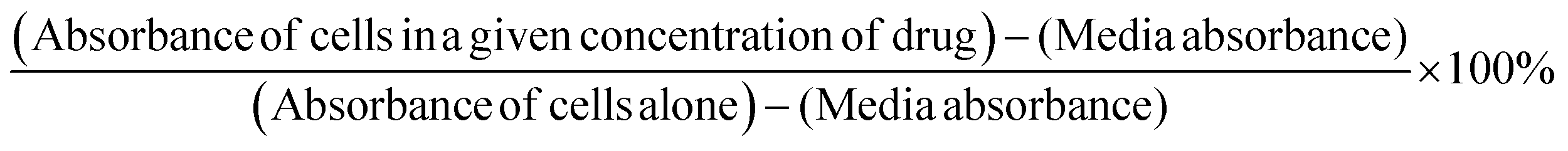DOI:
10.1039/C5BM00519A
(Paper)
Biomater. Sci., 2016,
4, 511-521
Oseltamivir-conjugated polymeric micelles prepared by RAFT living radical polymerization as a new active tumor targeting drug delivery platform†
Received
9th November 2015
, Accepted 11th January 2016
First published on 20th January 2016
Abstract
Targeted drug delivery using polymeric nanostructures has been at the forefront of cancer research, engineered for safer, more efficient and effective use of chemotherapy. Here, we designed a new polymeric micelle delivery system for active tumor targeting followed by micelle–drug internalization via receptor-induced endocytosis. We recently reported that oseltamivir phosphate targets and inhibits Neu1 sialidase activity associated with receptor tyrosine kinases such as epidermal growth factor receptors (EGFRs) which are overexpressed in cancer cells. By decorating micelles with oseltamivir, we investigated whether they actively targeted human pancreatic PANC1 cancer cells. Amphiphilic block copolymers with oseltamivir conjugated at the hydrophilic end, oseltamivir-pPEGMEMA-b-pMMA (oseltamivir-poly(polyethylene glycol methyl ether methacrylate)-block-poly(methyl methacrylate), were synthesized using reversible addition–fragmentation chain transfer (RAFT) living radical polymerization. Oseltamivir-conjugated micelles have self-assembling properties to give worm-like micellar structures with molecular weight of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Oseltamivir-conjugated water soluble pPEGMEMA, dose dependently, both inhibited sialidase activity associated with Neu1, and reduced viability of PANC1 cells. In addition, oseltamivir-conjugated micelles, labelled with a hydrophobic fluorescent dye within the micelle core, were subsequently internalized by PANC1 cells. Blocking cell surface Neu1 with anti-Neu1 antibody, reduced internalization of oseltamivir-conjugated micelles, demonstrating that Neu1 binding linked to sialidase inhibition were prerequisite steps for subsequent internalization of the micelles. The mechanism of internalization is likely that of receptor-induced endocytosis demonstrating potential as a new nanocarrier system for not only targeting a tumor cell, but also for directly reducing viability through Neu1 inhibition, followed by intracellular delivery of hydrophobic cytotoxic chemotherapeutics.
000 g mol−1. Oseltamivir-conjugated water soluble pPEGMEMA, dose dependently, both inhibited sialidase activity associated with Neu1, and reduced viability of PANC1 cells. In addition, oseltamivir-conjugated micelles, labelled with a hydrophobic fluorescent dye within the micelle core, were subsequently internalized by PANC1 cells. Blocking cell surface Neu1 with anti-Neu1 antibody, reduced internalization of oseltamivir-conjugated micelles, demonstrating that Neu1 binding linked to sialidase inhibition were prerequisite steps for subsequent internalization of the micelles. The mechanism of internalization is likely that of receptor-induced endocytosis demonstrating potential as a new nanocarrier system for not only targeting a tumor cell, but also for directly reducing viability through Neu1 inhibition, followed by intracellular delivery of hydrophobic cytotoxic chemotherapeutics.
1. Introduction
Nanoscale polymeric drug delivery systems such as polymeric micelles and liposomes have been developed in the past two decades for targeted and sustained release of chemotherapeutics. Traditional chemotherapy agents, such as doxorubicin and paclitaxel, are typically highly hydrophobic small molecules designed to work intracellularly, targeting rapidly dividing cells. Administration of these drugs alone, complicated by insolubility in biological fluids and tissues, and an incapacity to effectively localize in metastasized tumors, results in adverse toxic side effects and prevents potent selective targeting.1 The enhanced permeability and retention effect is the property by which functionalized drug loaded nanocarriers tend to accumulate in tumor tissue much more so than they do in healthy tissues.2
The development of amphiphilic polymeric vesicles capable of encapsulating large quantities of a chemotherapeutic agent and improving its targeting efficiency and bioavailability are emerging new delivery platforms for nanomedicine in cancer research.1–3 More recently, active drug delivery platforms have been engineered for effective tumor targeting. Polymeric micelles have been modified by attaching ligand molecules to the outer shell of the micelle with specificity for unique proteins that are overly expressed on tumor cells. Such ligands such as peptides, antibodies, sugars and aptamers have enabled significant improvement in tumor selectivity and overall therapeutic efficiency of cancer treatments.3,4 The limitations of these specific “smart” delivery vesicles however, are low overtime stability especially with antibodies, immunogenic adverse effects and high preparation costs.
Amphiphilic copolymers prepared by living radical polymerization (LRP) techniques have been gaining interest in the field of polymeric micelle based delivery systems for cancer therapeutics and diagnostic tools. The ability to prepare complex polymeric structures with highly controlled molecular weights and defined architectures enables multiple functionalities such as hydrophobic/hydrophilic blocks needed for self-assembly, stimuli-responsive regions (responding to CO2, pH and temperature) for triggered drug release, and reactive groups for drug conjugation, cross-linking and ‘click’ chemistry.5 In the present study, we describe the design of an amphiphilic block polymer–drug conjugate using reversible addition–fragmentation chain transfer (RAFT) polymerization which is a subclass of LRP. The mechanism of RAFT involves a degenerative chain transfer reaction facilitated by a radical-reactive chain transfer agent (CTA), known as the RAFT agent. It produces an equilibrium between the propagating chain and a deactivated or dormant chain capped by the RAFT agent (CTA).5b,c With an equilibrium shifted heavily to the dormant polymer-CTA, the RAFT polymerization has minimum termination reactions, producing a polymer with a very narrow molecular weight distribution. The final polymer-CTA exists in a ‘living’ state, meaning it can be redissolved, and the chain can be extended with different monomers to produce complex polymeric architectures.5d
Reports on the preparation of amphiphilic block copolymers for drug delivery via RAFT have dramatically increased over the last 10 years due to the ability to synthesize highly complex water soluble structures with biologically important architectures, and a wide range of acrylic monomers that can be polymerized to form a multi-functional stimuli-responsive polymer backbone.5d,e Notably, Pascual et al.6 have prepared a number of nanostructures by RAFT polymerization potentially useful for biomedical and drug delivery applications, including polystyrene-b-poly(butyl acrylate) block copolymer using xanthate as a RAFT-CTA which served as a hydrophilic end-group to stabilize the forming particles during microemulsion polymerization in water and later used to conjugate the particle surface with functional macromolecules. Another relevant design by Jia et al.7 was the synthesis of multifunctional nano-worms/rods via RAFT-mediated emulsion polymerization. Preparation of polymer–drug conjugates as a prodrug formulation using RAFT polymerization has been reported previously by Wang et al.8 The report described the conjugation of the chemotherapeutic drug gemcitabine to a RAFT agent which was then polymerized using methyl methacrylate (MMA) to produce gemcitabine-pMMA polymeric micelles. Although the gemcitabine loaded micelles were well characterized, they lack the specific “smart” chemotherapeutic delivery platform for active tumor targeting.
We recently reported that oseltamivir phosphate (OP) can impede tumor neovascularization, growth and metastasis of human ovarian,9 triple-negative breast10 and pancreatic11 cancer cells in heterotopic xenografts of these tumors in RAGxCγ double mutant mice. The findings also disclosed a novel signaling paradigm that regulates epidermal growth factor receptors (EGFR).11,12 Mutations of the EGFR, resulting in hyperactivity of the receptor, is reported in over 90% of pancreatic cancer cases causing rapid, uncontrolled cellular growth, which contributes to the aggressive nature of pancreatic tumors.13 Chemotherapy designed to target EGFR hasn't been successful in the clinic since no single molecule was able to target the full range of EGFR mutations which limits the application of highly targeted chemotherapies.13,14 The role of Neu1 within the EGFR complex is that it is critical for EGF-induced receptor activation, the essential signalling platform on the cell surface. Neu1 involvement in this novel receptor signaling platform has been previously reported for a number of other receptors, all of which are known to play major roles in cancer. Oseltamivir, a synthetic analog of α-2,3-sialic acid, was shown to inhibit the Neu1 sialidase activity in complex with these receptors, and impede tumor neovascularization, growth and metastasis in a number of cancers.9–11,15
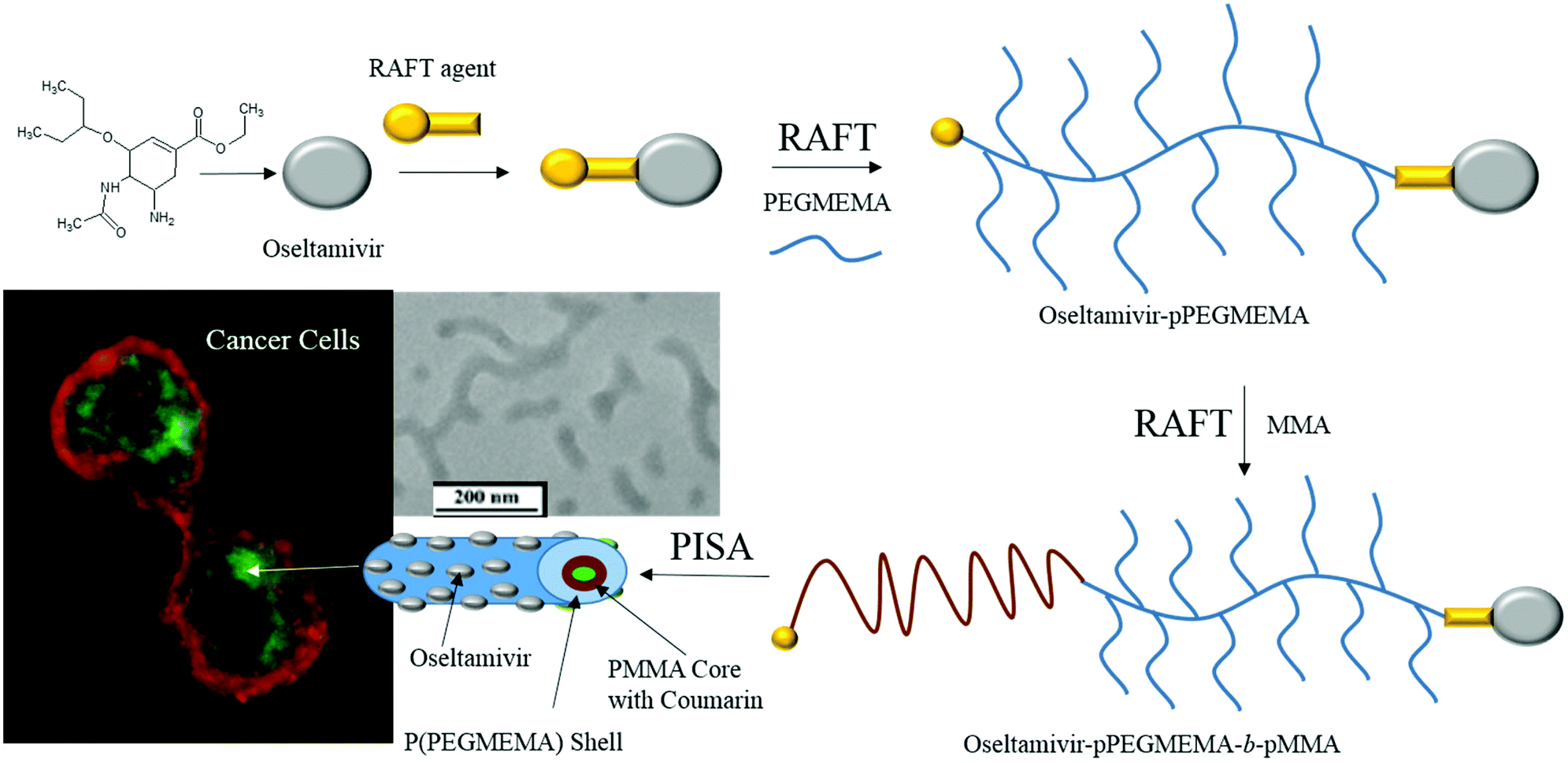
Based on these findings, an entirely new polymeric drug delivery system was designed for active tumor targeting. Micelles with oseltamivir grafted to the outer shell should provide three functions; (a) tumor cell targeting via Neu1 receptor binding, (b) anti-tumor effect induced through Neu1 inhibition, and (c) localized delivery of a chemotherapeutic drug carried by the polymeric micelle. As shown in Fig. 1, oseltamivir was first linked with the RAFT agent to facilitate polymerization of polyethylene glycol methyl ether methacrylate (PEGMEMA) producing a water soluble oseltamivir conjugated micellar shell. Methyl methacrylate was polymerized from the polymer living end to produce oseltamivir-p(PEGMEMA)-b-p(MMA) amphiphilic block copolymer which self-assembled during polymerization into a micelle with oseltamivir on the surface of the micellar shell.
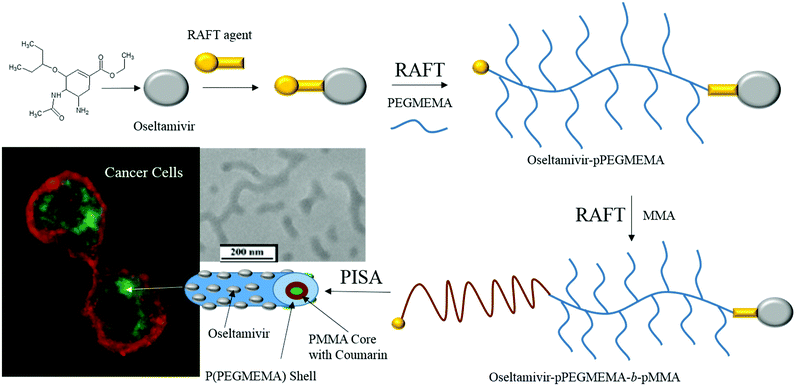 |
| | Fig. 1 Schematic representation of the synthetic steps and subsequent preparation of oseltamivir-conjugated micelle capable of triggering receptor-induced endocytosis in cancer cells. | |
Our findings indicate that oseltamivir-micelles demonstrated targeting to PANC1 cells, a reduction in sialidase activity associated with Neu1, reduction in tumor cell viability, and subsequent internalization of the micelles carrying a fluorescent label as a model drug compound. Neu1 binding was shown to be a prerequisite step toward micelle internalization since it was blocked by anti-Neu1 antibodies. The ability to both target and halt the growth of a tumor cell using a newly designed nanocarrier system, combined with the internalization of the micelle loaded with a cytotoxic chemotherapeutic represents the novel aspect of this work.
2. Experimental section
2.1 Materials
Oseltamivir phosphate was purchased from Hangzhou Dayang Chem Co Ltd (Hangzhou City, PRC). 4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid N-succinimidyl ester (N-hydroxysuccinimide-protected RAFT agent or RAFT-NHS), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid >97%, poly(ethylene glycol) methyl ether methacrylate (Mn 300), methyl methacrylate (MMA) ≤99%, 2,2′-azobis(2-methylpropionitrile) (AIBN) 98%, triethylamine (TEA), coumarin 153 (C153) dye content 99% were purchased from Sigma-Aldrich. N,N-Dimethylformamide anhydrous DrySolv® was purchased from EMD Millipore-AA-Corp., toluene reagent A.S.C. was purchased from ACP Chemicals Montreal, QC. Human pancreatic cancer cells PANC1 (human epithelioid carcinoma, epithelial-like, ATCC® CRL-1469™) were purchased from the American Type Culture Collection. Cell culture medium contain 1× Dulbecco's Modified Eagle's Medium (DMEM) purchased from Gibco, 10% fetal bovine serum (FBS) purchased from HyClone and 5 μg mL−1 Plasmocin™ purchased from InvivoGen. WST-1 reagent was purchased from Roche Diagnostics. CellMask™ Deep Red plasma membrane stain was purchased from Life Technologies Inc. Antibody to human neuraminidase 1 was purchased from Santa Cruz Biotechnology Inc.
2.2 Characterization
1H and 13C NMR of all products in D2O, CDCl3 or DMSO-d6 were acquired with Bruker Avance 400 MHz. Dry samples were dissolved in a deuterated solvent at 15 mg mL−1 for 1H and 75 mg mL−1 for 13C NMR. Overtime polymerization samples were prepared by adding 3 drops of reaction solution to 0.5 mL of deuterated solvent. Molecular weight of the polymers was analyzed by a Viscotek 270max with dual detector, (mobile phase THF), calibrated with polymethyl methacrylate standards. GPC samples were prepared at 1–2 mg mL−1 or by addition of 5 drops of reaction solution to 1 mL THF. Micelle morphology was observed using Hitachi H-7000 transmission electron microscope (TEM) (75 kV). Micelle size distribution was measured by dynamic light scattering (DLS) at 25 °C using a Malvern Zetasizer Nano Series (Nano ZS). Thermo Scientific Orbitrap Velos Pro mass spectrometer with heated electrostatic spray ionization source (ESI-MS) was used to confirm the synthesis of the oseltamivir-RAFT agent.
2.3 Synthesis of oseltamivir-RAFT agent
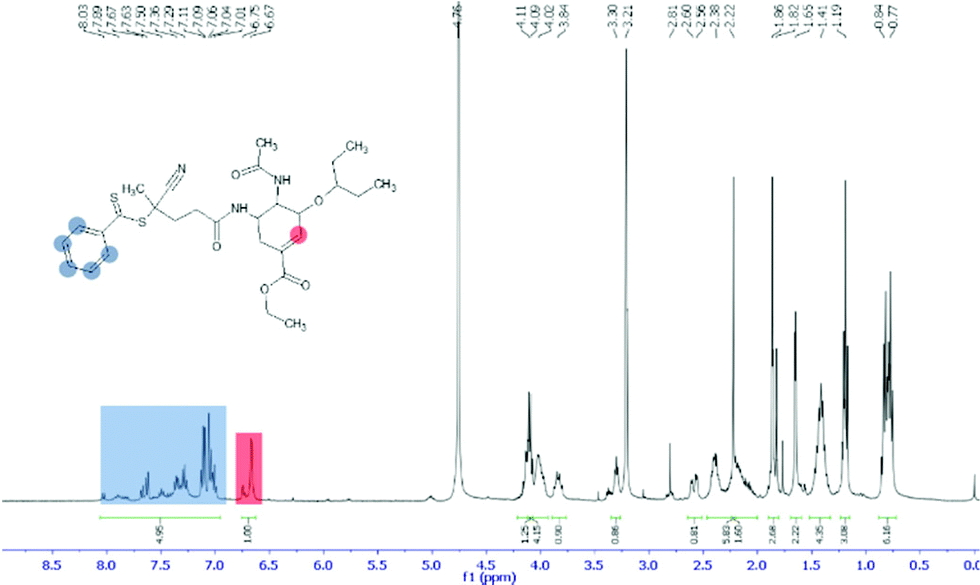
A molar excess of oseltamivir phosphate (0.8 g, 2 mmol) was dissolved in DMF (10 mL) with addition of an equimolar amount of triethylamine (TEA). RAFT-NHS (0.46 g, 1 mmol) was dissolved in DMF (5 mL) then added to the reaction flask. The solution was allowed to react for 4 h at room temperature under stirring. After the reaction was completed, DMF was removed using a rotary evaporator. The remaining solid was redissolved in toluene and washed with three aliquots of water (acidified to pH 2.5 with phosphoric acid) to remove unreacted oseltamivir. The final product was then air dried to give orange solid (0.532 g, 92.4% yield) and characterized by NMR and LC-MS. 1H NMR (400 MHz, CD3OD): 7.6 (d, 2H, J = 7.3 Hz, o-ArH), 7.4 (t, 1H, p-ArH), 7.1 (m, 2H, m-ArH), 6.77 (d, 1H, (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–)), 4.09 (q, 2H, (–CH2CH3)), 3.84 (q, 1H, (–CH–NHAc)), 3.30 (quint, 1H, (–CH(CH2CH3)2), 2.80 (m, 1H, (oseltamivir-CHNHCO-RAFT)), 2.58 (dd, 1H, (–CH2–)), 2.2–2.6 (m, 4H RAFT-(CH2CH2)CONH-oseltamivir), 2.2 (s, 3H, (–NH–C(O)CH3)), 2.11–2.18 (m, 2H, (–CH2–)), 1.86 (s, 3H, SC(CH3)CN), 1.41 (m, 4H, (–C(CH2CH3)2)), 1.19 (t, 3H, (–CH2CH3)), 0.84 (t, 3H, (–C(CH2CH3)2), 0.77 (t, 3H, (–C(CH2CH3)2). 13C NMR (CDCl3) (δ, ppm): 8.0, 8.5, 13.1, 20.4, 22.6, 24.6, 25.2, 29.4, 31.5, 32.9, 45.2, 47.6, 52.8, 59.8, 75.7, 81.2, 124.3, 126.0, 127.2, 128.0, 130.1, 136.8, 144.4, 164.6, 170.0, 170.4. ESI-MS: m/z calc. for C29H40N3O5S2 (M − H+) 574.24; found 574.24.
C–)), 4.09 (q, 2H, (–CH2CH3)), 3.84 (q, 1H, (–CH–NHAc)), 3.30 (quint, 1H, (–CH(CH2CH3)2), 2.80 (m, 1H, (oseltamivir-CHNHCO-RAFT)), 2.58 (dd, 1H, (–CH2–)), 2.2–2.6 (m, 4H RAFT-(CH2CH2)CONH-oseltamivir), 2.2 (s, 3H, (–NH–C(O)CH3)), 2.11–2.18 (m, 2H, (–CH2–)), 1.86 (s, 3H, SC(CH3)CN), 1.41 (m, 4H, (–C(CH2CH3)2)), 1.19 (t, 3H, (–CH2CH3)), 0.84 (t, 3H, (–C(CH2CH3)2), 0.77 (t, 3H, (–C(CH2CH3)2). 13C NMR (CDCl3) (δ, ppm): 8.0, 8.5, 13.1, 20.4, 22.6, 24.6, 25.2, 29.4, 31.5, 32.9, 45.2, 47.6, 52.8, 59.8, 75.7, 81.2, 124.3, 126.0, 127.2, 128.0, 130.1, 136.8, 144.4, 164.6, 170.0, 170.4. ESI-MS: m/z calc. for C29H40N3O5S2 (M − H+) 574.24; found 574.24.
2.4 Synthesis of oseltamivir-pPEGMEMA via RAFT polymerization
PEGMEMA (9.0 g, Mn 300 g mol−1, 30 mmol), oseltamivir-RAFT (0.10 g, 575.78 g mol−1, 0.17 mmol) and AIBN (0.008 g, 0.05 mmol) (monomer/CTA/initiator 150:1:0.25) were dissolved in 25 mL of toluene and purged with nitrogen for 30 min at room temperature. The solution temperature was then raised to 70 °C to thermally initiate polymerization. Samples were taken every hour and analyzed by GPC for molecular weight and NMR for conversion. In the end, reaction solution was precipitated in cold diethyl ether, and the final polymer isolated by centrifugation and cleaned by dialysis against water. 1H NMR (400 MHz): 4.1 ppm 2H (–C(O)–O–CH2–), 3.5–3.7 ppm 16H (–C(O)–O–CH2–CH2–O–(CH2–CH2–O)3/4–CH3), 3.3 ppm 3H (–O–CH3), 1.7–2.1 ppm 2H (–CH2–), 0.7–1.1 ppm 3H (a-CH3). GPC: 35000 g mol−1.
2.5 Preparation of oseltamivir-conjugated micelle by RAFT PISA
Methyl methacrylate (MMA) (0.94 g, 9.4 mmol), oseltamivir-p(PEGMEMA)-RAFT (0.20 g, 36000 g mol−1, 0.006 mmol), AIBN (0.001 g, 0.006 mmol) (monomer/CTA/initiator 1500:1:1) were dissolved in 6 mL of water/methanol 1:1 v/v. The polymerization was allowed to proceed until the onset of PISA which typically occurred after 6–8 h, with the final micellar solution appearing as milky-white colloidal suspension. Samples were taken at the beginning and the end of reaction for molecular weight analysis by GPC. The micellar solution was then dialyzed against water, while changing the dialysis solvent 3 times every 3 h. After cleaning, micelle size was measured by DLS and morphology observed by TEM. A small aliquot was lyophilized for structure characterization by NMR. Micelles without oseltamivir were prepared by precisely following the two RAFT polymerization protocols described above, but using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid as a CTA instead of oseltamivir-RAFT.
2.6 Sialidase activity assay
A series of solutions (0–1 mg mL−1) of oseltamivir-pPEGMEMA (4000 g mol−1) were prepared in sodium acetate with pH 5. To each solution, 40 μL of 9.9 unit mL−1 α(2 → 3) neuraminidase from Streptococcus pneumoniae (α-2,3-Neu, 50 mU mL−1) (Sigma, St. Louis, MO) was used at predetermined optimal dosage and samples were incubated for 2 h at 37 °C. Then 5 μL of 12 mM 4MU-NeuAc were added to each sample and further incubated for 1 h at 37 °C. Reaction was then terminated by addition of 0.25 M glycine–NaOH at pH10 and fluorescence intensity measured at 448 nm emission and 365 nm excitation.
2.7 Cell line
PANC1 (human pancreatic carcinoma, epithelial-like, ATCC® CRL-1469™) cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were grown in a 5% CO2 incubator at 37 °C in culture containing 1 × DMEM (Gibco, Rockville, MD, USA) supplemented with 10% fetal calf serum (HyClone, Logan, UT, USA) and 5 μg mL−1 Plasmocin™ (InvivoGen, San Diego, CA, USA). When the cells reached ∼80% confluence, they were passaged at least five times for use in the experiments.
2.8 WST-1 cell viability assay
PANC1 cells were plated in 96-multiwell plates at a density of 5000 cells per well and incubated at 37 °C overnight. The following day, cells were exposed to 30 mg mL−1 pPEGMEMA, 7.5, 15, or 30 mg mL−1 oseltamivir-pPEGMEMA, or left untreated as a control for 0, 24, 48, and 72 h. pPEGMEMA and oseltamivir-pPEGMEMA were diluted in culture medium. 100 μL of WST-1 reagent diluted 1:10 in culture medium was added to wells for 2 h prior to reading of absorbance at 420 nm at each time point. Cell viability was presented as a percentage of control using GraphPad Prism software. The following formula was used to determine cell viability as a percent of control for each time point and treatment:
2.9 Fluorescent microscopy with coumarin-loaded micelles
PANC1 cells were plated on 12 mm glass coverslips in 24 multiwell plates at a density of 15000 cells per well and incubated overnight at 37 °C. Cells were washed once with phosphate-buffered saline pH 7.4 (PBS), incubated in CellMask™ Deep Red plasma membrane stain diluted 1:1000 in PBS at 37 °C for 7 min, then washed 3 times with PBS. Cells were treated with oseltamivir-pPEGMEMA or pPEGMEMA, or untreated as a control for 1 h at 37 °C. Cells were washed 3 times with PBS, then mounted on glass slides and visualized using a Zeiss M2 fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) at 200 and 400× magnification. The experiment was repeated, but PANC1 were treated with 25 μg mL−1 anti-Neu1 antibody for 1 h at 37 °C prior to membrane staining.
3. Results and discussion
3.1 Synthesis of oseltamivir-RAFT agent
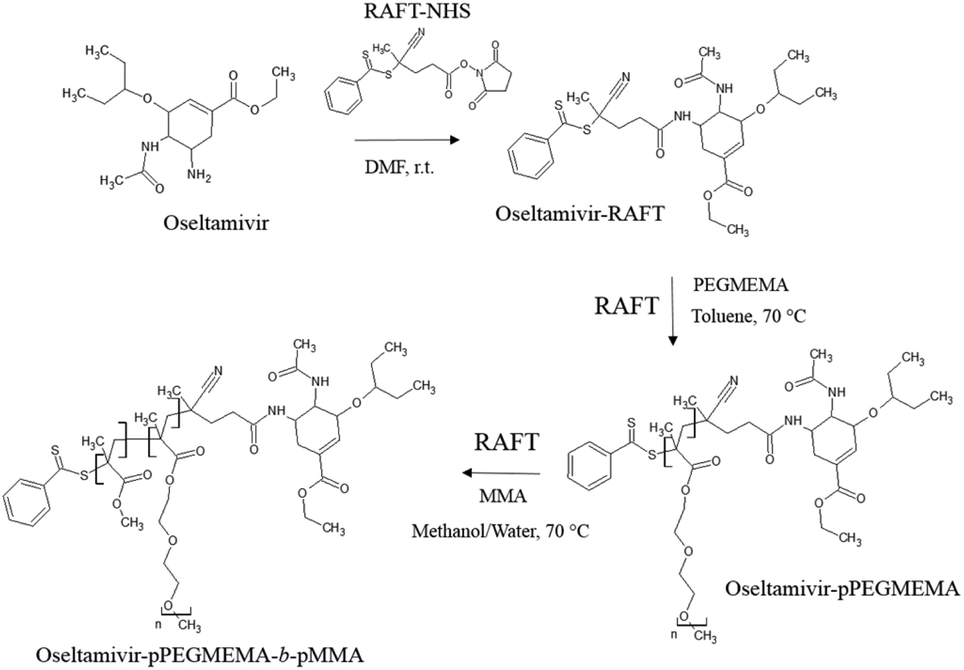
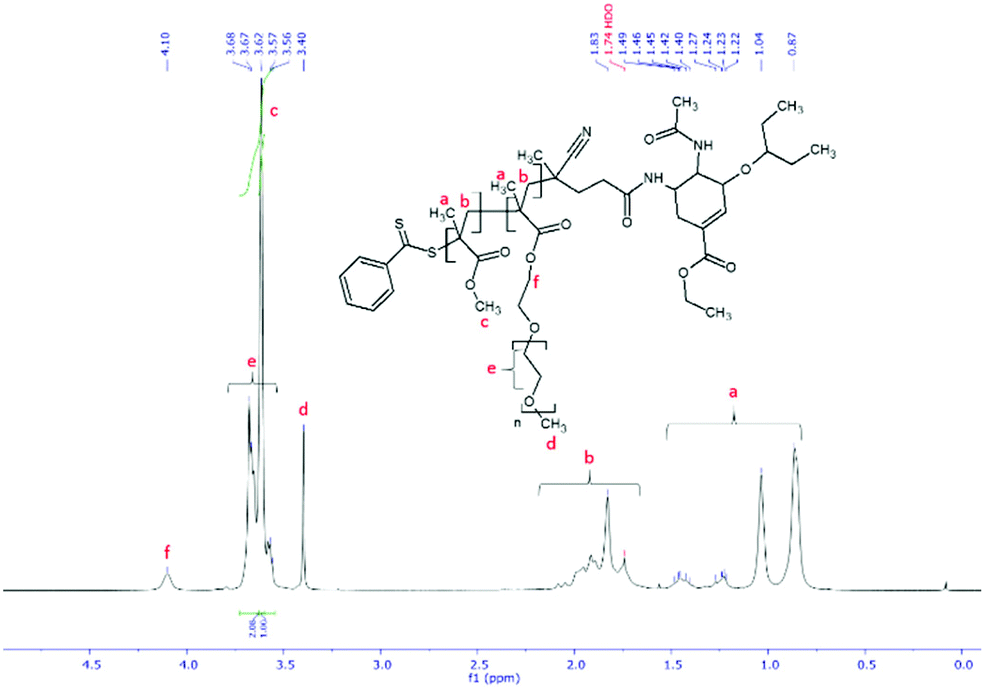
The first step in preparation of oseltamivir conjugated polymeric micelles was to attach the drug to the chain transfer agent (CTA) to be used in the RAFT polymerization, shown as the first step in Fig. 2. CTA selected was 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid due to its proven robustness in a range of solvents and an available carboxylic acid for amidation reaction with the amine group on oseltamivir. The carboxylic acid activated N-succinimidyl ester form of this CTA was reacted with excess oseltamivir to maximize the conversion of RAFT agent to a final amide conjugate (oseltamivir-RAFT) with 92% yield. 1H NMR of oseltamivir-RAFT is shown in Fig. 3. A complex set of peaks appearing in the phenylic ppm range is likely due to a side reaction of the CTA thioester bond with an ammonia form of oseltamivir and phosphoric acid during the acid wash step, resulting in partial decomposition of CTA, and therefore additional signals on NMR. However, the overall ratio of phenyl hydrogens to a hydrogen located at the double bond of the oseltamivir ring was 5 to 1, as determined by integration of corresponding peaks which is in agreement with the molecular structure of the new compound (oseltamivir-RAFT).
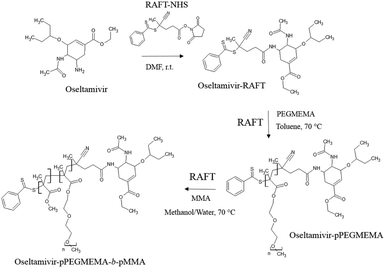 |
| | Fig. 2 Synthetic route for the preparation of oseltamivir-conjugated amphiphilic copolymer by RAFT polymerization. | |
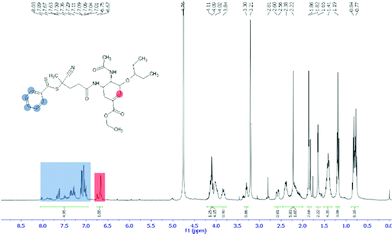 |
| | Fig. 3
1H NMR spectrum of oseltamivir-RAFT agent conjugate in methanol-d4. | |
3.2 Synthesis of oseltamivir-pPEGMEMA via RAFT polymerization
The 1H NMR spectrum of oseltamivir-pPEGMEMA as seen in Fig. S1 (ESI†) shows resonance signals at 1.7–2.1 ppm due to the main chain methylene protons, and a-methyl protons appearing at 0.7–1.1 ppm. The side chain methylene protons –C(O)–O–CH2– and –C(O)–O–CH2–CH2–O–(CH2–CH2–O)3/4–CH3 appeared at 4.1 and 3.5–3.7 ppm, respectively. The peak centered at 3.3 ppm was assigned to the –O–CH3 protons. Very weak signals, shown on Fig. S2,† at 7.3–7.9 ppm and 6.8 ppm corresponded to RAFT-phenyl and oseltamivir-(–CCH–) hydrogens respectively (still at 5:1 ratio) located at different terminals of pPEGMEMA. The retained 1:1 molar ratio of RAFT CTA to oseltamivir suggests no loss of drug during the RAFT polymerization of PEGMEMA. Two faint peaks at 5.6 ppm and 6.2 ppm are acrylic hydrogens coming from the traces of residual monomer.
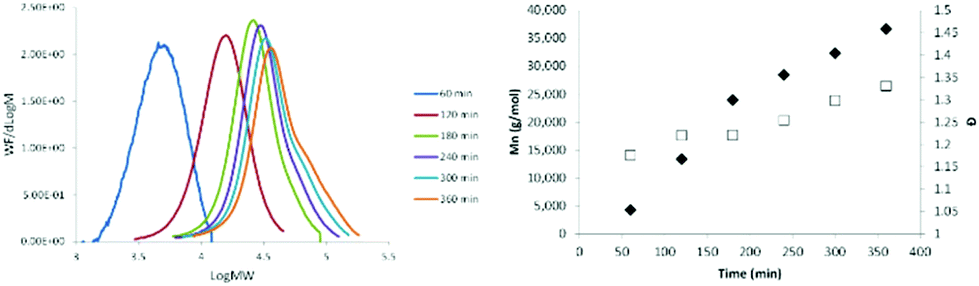
The progression with time of the molecular weight of oseltamivir-pPEGMEMA by RAFT LRP determined by GPC is presented in Fig. 4, and its progression over conversion (determined by 1H NMR) in Fig. S3.† After 6 h polymerization, conversion reached ∼85% (Fig. 4, right) and over time GPC traces showed polymers retaining low molecular weight dispersity (Đ 1.15–1.35), which is indicative of LRP. As seen from the GPC curves (Fig. 4, left), there is a slight broadening of the right (high molecular weight) shoulder of the molecular weight distributions after 3 h reaction time and at 60% conversion, probably due to increased termination side-reactions, which is also reflected by slightly increasing Đ as polymerization approaches full conversion. Well controlled RAFT-LRP was also demonstrated by observing first-order kinetic behavior presented in Fig. S3,† left. Linear dependence of the log of monomer concentration with time means that the number of active propagating species remains constant. Finally, number average molecular weight (Mn) of pPEGMEMA increased as a linear function of monomer conversion (Fig. S3,† right), yet again confirming constant number of chains during polymerization. Based on both first-order kinetic behavior and linear Mn progression over conversion, we have concluded that newly synthesized drug-conjugated RAFT CTA can be used for a well-controlled living polymerization.
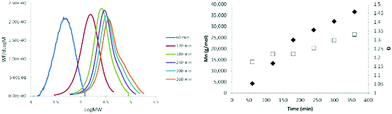 |
| | Fig. 4 Molecular weight distributions determined by GPC showing molecular weight progression (left) and molecular weight and dispersity (Đ) plotted against reaction time (right) (Mn ◆; Đ □) during RAFT polymerization of PEGMEMA from oseltamivir-RAFT agent. MW/dlogM – normalized weight fraction divided by log of normalized mole fraction, calculated from changes in refractive index intensity inside GPC detector. GPC: mobile phase THF, calibrated with PMMA standards. | |
3.3 Preparation of oseltamivir-conjugated micelle by RAFT PISA
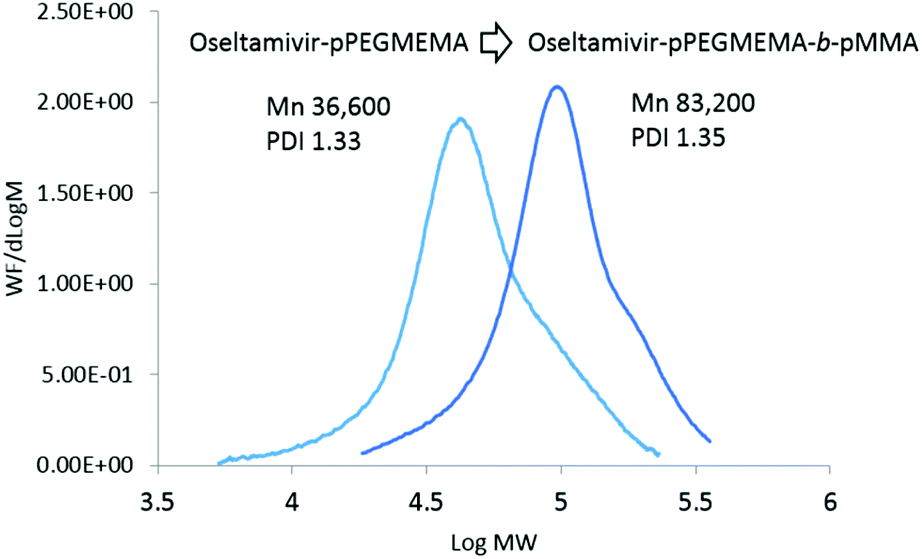
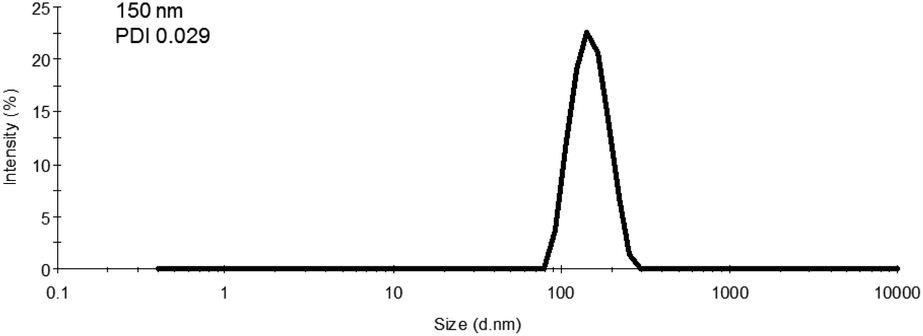
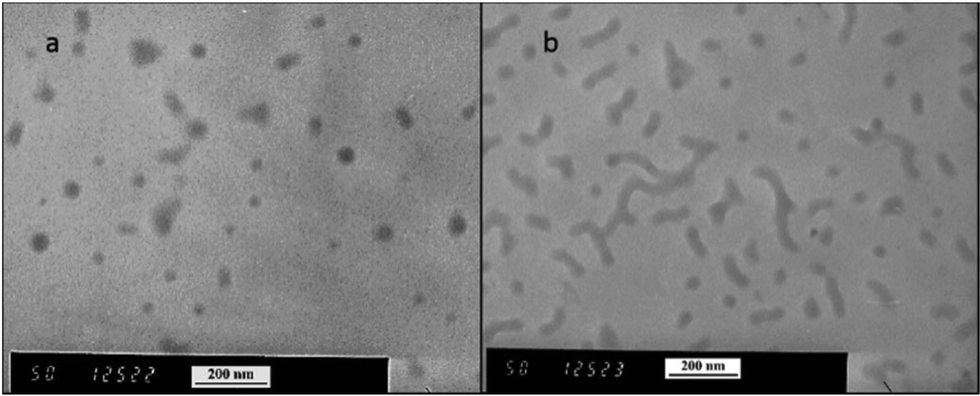
As the final synthetic step toward the preparation of oseltamivir-conjugated micelles, a hydrophobic block was grown from oseltamivir-pPEGMEMA by RAFT polymerization of methyl methacrylate (MMA) in a mixture of water/methanol to give oseltamivir-pPEGMEMA-b-pMMA amphiphilic block copolymer. A 1:1 v/v ratio of water/methanol was found to be suitable, with enough methanol to solubilize the required volumes of monomer (water insoluble MMA) and water for aqueous conditions used in the PISA stage. Monomer/CTA/initiator ratios were similar to RAFT polymerization of PEGMEMA but with slightly increased initiator concentration, just enough to increase the rate of polymerization without negatively affecting the molecular weight dispersity. PISA has occurred after 6 h of polymerization (29% conversion gravimetrically) and was visually observed as the polymerization solution turned milky-white. Formation of block copolymer was confirmed by 1H NMR and is shown in Fig. 5. Addition of pMMA to pPEGMEMA can be detected by the newly formed peak at 3.6 ppm (c), showing resonance of the methyl ester hydrogens. Integration of the methyl ester hydrogens (c, 3H) to ethylene glycol hydrogens (e, 4H) was translated to a roughly 1:1 ratio of pMMA to pPEGMEMA, which is in agreement with GPC data showing a ∼2× increase in molecular weight. The molecular weight of the polymer increased from 36600 g mol−1 at time zero to 83200 g mol−1 at the onset of PISA, as determined by GPC (Fig. 6). Oseltamivir-pPEGMEMA-b-pMMA molecular weight distribution remained narrow with low dispersity, showing only a slight increase (from Đ 1.33 to 1.35).The size of the self-assembled particles from RAFT-PISA, as determined by DLS, was 150 nm in diameter with very low size polydispersity (PDI 0.03) and is presented in Fig. 7. TEM imaging of pPEGMEMA-b-pMMA (Fig. 8a) and oseltamivir-pPEGMEMA-b-pMMA (Fig. 8b) micelles revealed that oseltamivir-conjugated polymers self-assembled into worm like micellar structures, while drug free polymers produced typical spherical micelles. It was previously reported that attaching pendant groups such as targeting ligands and prodrugs to amphiphilic block copolymers1,2 has an effect on the final morphology of self-assembled structures favouring formation of worm-like micelles due to steric effects of the pendant molecules on the outershell. In our case, worm-like micellar structures have only appeared in samples with oseltamivir-conjugated copolymers and never in drug free copolymers for this range of molecular weights of pPEGMEMA and pMMA blocks. Therefore, we concluded that existence of oseltamivir located at the pPEGMEMA end of pPEGMEMA-b-pMMA copolymers has a steric effect on the self-assembly into a micelle during RAFT-PISA. Fig. 8b shows worm-like micelles 100–200 nm in length and also some smaller spherical structures, possibly with lower oseltamivir content, 30–50 nm in diameter. This range of sizes is in agreement with DLS measurements which showed an average particle size of 150 nm.
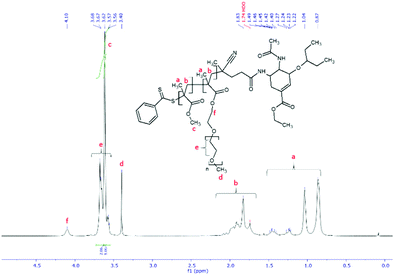 |
| | Fig. 5
1H NMR spectrum of oseltamivir-pPEGMEMA-b-pMMA in CDCl3. | |
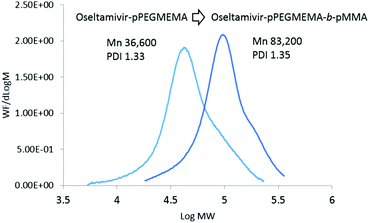 |
| | Fig. 6 Molecular weight distributions of oseltamivir-pPEGMEMA and oseltamivir-pPEGMEMA-b-pMMA after PISA. | |
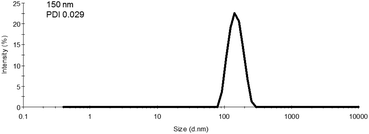 |
| | Fig. 7 Size measurement of oseltamivir-pPEGMEMA-b-pMMA micelles determined by DLS. The calculation of diameter from DLS assumes a spherical particle, and therefore the reported values for worm-like micelles are approximated to spherical equivalent diameters. | |
 |
| | Fig. 8 TEM images of pPEGMEMA-b-pMMA micelles (a) and oseltamivir-pPEGMEMA-b-pMMA micelles (b). | |
The 1:1 water–methanol mixture was selected to solvate all reagents initially (oseltamivir-pPEGMEMA, MMA and initiator) and also to induce self-assembly later in polymerization. The nature and composition of the solvent or solvent system could potentially affect the self-assembly and micellar shape. However, given that the intended use of the micelles is in aqueous environment, oseltamivir-conjugated amphiphilic polymers will ultimately rearrange into more thermodynamically preferred structures as demonstrated by TEM of micelles in water.
3.4 Oseltamivir-pPEGMEMA inhibits sialidase activity
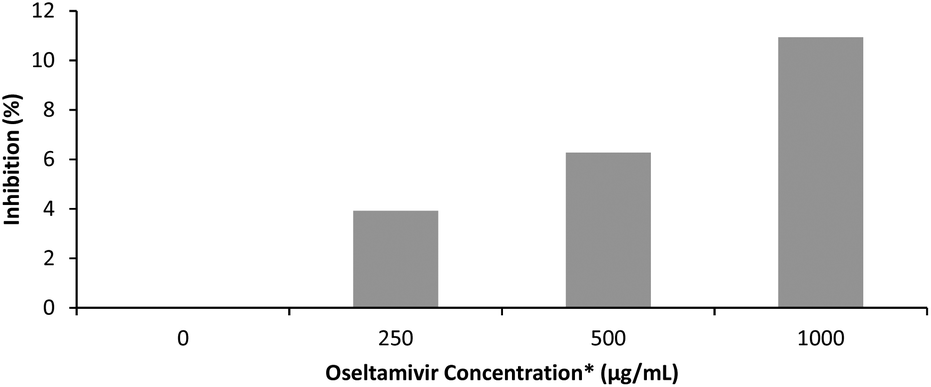
The effectiveness of oseltamivir in a polymer-conjugated form was assayed for Neu1 sialidase enzyme inhibition, simulated using a microbial α(2 → 3) neuraminidase from Streptococcus pneumoniae. This enzyme, like the human Neu1, cleaves sialic acid and is inhibited competitively by free oseltamivir which is an α(2 → 3) sialic acid analog. The inhibitory activity of oseltamivir-pPEGMEMA was measured using 4MU-NeuAc as substrate which gives a fluorescent product after being cleaved with α(2 → 3) neuraminidase. Because of the hydrophobicity of the fluorescent molecule produced from cleavage of 4MU-NeuAc by α(2 → 3) neuraminidase, it was decided not to conduct free sialidase activity assay with oseltamivir-conjugated micelles, due to potential diffusion of the fluorescent product into the hydrophobic core of the micelle, interfering with the fluorescent readings. Table 1 shows fluorescence intensity following reaction in the presence of three different concentrations of oseltamivir-pPEGMEMA. A concentration dependent decrease in fluorescence was observed and converted to concentration-dependent inhibition of α-2,3-neuraminidase by the oseltamivir-conjugated micelles, as depicted in Fig. 9. The conversion from fluorescence intensity to inhibition was done by taking the sample with only enzyme and substrate as representing 100% activity and 0% inhibition, and calculating the percent inhibition relative to that sample. Therefore, it was shown that oseltamivir exerts Neu1 sialidase inhibitory activity in a micelle-conjugated form. Inhibition suggests a competitive binding of oseltamivir-conjugate to the neuraminidase active site, and thus shows potential for binding to cell surface Neu1 within the EGFR complex.
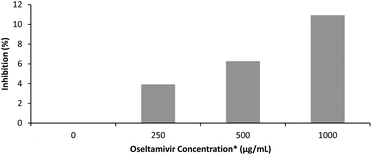 |
| | Fig. 9 Inhibitory effect of oseltamivir-pPEGMEMA on α(2 → 3) neuraminidase from Streptococcus pneumoniae measured by fluorescence. *oseltamivir concentration is calculated from an estimated oseltamivir content in micelle–drug conjugate determined by 1H NMR. | |
Table 1 Fluorescence intensity measurements from free α-2,3-sialidase activity assay
| [Oseltamivir-pPEGMEMA] (mg mL−1) |
Estimated [Oseltamivir] |
Fluorescence intensity |
| 0 |
0 μg mL−1 |
18490 |
| 3.75 |
250 μg mL−1 |
17770 |
| 7.5 |
500 μg mL−1 |
17330 |
| 15 |
1000 μg mL−1 |
16470 |
| Buffer |
|
0.515 |
3.5 Oseltamivir-pPEGMEMA reduces PANC1 cell viability
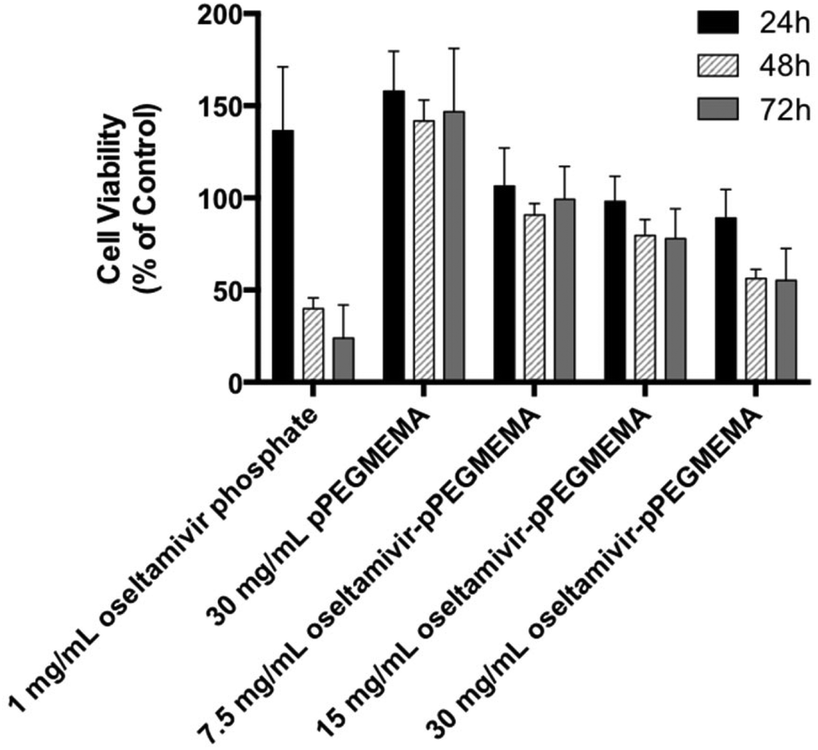
Since neuraminidase inhibition requiring oseltamivir-conjugate association was demonstrated using free enzyme, the potential for cell Neu1 binding was determined by the effect of conjugate on viability of PANC1 cells, which had previously been shown susceptible to free oseltamivir phosphate. Cell viability was determined using the WST-1 cell proliferation assay.16 The cell viability assay could not be conducted with oseltamivir-conjugated micelles due to the turbidity of the micellar solution interfering with the absorbance reading of the metabolite being quantified. Therefore, this assay was limited to water-soluble oseltamivir-pPEGMEMA conjugates. The data shown in Fig. 10 indicate that treatment of PANC1 cells with oseltamivir-conjugated polymer reproducibly and dose-dependently decreased cell viability expressed as a percent of the untreated control, with up to 45% viability reduction after 24, 48, and 72 h of incubation. The control polymer without oseltamivir did not impede growth of PANC1 cells when used at same polymer concentration (30 mg mL−1).
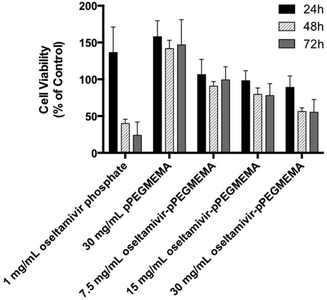 |
| | Fig. 10 Cell viability as a percent relative to untreated PANC1 cells (control), showing effect of free oseltamivir phosphate, oseltamivir-free polymer, and oseltamivir–polymer conjugate (∼3% w/w oseltamivir). n = 3, with error bars – mean ± standard error. | |
A direct comparison may be made between oseltamivir phosphate in the free form (1 mg mL−1) to the same amount of oseltamivir, but in the conjugate form, which is the case for 30 mg mL−1 oseltamivir-pPEGMEMA polymer. It is noteworthy that the pure oseltamivir phosphate showed a stronger reduction in cell viability than the polymer conjugated form which was likely due to increased steric hindrance around oseltamivir reducing access to the Neu1 active site. However, overall the results indicate that oseltamivir-conjugated polymer shows anti-PANC1 cell activity and oseltamivir therefore can potentially exert anti-tumor effect in micelle-conjugated form.
3.6 Interaction of oseltamivir-conjugated micelles with PANC1 cells
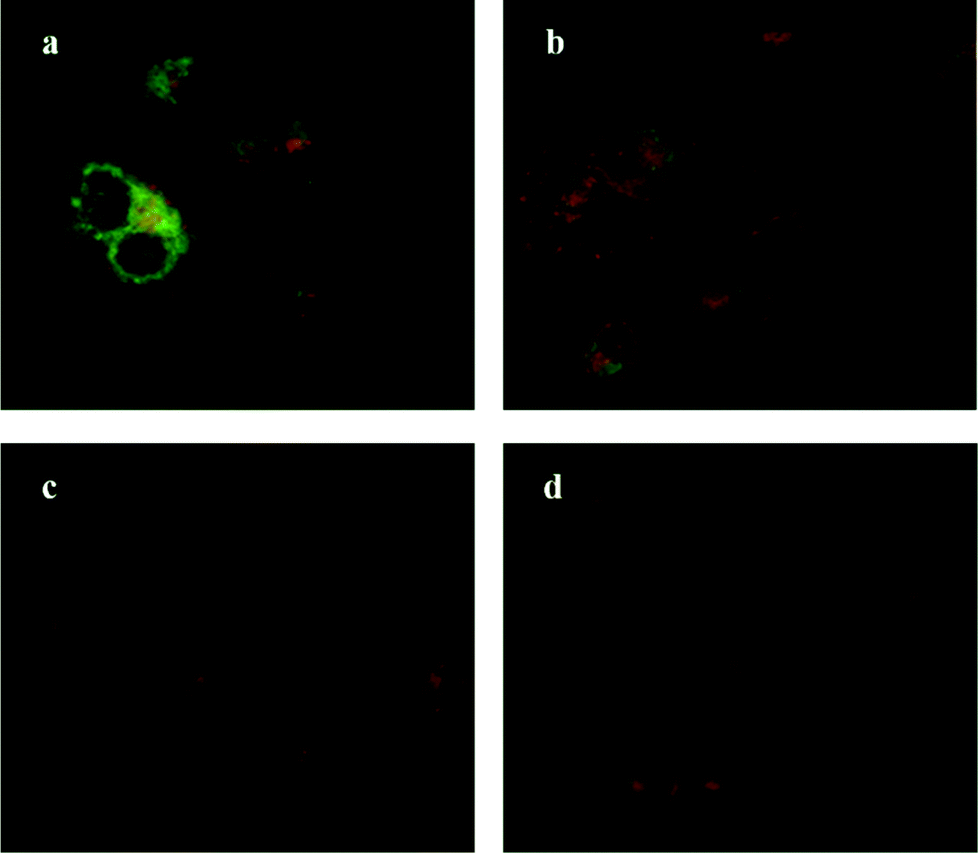
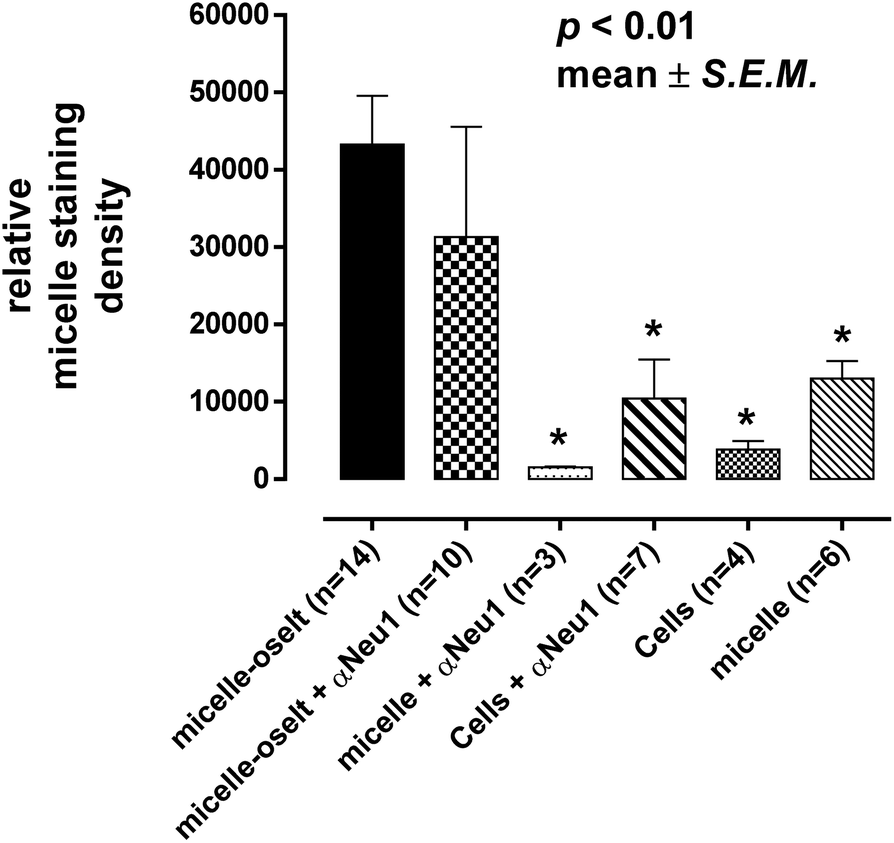
Oseltamivir-conjugate shows neuraminidase (Neu1) inhibition, and negatively affects viability of PANC1 cells, in a manner similar to free oseltamivir phosphate. Thus both Neu1 binding and cell inhibition can be anticipated with the new drug polymer conjugate. The potential then for not only the targeting of tumor cells, but the concomitant delivery of an oseltamivir-micelle filled chemotherapeutic was examined on PANC1 cells pre-stained with plasma membrane stain to potentially visualize surface Neu1-bound oseltamivir-micelles. To simulate the delivery of a hydrophobic chemotherapeutic, oseltamivir-micelles were loaded with the fluorescent probe coumarin-153 (C153), to serve as a hydrophobic drug model. The images depicted in Fig. 11 show red fluorescence for plasma membrane stain and green fluorescence for micelle loaded C153. Fig. 11a shows internalization of C153 loaded oseltamivir conjugated micelles within the plasma membrane of PANC1 cells. In contrast, C153 loaded micelles without oseltamivir did not show internalized C153, as seen in Fig. 11c, comparable to the micelle-free cell control seen in Fig. 11d. Thus it appears that oseltamivir conjugated to the outer surface of the micelles is necessary both for Neu1-EGFR binding, but also for cellular internalization of the micelles. To verify whether oseltamivir-conjugated micelles target Neu1, PANC1 cells were pre-treated with anti-Neu1 blocking antibodies followed by treatment with C153 loaded oseltamivir-conjugated micelles. Fig. 11b shows that anti-Neu1 blocking antibody resulted in a reduction of visible label within the cell cytoplasm. Fluorescent intensity was measured from a number of images for each condition and plotted as relative C153 staining density as shown in Fig. 12. Oseltamivir-conjugated micelles produced a 3.3-fold increased internalization compared to oseltamivir-free micelles, while the blocking action of anti-Neu1 antibody resulted in a 1.4-fold decrease in the internalization of C153 loaded oseltamivir-conjugated micelles. The extent of the effect of addition of anti-Neu1 antibody will greatly depend on the total population of Neu1 membrane protein in a given cell culture. Neu1 is also found in other membrane receptors which could in turn interfere with the output from this particular experiment. In our case, a fixed amount of antibody was added to determine whether or not there is any reduction in cellular uptake of micelles. We suggest that increasing the amount of antibody should further decrease the uptake of micelles.
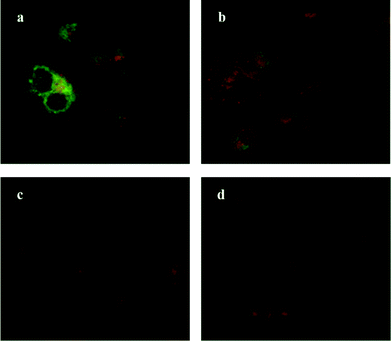 |
| | Fig. 11 Fluorescent microscopy images of pancreatic tumor cells, stained with deep red cell mask, incubated with (a) C153-loaded oseltamivir-conjugated micelle, (b) C153-loaded oseltamivir-conjugated micelle plus anti-Neu1 antibody, (c) C153-loaded micelles without oseltamivir and (d) media (no micelles). | |
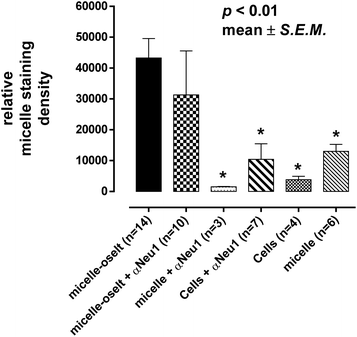 |
| | Fig. 12 Quantitative analysis of cell uptake of micelle-conjugated oseltamivir (oselt) loaded with coumarin 153 by measuring relative fluorescence staining density from microscopy images. *Number of samples n = number of individual cells from one or more images from each experiment. Oselt – oseltamivir; αNeu1 – anti-Neu1 antibody. | |
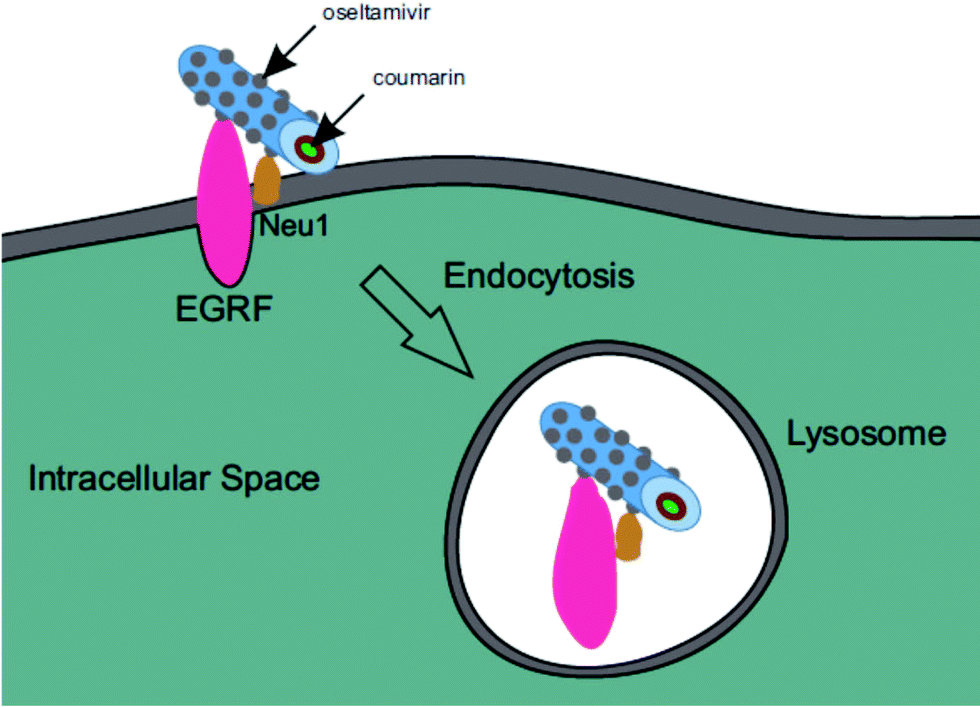
Oseltamivir-conjugated micelles are thus shown to be binding to Neu1 in complex with EGFR. A mechanism that can explain micelle internalization carrying C153 as a drug model is receptor-induced endocytosis (RIE). Endocytic mechanisms have been previously explored for targeted drug delivery by attaching high affinity ligands on vesicles to bind with clathrin, transferrin and folate receptors.17 Endocytosis of EGFR is a part of the receptor recycling process triggered by extracellular signal molecules followed by internalization of receptor bound molecule complex into lysosomes for degradation.18 It has been reported that binding of EGF to EGFR induces the internalization of the receptor into lysosomes for intracellular degradation.19 Although it was found that Neu1 inhibition by free oseltamivir did not result in receptor internalization,11 it is possible that the oseltamivir decorated micelle exerts additional steric and/or conformational changes to the receptor that now triggers endocytosis, as illustrated in Fig. 13. Thus oseltamivir conjugated micelles could induce endocytosis of EGFR via binding to the receptor's Neu1 sialidase located at the ectodomain of EGFR and through interaction with EGFR, triggers internalization. The exact mechanism of EGFR activation by the oseltamivir-micelle and subsequent internalization at the molecular level is presently the subject of further investigated. Nevertheless, this active targeting approach has potential for delivery of hydrophobic anti-cancer agents such as doxorubicin into the target cell cytoplasm.
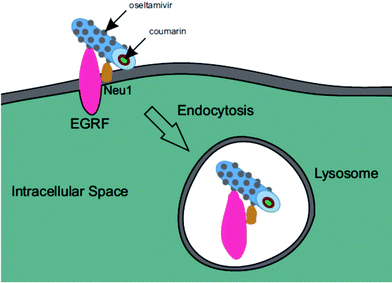 |
| | Fig. 13 Graphic representation of EGFR induced endocytosis of oseltamivir micelle via binding to Neu1. | |
Oseltamivir-conjugated micelles designed to target Neu1 in complex with EGFR offer a new and effective way of targeting, delivering and internalizing a chemotherapeutic agent via oseltamivir-Neu1 binding followed by receptor-induced endocytosis. Compared to other targeting ligands such as growth factors and antibodies used in active tumor targeting, which are typically expensive, unstable and potentially immunogenic, oseltamivir is a small, relatively inexpensive molecule which can be easily attached to the end of polymer via reactive amine group and is expected to be biologically stable when conjugated to a micelle. A synthetic route for oseltamivir-conjugated micelles via RAFT polymerization by linking the drug to the chain transfer agent is only one of many ways to prepare oseltamivir decorated nanocarriers, serving as a template for development of other oseltamivir-based cancer targeting platforms. Finally, as seen from sialidase enzyme activity assay, and PANC1 cell viability assays, oseltamivir, in the polymer conjugated form, shows both sialidase inhibition and anti-PANC1 cell activity of around 45% of its free form, oseltamivir phosphate. Therefore, oseltamivir-decorated nanocarriers can provide a triple-effect of targeting and exerting an anti-tumor cell effect directly from oseltamivir, combined with a subsequent internalization and delivery of a separate chemotherapeutic as demonstrated by the C153 as a model hydrophobic drug.
4. Conclusion
The synthesis and preparation of oseltamivir-conjugated micelles formed via RAFT polymerization was designed for active targeted delivery of a hydrophobic chemotherapeutic. This new drug delivery platform was designed to target the EGFR complex located and overexpressed on the surface of tumor cells, and more specifically bind to Neu1 within the EGFR complex. The synthetic route consisted of three major steps: conjugating oseltamivir with RAFT chain transfer agent, RAFT polymerization of a hydrophilic polymer block, and finally RAFT polymerization of a hydrophobic block. The resultant oseltamivir-conjugated block copolymer self-assembled into nanosize micelles with the oseltamivir ends orienting into the aqueous phase. Polymer-conjugated oseltamivir was shown to both inhibit neuraminidase activity representing sialidase activity of Neu1 within the EGFR, and reduce the viability of PANC1 cells, in a manner previously demonstrated for free oseltamivir phosphate. Further evidence for Neu1 cell surface binding was provided by demonstrating that C153 loaded oseltamivir conjugate was internalized by PANC1 cells but not so in the absence of the linked oseltamivir, or when cells had been pre-treated with anti-Neu1 blocking antibody. It is proposed that oseltamivir micelle conjugate cell internalization occurs as a result of binding to cell surface Neu1 within the EGFR complex, triggering receptor-induced endocytosis, as part of the EGFR recycling process. Therefore, oseltamivir conjugated to the surface of the micelle, acts as a targeting molecule, enabling internalization of the micelles. The addition of C153 loaded within the micelles as model hydrophobic chemotherapeutic, demonstrates the potential for targeted delivery and subsequent internalization of other hydrophobic chemotherapeutics. These newly designed micelles for active tumor targeting thus have dual functionality by exerting an anti-tumor cell effect, and at the same time delivering and internalizing a hydrophobic chemotherapeutic.
Acknowledgements
This work was supported in part by grants to M. R. Szewczuk and R. J. Neufeld from the Natural Sciences and Engineering Research Council of Canada, and the Ontario Ministry of the Environment through the Council of Ontario Universities to M.R. Cunningham.
V. Kapishon was the recipient of the R. Samuel McLaughlin Fellowship of the School of Graduate Studies of Queen's University. S. Allison is the recipient of the Queen's Graduate Award (QGA).
References
-
(a) E. Segal and R. Satchi-Fainaro, Design and development of polymer conjugates as anti-angiogenic agents, Adv. Drug Delivery Rev., 2009, 61(13), 1159–1176 CrossRef CAS PubMed;
(b) D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol., 2007, 2(12), 751–760 CrossRef CAS PubMed;
(c) M. E. Davis, Z. G. Chen and D. M. Shin, Nanoparticle therapeutics: an emerging treatment modality for cancer, Nat. Rev. Drug Discovery, 2008, 7(9), 771–782 CrossRef CAS PubMed;
(d) S. S. Suri, H. Fenniri and B. Singh, Nanotechnology-based drug delivery systems, J. Occup. Med. Toxicol, 2007, 2, 16 CrossRef PubMed.
- Y. Noguchi, J. Wu, R. Duncan, J. Strohalm, K. Ulbrich, T. Akaike and H. Maeda, Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues, Jpn. J. Cancer Res., 1998, 89(3), 307–314 CrossRef CAS PubMed.
-
(a) N. Nishiyama and K. Kataoka, Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery, Pharmacol. Ther., 2006, 112(3), 630–648 CrossRef CAS PubMed;
(b) C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: Promises, progress and prospects, Nano Today, 2012, 7(5), 467–480 CrossRef CAS.
-
(a) J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, Polymeric micelles drug delivery system in oncology, J. Controlled Release, 2012, 159(3), 312–323 CrossRef CAS PubMed;
(b) U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Advances in polymeric micelles for drug delivery and tumor targeting, Nanomedicine, 2010, 6(6), 714–729 CrossRef CAS PubMed;
(c) W. Xu, P. Ling and T. Zhang, Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs, J. Drug Delivery, 2013, 2013, 340315 Search PubMed;
(d) V. P. Torchilin, A. N. Lukyanov and Z. Gao, Papahadjopoulos-Sternberg, B., Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(10), 6039–6044 CrossRef CAS PubMed;
(e) F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(7), 2586–2591 CrossRef CAS PubMed.
-
(a) K. Matyjaszewski and N. V. Tsarevsky, Macromolecular engineering by atom transfer radical polymerization, J. Am. Chem. Soc., 2014, 136(18), 6513–6533 CrossRef CAS PubMed;
(b) G. Moad, E. Rizzardo and S. H. Thang, RAFT polymerization and some of its applications, Chem. – Asian J., 2013, 8(8), 1634–1644 CrossRef CAS PubMed;
(c) G. Moad, E. Rizzardo and S. H. Thang, Toward living radical polymerization, Acc. Chem. Res., 2008, 41(9), 1133–1142 CrossRef CAS PubMed;
(d) M. H. Stenzel, RAFT polymerization: an avenue to functional polymeric micelles for drug delivery, Chem. Commun., 2008,(30), 3486–3503 RSC;
(e) A. W. York, S. E. Kirkland and C. L. McCormick, Advances in the synthesis of amphiphilic block copolymers via RAFT polymerization: stimuli-responsive drug and gene delivery, Adv. Drug Delivery Rev., 2008, 60(9), 1018–1036 CrossRef CAS PubMed.
- S. Pascual, C. N. Urbani and M. J. Monteiro, Functionalization of Polymer Nanoparticles Formed by Microemulsion RAFT-Mediated Polymerization, Macromol. React. Eng., 2010, 4(3–4), 257–263 CrossRef CAS.
- Z. Jia, V. A. Bobrin, N. P. Truong, M. Gillard and M. J. Monteiro, Multifunctional Nanoworms and Nanorods through a One-Step Aqueous Dispersion Polymerization, J. Am. Chem. Soc., 2014, 136(16), 5824–5827 CrossRef CAS PubMed.
- W. Wang, C. Li, J. Zhang, A. Dong and D. Kong, Tailor-made gemcitabine prodrug nanoparticles from well-defined drug-polymer amphiphiles prepared by controlled living radical polymerization for cancer chemotherapy, J. Mater. Chem. B, 2014, 2(13), 1891–1901 RSC.
- S. Abdulkhalek, O. Geen, L. Brodhagen, F. Haxho, F. Alghamdi, S. Allison, D. Simmons, L. O'Shea, R. J. Neufeld and M. R. Szewczuk, Transcriptional factor Snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma, Clin. Transl. Med., 2014, 3 Search PubMed.
- F. Haxho, S. Allison, F. Alghamdi, L. Brodhagen, V. E. Kuta, S. Abdulkhalek, R. J. Neufeld and M. R. Szewczuk, Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma, Breast Cancer: Targets Ther., 2014, 6, 191–203 CAS.
- A. M. Gilmour, S. Abdulkhalek, T. S. Cheng, F. Alghamdi, P. Jayanth, L. K. O'Shea, O. Geen, L. A. Arvizu and M. R. Szewczuk, A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer, Cell. Signalling, 2013, 25(12), 2587–2603 CrossRef CAS PubMed.
- S. Abdulkhalek, M. Hrynyk and M. R. Szewczuk, A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease, Res. Rep. Biochem., 2013, 3, 17–30 Search PubMed.
- H. Q. Xiong and J. L. Abbruzzese, Epidermal growth factor receptor-targeted therapy for pancreatic cancer, Semin. Oncol., 2002, 29(5 Suppl 14), 31–37 CrossRef CAS PubMed.
-
(a) H. Safran, T. Dipetrillo, D. Iannitti, D. Quirk, P. Akerman, D. Cruff, W. Cioffi, S. Shah, N. Ramdin and T. Rich, Gemcitabine, paclitaxel, and radiation for locally advanced pancreatic cancer: a Phase I trial, Int. J. Radiat. Oncol., Biol., Phys., 2002, 54(1), 137–141 CrossRef CAS;
(b) H. Safran and R. Rathore, Paclitaxel as a radiation sensitizer for locally advanced pancreatic cancer, Crit. Rev. Oncol. Hematol., 2002, 43(1), 57–62 CrossRef PubMed.
-
(a) L. K. O'Shea, S. Abdulkhalek, S. Allison, R. J. Neufeld and M. R. Szewczuk, Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu(R)) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance, OncoTargets Ther., 2014, 7, 117–134 Search PubMed;
(b) M. Hrynyk, J. P. Ellis, F. Haxho, S. Allison, J. A. Steele, S. Abdulkhalek, R. J. Neufeld and M. R. Szewczuk, Therapeutic designed poly (lactic-co-glycolic acid) cylindrical oseltamivir phosphate-loaded implants impede tumor neovascularization, growth and metastasis in mouse model of human pancreatic carcinoma, Drug Des., Dev. Ther., 2015, 9, 4573–4586 Search PubMed.
- A. S. Tan and M. V. Berridge, Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: a simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents, J. Immunol. Methods, 2000, 238(1–2), 59–68 CrossRef CAS PubMed.
-
(a) S. Xu, B. Z. Olenyuk, C. T. Okamoto and S. F. Hamm-Alvarez, Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances, Adv. Drug Delivery Rev., 2013, 65(1), 121–138 CrossRef CAS PubMed;
(b) K. G. Rothberg, Y. S. Ying, J. F. Kolhouse, B. A. Kamen and R. G. Anderson, The glycophospholipid-linked folate receptor internalizes folate without entering the clathrin-coated pit endocytic pathway, J. Cell Biol., 1990, 110(3), 637–649 CrossRef CAS PubMed;
(c) J. J. Turek, C. P. Leamon and P. S. Low, Endocytosis of folate-protein conjugates: ultrastructural localization in KB cells, J. Cell Sci., 1993, 106(Pt 1), 423–430 CAS;
(d) P. Dalhaimer, A. J. Engler, R. Parthasarathy and D. E. Discher, Targeted worm micelles, Biomacromolecules, 2004, 5(5), 1714–1719 CrossRef CAS PubMed;
(e) E. J. Oh, K. Park, K. S. Kim, J. Kim, J. A. Yang, J. H. Kong, M. Y. Lee, A. S. Hoffman and S. K. Hahn, Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives, J. Controlled Release, 2010, 141(1), 2–12 CrossRef CAS PubMed.
- L. K. Goh and A. Sorkin, Endocytosis of receptor tyrosine kinases, Cold Spring Harbor Perspect. Biol., 2013, 5(5), a017459 Search PubMed.
- L. Beguinot, R. M. Lyall, M. C. Willingham and I. Pastan, Down-regulation of the epidermal growth factor receptor in KB cells is due to receptor internalization and subsequent degradation in lysosomes, Proc. Natl. Acad. Sci. U. S. A., 1984, 81(8), 2384–2388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5bm00519a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence