
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodegradable liposome-encapsulated hydrogels for biomedical applications: a marriage of convenience
Santiago
Grijalvo
ab,
Judith
Mayr
c,
Ramon
Eritja
ab and
David Díaz
Díaz
*ac
aInstitute of Advanced Chemistry of Catalonia (IQAC-CSIC), Spain
bBiomedical Research Networking Center in Bioengineering, Biomaterials and Nanomedicine (CIBER BBN), Spain
cInstitute of Organic Chemistry, University of Regensburg, Universitätstrasse. 31, D-93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 941 943-4121; Tel: +49 941 943-4373
First published on 28th January 2016
Abstract
Hydrogels are hydrophilic three-dimensional networks with demonstrated potential for medical and pharmaceutical applications. Specifically, biopolymer-based hydrogels offer certain advantages over synthetic polymers in terms of biocompatibility and biodegradability. Because of their inherent properties, hydrogels are able to efficiently encapsulate and liberate in a controlled release manner, different hydrophobic and hydrophilic therapeutic molecules, including nucleic acids, proteins and antibodies. Several strategies have been reported in the literature to minimize the potential burst release of encapsulated drugs, thus preventing their local accumulation and consequent toxic responses. Within this context, liposomes embedded in hydrogels have emerged as an attractive strategy to reduce this undesirable effect. This tutorial review covers a selection of the most promising cationic, neutral and anionic biopolymer-based hydrogels containing liposomes, niosomes or vesicles for drug delivery or tissue engineering applications.
 Santiago Grijalvo | Santiago Grijalvo was born and grew up in Madrid. In 2000, he received his Bachelor's degree in Chemistry (with an emphasis in Organic Chemistry) from the Autonomous University of Madrid. After finishing his studies, he was hired as an intern at GlaxoSmithKline for one year. In 2002 he moved to Barcelona to join Dr Antonio Delgado's research group to do his PhD studies focusing on the synthesis of modified sphingolipids combinatorial libraries at the Institute of Research Chemistry (IIQAB-CSIC). In 2007 he joined Prof. Dr Ramon Eritja's research group as a post-doctoral researcher at the Institute of Advanced Chemistry (IQAC). Since then, he has participated in several research projects focusing on improving the cellular uptake of nucleic acids using either covalent or electrostatic strategies. |
 Judith Mayr | Judith Mayr studied Chemistry at the University of Regensburg, receiving her Bachelor′s degree in 2011 and her Master degree in 2013, respectively. Currently she is doing her PhD in the group of Prof. Díaz Díaz. Her main research interest lies in the use of soft material for the formulation and delivery of therapeutically active molecules. |
 Ramon Eritja | Ramon Eritja studied Chemistry and Pharmacy at the University of Barcelona, receiving his Ph.D. from the University of Barcelona in 1983. He did postdoctoral work in Dr Itakura's and Caruthers's groups. In 1990, he became group leader at CSIC and then at EMBL. He returned in 1999 to IQAC-CSIC and in 2012 he was appointed director of the institute (IQAC). He is a member of the CIBER-BBN network since 2006. His research focuses on oligonucleotide synthesis for biomedical and nanotechnological applications. |
 David Díaz Díaz | David Díaz Díaz (born in 1974, Tenerife) received his PhD in Chemistry at the University of La Laguna. In 2002, he joined Prof. Finn's group as postdoc at The Scripps Research Institute (San Diego, USA). Since 2006, he has held various positions in academia and industry (‘Ramón y Cajal’ Researcher, Autonomous University of Madrid, Spain, 2006; Sr. Chemist, The Dow Chemical Company, Switzerland, 2007; Tenured Scientist, CSIC, Spain, 2009; Alexander von Humboldt Experienced Researcher, University of Regensburg, Germany, 2010). In 2013, he was named recipient of the DFG Heisenberg Professorship. He has received the Young Investigator Award from the Polymer Network Group (Japan, 2014) and he is Editor-in-Chief of Gels. His main research interest focuses on the development of advanced functional soft materials. |
1. Introduction
Over the past few decades, synthetic progress towards fabrication of new polymeric materials that respond to external stimuli (e.g., temperature, light, pH)1–4 has stimulated the development of novel approaches for drug delivery,5,6 tissue engineering7 and nanobiotechnology.8,9 This environmental responsiveness triggers major changes in both the physicochemical and self-assembling properties of such macromolecular systems, which can be used to favour the encapsulation/release of active molecules. Indeed, there are numerous examples in the literature where synthetic polymers have been used either as therapeutic macromolecules or as drug delivery vehicles upon combination with small drugs.10–12However, for real applications, synthetic polymers must fulfill a series of requirements such as being non-toxic, non-immunogenic and water-soluble together with safe excretion properties, which in most cases become very difficult challenges. It is at this point where biocompatible and biodegradable natural polymers represent a versatile and renewable alternative to synthetic polymers. In general, biopolymer-based materials are mainly made of polysaccharides and are usually key components in food industrial formulations acting as emulsifying, gelling and/or stabilizing agents.13 Furthermore, they have also been proven to be promising materials for biomedical applications including drug delivery and regenerative medicine.14–17
1.1. Hydrogels
Polymeric hydrogels are hydrophilic 3D polymer networks capable of absorbing large amounts of water in a similar way to body tissues. This property allows hydrogels to encapsulate therapeutic molecules and protect them from rapid degradation,18–20 making them useful materials for pharmaceutical and medical purposes.18,19In 1960, Wichterle and Lim21 reported the use of poly-2-hydroxymethacrylate (PHEMA)-based hydrogels for contact lens applications. Since then, the extensive research carried out in the field of hydrogels has allowed the development of not only polymeric networks based on synthetic polymers such as poly(vinyl alcohol),22,23 poly(ethyleneimine)24 and poly(ethylene oxide),25 among others, but also based on natural polymers such as alginate26 and chitosan.27
Another important feature of many polymeric hydrogels is their ability to release entrapped therapeutic molecules in a well-controlled manner. This property is usually governed by passive diffusion mechanisms and can also depend on additional factors (e.g., cross-linking degrees, hydrogel mesh sizes, stimuli-sensitive hydrogel capacity, etc.). One major aspect to take into account during release experiments is the possibility of obtaining undesired initial liberation of the drug immediately upon contact with the release medium. Although this undesirable effect could be positive in exceptional therapeutic strategies, it usually causes an unexpected in vivo toxicity due to the local presence of large amounts of drug (“dose-dumping”). Such a process is known as burst release effect.28
Although the exact mechanism and prediction of burst release in hydrogel/drug systems has not been elucidated yet,29,30 several strategies have been reported to minimize this effect. These include, for example, increasing the cross-linking density of the material, coating additional drug-free layers or using drug surface extractions prior to in vivo usages.31–33
1.2 Hydrogel classification and basic structure
Hydrogels are classified according to several characteristics. For example, they can be classified on the basis of ionic charges (neutral, anionic, cationic or ampholytic hydrogels), the nature of side groups (e.g., neutral or ionic), their physical structure (e.g. amorphous, semi-crystalline, hydrogen-bonded structures, super-molecular structures and hydrocolloidal aggregates), the crosslink nature (e.g. chemical or physical) and their preparation method (e.g., homo- or co-polymers).34Hydrogels can also have many different physical forms and have been used in varying therapeutic applications. Some examples include solid molded forms (e.g. contact lenses), pressed powder matrices (e.g. pills and capsules), beads (e.g., drug delivery), microparticles (e.g. wound treatments), coatings (e.g. implants or catheters) and membranes or sheets (e.g., a reservoir in a transdermal delivery patch).
An important issue to consider for drug delivery applications is to understand the transport mechanisms of the drug through the hydrogel matrix, which is usually governed by several parameters such as polymer composition, porosity, degradation rate, size and gel swelling.35 The mesh size is an important property that is related with degradability, mechanical strength and drug diffusion. Thus, as the gel swells, its mesh size increases and thus facilitates the corresponding drug release in solution. As a consequence, a good number of factors has to be considered and this requires a high degree control over some properties when designing hydrogels. Hence, hydrogel crosslinking degree, stimuli (pH, temperature) or monomer chemical structures are important parameters to design appropriate diffusion patterns of model drugs.36 Most hydrogels have the possibility of tuning their mesh sizes with varying dimensions and patterns that can range from micrometers to nanometers (5 to 100 nm) approximately in their swollen state. Specifically, microgels have been designed as novel platforms for both local sustained drug release and their ability to mimic the native tissue mechanical properties (3D-cell culture environment) as well as a platform for local protein release. This application has allowed both a rational design and the development of microfabrication strategies that have helped to provide new insights into regeneration and tissue repair applications.37
Nanogels have also become promising materials to be mostly used in systemic drug delivery applications. Moreover, they have shown certain advantages over other existing drug delivery systems. Therefore, a decrease in the hydrogel size may produce a faster response for gel swelling and thus increasing the material response and, consequently, the anticipated drug action.38 The recent advances of click chemistries affording the effective introduction of targeting ligands combined with the use of either polysaccharide-based crosslinked agents or cross-linkers containing biodegradable linkages have allowed the preparation of functional nanogels with narrow size distributions that have facilitated both cellular uptake and have avoided undesirable aggregation processes.39,40 Such control of microgel and nanogel properties has allowed extensive research in the development of synthetic strategies for the preparation of such materials. For example, a variety of processes like photolithographic, microfluidic techniques, micromolding methods as well as the use of heterogeneous solution, aqueous homogeneous gelation, spray drying methods, chemical crosslinking of dextranes or heterogeneous radical polymerizations are the most representative methods that have been described and revised in the literature.41
1.3. Liposomes and preparation methods
Liposomes (self-assembled lipid vesicles) are one of the most used nanosystems to encapsulate drug molecules. Because of their nanometer size, these kinds of non-viral carriers have become one of the most studied drug delivery systems for clinical applications to date.42–44 Since their beginning in the 60s, the field of liposomes has undergone a major change with respect to the development of novel responsive vesicles with at least one lipid bilayer45,46 and their applications in hyperthermia processes for cancer treatment.47,48A good number of encapsulation strategies where liposomes are used have been reported to date. For instance, an elegant and recent approach consists of introducing different types of nanoparticles (e.g., gold and silver nanoparticles, SPIOs or lipid vesicles) and obtaining the corresponding “liposome–nanoparticle” hybrids using liposomes as templates.49,50 The methodology involved in the synthesis of these constructs has helped us to obtain systems that have enhanced their biocompatibility and colloidal stability. Other methods have also been developed for the preparation of “liposome–virus”51 and “liposome–quantum dot” hybrids.52 These novel approaches have allowed the use of liposomes in therapeutic, diagnostic and theranostic applications.
There is a wide range of possibilities for preparing liposomes42 (Table 1). Therefore, depending on the procedure, properties like lamellarity, size, morphology and composition of liposomes can be different. Although most of these processes are standardized and optimized for laboratory scale and preparation is straightforward, there are some limitations like the length of procedures, polydispersity and low drug encapsulation, which are the main drawbacks for conventional liposome preparation methods.
| Conventional methods | ||||
|---|---|---|---|---|
| Preparation method | Particle size (nm) | Advantages | Disadvantages | Ref. |
| Thin-lipid film hydration | 100–1000 | Most widely used method | Low encapsulation efficiencies, sonication, temperature exposure, heterogeneous size distribution | 53 |
| Reverse-phase evaporation | 100–1000 | High encapsulation efficiency | Organic solvent traces | 56 |
| Solvent injection (ether or ethanol) | 70–200 | Ability to control vesicle size | Dilution of liposomes, heterogeneous population, use of high temperature | 57 |
| Novel methods | ||||
|---|---|---|---|---|
| Preparation method | Particle size (nm) | Advantages | Disadvantages | Ref. |
| Microfluidic technology | 100–300 | Synthesis of monodisperse liposomes, high encapsulation efficiency | Fabrication could be complex and needs optimization | 58 and 59 |
| Supercritical reverse phase evaporation | 100–1200 | Environmentally-friendly process, high encapsulation efficiencies | Elevated pressures and temperatures | 60 |
| Spray drying | 100–1000 | Control over particle formation, easily translated to large-scale production | Expense and time-required | 61 |
| Membrane contactor technology | ∼100 | Liposomes have homogeneous and small size, high encapsulation efficiency, simplicity for scaling-up | Hydrophilic drug encapsulation needs optimization | 62 |
| Crossflow injection | ∼50 | Liposomes of defined size | Vesicle stability due to residual solvent | 63 |
To address these issues, more efficient novel procedures have been described. Conventional liposome preparations are as follows: (a) the thin-lipid film hydration approach was the first methodology developed and the most common method for preparing liposomes.53 Thus, multilamellar (MLVs), giant unilamellar (GUVs) or small unilamellar (SUVs) vesicles may be obtained which are different from each other according to the preparation technique used.54,55 However, some limitations can clearly be identified using the technique such as possible liposome degradation upon sonication, heterogeneous size distribution, high temperature exposure or low drug encapsulation efficiencies. (b) Reverse-phase evaporation56 is another technique that consists of obtaining suspension of large unilamellar vesicles by removing the organic solvent under reduced pressure. This technique involves water-in-oil formation from a mixture of surfactants and lipids as well as an aqueous solution containing the drug. The presence of organic traces in the final formulation could influence the vesicle stability. (c) The solvent injection technique is another conventional method used to prepare liposomes which is based on injection of phospholipids dissolved in an organic solvent like ether or ethanol into an aqueous phase that contains a particular drug and is maintained at a temperature above the organic solvent boiling point.57 Although the advantage of this technology is the ability to control the vesicle size, the presence of organic solvent traces in the final formulation, dilution of the liposomes represent a clear disadvantage for this type of approach which reduces the efficiency of this approach.
More recently, novel methods have been described for preparing liposomes. Microfluidic technology has been used for preparing monodisperse liposomes on both laboratory and clinical scales and are easily reproducible.58,59 Optimizing the parameters such as the flow rate ratio or the size of microchannels provides liposomes with varying and more accurate control sizes. Efficient liposomal distribution, higher encapsulation efficiencies and the reduced loss of reagents are considered as being the advantages of this process. However, this microfluidic device fabrication may be considered a difficult and complex since it requires optimization of multiple fluid inputs and different fluid phases; as a consequence, liposome scale-up production becomes challenging.
On the other hand, there are also techniques that have been developed on an industrial scale which include supercritical reverse phase evaporation,60 which employs supercritical CO2 in order to dissolve phospholipids. From an industrial point of view, the use of CO2 creates an environmentally-friendly process and is a good organic solvent substitute for preparing liposomes as it is non-toxic and not inflammable. Moreover, this technology often yields higher encapsulation efficiencies compared with conventional procedures. Despite these advantages, this method uses elevated pressures (around 20–25 MPa) and temperatures (333 K). Spray drying,61 membrane contactor technology62 or crossflow injection63 techniques are other alternative methods that have also been described for industrial scale-up in liposome production because of their cost efficiency and short-duration processes. The advantages and disadvantages are both covered in Table 1.
Liposome-based technology has revealed serious shortcomings such as instability, a short life and rapid degradation. To address these issues, extensive research has been carried out in order to increase and improve its efficacy. In addition to the synthesis of second generation liposomal drugs described above, the integration of liposomes and polymeric matrices (“liposomes-in-hydrogels” or lipo-beads) may represent a promising approach for minimizing burst release and improving tissue localization, especially in topical drug delivery applications including wound therapy and sustained drug delivery. The nature of the polymer may also modify the properties of the encapsulated liposomes (membrane integrity and mechanical stability). This property, together with the liposome concentration, lipid composition as well as liposome interaction with hydrogels, which are also related to the swelling/deswelling properties of the hydrogels, may affect their rheology properties64 and thereby modulate the release of a therapeutic drug.
The preparation methods described for liposomes-in-hydrogels are mainly governed by incubating the anticipated liposomes with the pre-formed hydrogel beads. Although the encapsulation process of the resultant liposomes within the polymeric matrix takes place and has been experimentally confirmed, some concerns regarding phospholipids self-assembling and bilayer stabilities should also be considered.65 This combination has successfully allowed the incorporation of a wide number of liposomes into both synthetic and natural polymer-based hydrogels, obtaining the resultant stimuli-responsive hybrid materials.
Although a number of synthetic polymer-based hydrogels containing liposomes have been described to date,66–68 many of them may present limitations for clinical applications due to cytotoxic effects. Thus, the use of biocompatible and biodegradable hydrogel matrices has gained increased scientific attention over the last decades. The first liposomal biohydrogel system was described by Weiner and co-workers in 1985.69 In this work, a liposomal formulation containing two peptide hormones (insulin and growth hormone) was introduced into a collagen gel matrix. Slow release rates of the hormones were obtained when this liposome–collagen hybrid was used. Furthermore, the authors also observed important differences in the release when comparing these liposomal formulations in the absence and in the presence of the hydrogel. Since then the “liposomes-in-hydrogels” strategy has emerged as a promising approach for obtaining self-regulated drug delivery systems.
2. Liposomes in biopolymer-based hydrogels
In principle, the combination of liposomes and biopolymer-based hydrogels could offer important advantages with respect to synthetic polymers for biomedical applications70 by improving both drug formulation stabilities and drug administration routes.This review describes the most relevant examples of liposomes encapsulated in biopolymer-based hydrogels for biomedical applications. The use of synthetic polymers66–68 instead of biopolymers is out of the scope of this review. The manuscript is organized in subsections according to the type of biopolymer used for the preparation of the hydrogel matrix. Particular emphasis is given to the characterization of these hybrid materials and the release profile of encapsulated model drugs or therapeutic molecules (Fig. 1).
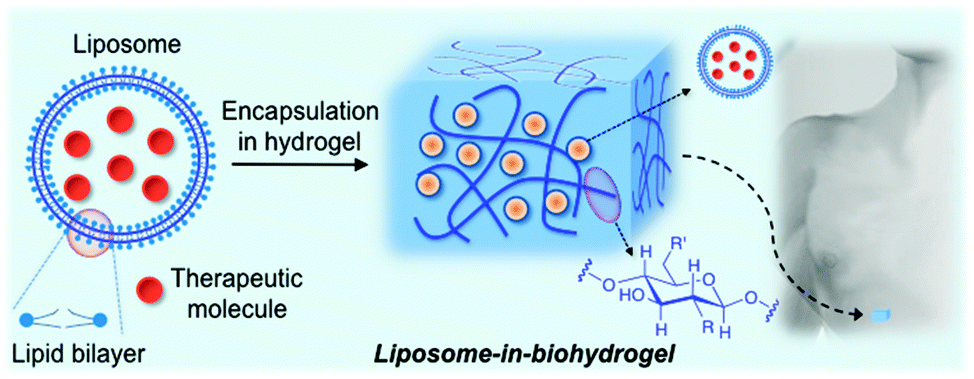 | ||
| Fig. 1 Schematic concept covered by this review. | ||
2.1. Chitosan
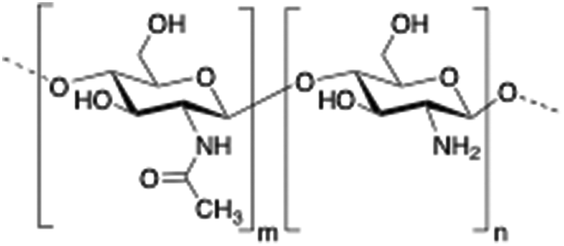
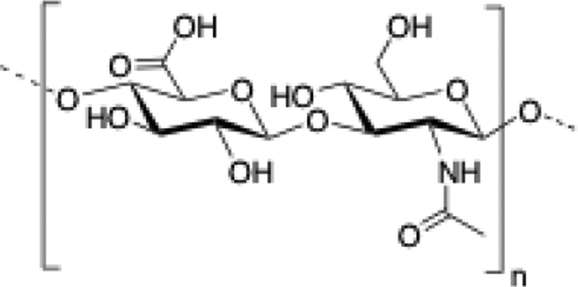
Chitosan is a cationic, biodegradable, biocompatible and mucoadhesive biopolymer composed of randomly distributed β(1 → 4)-linked D-glucosamine (deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit). Chitosan-based hydrogels are one of the most studied systems for applications in biomedical fields such as enzyme immobilization, tissue engineering, wound therapy and drug delivery.27,71–73
In 2002, Leroux and co-workers74 described for the first time the encapsulation of liposomes within chitosan polymeric matrices. Rheological experiments showed that the presence of liposomes in a chitosan-β-glycerophosphate formulation slightly increased both the viscoelastic properties and the gel strength. Additionally, a significant increase in the in vitro release time was achieved when liposomes containing model hydrophilic molecules (i.e., 5- and 6-carboxyfluorescein) were embedded in the hydrogel matrix. It is worth mentioning that sustained drug liberation over 2 weeks was obtained with this liposomal formulation. As a control experiment, the time of the release in the absence of liposomes was also evaluated, confirming the complete drug release within 24 h.
Among the variables that can affect control release processes, liposomal composition, vesicle size and surface charge have been studied in order to find a suitable liposomal formulation for application in wound therapy. For example, Škalko-Basnet and co-workers carried out detailed studies using two pH-dependent rhodamine derivatives as model drugs embedded into chitosan-based hydrogels.75 The results demonstrated that liposome sizes had a minor effect on the in vitro drug release whereas both liposomal surface charge and drug lipophilicity considerably affected the release kinetics.
Similar systematic studies have been recently carried out by Ladavière and co-workers.76 Phosphatidylcholine liposomes made of a mixture of DPPC and a fluorescent lipid at a 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, were mixed in chitosan solutions (Fig. 2A). This mixture did not affect the final process of the hydrogel formation and rheological studies confirmed that introducing phosphatidylcholine liposomes within the chitosan matrix did not affect the viscoelastic properties of the system either. Additionally, fluorescent liposomes were introduced within the polymeric network and the complexes were characterized by fluorescence and cryo-SEM microscopy. In vitro release experiments using carboxyfluorescein as a model drug showed delayed drug release compared to that in the absence of the liposomes (Fig. 2B).
:1 molar ratio, were mixed in chitosan solutions (Fig. 2A). This mixture did not affect the final process of the hydrogel formation and rheological studies confirmed that introducing phosphatidylcholine liposomes within the chitosan matrix did not affect the viscoelastic properties of the system either. Additionally, fluorescent liposomes were introduced within the polymeric network and the complexes were characterized by fluorescence and cryo-SEM microscopy. In vitro release experiments using carboxyfluorescein as a model drug showed delayed drug release compared to that in the absence of the liposomes (Fig. 2B).
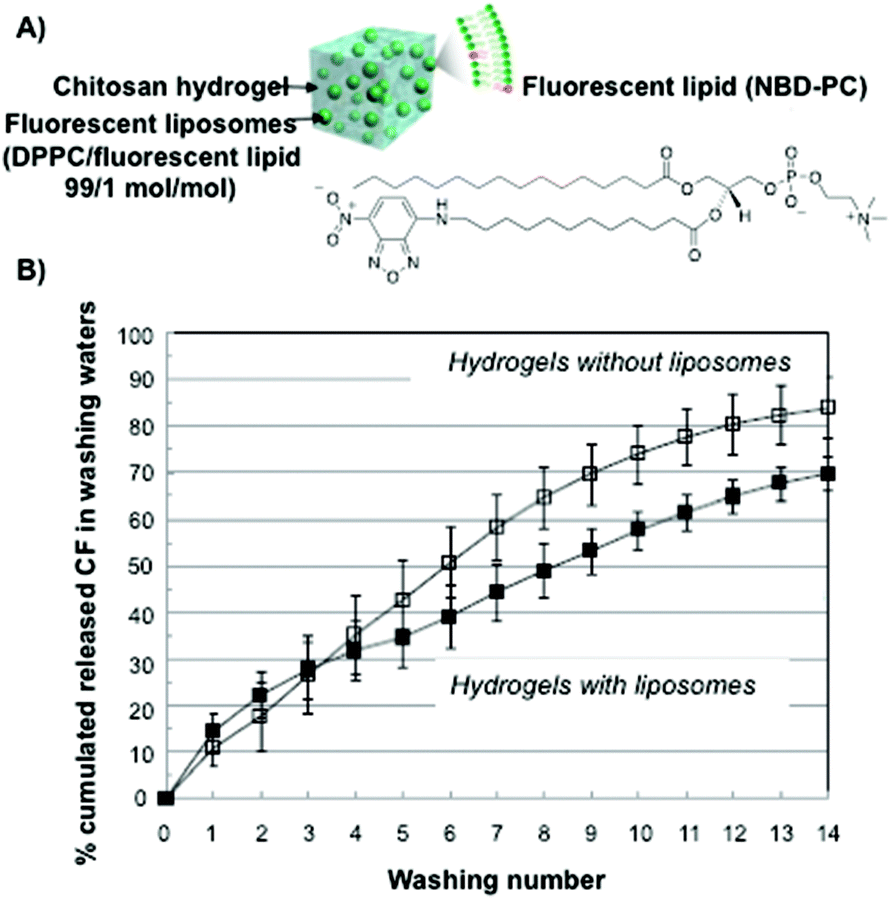 | ||
| Fig. 2 (A) Schematic representation and the chemical structure of fluorescent liposomes in chitosan hydrogels. (B) Carboxyfluorescein cumulative release percentage for chitosan hydrogels in the absence (open symbols) and in the presence (full symbols) of liposomes. Adapted with permission from ref. 76. Copyright © 2015 Elsevier. | ||
Rouini and co-workers77 performed both in vitro and in vivo experiments with a series of radiolabelled liposomes embedded into chitosan-β-glycerophosphate hydrogels. The studied vesicles varied either on the acyl chain length (DMPC, DPPC or DSPC) or on the surface charge (DSPG and DOTAP). The authors found that liposomes made of DSPC showed better release profiles than the other ones when injecting the corresponding liposomal formulations in mice models. Furthermore, liposomes made of DSPC showed a higher transition temperature, which generated stronger bilayers than the other liposomes tested. This stability effect induced liposome retention for a long period of time in the peritoneum after injection. An opposite effect was observed for liposomal dispersions made of negatively charged DSPG liposomes. These vesicular carriers left the peritoneum rapidly after injection. Interestingly, the authors were able to prolong this liposomal release at the highest level when entrapping the same vesicular dispersions within chitosan hydrogels. This change in the release behaviour was attributed to the presence of interactions between the matrix polymer and phospholipids.
Besides studying the parameters that can govern control release processes such as size, charge or lipid composition by using model drugs, the use of liposomal formulations entrapped into chitosan hydrogels containing either therapeutic molecules or macromolecules has been successfully reported in the last few years. For example, Škalko-Basnet and co-workers78,79 were able to entrap Mupirocin, a promising antimicrobial drug, in phosphatidylcholine liposomes of various sizes. Cytotoxicity and both in vitro and in vivo experiments were also reported. The results confirmed an antimicrobial potential activity against Staphylococcus aureus together with an enhancement in wound healing over 28 days in vivo. Furthermore, a superior bio-adhesiveness was also exhibited when compared to conventional treatments in burn wound healing rat models.
Ofloxacin, a second-generation fluoroquinolone derivative used for treating severe eye infections, usually suffers major drawbacks (e.g., necessity of frequent administration, low solubility) when administered in solution. However, Hosny and co-workers80 have recently shown promising therapeutic effects (i.e., prolonged release time and improved stability) when carrying out in vitro transcorneal permeation assays by using chitosan-based hydrogels as carriers.
Certain hydrogels are injectable due to the fact that they can exist in a low-viscosity form either prior to or during injection.81 This property has been applied for site-specific drug delivery applications. Interesting results were obtained in the case of measuring the release kinetics of doxorubicin (Dox), which was encapsulated in the interior of small unilamellar vesicles from a hydrophobically modified chitosan (hmC) gel (Fig. 3).70 The connection between the liposomes and the hydrogel network was achieved by hydrophobic interactions between polymer chains and liposome bilayers. Steady-shear rheology studies confirmed the properties of injectability of the liposomal Dox–hmC hybrid systems (Fig. 3A). The results showed that the combined system of liposomes and hmC hydrogel could act as an efficient barrier, promoting sustained release of the drug over a week (Fig. 3B).
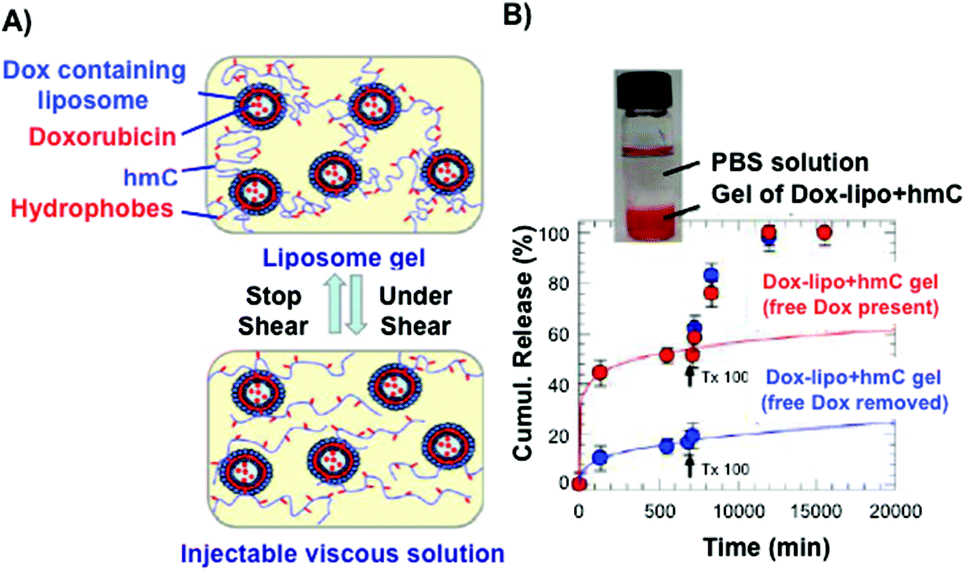 | ||
| Fig. 3 (A) Pictures of an injectable liposome gel at rest (top) and under shear (bottom). Modified chitosan hydrogels are represented in blue and the grafted hydrophobes in red. (B) Cumulative release of Dox through modified chitosan matrices. Dox release from hydrogels containing liposomes is slower than those in the absence of liposomes. Adapted with permission from ref. 70. Copyright © 2012 American Chemical Society. | ||
Recently, Duffy and Ruiz-Hernández82 have reported the use of chitosan-β-glycerophosphate matrices as carrier systems for local chemotherapy treatment. In this work, a mixture of non-toxic thermosensitive liposomes made of dipalmitoyl phosphatidyl choline (DPPC), monostearoyl-phosphatidyl choline (MSPC) and distearoylphosphatidylethanol-aminepoly(ethylene)glycol (DSPE-PEG2000) at a 85.3:9.7:5.0 ratio, respectively, was used to entrap Dox (Fig. 4). Interestingly, the cumulative Dox release from the polymer network was enhanced after using hyperthermic pulses, which reduced dsDNA levels when compared to the corresponding non-pulsed samples (Fig. 4B and C).
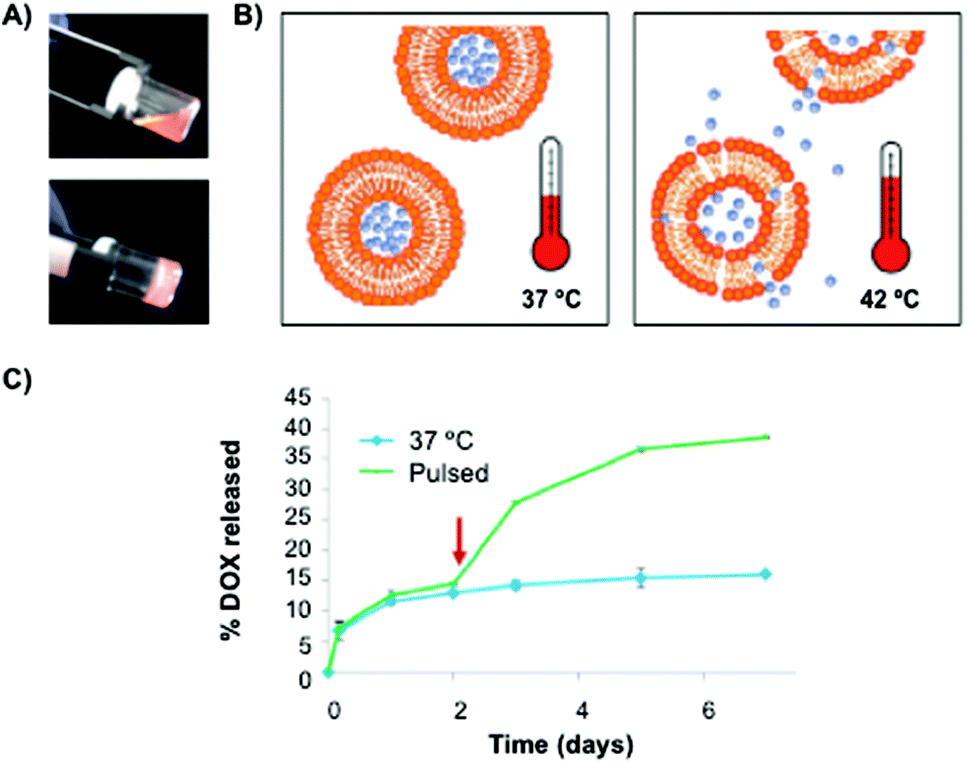 | ||
| Fig. 4 (A) Pictures of hybrid chitosan hydrogels before (20 °C) and after (37 °C) gelation. (B) Liposome release through the polymeric matrix by using a hyperthermia approach. (C) Cumulative Dox release profiles from the polymeric matrix with and without a 1 h pulse at 42 °C. Adapted with permission from ref. 82. Copyright © 2014 Wiley-VCH. | ||
Besides using chitosan hydrogels for the release studies of low molecular weight molecules, the release of macrobiomolecules has also been studied. In this context, ovalbumin (OVA) and the immunopotentiator Quil A (QA) were entrapped into both cationic nanosized liposomes and cubosomes, a more stable lipid vesicle system.83 A nice sustained release of the model antigen (OVA) was observed in vivo when liposomes were entrapped into chitosan hydrogels. Although OVA-specific antibodies were detected, the system evidenced certain instability even for cubosomes. Unfortunately, OVA and QA in solution forms exhibited similar immunogenicity responses to liposomes when the same thermosensitive chitosan hydrogels were used.
Chitosan-based hydrogels containing liposomes have been further modified by adding additional biopolymers in order to generate novel bio-systems capable of both protecting liposomal formulations and obtaining prolonged, controlled release kinetics. For example, Peptu and co-workers84 prepared hydrogels made of a mixture of chitosan and gelatin. Both biopolymers were crosslinked with glutaraldehyde and sodium/sulphate tripolyphosphate. A hydrophilic model drug (calcein) was encapsulated in both small unilamellar vesicles (SUVs) and multilamellar vesicles (MLVs) and finally entrapped within the mixture of the aforementioned hydrogels with variable compositions. In vitro control release experiments showed improvement in the liposomal stability together with prolonged calcein release from several days to weeks. Interestingly, chitosan/gelatin hydrogels containing MLVs led to better sustained and prolonged release kinetics than those obtained with polymeric matrices containing SUVs. Although the calcein release mechanism from the two biopolymer-based hydrogels is complex, the authors suggested the existence of two steps until the drug reaches the hydrogel surface, consisting of liposome diffusion through the matrix followed by destabilization of the vesicles produced before leaving the hydrogel.
2.2. Gelatin
Gelatin is a biodegradable, thermally denatured protein derived from collagen, a natural protein that is present in mammal tissues, tendons and ligaments. The isoelectric point of gelatine at physiological pH can be modified during its extraction to afford either positively charged basic gelatin (classified as gelatin A) by acidic treatment, or negatively charged acidic gelatin (classified as gelatin B) by alkaline treatment. This versatility has allowed the preparation of numerous polyplexes for drug delivery by combination with a variety of charged biomolecules.85–87There are several examples in which gelatin-based hydrogels have been used for controlled release studies. In most cases, gelatin hydrogels have been chemically modified by adding cross-linking agents such as glutaraldehyde84 and genipin,88 among others,89–91 in order to modify the drug release rates. However, the encapsulation of liposomes in gelatin hydrogels has been only scarcely explored. In 2009, Raghavan and co-workers92 described the entrapment of vesicles made of sodium oleate (NaOA) in a gelatin matrix at slightly alkaline pH (8.3). It is well-known that these vesicles have the property to self-assemble into spherical micelles in solution at a pH higher than 10 (Fig. 5A and B).93 Interestingly, this pH-responsive property was observed when these vesicles were loaded into gelatin hydrogels at pH 8.3 and 10. When calcein was embedded into the liposomes, a more gradual release was observed at pH 8.3, compared to the control. As expected, this release was accelerated when the pH solution was raised to pH 10 (Fig. 5C). This rapid release was attributed to vesicle-to-micelle transitions, which reduced the transport resistance in the drug diffusion from the hydrogel to the external release solution.
 | ||
| Fig. 5 (A) Micellar movement through gelatin hydrogels due to diffusion of pH 10 buffer. NaOA vesicles are converted into NaOA micelles at different times. (B) Pictures of gelatin hydrogels, NaO vesicles and gelatin-loaded with NaOA vesicles. (C) Calcein release from pH-responsive hydrogels. The release was more gradual at pH 8.3 than at pH 10. Adapted with permission from ref. 92. Copyright © 2009 American Chemical Society. | ||
2.3. Dextran
Dextran is a neutral, biodegradable, branched polysaccharide made of glucose molecules composed of chains of varying lengths (from 10 to 150 kDa). From a structural point of view, the native straight chain consists of α-1,6-glycosidic linkages between glucose molecules whereas branches begin from α-1,2, α-1,3 and α-1,4 linkages. Dextran-based hydrogels have been fine-tuned either by adding cross-linking agents such as lactic acid,94 diisocyanate,95 polyamines,96 a mixture of biopolymers97 or by free radical polymerization using glycidyl methacrylate.98 These kinds of hydrogels have been explored in a wide variety of biomedical applications such as detection of small molecule drugs99 and non-viral carriers for proteins.100
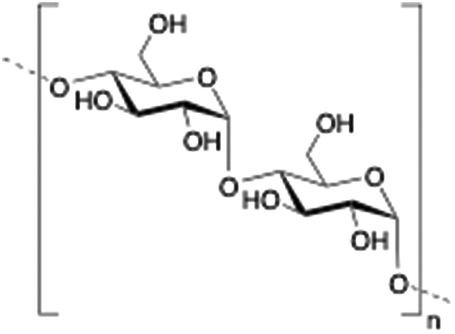
De Smedt and co-workers reported the coating of hydroxyethyl methacrylated dextran (dex-HEMA) nanogels with SOPC/DOTAP liposomes by either using UV polymerization101 or by electrostatic interactions between dextran's charged surface and oppositely charged lipid vesicles.102 DLS and AFM measurements of dex-HEMA nanogels containing lipid particles showed sizes of ca. 350 nm. To confirm the presence of the lipid composition in dex-HEMA nanogels, Triton-X100 (TX100) was added to the dispersions in order to solubilize the lipid coating. This resulted in a mixture of naked dex-HEMA and micelles that were also detected by DLS experiments. Alternatively, removal of the lipid coating in dex-HEMA nanogels was corroborated by AFM measurements. As illustrated in Fig. 6, nanogel particles without liposomes (Fig. 6B) displayed a softer surface than those containing SOPC/DOTAP liposomes (Fig. 6A). DLS measurements revealed a relationship between cross-linking density and stability of the particles. Specifically, highly substituted dex-HEMA nanogels showed better stabilities in PBS buffer at 37 °C, which ranged from few days to 2 weeks. However, poorly substituted dextran hydrogels were unstable and degraded quickly. Finally, transfection studies were successfully carried out and confirmed that dex-HEMA nanogels containing SOPC/DOTAP liposomes were able to impart cellular uptake in VERO-1 cells, showing potential as drug delivery carriers (Fig. 6C).101
 | ||
| Fig. 6 (A) AFM images of SOPC/DOTAP coated dex-HEMA nanogels. (B) AFM images of native dex-HEMA nanogels when TX100 was added to the dispersion displayed in panel (A). (C) Image of VERO-1 cells incubated with lipid coated dex-HEMA nanogels. Adapted with permission from ref. 101. Copyright © 2005 American Chemical Society. | ||
Another interesting application involving covalently cross-linked dextran-polyethyleneglycol (Dex-PEG) hydrogels was developed by Kros and co-workers.103 This method is based on the formation of giant unilamellar vesicles (GUVs) by using Dex-PEG hydrogels which were incorporated on glass surfaces via a Michael addition reaction. Once Dex-PEG polymer was deposited on the surface, several lipid mixture compositions were added following the film-hydration method by using both PBS and HEPES potassium chloride saline buffers in order to study the effect of ionic strength on the vesicle growth. Interestingly, a nice correlation between Dex-PEG cross-linking density and vesicle size distributions was observed. In principle, the possibility of being able to grow and vary the size of the vesicles could make this system a promising tool to be used in numerous applications such as drug delivery and molecular recognition, among others.
2.4. Pullulan
Pullulan is a highly water-soluble biodegradable polysaccharide made of maltotriose repeating units, containing α-(1 → 4) and α-(1 → 6) glycosidic linkages in a 2
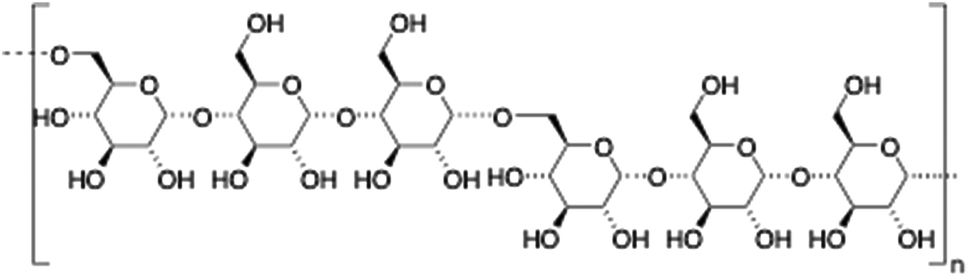 :1 ratio. This polysaccharide has been widely used in coating applications104 and drug delivery.105
:1 ratio. This polysaccharide has been widely used in coating applications104 and drug delivery.105
Akiyoshi and co-workers106 synthesized stable cholesterol-pullulan (CHP) nanogels of ca. 30 nm in size based on specific hydrophobic interactions107 between cholesteryl moieties and pullulan biopolymer in aqueous solutions (Fig. 7).108,109 So formed CHP particles showed also the ability to form stable hydrogels in the presence of liposomes and using PEG as a cross-linker (Fig. 7A). The hydrogel could help us to increase the colloidal stability, thereby avoiding undesirable aggregation processes. The final CHP hybrid systems were characterized in terms of TEM microscopy and particle-tracking micro-rheology which indicated that both the strength and the gelation time could be tuned depending on the CHP:liposome ratio. Specifically, liposomal release from CHP nanogels was monitored by coating the aforementioned CHP with liposomes based on a mixture of dimyristoylphosphatidylcholine (DMPC) and cholesterol at a 3:1 ratio. The authors observed a sequential dual release profile in which CHP nanogel was first liberated followed by the release of nanogel-coated liposome complexes under physiological conditions, which was fully liberated within 40 days. Interestingly, a clear dependence of the release with the pH was observed between pH 7.4 and 8.0 in which the liposome complex was released within 20 days (Fig. 7B).
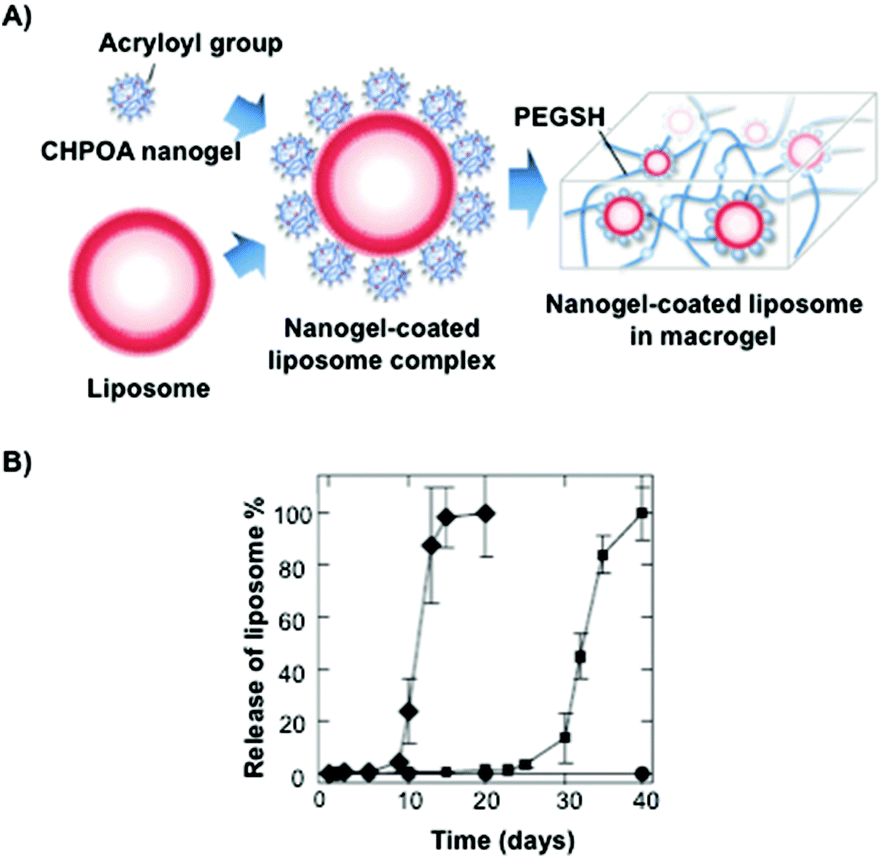 | ||
| Fig. 7 (A) Illustration of a nanogel-coated liposome complex and PEGSH used as a crosslinking agent. (B) Release profiles of the nanogel-coated liposome from the hydrogel at different pH values (pH 7.4 in squares and pH 8.0 in diamonds). Adapted with permission from ref. 106. Copyright © 2012 Wiley-VCH. | ||
2.5. Hyaluronic acid
Hyaluronic acid (HA) is a linear and negatively charged polysaccharide made of disaccharide repeating units of D-glucuronic acid and N-acetyl-D-glucosamine which are linked by β-(1,3) and β-(1,4) glycosidic bonds. HA is involved in several important physiological and biological functions including protein transport110 and regulation of water homeostasis.111 Moreover, HA displays anti-inflammatory properties112 and has been widely used as surgical aid in wound healing.113 HA has the ability to form robust hydrogel networks and this property has also been exploited, for instance, as a drug delivery system in combination with other biopolymers such as hydroxypropyl methylcellulose (HPMC), polyethylenimine (PEI) or liposomes.114–117
Several articles have also described the use of liposomes in combination with HA hydrogels (Fig. 8) for different biomedical applications. For example, the effect promoted by a series of liposomes that depended on their net charge (i.e., neutral, positive or negatively charged), size, lipid concentration and lipid composition was thoroughly studied in HA hydrogels by Agnely and co-workers.115 The highest viscosity and elasticity in HA hydrogels were observed when liposomes were covered with polyethylene glycol chains. This induced fast immobilization and a long residence time after injection. Interestingly, despite this viscosity detected in these kinds of hydrogels, all formulations were easily injectable by local administration due to the shear-thinning behavior of the formulation.
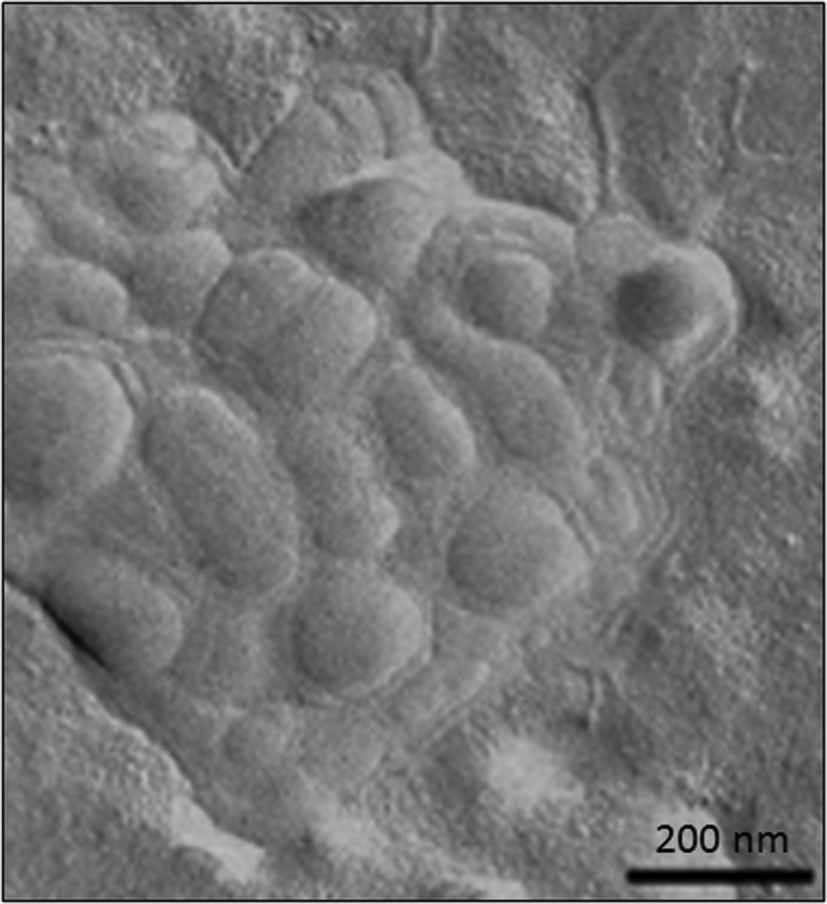 | ||
| Fig. 8 TEM micrograph of freeze-fractured hyaluronic acid hydrogels containing liposomes. Adapted with permission from ref. 115. Copyright © 2015 Elsevier. | ||
The combination between the HA polymer matrix and liposomes has also contributed to the advance of some disease treatments. For example, Miao and Huang118 improved osteoarthritis (OA) therapy by combining celecoxib (Clx), an analgesic and anti-inflammatory drug, with a mixture of liposomes (SPC and Chol) that were embedded in HA hydrogels. This formulation showed high encapsulation efficiencies and reduced the adverse effects induced by using Clx alone, such as gastrointestinal toxicities and high risk for cardiovascular events at high doses. In vitro experiments confirmed the delay in the liposomal Clx release when liposomes were part of the HA structural framework. These preliminary results were also confirmed in vivo showing better effectiveness of the liposomal formulation than current Clx-treatments in both pain control and cartilage protection.
HA is also a natural component of eye tissue, making it ideal as a carrier for ocular drug delivery. In this area, Bochot and co-workers119 developed a novel formulation consisting of liposomes embedded in HA hydrogels for human uveitis, an intraocular inflammatory disease of the uvea causing loss of vision. In particular, the authors demonstrated that embedding a vasoactive intestinal peptide (VIP), an immunosuppressive agent, within a mixture of liposomes (ratio PG:Chol:PEG:DSPE = 50:10:35:5) remarkably reduced ocular inflammation signs in rats. Although the treatment with VIP loaded in liposomes resulted to be less invasive than surgical implantation, the liposomal formulation leakage could reduce the effectiveness for long-term treatments. However, sustained drug release was clearly improved when liposomal VIP was incorporated into HA hydrogels. This effect was observed in both in vitro and in vivo experiments with the naked eye, thereby became a promising strategy for local delivery.
In 2013, Venkatraman and co-workers120 studied the liposomal release of latanoprost (Ltp), a drug used to reduce the ocular pressure, by using two HA-based hydrogels. Before loading the selected liposomes (EPC) in HA hydrogels, the biopolymer was first cross-linked with adipic dihydrazide (ADH) and methacrylic anhydride (MA). The mechanical properties of both systems were controlled by varying the cross-linking degree, which clearly affected the rheological and swelling properties of the hybrid systems. Remarkably, sustained release of liposomal Ltp was accomplished over 90 days with a single administration.
In the area of tissue engineering, Ying and co-workers121 have recently developed room temperature-liquid injectable HA hydrogels based on a HA–tyramine (Tyr) conjugate containing horseradish peroxidase (HRP). HRP was embedded in a mixture of two thermoresponsive liposomes (i.e., dipalmitoylphosphatidylcholine (DPPC) and dimyristoylphosphatidyl choline (DMPC)). Cholesterol was also introduced into the formulation in order to modify the fluidity and reduce the undesirable HRP release at room temperature. The gelation kinetic of the hybrid was found to depend on several factors including lipid content, HA–Tyr concentration, sonication time and composition of the liposome washing solution. Injectability studies were carried out subcutaneously in a mouse model. While the HA–Tyr/H2O2/HRP hybrid system remained as a liquid at room temperature, the HRP release upon injection promoted a reaction between HA–Tyr and H2O2, inducing the formation of the cross-linked hydrogel scaffold.
2.6. Alginate
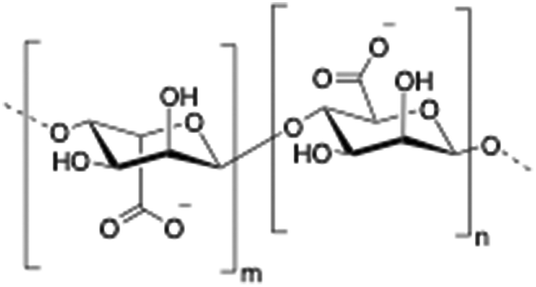
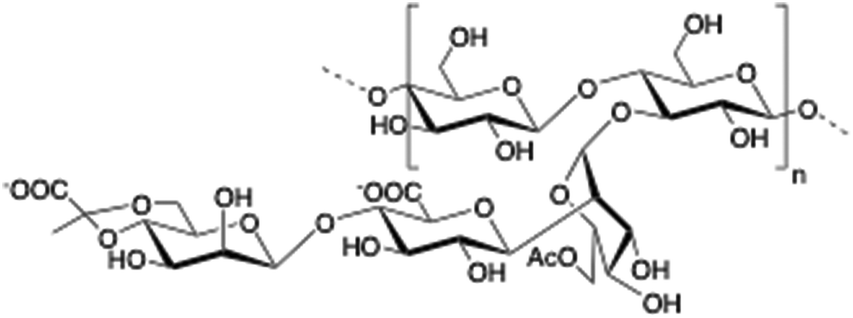
Alginate (salt of alginic acid) is an anionic, high molecular weight (500–2000 kDa) polysaccharide comprised of β-D-mannuronate (M) and its C5-epimer α-L-glucuronate (G) residues, which are linked by β-1,4 glycosidic bonds. Alginate has the ability to form hydrogel networks in the presence of divalent cations and other cross-linking agents.122 A wide number of biomedical applications including wound healing, drug delivery or tissue engineering have been extensively reported.123
In 1995, Rudolph and Monshipouri reported the first example in which liposomes were encapsulated within an alginate hydrogel.124 The authors used dipalmitoyl phosphatidylcholine liposomes as reaction sites for alginate hydrogelation. As a model compound, cytochrome-c was embedded in these large unilamellar liposomes and the release of liposome-encapsulated alginate and calcium alginate beads was evaluated. The authors found rapid drug release for both formulations followed by a slower release rate for alginate beads containing encapsulated liposomes. Around the same time, Yotsuyanagi and co-workers125 described the factors affecting the loading and release of drug-containing liposomes in alginate beads. The results obtained using 5(6)-carboxyfluorescein (CF) as a model drug in liposomes of egg phosphatidylcholine (EPC) and EPC/cholesterol (EPC/Cho) demonstrated that alginate interacts with the liposomes causing a significant change of permeability. Moreover, the presence of calcium ions seemed to induce a higher drug-leakage than sodium ions. Interestingly, a faster liposome release was observed when using washed gel beads instead of fully-cured beads, showing a proportional relationship between the release and the erosion of the gel at high liposome concentrations.126 These results suggested that the liposomes were not homogeneous distributed through the gel beads but rather concentrated in the central part. Such dependence between drug release and the erosion of the alginate gel matrix was also observed by Rongqing and co-workers.127 This group performed in vitro and in vivo studies on the release of liposomes of L-α-phosphatidylcholine containing bee venom peptide, which were encapsulated into calcium alginate gel beads coated with Eudragit S100 (methacrylic acid copolymer B). The results demonstrated the potential of this formulation for colonic drug delivery.
In 2006, Wang and co-workers described128 the use of alginate hydrogel microcapsules for the delivery of bovine serum albumin (BSA) previously encapsulated in multivesicular liposomes (MVLs) via a double emulsification process (water-in-oil-in-water). In vitro release experiments indicated gradual liberation of the BSA protein over a period of approximately 2 weeks without detecting any burst release effect.
In 2010, Smith and co-workers reported the preparation of alginate-loaded liposomes as a vehicle for the oral delivery of bioactive proteins (i.e., alkaline phosphatase (ALP) as model).129 The vesicles were prepared from the phospholipid dipalmitoyl phosphatidylcholine using a simple dry film hydration technique. On the other hand, the incorporation of alginate in the liposomes enhanced their stability at gastric pH.
More recently, Štěpánek's group130 described the preparation of composite microparticles consisting of a calcium alginate gel matrix with embedded liposomes made from cholesterol:DPPC (dipalmitoylphosphatidylcholine) containing 5(6)-carboxyfluorescein. The authors demonstrated the “on-demand” thermo-responsive release from liposomes when embedded within the alginate hydrogel particles. The sensitivity of the thermo-responsive system was found to depend on the DPPC:cholesterol ratio and it was closely related to the phase behavior of the lipid bilayer that formed the liposome.
The same group131 also prepared hydrogel microcapsules (61 μm) containing fluorescent labelled liposomes having yeast cells that worked as a biological trigger for controlled opening of the microcapsules and consequent liberation of sub-micrometer objects (dye). A similar formulation containing iron-oxide nanoparticles allowed the controlled release of the dye by means of an alternating magnetic field (AMF) (Fig. 9).132 A temperature increase above the phase transition of the phospholipid bilayer was a necessary condition for the functionality of the system. In spite of high SAR values of the nanoparticles used, the rate of heat loss by conduction from an individual microgel bead was too high to allow any significant temperature rise to take place. The required temperature rise could only be achieved in cases where the beads were more concentrated or accumulated in a small volume.
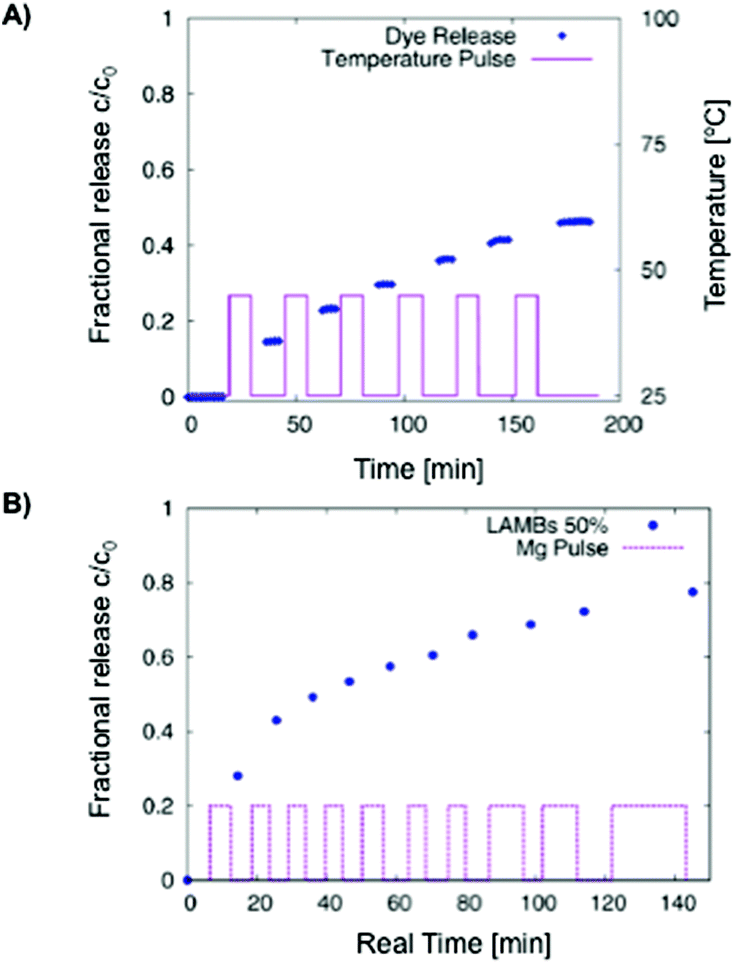 | ||
| Fig. 9 (A) Carboxyfluorescein diffusion from LAMBs during an on–off temperature program. DPPC:Chol ratio of 2:1. (B) AMF-induced release of carboxyfluorescein from LAMBs in water at 50% dilution. Adapted with permission from ref. 132. Copyright © 2013 The American Chemical Society. | ||
Kim and co-workers133 reported the preparation of Janus-compartmental alginate microbeads having two divided phases of sensory polydiacetylene (PDA) liposomes and magnetic nanoparticles. The sensory liposomes were composed of PDA for label-free signal generation and 1,2-dipalmitoyl-sn-glycero-3-galloyl (DPGG) lipids whose galloyl headgroup has specific interactions with lead(II) forming phenolic–metal complexes, sensing this metal at concentrations as low as 0.1 mM (∼20.7 ppm). In addition, about 45% of lead(II) ions could be absorbed by hundred Janus microbeads in 4 h mainly due to the interactions with the carboxylic acids of the alginate matrix (they are around 200 times more present than those of the lipids). The results show the recognition of lead(II) at the PDA liposome surface by DPGG induced distortion of the conjugated yne–ene main chain of PDA, causing a color change from blue to red as well as the development of red fluorescence (Fig. 10). These microbeads have the additional advantage of easy manipulation and convenient collection by applying an external magnetic field.
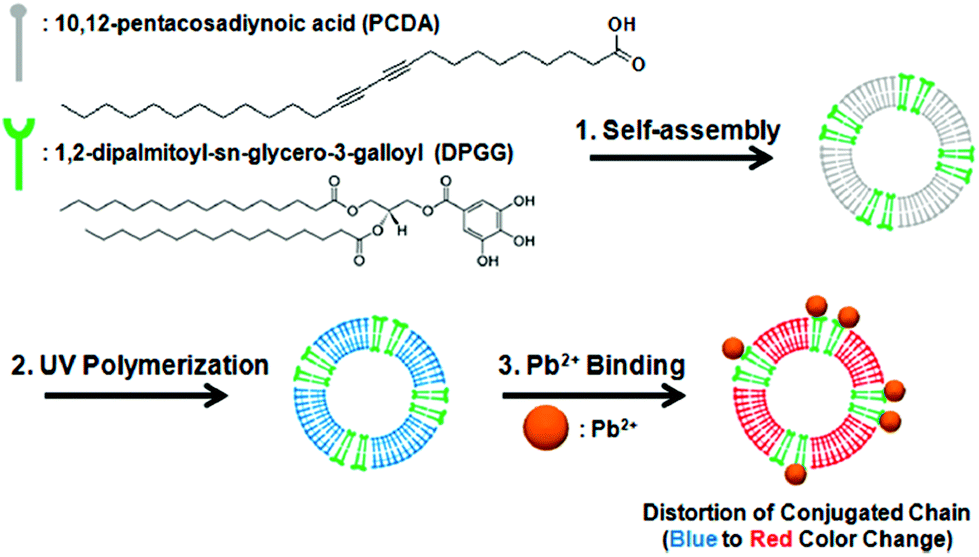 | ||
| Fig. 10 Co-assembly of PCDA with DPGG to form PDA-DPGG liposomes and a schematic illustration of the colorimetric lead(II) detection. Reprinted with permission from ref. 133. Copyright © 2014 The American Chemical Society. | ||
The release study of a recombinant hepatitis B surface antigen (HBsAg) was investigated by Cohen and co-workers.134 HBsAg was first encapsulated in liposomes made of a mixture of phosphatidylcholine and cholesterol at a 1:1 molar ratio. This carrier was successfully used as an adjuvant for BSA. Liposomes containing HBsAg antigen were confirmed by cryo-TEM microscopy and were entrapped into alginate-poly-(L-lysine) (PLL) hydrogel polymeric microspheres. Both in vitro and in vivo experiments showed clear dependency of the liposome release rate on the PLL molecular weight (i.e., liposomes coated with PLL of 214 kDa produced higher liposomal release than PLL of 25 kDa). These findings suggest the efficiency of these hybrids as potential immunoadjuvants because they reduce the amount of antigens and, hence, may eliminate the number of shots necessary for optimal vaccinations.
Oral delivery is another application in which alginate hydrogels have shown to be efficient and robust systems even in the presence of acid environments located in gastrointestinal tracts. This robustness has been recently reported by Yuasa and co-workers.135 These authors developed a liposomal formulation made of DMPC, cholesterol and non-ionic surfactant tween-20 at a 288:72:40 molar ratio, respectively, to entrap manganese porphyrin (Mn-por), a superoxide dismutase mimic that maintains the appropriate reactive oxygen species (ROS) levels in cells (Fig. 11A). In order to protect the drug from the gastrointestinal tract acid environment, the liposomal formulation was embedded in alginate hydrogel fibers obtaining hybrids of ca. 100 μm in diameter. Although encapsulation efficiencies of Mn-por in liposomes were high (71%), this efficiency diminished dramatically when liposome formulation was embedded in the alginate matrix (3%). In vitro experiments confirmed that both alginate hydrogels and liposomes were able to maintain their physical properties under acidic pH, allowing the delivery of Mn-por to the intestine retaining the O2˙− inhibitory activity. The efficacy of this system was also evaluated in vivo by monitoring the tumor size in mice (Fig. 11B). Interestingly, there was an inhibition of the tumor growth when alginate fibers containing liposomes were orally administered.
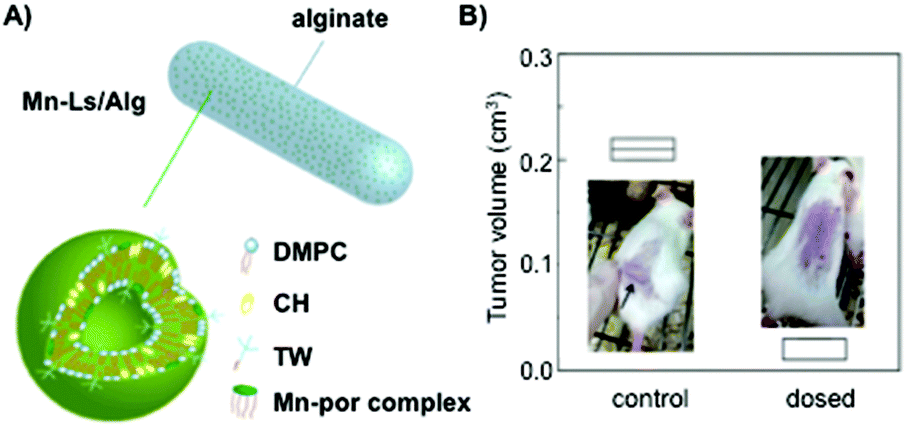 | ||
| Fig. 11 (A) Mn-Ls/Alg represents an alginate hydrogel matrix containing liposomes with embedded manganese porphyrin (Mn-Ls). (B) In vivo anti-tumor efficacy when Mn-Ls/Alg was administered orally. Comparison of the tumor volume in mice transplanted with Colon-26-cells. Adapted with permission from ref. 135. Copyright © 2015 The Royal Society of Chemistry. | ||
Very recently Deckers and co-workers136 have described the preparation of barium crosslinked alginate microspheres loaded with temperature sensitive liposomes (TSL/TSL-Ba-ms) for embolization of blood vessels of tumors under mild hyperthermia conditions. TSLs contained anti-cancer doxorubicin and [Gd(HPDO3A)(H2O)] as contrast agents, which were combined with holmium-containing microspheres (Ho-ms) as a tracer agent (Fig. 12). In vivo monitoring was carried out in the auricle VX2 tumor of a rabbit after intratumoral injection of a dispersion containing TSL-Ba-ms as well as Ho-ms. The results showed that [Gd(HPDO3A)(H2O)] was only released after applying mild hyperthermia (42 °C), indicating that the TSLs are stable at 37 °C and do not leak their loading.
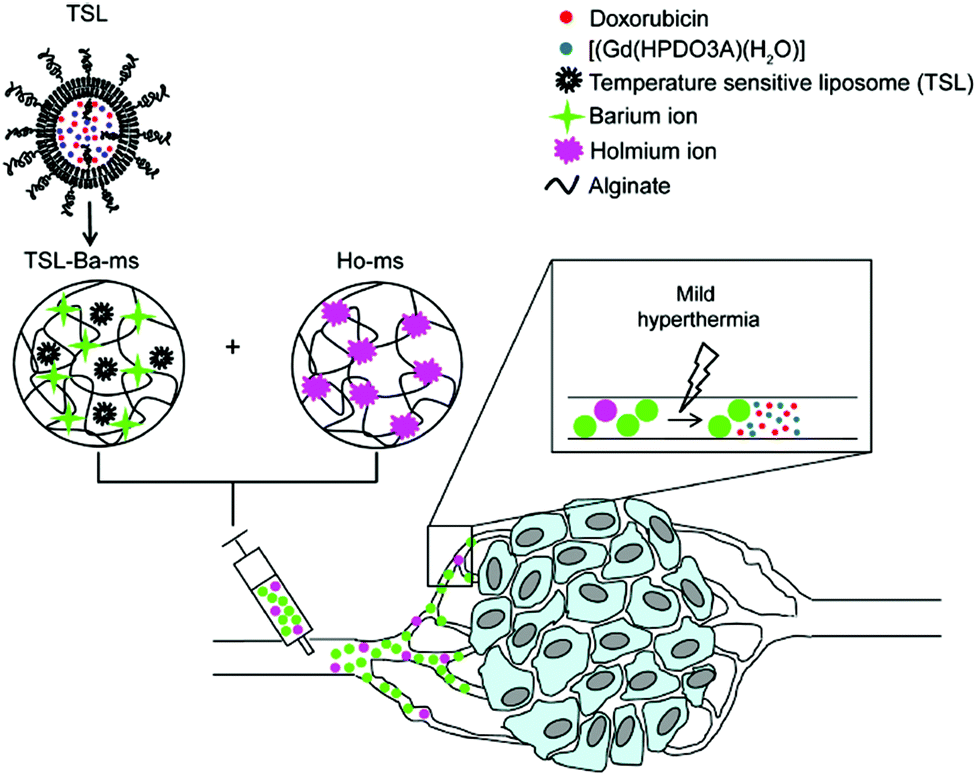 | ||
Fig. 12 Schematic representation of temperature sensitive liposomes (TSL) loaded in alginate microspheres crosslinked with barium ions (TSL Bams). The TSLs are loaded with doxorubicin (DOX) and [Gd(HPDO3A)(H2O)] (T1 MRI contrast agent). The DOX and [Gd(HPDO3A)(H2O)] are released from the TSL-Ba-ms during mild hyperthermia. The release of [Gd(HPDO3A)(H2O)] can be monitored by MRI. Empty alginate microspheres crosslinked with holmium ions (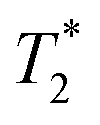 MRI contrast agent, Ho-ms) are co-injected with TSL-Ba-ms to allow microsphere visualization by MRI. Reprinted with permission from ref. 136. Copyright © 2015 Public Library of Science. MRI contrast agent, Ho-ms) are co-injected with TSL-Ba-ms to allow microsphere visualization by MRI. Reprinted with permission from ref. 136. Copyright © 2015 Public Library of Science. | ||
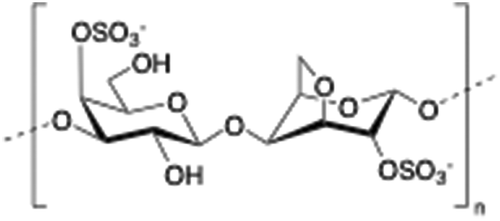
2.7. Carrageenan
Carrageenan is a family of linear sulphated polysaccharides made up of galactose repeating units and 3,6-anhydrogalactose (3,6-AG) connected by α-1,3 and β-1,4 glycosidic linkages. Some of these flexible macromolecules such as kappa (κ) and iota (τ) carrageenans can adopt helical structures and promote the formation of hydrogels in the presence of cations. Besides its wide use in food industry as a thickening agent,137 a good number of biomedical applications138 have also been described involving carrageenan alone or in combination with additional biopolymers like chitosan,139 alginate140 or gelatin.141 However, the incorporation of liposomes into carrageenan hydrogels has been scarcely explored to date.
Recently, Kulkarni and co-workers142 have described the combination of lipid vesicles and κ-carrageenan hydrogels for sustained drug release applications. The strategy involved the loading of a therapeutic drug in a liposomal formulation and subsequent homogenization within the κ-carrageenan hydrogel. The final hybrid system was dehydrated to afford hybrid drug-loaded films (Fig. 13A), which were fully characterized by Fourier transform infrared (FITR) spectroscopy and small-angle X-ray scattering (SAXS). The release kinetics from both native and lipid particles in hydrogel films were investigated and monitored by UV-Vis spectroscopy. The results showed that the drug encapsulation within the hydrogel allowed for more efficient protection of the drug and sustained release in comparison to the native hydrogel in the absence of liposomes (Fig. 13B).
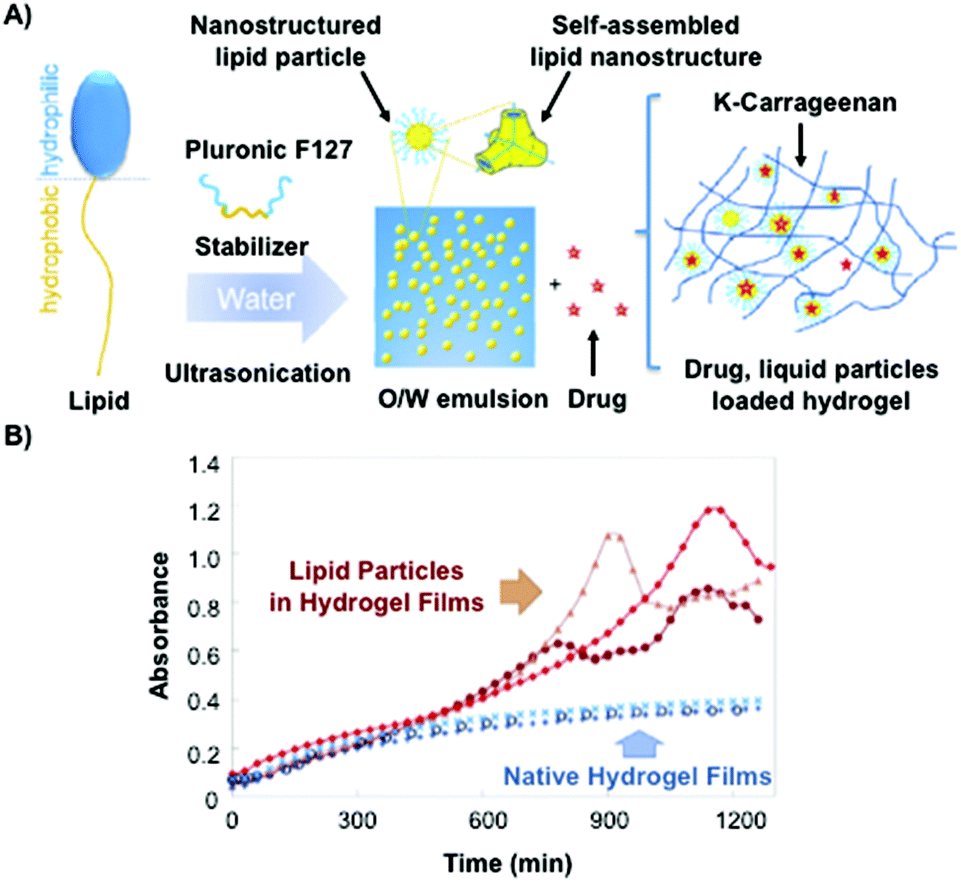 | ||
| Fig. 13 (A) Lipid molecule made of a hydrophobic and a hydrophilic part is stabilized by using Pluronic F127 upon ultrasonication. This mixture is loaded containing a drug within the κ-carrageenan hydrogel. (B) Release kinetic of the drug was monitored by UV-vis spectroscopy. Drug release from native hydrogel films (blue) and lipid particles within hydrogel films (red). Adapted with permission from ref. 142. Copyright © 2015 Elsevier. | ||
In 2013, El-Menshawe and co-workers143 developed an alternative to oral delivery systems by using topical administration to improve anti-inflammatory activities and reduce undesirable side effects. Specifically, the authors described the encapsulation of Meloxicam (MX), a non-steroidal anti-inflammatory drug, in niosomes based on a non-ionic surfactant molecule and cholesterol at a 6:4 molar ratio. The corresponding niosomal formulation was entrapped into two hydrogels: carrageenan and carboxymethyl-cellulose (CMC) hydrogels. The results of the anti-inflammatory activities in vivo clearly showed a decrease in the permeability when increasing the cholesterol molar ratio. Consequently, this enhanced the bioavailability of MX and, thereby, provided better skin permeation.
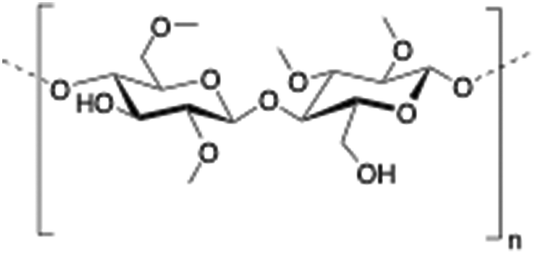
2.8. Methylcellulose
Methylcellulose (MC) is a neutral derivative of cellulose, the most abundant natural polysaccharide composed of (1 → 4) linked β-D-glucopyranosyl units. In MC, methoxy groups substitute the hydroxyls at the C-2, C-3 and/or C-6 positions of anhydro-D-glucose units. MC is able to form hydrogels under isothermal conditions when it is dissolved in hot water,144 being commonly used in food industry as a thickener and an emulsifier as well as in several biomedical applications.145,146
Within the context of this review, Kapadia and co-workers147 developed a novel approach to deliver acyclovir by using niosomes that were entrapped into a mixture of MC and carbopol. The final goal was to improve the poor bioavailability exhibited by conventional ophthalmic solutions for treatment of herpes simplex keratitis. Thus, acyclovir was encapsulated in two niosomes based on two non-ionic surfactants (span 20 and span 60) and cholesterol at different molar ratios. The results showed that the drug encapsulation efficiency was higher in the case of span 60. In vitro release experiments indicated the suitability of the MC hydrogel as an efficient vehicle to deliver the drug in a controlled manner to the site of action due to the presence of bioadhesive polymers in the formulation. Moreover, the use of MC hydrogels containing niosomes helped us to reduce the toxicity that can be observed in conventional therapies.
2.9. Xanthan gum
Xanthan gum (XG) is a polysaccharide composed of pentasaccharide repeating units of glucose, mannose and glucuronic acid in a 2
 :2:1 molar ratio. Besides its applications in food industry as a stabilizer and thickening agent, XG has also been used in some biomedical applications148 as a drug release modifier due to its pH-responsive properties.
:2:1 molar ratio. Besides its applications in food industry as a stabilizer and thickening agent, XG has also been used in some biomedical applications148 as a drug release modifier due to its pH-responsive properties.
The mechanical properties of XG-based hydrogels can be modulated upon blending with other biopolymers such as locust bean gum (LBG) in a weight ratio of 1:1.149 The polymeric system LBG/XG was described by Carafa and co-workers150 as a convenient drug delivery system for ibuprofen and caffeine as model drugs. Firstly, these drugs were encapsulated in non-ionic surfactant niosomes based on Tween-20 and cholesterol at a 1:1 molar ratio. As expected, a slight increase in size was observed when model drugs were incorporated into niosomes. However, no significant change in the surface charge was detected. Interestingly, a different behaviour in the in vitro release was observed from both the niosome-hydrogel matrix and the LBG/XG hydrogel without containing vesicles. Consequently, caffeine, the more hydrophilic drug, was delivered completely and much faster than ibuprofen, which was capable to diffuse only 60% after 48 h. These findings revealed the suitability of niosomes and LBG/XG hydrogels to be used in topical applications due to the slow diffusion of the drugs from the matrix and the protection of the noisome by the hydrogel.
3. Summary and outlook
Biohydrogels (biopolymer-based hydrogels) have found numerous uses across a wide range of food, pharmaceutical and biomedical industries, where their inherent properties of biocompatibility and biodegradability play a key role. In the area of drug release applications, many therapeutic molecules have been successfully entrapped into different biohydrogel carriers (Table 2). However, one of the major concerns is the rapid and uncontrolled release of the drug from the polymer network (burst release), which may induce certain toxicity and undesirable side effects in biological systems. Over the last few years, several strategies have been described to minimize the burst release effect including, among others, the use of liposomes. In this particular approach, and before obtaining the corresponding drug-in-liposome system, a number of parameters need to be optimized such as lipid composition, particle size, morphology and surface charge in order to efficiently encapsulate hydrophilic and/or hydrophobic molecules. Remarkably, the protecting effect provided by liposomes to drugs can be further enhanced through their encapsulation into hydrogel networks. In addition, the possibility of tuning and obtaining either liposomes and/or hydrogels that can respond to environmental stimuli (e.g., light, temperature, pH) makes them ideal candidates for the fabrication of novel drug delivery systems. Several kinds of biopolymer-based hydrogels containing liposomes discussed in this review have proven their efficacy in controlled drug delivery and tissue engineering applications.| Entry | Biopolymer | Chemical structure | Ref. |
|---|---|---|---|
| 1 | Chitosan |
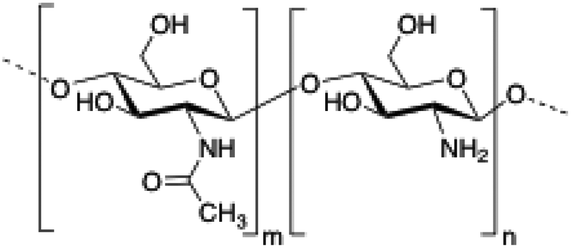
|
70 and 74–84 |
| 2 | Gelatin | Mixture of peptides and proteins produced by partial hydrolysis of collagen | 85–93 |
| 3 | Dextran |
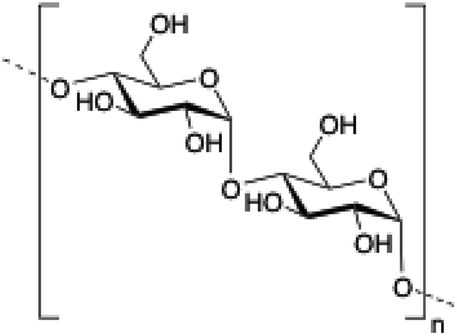
|
101–103 |
| 4 | Pullulan |
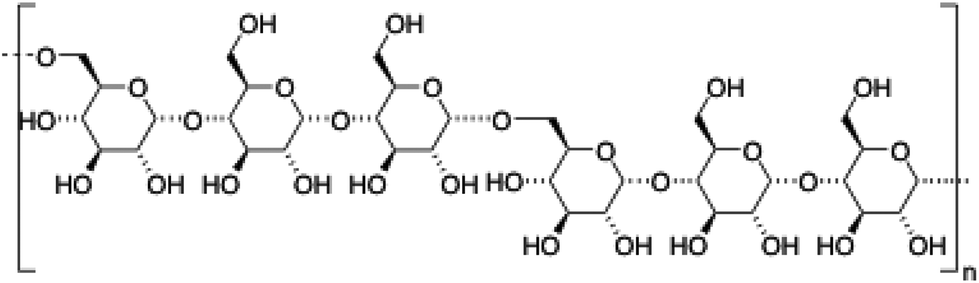
|
106–109 |
| 5 | Hyaluronic acid |
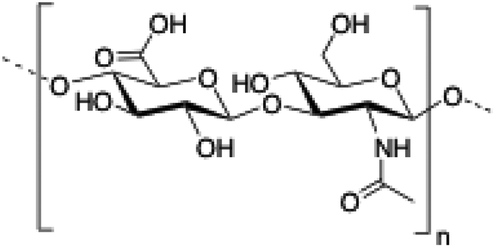
|
115–121 |
| 6 | Alginate |
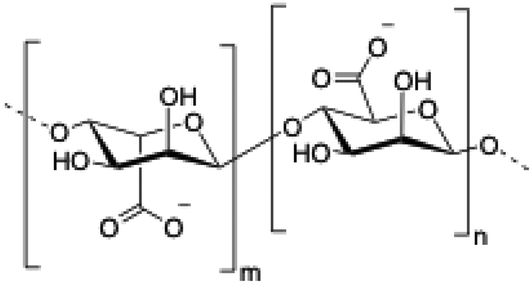
|
124–136 |
| 7 | Carrageenan |
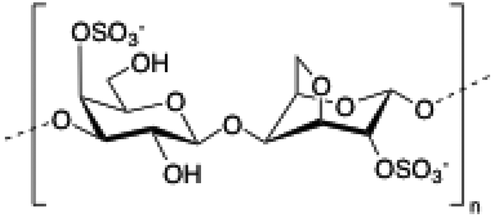
|
142–143 |
| 8 | Methylcellulose |
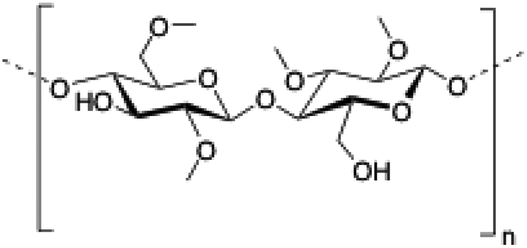
|
147 |
| 9 | Xanthan gum |
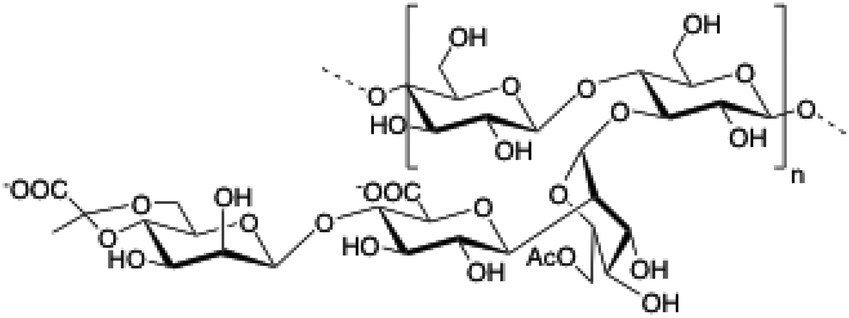
|
149 and 150 |
These promising results may guide future development of advanced mathematical models of controlled and burst release profiles, providing a platform for the design of more efficient liposomal formulations for biomedical applications.
Acknowledgements
This work is supported by the University of Regensburg, the DFG (DI 1748/3-1), the Spanish Ministry of Education (Grant CTQ2014-52588-R, RTC-2014-2038-1), the Generalitat de Catalunya (2014/SGR/624), the Instituto de Salud Carlos III (CB06_01_0019) and the Spanish Ministry of Education, Culture and Sports. S.G. thanks the Spanish Ministry of Education, Culture and Sports for a “Jose Castillejo” grant within the researchers mobility program (Programa Estatal de Promoción del Talento y su Empleabilidad en I + D + i, Subprograma Estatal de Movilidad, del Plan Estatal de Investigación Científica y Técnica y de Innovación 2013–2016). D. D. D. thanks the Deutsche Forschungsgemeinschaft (DFG) for the Heisenberg Professorship Award.Notes and references
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Thermally responsive hydrogel blends. A general drug carrier model for controlled drug release, Angew. Chem., Int. Ed., 2015, 54(25), 7376–7380 CrossRef CAS PubMed.
- N. K. Singh, Q. V. Nguyen, B. S. Kim and D. S. Lee, Nanostructure controlled sustained delivery of human growth hormone using injectable, biodegradable, Ph/temperature responsive nanobiohybrid hydrogel, Nanoscale, 2014, 7, 3043–3054 RSC.
- A. Klymenko, T. Nicolai, L. Benyahia, C. Chassenieux, O. Colombani and E. Nicol, Multiresponsive hydrogels formed by interpenetrated self-assembled polymer networks, Macromolecules, 2014, 47(23), 8386–8393 CrossRef CAS.
- M. Joglekar and B. G. Trewyn, Polymer-based stimuli-responsive nanosystems for biomedical applications, Biotechnol. J., 2013, 8(8), 931–945 CrossRef CAS PubMed.
- C. Álvarez-Lorenzo and A. Concheiro, Smart drug delivery systems: From fundamentals to the clinic, Chem. Commun., 2014, 50(58), 7743–7765 RSC.
- S. J. Buwalda, K. W. M. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennik, Hydrogels in a historical perspective: From simple networks to smart materials, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS PubMed.
- R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, Advances in polymeric systems for tissue engineering and biomedical applications, Macromol. Biosci., 2012, 12(3), 286–311 CrossRef CAS PubMed.
- M. S. Desai and S.-W. Lee, Protein-based functional nanomaterial design for bioengineering applications, WIREs Nanomed. Nanobiotechnol., 2015, 7(1), 69–97 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Hydrogels for protein delivery, Chem. Rev., 2012, 112(5), 2853–2888 CrossRef CAS PubMed.
- J. V. John, R. P. Johnson, M. S. Heo, B. K. Moon, S. J. Byeon and I. Kim, Polymer-block-polypeptides and polymer-conjugated hybrid materials as stimuli-responsive nanocarriers for biomedical applications, J. Biomed. Nanotechnol., 2015, 11(1), 1–39 CrossRef CAS PubMed.
- R. J. Gübeli, M. Ehrbar, M. Fussenegger, C. Friedrich and W. Weber, Synthesis and characterization of PEG-based drug responsive biohybrid hydrogels, Macromol. Rapid Commun., 2012, 33(15), 1280–1285 CrossRef PubMed.
- A. Kutikov and J. Song, Biodegradable PEG-based amphiphilic block copolymers for tissue engineering applications, ACS Biomater. Sci. Eng., 2015, 1(7), 463–480 CrossRef CAS.
- E. Bouyer, G. Mekhloufi, V. Rosilio, J.-L. Grossiord and F. Agnely, Proteins, polysaccharides and their complexes used as stabilizers for emulsions: Alternatives to synthetic surfactants in the pharmaceutical field?, Int. J. Pharm., 2012, 436(1–2), 359–378 CrossRef CAS PubMed.
- D. Singh, H. Deepti, S. Sung and E. J. Shin, Polysaccharides as nanocarriers for therapeutic applications, J. Biomed. Nanotechnol., 2014, 10(9), 2149–2172 CrossRef CAS PubMed.
- L. S. Liu, J. Kost, F. Yang and R. C. Spiro, Hydrogels from biopolymer hybrid for biomedical, food and functional food applications, Polymers, 2012, 4(2), 997–1011 CrossRef.
- S. K. Nitta and K. Numata, Biopolymer-based nanoparticles for drug/gene delivery and tissue engineering, Int. J. Mol. Sci., 2013, 14(1), 1629–1654 CrossRef CAS PubMed.
- J. K. Oh, D. I. Lee and J. M. Park, Biopolymer-based microgels/nanogels for drug delivery applications, Prog. Polym. Sci., 2009, 34(12), 1261–1282 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Hydrogels in biology and medicine: From molecular principles to bionanotechnology, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Hydrogels in pharmaceutical formulations, Eur. J. Pharm. Biopharm., 2000, 50(1), 27–46 CrossRef CAS PubMed.
- E. Caló and V. V. Khutoryanskiy, Biomedical applications of hydrogels: A review of patents and commercial products, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- O. Wichterle and D. Lim, Hydrophilic gels for biological use, Nature, 1960, 185, 117–118 CrossRef.
- Q. An, C. Beh and H. Xiao, Preparation and characterization of thermo-sensitive poly(vinyl alcohol)-based hydrogel as drug carrier, J. Appl. Polym. Sci., 2014, 131(1), 39720–39729 Search PubMed.
- L. F. Gudeman and N. A. Peppas, Preparation and characterization of pH-sensitive, interpenetrating networks of poly(vinyl alcohol) and poly(acrylic acid), J. Appl. Polym. Sci., 1995, 55(6), 919–928 CrossRef CAS.
- H. N. Yang, J. S. Park, S. Y. Jeon and K.-H. Park, Carboxymethylcellulose (CMC) formed nanogels with branched poly(ethyleneimine) (bPEI) for inhibition of cytotoxicity in human MSCs as a gene delivery vehicles, Carbohydr. Chem., 2015, 122, 265–275 CrossRef CAS PubMed.
- C.-C. Lin, Recent advances in crosslinking chemistry of biomimetic poly(ethylene glycol) hydrogels, RSC Adv., 2015, 5(50), 39844–39853 RSC.
- S. J. Bidarra, C. C. Barrias and P. L. Granja, Injectable alginate hydrogels for cell delivery in tissue engineering, Acta Biomater., 2014, 10(4), 1646–1662 CrossRef CAS PubMed.
- S. K. Shukla, A. K. Mishra, O. A. Arotiba and B. B. Mamba, Chitosan-based nanomaterials: a state-of-the-art review, Int. J. Biol. Macromol., 2013, 59, 46–58 CrossRef CAS PubMed.
- F. Wu and T. Jin, Polymer-based sustained-release dosage forms for protein drugs, challenges and recent advances, AAPS PharmSciTech, 2008, 9(4), 1218–1229 CrossRef CAS PubMed.
- S. D. Allison, Analysis of initial burst in PGLA microparticles, Expert Opin. Drug Delivery, 2008, 5(6), 615–628 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, On the importance and mechanisms of burst release in matrix-controlled drug delivery systems, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- A. W. Martínez, J. M. Caves, L. W. Swathi and E. L. Chaikof, Effects of crosslinking on the mechanical properties, drug release and cytocompatibility of protein polymers, Acta Biomater., 2014, 10(1), 26–33 CrossRef PubMed.
- J. Sankaranarayanan, A. E. Mahmoud, G. Kim, J. M. Morachis and A. Almutairi, Multiresponse strategies to modulate burst degradation and release from nanoparticles, ACS Nano, 2010, 4(10), 5930–5936 CrossRef CAS PubMed.
- Z. Xiang, P. Sarazin and B. D. Favis, Controlling burst and final release times from porous polylactide devices derived from co-continuous polymer blends, Biomacromolecules, 2009, 10(8), 2053–2066 CrossRef CAS PubMed.
- E. M. Ahmed, Hydrogel: Preparation, characterization and applications: A review, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- C. C. Lin and A. T. Metters, Hydrogels in controlled release formulations: Network design and mathematical modelling, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed.
- N. A. Peppas, K. B. Keys, M. Torres-Lugo and A. M. Lowman, Poly(ethylene glycol)-containing hydrogels in drug delivery, J. Controlled Release, 1999, 62, 81–87 CrossRef CAS PubMed.
- R. Gauvin, R. Parenteau-Bareil, M. R. Dokmeci, W. D. Merryman and A. Khademhosseini, Hydrogels and microtechnologies for engineering the cellular microenvironment, WIREs Nanomed. Nanobiotechnol., 2012, 4, 235–246 CrossRef CAS PubMed.
- T. Tanaka and D. J. Fillmore, Kinetics of swelling of gels, J. Chem. Phys., 1979, 70, 1214–1218 CrossRef CAS.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- Y. Wen and J. K. Oh, Recent strategies to develop polysaccharide-based nanomaterials for biomedical applications, Macromol. Rapid Commun., 2014, 35, 1819–1832 CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, The development of microgels/nanogels for drug delivery applications, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- B. S. Pattni, V. V. Chupin and V. P. V. P. Torchilin, New developments in liposomal drug delivery, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- G. Bozzuto and A. Molinari, Liposomes as nanomedical devices, Int. J. Nanomed., 2015, 10, 975–999 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Liposomal drug delivery systems: from concept to clinical applications, Adv. Drug Delivery, 2013, 65(1), 36–48 CrossRef CAS PubMed.
- F. Movahedi, R. G. Hu, D. L. Becker and C. Xu, Stimuli-responsive liposomes for the delivery of nucleic acid therapeutics, Nanomedicine, 2015, 11(6), 1575–1584 CrossRef CAS PubMed.
- T. Ta and T. M. Porter, Thermosensitive liposomes for localized delivery and triggered release of chemotherapy, J. Controlled Release, 2013, 169(1–2), 112–125 CrossRef CAS PubMed.
- D. K. Kirui, C. Celia, R. Molinaro, S. S. Bansal, D. Cosco, M. Fresta, H. Shen and M. Ferrari, Mild hyperthermia enhances transport of liposomal gemcitabine and improves in vivo therapeutic response, Adv. Healthcare Mater., 2015, 4(7), 1092–1103 CrossRef CAS PubMed.
- R. Di Corato, G. Bealle, J. Kolosnjaj-Tabi, A. Espinosa, O. Clement, A. K. A. Silva, C. Menager and C. Wilhelm, Combining magnetic hyperthermia and photodynamic therapy for tumor ablation with photoresponsive magnetic liposomes, ACS Nano, 2015, 9(3), 2904–2916 CrossRef CAS PubMed.
- N. Lozano, W. T. Al-Jamal, A. Taruttis, N. Beziere, N. C. Burton, J. Van den Bossche, M. Mazza, E. Herzog, V. Ntziachristos and K. Kostarelos, Liposome-gold nanorods hybrids for high-resolution visualization Deep in tissues, J. Am. Chem. Soc., 2012, 134(32), 13256–13258 CrossRef CAS PubMed.
- W. T. Al-Jamal and K. Kostarelos, Liposome-nanoparticle hybrids for multimodal diagnostic and therapeutic applications, Nanomedicine, 2007, 2(1), 85–98 CrossRef CAS PubMed.
- R. K. Keswani, I. M. Pozdol and D. W. Pack, Design of hybrid lipid/retroviral-like particle gene delivery vectors, Mol. Pharm., 2013, 10(5), 1725–1735 CrossRef CAS PubMed.
- J. Zhou, Q.-x. Wang and C.-Y. Zhang, Liposome-quantum dot complexes enable multiplexed detection of attomolar DNAs without target amplification, J. Am. Chem. Soc., 2013, 135(6), 2056–2059 CrossRef CAS PubMed.
- A. Bangham, J. De Gier and G. Greville, Osmotic properties and water permeability of phospholipid liquid crystals, Chem. Phys. Lipids, 1967, 1, 225–246 CrossRef CAS.
- L. Saunders, J. Perrin and D. Gammack, Ultrasonic irradiation of some phospholipid sols, J. Pharm. Pharmacol., 1962, 14, 567–572 CrossRef CAS PubMed.
- R. A. Parente and B. R. Lentz, Phase behavior of large unilamellar vesicles composed of synthetic phospholipids, Biochemistry, 1984, 23, 2353–2362 CrossRef CAS PubMed.
- J. Szoka and D. Papahadjopoulos, Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef.
- D. W. Deamer, Preparation and properties of ether-injection liposomes, Ann. N. Y. Acad. Sci., 1978, 308, 250–258 CrossRef CAS PubMed.
- A. Jahn, W. N. Vreeland, M. Gaitan and L. E. Locascio, Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing, J. Am. Chem. Soc., 2004, 126, 2674–2675 CrossRef CAS PubMed.
- D. van Swaay and A. deMello, Microfluidic methods for forming liposomes, Lab Chip, 2013, 13, 752–767 RSC.
- K. Otake, T. Imura, H. Sakai and M. Abe, Development of a new preparation method of liposomes using supercritical carbon dioxide, Langmuir, 2001, 17, 3898–3901 CrossRef CAS.
- N. Škalko-Basnet, Z. Pavelic and M. Becirevic-Lacan, Liposomes containing drug and cyclodextrin prepared by the one-step spray-drying method, Drug. Dev. Ind. Pharm., 2000, 26, 1279–1284 CrossRef PubMed.
- C. Jaafar-Maalej, C. Charcosset and H. Fessi, A new method for liposome preparation using a membrane contactor, J. Liposome Res., 2011, 21, 213–220 CrossRef CAS PubMed.
- R. Peschka, T. Purmann and R. Schubert, Cross-flow filtration – an improved detergent removal technique for the preparation of liposomes, Int. J. Pharm., 1998, 162, 177–183 CrossRef CAS.
- S. Thirumaleshwar, P. K. Kulkarni and V. Devegowda, Liposomal hydrogels: a novel drug delivery system for wound dressing, Curr. Drug Ther., 2012, 7(3), 212–218 CrossRef CAS.
- S. Kazakov and K. Levon, Liposome-Nanogel structures for future pharmaceutical applications, Curr. Pharm. Des., 2006, 12, 4713–4728 CrossRef CAS PubMed.
- A. Suri, R. Campos, D. G. Rackus, N. J. S. Nicholas, C. Richardson, L.-O. Palsson and R. Kataky, Liposome-doped hydrogel for implantable tissue, Soft Matter, 2011, 7(15), 7071–7077 RSC.
- S. Y. An, M.-P. N. Minh, J. Yun, K. N. Han, C. A. Li, J. Choo, E. K. Lee, S. Katoh, Y. Kumada and G. H. Yoichi, Preparation of monodisperse and size-controlled poly(ethylene glycol) hydrogel nanoparticles using liposome templates, J. Colloid Interface Sci., 2009, 331(1), 98–103 CrossRef CAS PubMed.
- J. N. Patton and A. F. Palmer, Physical properties of hemoglobin-poly(acrylamide) hydrogel-based oxygen carriers: Effect of reaction pH, Langmuir, 2006, 22(5), 2212–2221 CrossRef CAS PubMed.
- A. L. Weiner, S. S. Carpenter-Green, E. C. Soehngen, R. P. Lenk and M. C. Popescu, Liposome-collagen gel matrix: A novel sustained drug delivery system, J. Pharm. Sci., 1985, 74(9), 922–925 CrossRef CAS PubMed.
- J.-H. Lee, H. Oh, U. Baxa, S. R. Raghavan and R. Blumenthal, Biopolymer-connected liposome networks as injectable biomaterials capable of sustained local drug delivery, Biomacromolecules, 2012, 13(10), 3388–3394 CrossRef CAS PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications, Eur. J. Pharm. Biopharm., 2004, 57(1), 19–34 CrossRef CAS PubMed.
- S. K. Shukla, A. K. Mishra, O. A. Arotiba and B. B. Mamba, Chitosan-based nanomaterials: A state-of-the-art review, Int. J. Biol. Macromol., 2013, 59, 46–58 CrossRef CAS PubMed.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Chitosan-a versatile semi-synthetic polymer in biomedical applications, Prog. Polym. Sci., 2011, 36(8), 981–1014 CrossRef CAS.
- E. Ruel-Gariepy, G. Leclair, P. Hildgen, A. Gupta and J.-C. Leroux, Thermosensitive chitosan-based hydrogel containing liposomes for the delivery of hydrophilic molecules, J. Controlled Release, 2002, 82(2–3), 373–383 CrossRef CAS PubMed.
- J. Hurler, S. Žakelj, J. Mravljak, S. Pajk, A. Kristl, R. Schubert and N. Škalko-Basnet, The effect of lipid composition and liposome size on the release properties of liposomes-in-hydrogel, Int. J. Pharm., 2013, 456(1), 49–57 CrossRef CAS PubMed.
- A. Bilard, L. Pourchet, S. Malaise, P. Alcouffe, A. Montembault and C. Ladavière, Liposome-loaded chitosan physical hydrogel: Toward a promising delayed-release biosystem, Carbohydr. Polym., 2015, 115, 651–657 CrossRef PubMed.
- A. Alinaghi, M. R. Rouini, F. J. Daha and H. R. Moghimi, The influence of lipid composition and surface charge on biodistribution of intact liposomes releasing from hydrogel-embedded vesicles, Int. J. Pharm., 2014, 459(1–2), 30–39 CrossRef CAS PubMed.
- J. Hurler, O. A. Berg, M. Skar, A. H. Conradi, P. J. Johnsen and N. Škalko-Basnet, Improved burns therapy: Liposomes-in-hydrogel delivery system for mupirocin, J. Pharm. Sci., 2012, 101(10), 3906–3915 CrossRef CAS PubMed.
- J. Hurler, K. K. Sorensen, A. Fallarero, P. Vuorela and N. Škalko-Basnet, Liposomes-in-hydrogel delivery system with mupirocin: In vitro antibiofilm studies and in vivo evaluation in mice burn model, BioMed Res. Int., 2013, 498485 Search PubMed.
- K. M. Hosny, Preparation and evaluation of thermosensitive liposomal hydrogel for enhanced transcorneal permeation of ofloxacin, AAPS PharmSciTech, 2009, 10(4), 1336–1342 CrossRef CAS PubMed.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, Hydrogels for tissue engineering and regenerative medicine, J. Mater. Chem. B, 2014, 2(33), 5319–5338 RSC.
- A. López-Noriega, C. L. Hastings, B. Ozbakir, K. El O'Donnel, F. J. O'Brien, G. Storm, W. E. Hennink, G. P. Duffy and E. Ruiz-Hernández, Hyperthermia-induced drug delivery from thermosensitive liposomes encapsulated in an injectable hydrogel for local chemotherapy, Adv. Healthcare Mater., 2014, 3(6), 854–859 CrossRef PubMed.
- S. Gordon, K. Young, R. Wilson, S. Rizwan, R. Kemp, T. Rades and S. Hook, Chitosan hydrogels containing liposomes and cubosomes as particulate sustained release vaccine delivery systems, J. Liposome Res., 2012, 3, 193–204 CrossRef PubMed.
- B. C. Ciobanu, A. N. Cadinoiu, M. Popa, J. Desbrières and C. A. Peptu, Modulated release from liposomes entrapped in chitosan/gelatin hydrogels, Mater. Sci. Eng., C, 2014, 43, 383–391 CrossRef CAS PubMed.
- J. Song and S. C. G. Leeuwenburgh, Sustained delivery of biomolecules from gelatin carriers for applications in bone regeneration, Ther. Delivery, 2014, 5(8), 943–958 CrossRef CAS PubMed.
- M. Santoro, A. M. Tatara and A. G. Mikos, Gelatin carriers for drug and cell delivery in tissue engineering, J. Controlled Release, 2014, 190, 210–218 CrossRef CAS PubMed.
- A. O. Elzoghby, Gelatin-based nanoparticles as drug and gene delivery systems: Reviewing three decades of research, J. Controlled Release, 2013, 172, 1075–1091 CrossRef CAS PubMed.
- T. Wu, Q. Zhang, W. Ren, X. Yi, Z. Zhou, X. Peng, X. Yu and M. Lang, Controlled release of gentamicin from gelatin/genipin reinforced beta-tricalciumphosphate scaffold for the treatment of osteomyelitis, J. Mater. Chem. B, 2013, 1(26), 3304–3313 RSC.
- B. Sung, C. Kim and M.-H. Kim, Biodegradable colloidal microgels with tunable thermosensitive volume phase transitions for controllable drug delivery, J. Colloid Interface Sci., 2015, 450, 26–33 CrossRef CAS PubMed.
- D. M. G. Narayanan, H. L. Geena, M. Koyakutty, S. Nair and D. Menon, Poly-(ethylene glycol) modified gelatin nanoparticles for sustained delivery of the anti-inflammatory drug ibuprofen-sodium: an in vitro and in vivo analysis, Nanomedicine, 2013, 9(6), 818–828 CrossRef CAS PubMed.
- F. K. Kasper, E. Jerkins, K. Tanahashi, M. A. Barry, Y. Tabata and A. G. Mikos, Characterization of DNA release from composites of oligo(poly(ethylene glycol)fumarate) and cationized gelatin microspheres in vitro, J. Biomed. Mater. Res., Part A, 2006, 78(4), 823–835 CrossRef PubMed.
- M. B. Dowling, J. H. Lee and S. R. Raghavan, pH-responsive jello: gelatin gels containing fatty acid vesicles, Langmuir, 2009, 25(15), 8519–8525 CrossRef CAS PubMed.
- D. P. Cistola, J. A. Hamilton, D. Jackson and D. M. Small, Ionization and phase behavior of fatty acids in water: application of the Gibbs phase rule, Biochemistry, 1988, 27(6), 1881 CrossRef CAS PubMed.
- G. W. Bos, W. E. Hennink, L. A. Brouwer, W. den Otter, T. F. J. Veldhuis, C. F. van Nostrum and M. J. A. van Luyn, Tissue reactions of in situ formed dextran hydrogels crosslinked by stereocomplex formation after subcutaneous implantation in rats, Biomaterials, 2005, 26(18), 3901–3909 CrossRef CAS PubMed.
- L. Hovgaard and H. Brondsted, Dextran hydrogels for colon-specific drug delivery, J. Controlled Release, 1995, 36(1–2), 159–166 CrossRef CAS.
- N. A. O'Connor, A. Abugharbieh, F. Yasmeen, E. Buabeng, S. Mathew, D. Samaroo and H.-P. Cheng, The crosslinking of polysaccharides with polymanines and dextran-polyallylamine antibacterial hydrogels, Int. J. Biol. Macromol., 2015, 3(72), 88–93 CrossRef PubMed.
- J. D. Cabral, M. A. McConnell, C. Fitzpatrick, S. Mros, G. Williams, P. J. Wormald, S. C. Moratti and L. R. Hanton, Characterization of the in vivo host response to a bi-labeled chitosan-dextran based hydrogel for postsurgical adhesion prevention, J. Biomed. Mater. Res., Part A, 2015, 103(8), 2611–2620 CrossRef CAS PubMed.
- F. Xiao, L. Chen, R.-F. Xing, Y.-P. Zhao, J. Dong, G. Guo and R. Zhang, In vitro cyto-biocompatibility and cell detachment of temperature-sensitive dextran hydrogel, Colloids Surf., B, 2009, 71(1), 13–18 CrossRef CAS PubMed.
- S. Li, M. Yang, W. Zhou, J. Wenfei, G. Trevor, R. Wang and J. Zhu, Dextran hydrogel coated surfaced plasmon resonance imaging (SPRi) sensor for sensitive and label-free detection of small molecule drugs, Appl. Surf. Sci., 2015, 355, 570–576 CrossRef CAS.
- S. Parcelli, P. Paolicelli and M. A. Casadei, New biodegradable dextran-based hydrogels for protein delivery: synthesis and characterization, Carbohydr. Polym., 2015, 126, 208–214 CrossRef PubMed.
- T. G. van Thienen, B. Lucas, F. M. Flesch, C. F. van Nostrum, J. Demeester and S. C. De Smedt, On the synthesis and characterization of biodegradable dextran nanogels with tunable degradation properties, Macromolecules, 2005, 38(20), 8503–8511 CrossRef CAS.
- B. G. De Geest, B. G. Stubbe, A. M. Jonas, T. van Thienen, W. L. J. Hinrichs, J. Demeester and S. C. De Smedt, Self-exploding lipid-coated microgels, Biomacromolecules, 2006, 7(1), 373–379 CrossRef PubMed.
- N. López Mora, J. S. Hansen, Y. Gao, A. A. Ronald, R. Kieltyka, N. Malmstadt and A. Kros, Preparation of size tunable giant vesicles from cross-linked dextran(ethylene glycol) hydrogels, Chem. Commun., 2014, 50, 1953–1955 RSC.
- A. San Juan, M. Bala, H. Hlawaty, P. Portes, R. Vranckx, L. J. Feldman and D. Letourneur, Development of a functionalized polymer for stent coating in the arterial delivery of small interfering RNA, Biomacromolecules, 2009, 10(11), 3074–3080 CrossRef CAS PubMed.
- M. Fujioka-Kobayashi, M. S. Ota, A. Shimoda, K.-i. Nakahama, K. Akiyoshi, Y. Miyamoto and S. Iseki, Cholesteryl group- and cryloyl group bearing pullulan nanogel to deliver BMP2 and FGF18 for bone tissue engineering, Biomaterials, 2012, 33(30), 7613–7620 CrossRef CAS PubMed.
- Y. Sekine, Y. Moritani, T. Ikeda-Fukazawa, Y. Sasaki and K. Akiyoshi, A hybrid hydrogel biomaterial by nanogel engineering: Bottom-up design with nanogel and liposome building blocks to develop a multidrug delivery system, Adv. Healthcare Mater., 2012, 1(6), 722–728 CrossRef CAS PubMed.
- K. Akiyoshi, N. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Self-aggregates of hydrophobized polysaccharides in water. Formation and characteristics of nanoparticles, Macromolecules, 1993, 26(12), 3062–3068 CrossRef CAS.
- T. Ueda, S. J. Lee, Y. Nakatani, G. Ourisson and J. Sunamoto, Coating of POPC giant liposomes with hydrophobized polysaccharide, Chem. Lett., 1998, 5, 417–418 CrossRef.
- K. Akiyoshi, S. Kobayashi, S. Shichibe, D. Mix, M. Baudys, S. W. Kim and J. Sunamoto, Self-assembled hydrogel nanoparticle of cholesterol-bearing pullulan as a carrier of protein drugs: Complexation and stabilization of insulin, J. Controlled Release, 1998, 54(3), 313–320 CrossRef CAS PubMed.
- J. B. Leach and C. E. Schmidt, Characterization of protein release from photocrosslinkable hyaluronic acid-polyethylene glycol hydrogel tissue engineering scaffolds, Biomaterials, 2005, 26(2), 125–135 CrossRef CAS PubMed.
- A. Sannino, M. Madaghiele, F. Conversano, G. Mele, A. Maffezzoli, P. A. Netti, L. Ambrosio and L. Nicolais, Cellulose derivative-hyaluronic acid-based microporous hydrogels cross-linked through divinyl sulfone (DVS) to modulate equilibrium sorption capacity and network stability, Biomacromolecules, 2004, 5(1), 92–96 CrossRef CAS PubMed.
- T. Ito, I. P. Fraser, Y. Yeo, C. B. Highley, E. Bellas and D. S. Kohane, Anti-inflammatory function of an in situ cross-linkable conjugate hydrogel of hyaluronic acid and dexamethasone, Biomaterials, 2007, 28(10), 1778–1786 CrossRef CAS PubMed.
- R. D. Price, S. Myers, I. M. Leigh and H. A. Navsaria, The role of hyaluronic acid in wound healing: assessment of clinical evidence, Am. J. Clin. Dermatol., 2005, 6(6), 393–402 CrossRef PubMed.
- S. S. Kwon, B. J. Kong and S. N. Park, Physicochemical properties of pH-sensitive hydrogels based on hydroxyethyl cellulose-hyaluronic acid and for applications as transdermal delivery systems for skin lesions, Eur. J. Pharm. Biopharm., 2015, 92, 146–154 CrossRef CAS PubMed.
- N. El Kechai, A. Bochot, N. Huang, Y. Nguyen, E. Ferrary and F. Agnely, Effect of liposomes on rheological and syringeability properties of hyaluronic acid hydrogels intended for local injection of drugs, Int. J. Pharm., 2015, 487(1–2), 187–196 CrossRef CAS PubMed.
- M. R. El-Assar, G. F. El Fawal, E. A. Kamoun and M. M. G. Fouda, Controlled drug release from cross-linked K-carrageenan/hyaluronic acid membranes, Int. J. Biol. Macromol., 2015, 77, 322–329 CrossRef PubMed.
- L. Li, Y. Wei and C. Gong, Polymeric nanocarriers for non-viral gene delivery, J. Biomed. Nanotechnol., 2015, 11, 739–770 CrossRef CAS PubMed.
- J. Dong, D. Jiang, Z. Wang, G. Wu, L. Miao and L. Huang, Intra-articular delivery of liposomal celecoxib-hyaluronate combination for the treatment of osteoarthritis in rabbit model, Int. J. Pharm., 2013, 441(1–2), 285–290 CrossRef CAS PubMed.
- L. Lajavardi, S. Carmelo, F. Agnely, W. Luo, B. Goldenberg, M.-C. Naud, F. Behar-Cohen, Y. de Kozak and A. Bochot, New formulations of vasoactive intestinal peptide using liposomes in hyaluronic acid gel for uveitis, J. Controlled Release, 2009, 139(1), 22–30 CrossRef CAS PubMed.
- L. K. Widjaja, M. Bora, H. Paul Ng Poh, V. Lipik, T. T. L. Wong and S. S. Venkatraman, Hyaluronic acid-based nanocomposite hydrogels for ocular drug delivery applications, J. Biomed. Mater. Res., Part A, 2014, 102(9), 3056–3065 CrossRef PubMed.
- C. D. Ren, M. Kurisawa, J. E. Chung and J. Y. Ying, Liposomal delivery of horseradish peroxidase for thermally triggered injectable hyaluronic acid-tyramine hydrogel scaffolds, J. Mater. Chem. B, 2015, 3, 4663–4670 RSC.
- W. R. Gombotz and S. F. Wee, Protein release from alginate matrices, Adv. Drug Delivery Rev., 2012, 64, 194–205 CrossRef.
- K. Y. Lee and D. J. Mooney, Alginate: Properties and biomedical applications, Prog. Polym. Sci., 2012, 37(1), 106–126 CrossRef CAS PubMed.
- M. Monshipouri and A. S. Rudolph, Liposome-encapsulated alginate: Controlled hydrogel particle formation and release, J. Microencapsulation, 1995, 12(2), 117–127 CrossRef CAS PubMed.
- I. Takagi, H. Shimizu and T. Yotsuyanagi, Applications of alginate gel as a vehicle for liposomes. I. Factors affecting the loading of drug-containing liposomes and drug release, Chem. Pharm. Bull., 1996, 44, 1941–1947 CrossRef CAS PubMed.
- I. Takagi, H. Nakashima, M. Takagi, T. Yotsuyanagi and K. Ikeda, Applications of alginate gel as a vehicle for liposomes. II. Erosion of alginate gel beads and the release of loaded liposomes, Chem. Pharm. Bull., 1997, 45, 389–393 CrossRef CAS PubMed.
- L. Xing, C. Dawei, X. Liping and Z. Rongqing, Oral colon-specific drug delivery for bee venom peptide: development of a coated calcium alginate gel beads-entrapped liposomes, J. Controlled Release, 2003, 93, 293–300 CrossRef PubMed.
- C. Dai, B. Wang, H. Zhao, B. Li and J. Wang, Preparation and characterization of liposomes-in-alginate (LIA) for protein delivery system, Colloids Surf., B, 2006, 47(2), 205–210 CrossRef CAS PubMed.
- A. M. Smith, M. R. Jaime-Fonseca, L. M. Grover and S. Bakalis, Alginate-loaded liposomes can protect encapsulated alkaline phosphatase functionality when exposed to gastric pH, J. Agric. Food Chem., 2010, 58, 4719–4724 CrossRef CAS PubMed.
- M. Ullrich, J. Hanuš, J. Dohnal and F. Štěpánek, Encapsulation stability and temperature-dependent release kinetics from hydrogel-immobilised liposomes, J. Colloid Interface Sci., 2013, 394, 380–385 CrossRef CAS PubMed.
- J. Čejková, P. Haufová, D. Gorný, J. Hanuš and F. Štěpánek, Biologically triggered liberation of sub-micron particles from alginate microcapsules, J. Mater. Chem. B, 2013, 1, 5456–5461 RSC.
- J. Hanuš, M. Ullrich, J. Dohnal, M. Singh and F. Stěpánek, Remotely controlled diffusion from magnetic liposome microgels, Langmuir, 2013, 29, 4381–4387 CrossRef PubMed.
- D. H. Kang, H. S. Jung, N. Ahn, S. M. Yang, S. Seo, K. Y. Suh, P. S. Chang, N. L. Jeon, J. Kim and K. Kim, Janus-compartmental alginate microbeads having polydiacetylene liposomes and magnetic nanoparticles for visual lead(II) detection, ACS Appl. Mater. Interfaces, 2014, 6, 10631–10637 CAS.
- M. Machluf, R. N. Apte, O. Regev and S. Cohen, Enhancing the immunogenicity of liposomal hepatitis B surface antigen (HBsAg) by controlling its delivery from polymeric microspheres, J. Pharm. Sci., 2000, 89(12), 1550–1557 CrossRef CAS PubMed.
- T. Aikawa, S. Ito, M. Shinohara, M. Kaneko, T. Kondo and M. Yuasa, A drug formulation using an alginate hydrogel matrix for efficient oral delivery of the manganese porphyrin-based superoxide dismutase mimic, Biomater. Sci., 2015, 3(6), 861–869 RSC.
- M. Van Elk, B. Ozbakir, A. D. Barten-Rijbroek, G. Storm, F. Nijen, W. E. Hennink, T. Vermonden and R. Deckers, Alginate microspheres containing temperature sensitive liposomes (TSL) for MR-guided embolization and triggered release of doxorubicin, PLoS One, 2015, 10, 1–19 Search PubMed.
- V. D. Prajapati, P. M. Maheriya, G. K. Jani and H. K. Solanki, Carrageenan: A natural seaweed polysaccharide and its applications, Carbohydr. Polym., 2014, 105, 97–112 CrossRef CAS PubMed.
- L. L. Rui Ni, Y. Shao and S. Mao, Carrageenan and its applications in drug delivery, Carbohydr. Polym., 2014, 103, 1–11 CrossRef PubMed.
- A. Grenha, M. E. Gomes, M. Rodrigues, V. E. Santo, J. F. Mano, N. M. Neves and R. L. Reis, Development of new chitosan/carrageenan nanoparticles for drug delivery applications, J. Biomed. Mater. Res., Part A, 2009, 92(4), 1265–1272 Search PubMed.
- Z. Mohamadnia, M. J. Zohuriaan-Mehr, K. Kabiri, A. Jamshidi and H. Mobedi, pH-sensitive IPN hydrogel beads of carrageenan-alginate for controlled drug delivery, J. Bioact. Compat. Polym., 2007, 22(3), 342–356 CrossRef CAS.
- J. S. Varghese, N. Chellappa and N. N. Fathima, Gelatin-carrageenan hydrogels: Role of pore size distribution on drug delivery process, Colloids Surf., B, 2014, 113, 346–351 CrossRef CAS PubMed.
- C. V. Kulkarni, Z. Moinuddin, Y. Patil-Sen, R. Littlefield and M. Hood, Lipid-hydrogel films for sustained drug release, Int. J. Pharm., 2015, 479(2), 416–421 CrossRef CAS PubMed.
- S. F. El-Menshawe and A. K. Hussein, Formulation and evaluation of meloxicam niosomes as vesicular carriers for enhanced skin delivery, Pharm. Dev. Technol., 2013, 18(4), 779–786 CrossRef CAS PubMed.
- S. C. Joshi, I. C. Su, C. M. Liang and Y. C. Lam, Gelation of methylcellulose hydrogels under isothermal conditions, J. Appl. Polym. Sci., 2008, 107(4), 2101–2108 CrossRef CAS.
- X. Bourges, P. Weiss, G. Daculsi and G. Legeay, Synthesis and general properties of silated-hydroxypropyl methylcellulose in prospect of biomedical use, Adv. Colloid Interface Sci., 2002, 99(3), 215–228 CrossRef CAS PubMed.
- A. P. Rokhade, N. B. Shelke, S. A. Patil and T. M. Aminabhavi, Novel interpenetrating polymer network microspheres of chitosan and methylcellulose for controlled release of theophylline, Carbohydr. Polym., 2007, 69(4), 678–687 CrossRef CAS.
- R. Kapadia, H. Khambete, R. Katara and S. Ramteke, A novel approach for ocular delivery of acyclovir via niosomes entrapped in situ hydrogel system, J. Pharm. Res., 2009, 2, 745–751 CAS.
- D. F. S. Petri, Xanthan gum: A versatile biopolymer for biomedical and technological applications, J. Appl. Polym. Sci., 2015, 132(23), 42035 CrossRef.
- V. D. Prajapati, G. K. Jani, N. G. Moradiya, N. P. Randeria and B. J. Nagar, Locust bean gum: A versatile biopolymer, Carbohydr. Polym., 2013, 94(2), 814–821 CrossRef CAS PubMed.
- C. Marianecci, M. Carafa, L. Di Marzio, F. Rinaldi, C. Di Meo, P. Matricardi, F. Alhaique and T. Coviello, A new vesicle-loaded hydrogel system suitable for topical applications: Preparation and characterization, J. Pharm. Pharm. Sci., 2011, 14(3), 336–346 CAS.
| This journal is © The Royal Society of Chemistry 2016 |