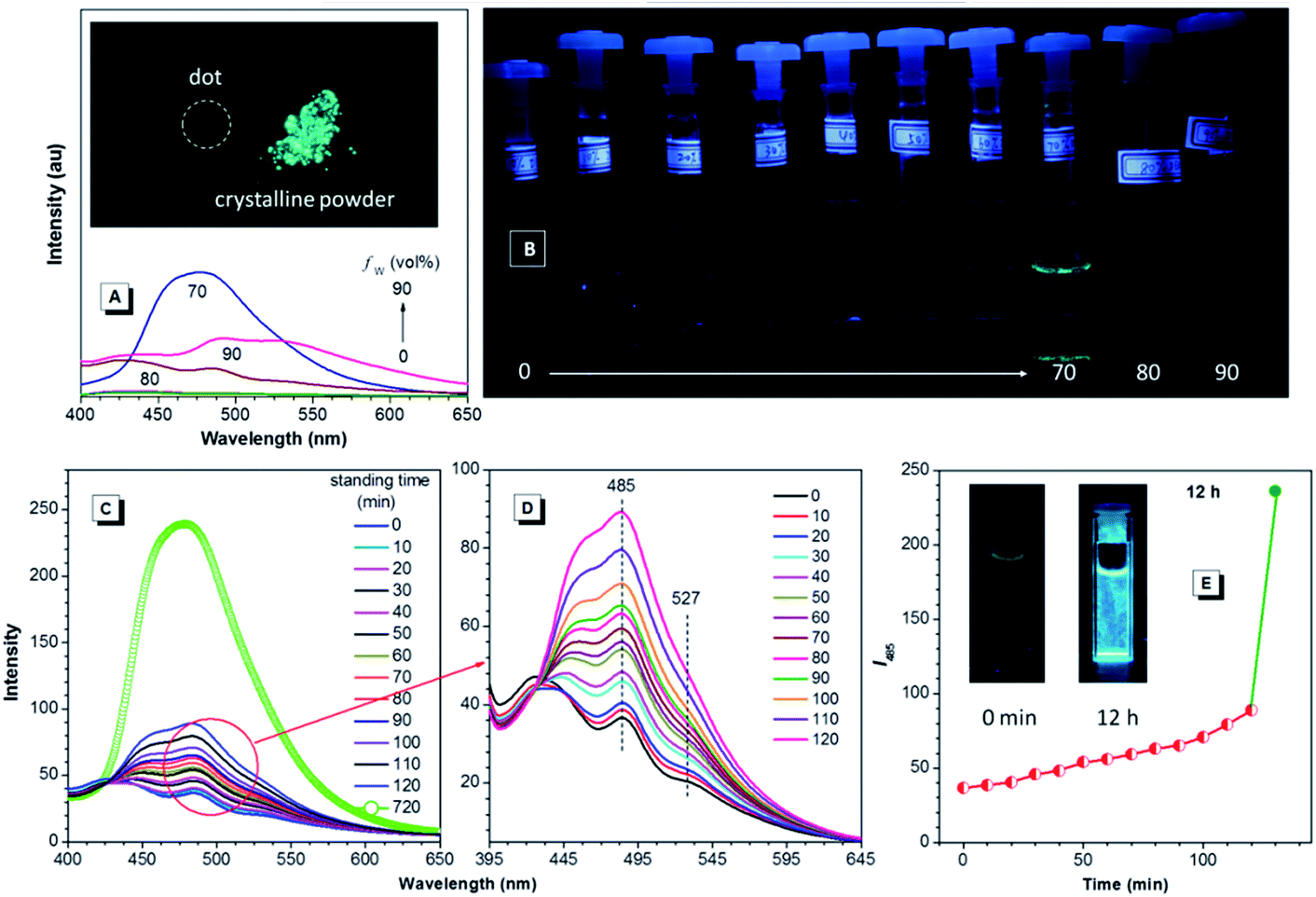 | ||
| Fig. 3 (A) PL spectra and (B) emission photographs of DEATPE in THF and THF–water mixtures with different fws. (C, D) Emission evolution and (E) emission intensity of DEATPE monitored at 485 nm in the 20/80 THF–water mixture. Concentration = 40 μM; excitation wavelength = 365 nm. The inset in (A) is the luminescent photograph of a dotted point (left) and crystalline powders (right) of DEATPE on a TLC plate. | ||
Considering the ICT nature, the emission peaks of DEATPE suspensions in the 80% and 90% aqueous media can be ascribed to the local excited (LE) and ICT emissions at 427/485 and 527 nm, respectively. It is noteworthy that only a weak signal peaked at ∼420 nm is recorded in THF (Fig. S6, ESI†), indicating more effective quenching of ICT emission compared to LE emission by free intramolecular rotations. In the media with fw ≤ 60%, luminogenic molecules are still molecularly dissolved, allowing for active intramolecular rotations, which efficaciously dissipate the exciton energies, thus making the molecules nonemissive. In the 70% mixture, the LE emission (477 nm) is enhanced and the ICT emission disappears, which should be ascribed to the formation of crystalline aggregates, in which intramolecular rotations and the ICT process are highly impeded.
Interestingly, the PL intensity of freshly prepared suspensions in 80% and 90% aqueous mixtures changes on standing with time. As exemplified by the former, the emission peak intensity at 485 nm becomes much stronger progressively as the time elapses; meanwhile, the ICT peak at 527 nm becomes more and more obscure (Fig. 3C–E). After standing for 12 h, only a predominant emission (477 nm) coinciding with that of the 70% aqueous mixture is observed. Dynamic light scattering (DLS) results (Fig. S7–S9, ESI†) reveal the continuously increasing mean diameter for the suspensions, which changes from 218.8 (freshly prepared) to 460.3 nm (standing for 12 h). Further, TEM observation and electron diffraction patterns (Fig. S10 and S11, ESI†) reveal essentially the amorphous and crystalline natures for the freshly prepared and standing (12 h) suspensions, respectively. Therefore, the emission changes of the suspensions should be attributed to the molecular rearrangement from the disordered amorphous state into a well ordered crystal lattice.
When fluorine atoms are introduced into DEATPE, the emission behaviour significantly varies. In THF and THF–water mixtures, PL intensity of DFDEATPE starts to increase at the fw of 60%, and reaches its maximum at fw = 70%, then decreases at fw = 80%. Afterwards, it reversely increases with further water addition. Though the PL intensity of aggregates in the 90% aqueous mixture is much lower than that in the 70% aqueous medium, it is still ∼1000-fold higher compared to that in THF (Fig. 4A and B). This emission change is also vividly depicted in Fig. 4C, disclosing the AIE inherent of DFDEATPE molecules, which is confirmed by the results obtained in the dimethyl formamide (DMF) and DMF–water system (Fig. S12, ESI†). In addition, much stronger light emission of the crystalline powders is observed compared to that from the dotted point on the TLC plate (Fig. 4B), suggesting its crystallization-induced emission enhancement (CIEE) attribute.
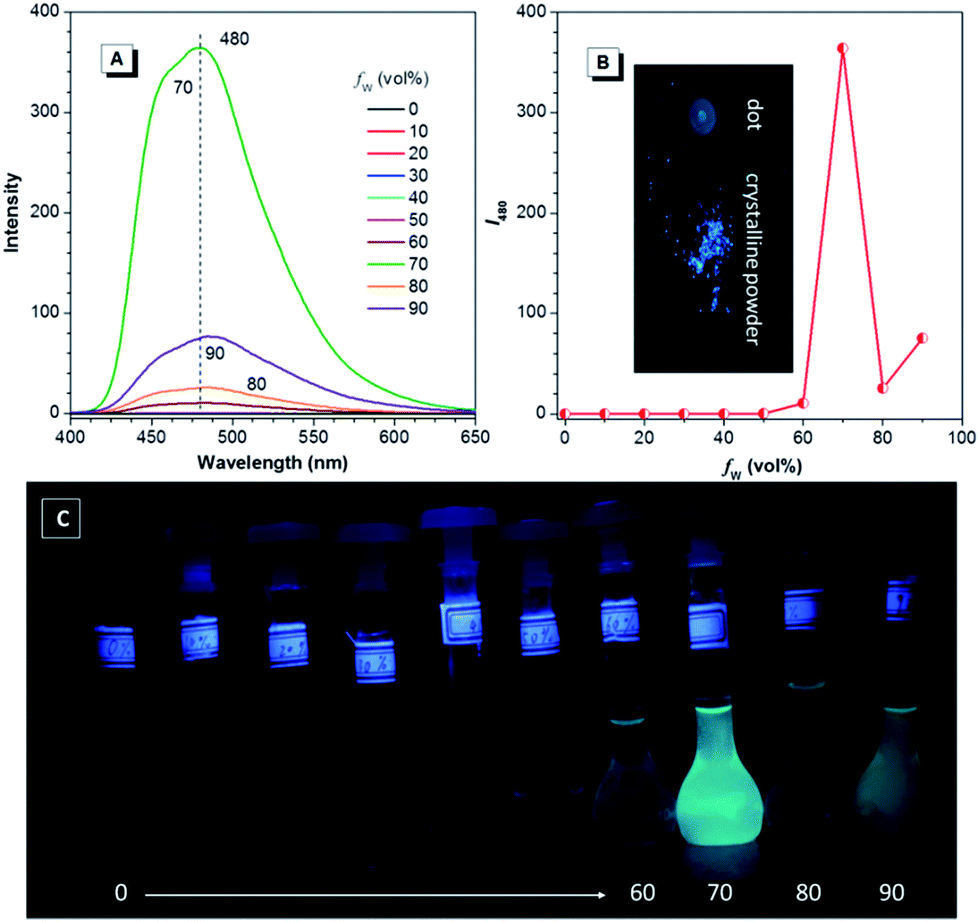 | ||
| Fig. 4 (A) PL spectra, (B) emission peak intensity at 480 nm and (C) luminescent photographs of DFDEATPE in THF and THF–water mixtures with different fws. Concentration = 40 μM; excitation wavelength = 365 nm. The inset in (B) is the luminescent photograph of a dotted point (left) and crystalline powders (right) of DFDEATPE on a TLC plate. | ||
A careful inspection into the emission profiles of different samples reveals hidden insights of the system. Normalized emission spectra of DFDEATPE in THF and THF–water mixtures (Fig. 5) show emission peaks/shoulders at around 432, 480 and 529 nm when fw ≤ 50%, which can be assigned to the LE (432/480 nm) and ICT (529 nm) emissions. However, in the 60% aqueous mixture, despite the intensified emission, no ICT emission is observed, displaying a single LE emission peak at 480 nm. This result indicates that aggregation has effectively blocked the ICT process. Further addition of water exerts little effect on the emission profile, which is significantly different from DEATPE, whose suspensions in the 90% aqueous mixture exhibit a distinct ICT emission. Apparently, introduction of fluorine atoms results in the restricted ICT process for the aggregates, which should be ascribed to the strong dipole–dipole interactions. It is thus envisioned that the ordered molecular packing with effective intermolecular interactions in the crystals may also restrain the ICT process. Indeed, both single crystals of DEATPE and DFDEATPE present predominantly LE emissions at 481 and 487 nm (Fig. S13, ESI†), respectively.
Based on the above results and taking into account of our previous systems,24,30 the emission peak and intensity changes of DFDEATPE in THF and THF–water mixtures can be rationalized as below: in the media with fw ≤ 50%, DFDEATPE is still molecularly dissolved, both LE and ICT emissions are pretty weak due to active intramolecular rotations. In the 60% aqueous mixture, the luminogenic molecules start to aggregate, inducing enhanced emission owing to the restricted intramolecular rotations; meanwhile, the ICT process is weakened because of effective dipole–dipole interactions in the aggregates. In the 70% aqueous mixture, the molecules are ready to orderly pack into crystalline nanosuspensions, thus generating boosted light emission. Further incorporation of water yields mainly amorphous aggregates, whose PL intensity is much lower compared to the crystalline counterparts because of the destruction of certain intramolecular interactions and thus less rigid conformations.
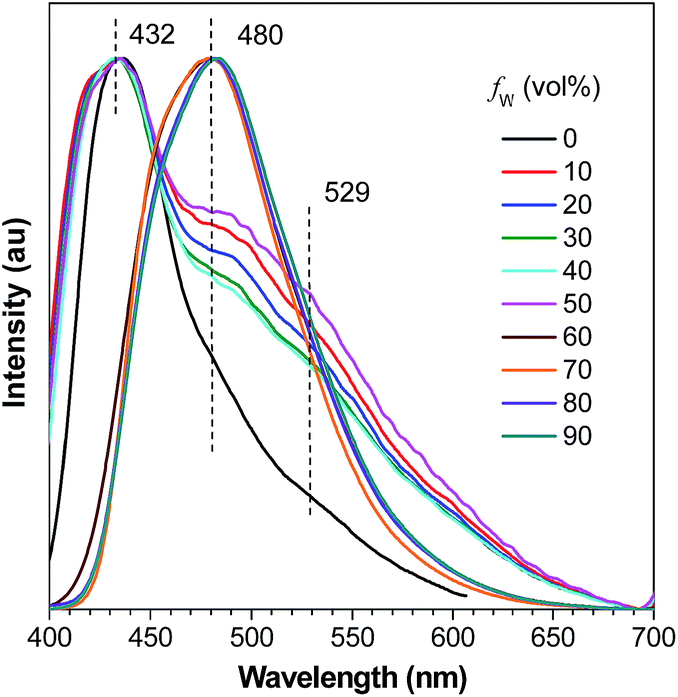 | ||
| Fig. 5 Normalized emission spectra of DFDEATPE in THF and THF–water mixtures with different fws. | ||
Single crystal structures of DEATPE and DFDEATPE are obtained to further decipher the CIE/AIE mechanism. As shown in Fig. 6, both molecules adopt highly twisted conformations. When they are dissolved in solvents, multiple phenyl blades and DEA units undergo active intramolecular rotations (Chart 2), which effectively dissipate the exciton energies from both LE and ICT states, thus making them nonemissive. When aggregated into crystalline solids, on one hand, plentiful intermolecular C–H⋯H–C (2.381 Å)/C–H⋯π (2.693, 2.721, 2.725, 2.729, 2.812, 2.852, 2.881, 2.892 Å) (Fig. 6B, C, E and F) contacts rigidify the molecular conformations; on the other hand, the propeller-like conformations prevent the formation of detrimental excimers or exciplexes, thus endowing the aggregates with intense emission. When aggregated into amorphous aggregates, however, such intermolecular interactions are destroyed, and intramolecular rotations and vibrations are activated, thus making the molecules less luminescent or almost nonemissive. Notably, abundant intermolecular interactions in the crystals also effectively restrain the ICT process, thus resulting in the disappearance of ICT emissions.
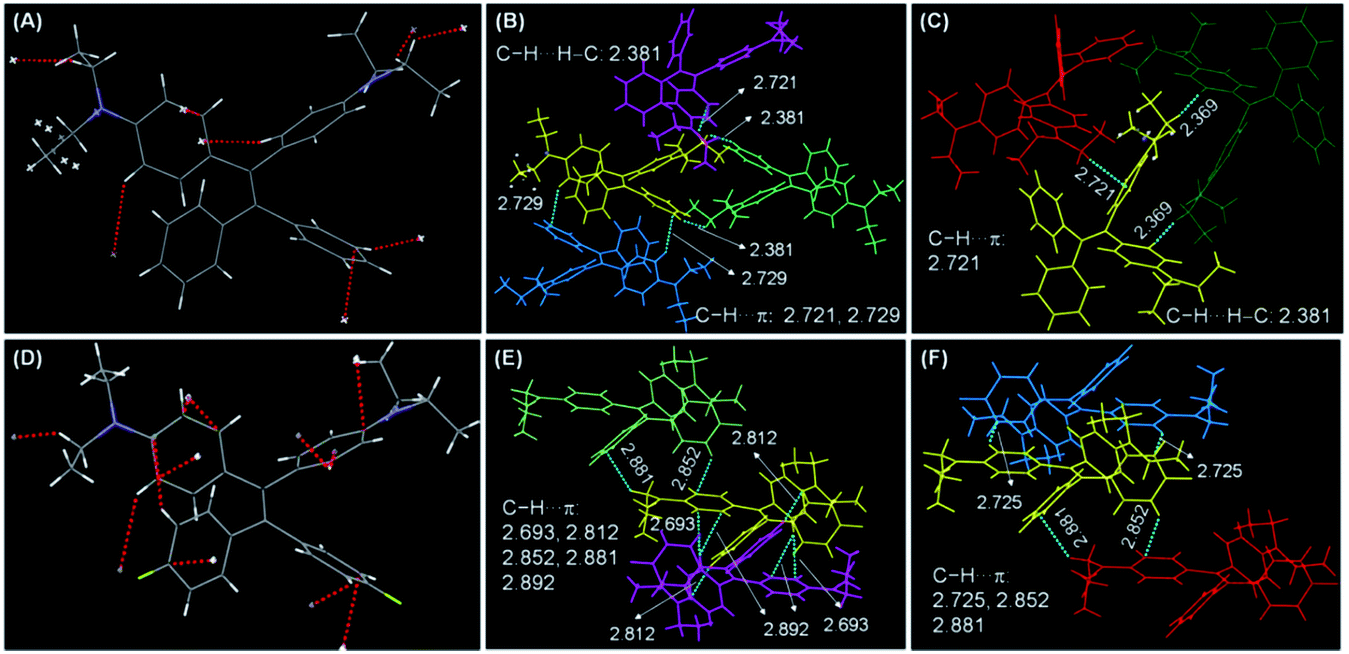 | ||
| Fig. 6 (A, D) Single crystal structures of (A) DEATPE and (D) DFDEATPE. (B, C, E, F) Intermolecular interactions in (B, C) DEATPE and (E, F) DFDEATPE crystals. The red dashed lines in (A) and (D) denote the contacts around one molecule in DEATPE and DFDEATPE crystals, respectively. | ||
For CNDEATPE, though no emission is observed for the solutions, it exhibits typical bathochromic shifts in various solvents, with emission maxima at 521, 567, 591 and 612 nm in n-hexane, THF, dichloromethane (DCM) and DMF (Fig. S14, ESI†), respectively. It seems that no evident LE emission exists in these solvents, which should be ascribed to the strong ICT effect and more efficient quenching of the LE emission than the ICT emission resulting from intramolecular rotations. However, in THF and THF–water mixtures, CNDEATPE is practically nonfluorescent when fw ≤ 60%, with almost unchanged PL intensity. Then it starts to increase when fw is increased to 70%, and swiftly gets enhanced afterwards (Fig. 7). The PL peak intensity at 588 nm is augmented by ∼50-fold in 10/90 THF–water compared to that in pure THF, indicating that it is AIE-active, which is confirmed by the photographs in Fig. 7B. In addition, besides the enhancement of ICT emission, a new peak at 486 nm is also observed at higher fws, which is assignable to the LE emission, therefore, aggregation has turned on both LE and ICT emissions.
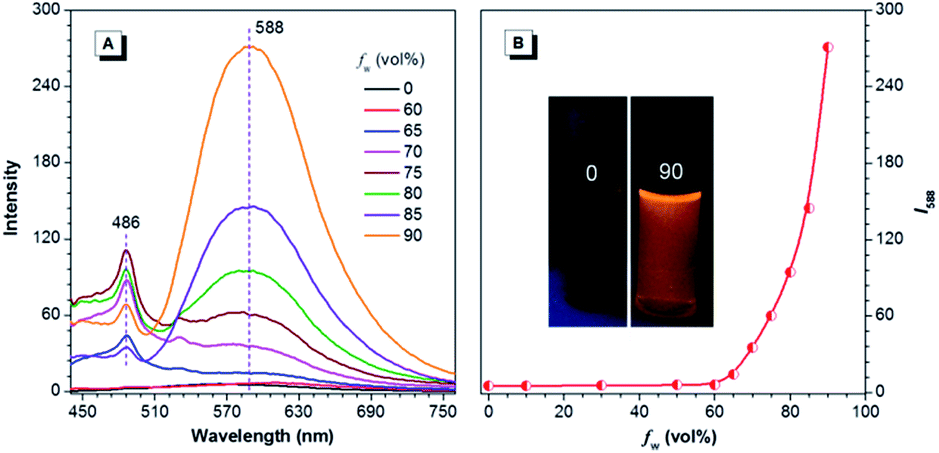 | ||
| Fig. 7 (A) PL spectra of CNDEATPE in THF and THF–water mixtures. (B) Plot of PL peak intensity at 588 nm vs. fw of the aqueous mixture. Concentration = 40 μM; excitation wavelength = 420 nm. Inset: photographs of CNDEATPE in the THF–water mixtures with fw of 0% and 90% taken under UV illumination. | ||
PL quantum efficiencies of the fluorogens in both solution (ΦF,s) and crystalline (ΦF,c) states are determined. While ΦF,s of the luminogens in THF are as low as 0.21%, 0.23% and 0.35%, their ΦF,c values are boosted to 44.8%, 100% and 17.9%, giving the corresponding augment factors (α = ΦF,c/ΦF,s) of 213, 437.8 and 51.1 for DEATPE, DFDEATPE and CNDEATPE (Chart 2), respectively.
Mechanochromic luminescence
The high efficiency, twisted conformation and effective intermolecular interactions of the crystalline solids of DEATPE, DFDEATPE and CNDEATPE render them promising mechanochromic materials.18 The as prepared crystalline powders of DEATPE and DFDEATPE (Fig. S15, ESI†) emit cyan light under UV irradiation, however, they emit yellow light at much lower intensity after grinding in an agate mortar (Fig. 8). Ground DEATPE solids rapidly get restored to the original emission without any external treatment within several seconds (Fig. 8A and B), exhibiting a distinct self-recovery property. Generally, emission restoration of the mechanochromic materials necessitates such external aiding as heating or solvent fuming.18–20 The fast self-recovery mechanochromism of DEATPE suggests highly active conformational adjustment and molecular rearrangement of the ground solids. It also indicates highly active molecular motions in the amorphous states, which can effectively quench the exciton energies, thus making the luminogenic molecules faintly luminescent or even nonemissive. This well explains the CIE phenomenon of DEATPE, and sheds light on understanding the CIE mechanism of other systems.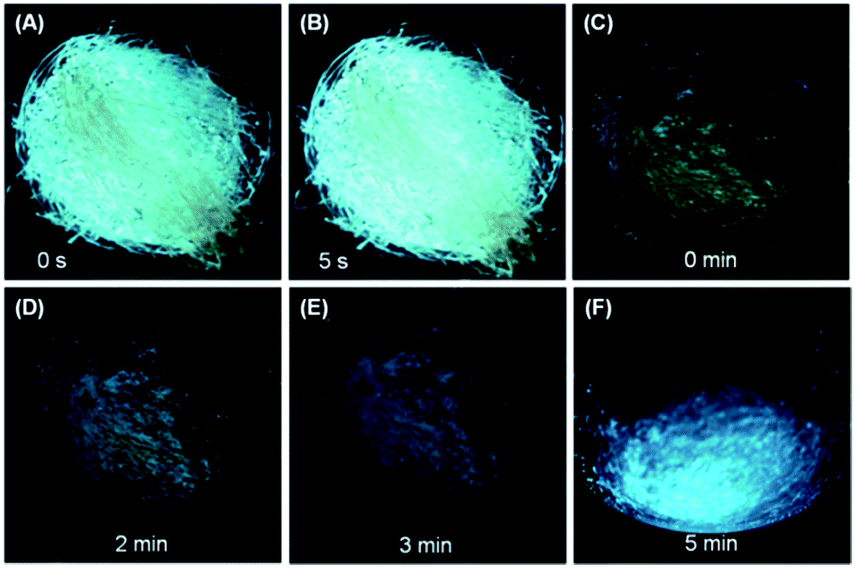 | ||
| Fig. 8 Fast self-recovery mechanochromism of (A, B) DEATPE and (C–F) DFDEATPE. Photographs were taken under 365 nm UV light irradiation. | ||
DFDEATPE shows a similar self-recovery feature in a much slower manner. It is found that the ground part with dim yellow light emission mostly returns to the original cyan light in 2–3 min, and is fully restored in 5 min (Fig. 8C–F). The slower self-recovery rate of DFDEATPE compared to DEATPE should be ascribed to its stronger dipole–dipole interactions, which slow down the molecular motions.
No meaningful emission profile is obtained for the ground DEATPE solids because of the rapid recovery process. For DFDEATPE, the ground powders (in mortar) luminesce at 490 nm, which is only slightly red-shifted compared to the as prepared crystalline solids (467, 481 nm) (Fig. 9). This small difference should be attributed to the fast recovery process. To shorten the sample transfer time, DFDEATPE was ground on a quartz plate for immediate measurement. Besides a broad emission peak at 498 nm, the other weak peak at 582 nm can be noticed, which might be associated with the excimer emission. The high contrast mechanochromism of crystalline DEATPE and DFDEATPE solids should be ascribed to the conformation planarization and destruction of intermolecular interactions, which lead to the enhanced effective conjugation length and more active molecular motions, thus resulting in red-shifted emissions with much lower intensity. Specifically, excimers constructed by phenyl blades via π–π stacking are formed upon mechanical grinding, which further red-shift and diminish the PL emission.
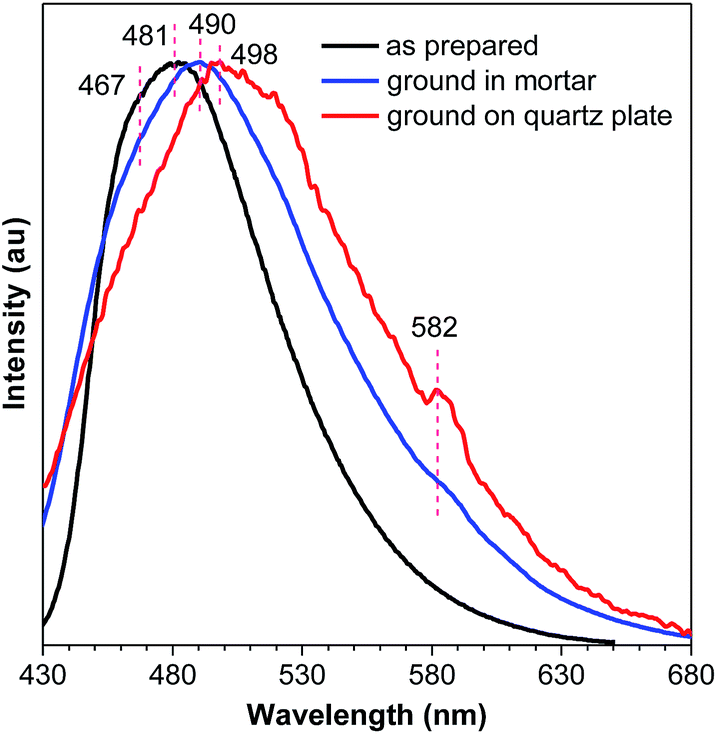 | ||
| Fig. 9 Emission spectra of DFDEATPE in different states. | ||
Though DEATPE and DFDEATPE are mechanochromic, TPE itself does not exhibit mechanochromic behaviors upon manual grinding (Fig. S16, ESI†), namely, no emission change is detectable before and after grinding. There are two possibilities for this phenomenon: (1) TPE molecules do not undergo conformational changes upon grinding; (2) the conformations and emissions are changed, however, it is undetectable owing to the prompt recovery process. Combined with the above results, the latter hypothesis is more likely the case. It is reasonable that intramolecular motions of TPE are much easier than those of DEATPE, owing to its smaller rotation barrier. The decoration with DEA groups, on one hand, offers extra rotatable units which can further consume the exciton energies, thus making DEATPE molecules less emissive in amorphous states; on the other hand, it increases the rotation barrier and thus prolongs the conformational relaxation time. This well explains why TPE is AIE-active and nonmechanochromic, whereas DEATPE is CIE-active and mechanochromic. For DFDEATPE, its stronger dipole–dipole interactions restrict the intramolecular rotations, which not only afford brighter emission than DEATPE, but also result in much slower recovery of the conformation and emission.
When further aromatic building block with stronger electron-accepting capacity is introduced, CNDEATPE shows mechanochromism without self-restoration within a short time. The as prepared solids emit yellow light at 558 nm, whereas the ground powders generate orange light with maxima at 588 and 598 nm (Fig. 10A and B). Such transformation is reversible upon solvent fuming or heating. XRD profiles of the as prepared and fumed solids show similar intense and sharp diffractions corresponding to regular molecular packing in crystals, whereas that of the ground powders merely exhibits a rather weak diffusion halo associated with the disordered molecular arrangement in the amorphous state (Fig. 10C). Therefore, the conversion between stable crystalline and metastable amorphous phases is essential for the reversible mechanochromism. Crystalline CNDEATPE becomes amorphous with less twisted (planarized) conformations upon grinding, moreover, excimers might be formed, thus yielding red-shifted emissions at 588 and 598 nm, respectively.
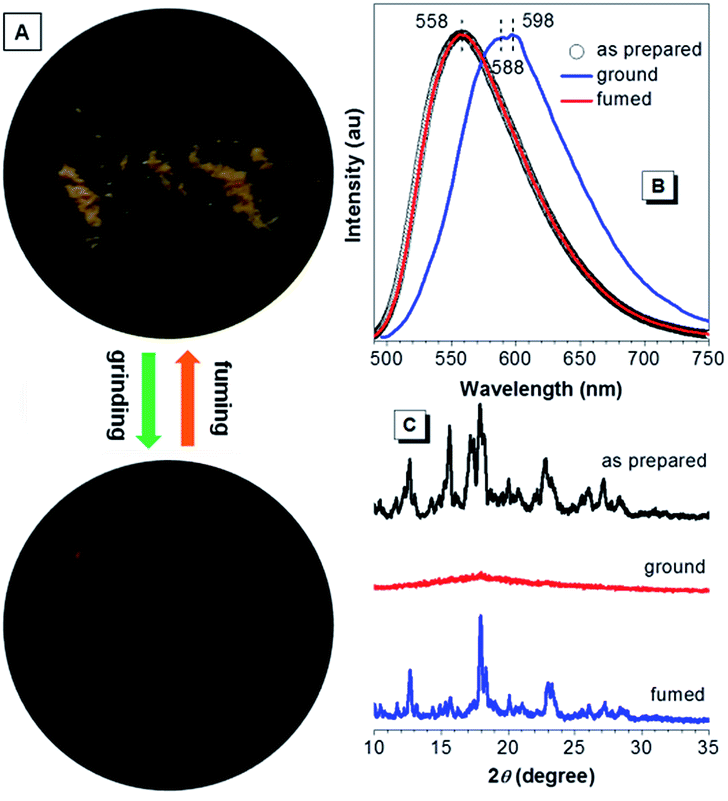 | ||
| Fig. 10 (A) Photographs of as prepared CNDEATPE powders before and after mechanical grinding. (B) Emission spectra and (C) XRD patterns of the as prepared, ground and fumed solids of CNDEATPE. | ||
Cell imaging
Besides the mechanochromic property, the presence of DEA groups may render these luminogens biocompatible, which was verified by our previous work.26 The AIE characteristics and good biocompatibility endow these luminogens highly applicable to bioimaging. Nanoaggregates of the luminogens are thus further utilized for cell imaging. Fig. 11 shows the phase contrast and fluorescence images of the HeLa cells treated with the nanoaggregates of DEATPE, DFDEATPE and CNDEATPE. Clearly, these nanoaggregates are able to readily stain the cells and realize fluorescence labelling under the standard cell incubation conditions. Closer scrutinization of the cell images reveals that these nanoparticles mainly stain the cytoplasmic regions of the cells and hardly enter the nucleus. We can thus conclude that these DEA-containing luminogens have crossed cell membranes to the interior of cells and concurrently enabled imaging of the HeLa cells. | ||
| Fig. 11 (A–C) Phase contrast and (D–F) fluorescent images of HeLa cells stained with (A, D) DEATPE, (B, E) DFDEATPE and (C, F) CNDEATPE, respectively. | ||
Conclusions
In summary, a group of DEA-bearing TPE derivatives are designed and synthesized. These luminogens possess high crystalline-state efficiency of up to unity due to the presence of the TPE unit. Moreover, photophysical properties of the luminogens can be facilely adjusted by the structural and electronic modulation: (1) incorporation of DEA groups yields extra absorption bands associated with the ICT transitions, which are absent in the TPE system; (2) while TPE is AIE-active, DEATPE is typically CIE-active, and DFDEATPE shows AIE and evident CIEE characteristics; further incorporation of the electron-accepting cyano moiety affords CNDEATPE with typical AIE and ICT characteristics; (3) TPE is nonmechanochromic, however, these luminogens exhibit high contrast mechanochromism with different recovery rates. The fast self-recovery of DEATPE and DFDEATPE luminogens suggests active molecular motions even in the amorphous solid state, which is indicative of the CIE mechanism.It is also noteworthy that the presence of DEA groups renders the molecules biocompatible, which is crucial for their biological applications. These unique properties of the DEA-containing TPE derivatives make them not only fundamentally interesting, but also promising for practical applications.
Acknowledgements
This work was financially supported by the National Science Foundation of China (21104044 and 51473092), the National Basic Research Program of China (973 Program, 2013CB834701) and the Ph.D. Programs Foundation of the Ministry of Education of China (20110073120040). W.Z.Y. thanks the SMC-Chenxing Young Scholar Program of Shanghai Jiao Tong University.Notes and references
- (a) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., 2014, 126, 2151 CrossRef; (b) L. Yao, B. Yang and Y. Ma, Sci. China: Chem., 2014, 57, 335 CrossRef CAS; (c) Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC; (d) W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed; (e) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- (a) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC; (b) M. Zhang, M. Yu, F. Li, M. Zhu, M. Li, Y. Gao, L. Li, Z. Liu, J. Zhang, D. Zhang, T. Yi and C. Huang, J. Am. Chem. Soc., 2007, 129, 10322 CrossRef CAS PubMed; (c) T. Han, X. Feng, B. Tong, J. Shi, L. Chen, J. Zhi and Y. Dong, Chem. Commun., 2012, 48, 416 RSC.
- (a) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed; (b) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed.
- (a) P. Sharma, S. Brown, G. Walter, S. Santra and B. Moudgil, Adv. Colloid Interface Sci., 2006, 123–126, 471 CrossRef CAS PubMed; (b) W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771 CrossRef CAS; (c) X. Zhang, X. Zhang, S. Wang, M. Liu, L. Tao and Y. Wei, Nanoscale, 2013, 5, 147 RSC.
- (a) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed; (b) J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, 1970 Search PubMed.
- Y. Huang, L.-P. Chen, C. Doyle, Y. Zhou and S.-T. Wu, Appl. Phys. Lett., 2006, 89, 111106 CrossRef PubMed.
- (a) S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751 RSC; (b) T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541 CrossRef CAS PubMed.
- (a) J. M. Lupton, L. R. Hemingway, I. D. W. Samuel and P. L. Burn, J. Mater. Chem., 2000, 10, 867 RSC; (b) J. Wang, Y. Zhao, C. Dou, H. Sun, P. Xu, K. Ye, J. Zhang, S. Jiang, F. Li and Y. Wang, J. Phys. Chem. B, 2007, 111, 5082 CrossRef CAS PubMed.
- (a) Z. Xie, B. Yang, F. Li, G. Cheng, L. Liu, G. Yang, H. Xu, L. Ye, M. Hanif, S. Liu, D. Ma and Y. Ma, J. Am. Chem. Soc., 2005, 127, 14152 CrossRef CAS PubMed; (b) F. He, H. Xu, B. Yang, Y. Duan, L. Tian, K. Huang, Y. Ma, S. Liu, S. Feng and J. Shen, Adv. Mater., 2005, 17, 2710 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- (a) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed; (b) J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675 RSC; (c) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329 CrossRef CAS; (d) W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518 CrossRef CAS.
- (a) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- (a) Y. Y. Gong, Y. Q. Tan, J. Mei, Y. R. Zhang, W. Z. Yuan, Y. M. Zhang, J. Z. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1178 CrossRef CAS; (b) Y. Y. Gong, Y. Q. Tan, H. Li, Y. R. Zhang, W. Z. Yuan, Y. M. Zhang, J. Z. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1183 CrossRef CAS PubMed; (c) W. Jia, P. Yang, J. Li, Z. Yin, L. Kong, H. Lu, Z. Ge, Y. Wu, X. Hao and J. Yang, Polym. Chem., 2014, 5, 2282 RSC.
- (a) M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834 CrossRef CAS PubMed; (b) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934 CrossRef CAS PubMed; (c) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273 CrossRef CAS PubMed.
- (a) Z. Yang, Z. Chi, T. Yu, X. Zhang, M. Chen, B. Xu, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2009, 19, 5541 RSC; (b) J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892 CrossRef CAS.
- (a) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed; (b) E. P. J. Parrott, N. Y. Tan, R. Hu, J. A. Zeitler, B. Z. Tang and E. Pickwell-MacPherson, Mater. Horiz., 2014, 1, 251 RSC.
- (a) B. Mi, Y. Dong, Z. Li, J. W. Y. Lam, M. Häußler, H. H. Y. Sung, H. S. Kwok, Y. Dong, I. D. Williams, Y. Liu, Y. Luo, Z. Shuai, D. Zhu and B. Z. Tang, Chem. Commun., 2005, 3583 RSC; (b) Y. Gong, J. Liu, Y. Zhang, G. He, Y. Lu, W. B. Fan, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Mater. Chem. C, 2014, 2, 7552 RSC.
- (a) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (b) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H.-Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed; (c) Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998 CrossRef CAS.
- (a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed; (b) A. Pucci and G. Ruggeri, J. Mater. Chem., 2011, 21, 8282 RSC; (c) K. Ariga, T. Mori and J. P. Hill, Adv. Mater., 2012, 24, 158 CrossRef CAS PubMed; (d) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119 CrossRef CAS; (e) Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372 RSC.
- (a) X. Luo, J. Li, C. Li, L. Heng, Y. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed; (b) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed; (c) C. Dou, L. Han, S. Zhao, H. Zhang and Y. Wang, J. Phys. Chem. Lett., 2011, 2, 666 CrossRef CAS; (d) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268 CrossRef CAS PubMed.
- (a) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed; (b) T. Tsukuda, M. Kawase, A. Dairiki, K. Matsumoto and T. Tsubomura, Chem. Commun., 2010, 46, 1905 RSC; (c) G.-G. Shan, H.-B. Li, H.-Z. Sun, D.-X. Zhu, H.-T. Cao and Z.-M. Su, J. Mater. Chem. C, 2013, 1, 1440 RSC.
- (a) Z. Li, Y. Dong, B. Mi, Y. Tang, M. Häussler, H. Tong, Y. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sung, K. S. Wong, P. Gao, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061 CrossRef CAS PubMed; (b) H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H.-Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287 CrossRef CAS; (c) H. Tong, Y. Dong, M. Häußler, J. W. Y. Lam, H. H.-Y. Sung, I. D. Williams, J. Sun and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef PubMed.
- (a) Y. Dong, J. W. Y. Lam, A. J. Qin, Z. Li, J. Z. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40 RSC; (b) Y. Dong, C. Li, W. Zhao, Y. Dong and B. Z. Tang, J. Mol. Eng. Mater., 2013, 1, 1340010 CrossRef.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS.
- Y. Yu, C. Feng, Y. Hong, J. Liu, S. Chen, K. M. Ng, K. Q. Luo and B. Z. Tang, Adv. Mater., 2011, 23, 3298 CrossRef CAS PubMed.
- (a) X. F. Duan, J. Zeng, J. W. Lü and Z. B. Zhang, J. Org. Chem., 2006, 71, 9873 CrossRef CAS PubMed; (b) S. Odabas, E. Tekin, F. Turksoy and C. Tanyeli, J. Mater. Chem. C, 2013, 1, 7081 RSC; (c) J. Liu, Y. Shi and Y. Yang, Adv. Funct. Mater., 2001, 11, 420 CrossRef CAS; (d) Z. Zhao, Z. Li, J. W. Y. Lam, J. L. Maldonado, G. Ramos-Ortiz, Y. Liu, W. Z. Yuan, J. Xu, Q. Miao and B. Z. Tang, Chem. Commun., 2011, 47, 6924 RSC.
- W. Z. Yuan, H. Zhao, X. Y. Shen, F. Mahtab, J. W. Y. Lam, J. Z. Sun and B. Z. Tang, Macromolecules, 2009, 42, 9400 CrossRef CAS.
- The calculation was conducted with DFT method using B3LYP/6-31G(d) basis set in Gaussian 09 program: M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) W. Z. Yuan, F. Mahtab, Y. Gong, Z.-Q. Yu, P. Lu, Y. Tang, J. W. Y. Lam, C. Zhu and B. Z. Tang, J. Mater. Chem., 2012, 22, 10472 RSC; (b) N. Zhao, M. Li, Y. Yan, J. W. Y. Lam, Y. L. Zhang, Y. S. Zhao, K. S. Wong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 4640 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, including experimental procedures, structural characterization data, crystal data, and high resolution mass spectra. CCDC 1024902 and 1024903. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc02161d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |