
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustained release of active chemotherapeutics from injectable-solid β-hairpin peptide hydrogel†
Jessie E. P.
Sun
a,
Brandon
Stewart
a,
Alisa
Litan
b,
Seung Joon
Lee
b,
Joel P.
Schneider
c,
Sigrid A.
Langhans
b and
Darrin J.
Pochan
*a
aDepartment of Materials Science & Engineering, University of Delaware, Newark, DE 19176, USA. E-mail: pochan@udel.edu
bNemours Center for Childhood Cancer Research, Nemours/A.I. duPont Hospital for Children, Wilmington, DE 19803, USA
cCenter for Cancer Research, National Cancer Institute at National Institute of Health, Frederick, MD 21702, USA
First published on 24th February 2016
Abstract
MAX8 β-hairpin peptide hydrogel is a solid, preformed gel that can be syringe injected due to shear-thinning properties and can recover solid gel properties immediately after injection. This behavior makes the hydrogel an excellent candidate as a local drug delivery vehicle. In this study, vincristine, a hydrophobic and commonly used chemotherapeutic, is encapsulated within MAX8 hydrogel and shown to release constantly over the course of one month. Vincristine was observed to be cytotoxic in vitro at picomolar to nanomolar concentrations. The amounts of drug released from the hydrogels over the entire time-course were in this concentration range. After encapsulation, release of vincristine from the hydrogel was observed for four weeks. Further characterization showed the vincristine released during the 28 days remained biologically active, well beyond its half-life in bulk aqueous solution. This study shows that vincristine-loaded MAX8 hydrogels are excellent candidates as drug delivery vehicles, through sustained, low, local and effective release of vincristine to a specific target. Oscillatory rheology was employed to show that the shear-thinning and re-healing, injectable-solid properties that make MAX8 a desirable drug delivery vehicle are unaffected by vincristine encapsulation. Rheology measurements also were used to monitor hydrogel nanostructure before and after drug encapsulation.
Introduction
A current strategy for chemotherapeutic delivery vehicles is to use injectable delivery vehicles that can directly deliver chemotherapeutics or other drug therapies. Injectable vehicles include nanoparticles,1,2 polymer gels,3–5 or micelles all loaded with chemotherapeutics.6,7 Many of these vehicles are surface modified or functionalized with ligands or protein sequences for better targeting.8,9 Presently, there are two types of injectable vehicles, those introduced intravenously and those introduced through site-specific local delivery. Intravenous delivery typically introduces a particle into the body, with modifiable targeting, drug encapsulation, and drug release methods.10,11 While useful for broad targets easily reached by the blood stream, in some cases the vehicles coalesce in the kidney or liver permanently.12,13 Site-specific, local delivery vehicles can be useful, reducing healthy tissue exposure to possibly toxic drugs. Once administered, the drug-encapsulated vehicles can continuously administer active drugs through controlled.One family of drug delivery vehicles with potential for effective and sustained release is the hydrogel. Hydrogels are water-based three-dimensional solid networks composed of polymer chains. One use of hydrogels are as platforms for local, injectable applications, with the capability to encapsulate and distribute a wide range of materials such as drugs,14,15 large proteins,16,17 and even cells.18–20 After injection for deposition, the hydrogel could continue to release chemotherapeutics while remaining in the desired location for a prolonged, desired period of time, reducing the need for more surgeries and invasive procedures. Ideally, hydrogels also possess shear-thinning and self-healing capabilities that allow for more specific injectable locations and fewer needs for additional surgeries for continuous care, carrying fewer risks for complications.21 The Pochan and Schneider groups have investigated extensively various β-hairpin forming peptide hydrogels that are able to intermolecularly self-assemble into nanofibrillar, physical hydrogels as a result of an intramolecular folding response.22–27 These β-hairpin peptide hydrogels display injectable-solid properties; solid hydrogels that exhibit shear-thinning flow during syringe injection but also exhibit immediate solid recovery after cessation of shear. In addition, the hydrogel material properties such as gelation time, stiffness, and network mesh size are tunable via molecule design as well as solution conditions that control the intermolecular self-assembly into a hydrogel network.
Currently, many injectable hydrogels are designed as precursor, low viscosity solutions ex vivo that then assemble in vivo when exposed to environmental triggers such as temperature,25,28 ions,29,30 pH,31,32 or ultraviolet (UV) radiation.30,32–38 External triggers such as UV radiation may damage nearby tissue, whereas introduction of non-physiological materials like iron oxide may lead to long term effects that greatly influence the body.39–43 The injectable solid hydrogel properties allow solid gel formation within a syringe, after which injection and deposition can occur without the need for further external interactions. From this family of β-hairpin peptides, MAX8 is a model candidate as a payload delivery vehicle. MAX8 has been studied for in vitro and in vivo studies because it self-assembles at physiological conditions and can successfully encapsulate many different types of payloads. Previous studies using MAX8 have shown successful, homogenous encapsulation of various particles,44 drugs,14 and cell lines.23,45–47 Branco et al. encapsulated dextran probes of neutral charge and varying sizes to better understand MAX8 network characteristics.44 The probes revealed the average pore sizes of the overall networks through their diffusion profiles from the hydrogel. Yan et al. encapsulated mesenchymal stem cells (MSC) to better understand effects of shear on the overall hydrogel system.46 Smaller molecules have also been studied. For example, MAX8 hydrogels have been utilized to encapsulate curcumin, a hydrophobic chemotherapeutic agent.14 Curcumin is a natural compound derived from the Indian spice turmeric and degrades after 8 hours in water.48 Altunbas et al. successfully encapsulated and released curcumin from MAX8. Despite the high water content of the MAX8 hydrogel, the continuously-released curcumin remained active and effective after 14 days of encapsulation.
Vincristine, the target drug, is a long accepted, intravenously delivered, and commonly used clinical chemotherapeutic).49 Vincristine alone, or in a combination, is usually administered to treat many types of cancers, including lymphoma (Hodgkin's and Non-Hodgkin's),50–52 leukemia,53,54 glioma,55,56 embryoma,57 lung cancer,58 and neuroblastoma.59 Vincristine disrupts cell division by binding to tubulin, poisoning the tubulin heterodimer, then incorporating itself into microtubule bundles to prevent further growth.60,61 However, the effectiveness of vincristine also leads to many adverse side effects such as organ toxicity, nausea/vomiting, and hair loss.50,51 Vincristine is unable to differentiate healthy cells from cancerous cells and will target any dividing cell indiscriminately.60,61 Several rounds of treatments are required in order to provide constant exposure of the cancerous cells to vincristine. Naturally, this prolonged exposure to the drug leads to an increase in detrimental side effects in patients.
In this work, vincristine is encapsulated within MAX8 hydrogel to show that the drug-hydrogel construct is a promising candidate as a site specific local delivery vehicle, with the potential to minimize overall invasiveness and damage to healthy tissue through the local, continuous release of the chemotherapeutic from the hydrogel. Importantly, the hydrogel provides a protective environment for the hydrophobic drug in the deposited area, so that released drug continues to be effective at killing cancer cells at month-long time scales. We first demonstrate, using oscillatory rheometry, that the presence of vincristine within the MAX8 network does not alter the general viscoelastic properties, and specific shear-thinning and self-healing properties, that make it attractive as a drug delivery vehicle. In addition, small-angle neutron scattering (SANS) measurements find that the structure of the MAX8 network (e.g., fibrillar character, porous network) is not altered significantly by the presence of vincristine and that the drug appears to be closely associated with the fibrillar nanostructure and not relegated to separate domains of drug within the fibrillar network. Vincristine release from the hydrogel was quantified using tritium-labeled vincristine, and release profiles confirm that vincristine is released continuously from the material for up to 28 days from encapsulation. Furthermore, in vitro studies demonstrate that vincristine remains biologically active after 28 days – over 20 times longer than its half-life in bulk water. The present work shows that in contrast to the current intravenous vincristine delivery method vincristine-loaded MAX8 hydrogels provide sustained, low, but effective release to a specific target and may be excellent candidates as drug delivery vehicles that exhibit minimal side effects and damage to healthy tissue.
Results and discussion
MAX8 hydrogel rheology and structure
The injectable solid properties of MAX8, or shear thinning and immediate solidification, make the material a desirable injection delivery vehicle. To ensure that the hydrogel retains these properties with drug included, the storage (G′) and loss (G′′) moduli of the system were measured with a frequency sweep for 0.5 wt% MAX8 hydrogel with or without 500 μM vincristine. The storage and loss moduli characterize the elastic and viscous behavior of the material.23,62 As shown in Fig. 1a, there is a negligible difference between G′ and G′′ with and without vincristine for the MAX8 hydrogel showing that the presence of the drug does not alter the material properties of the hydrogel. Moreover, these data show that once deposited, the drug-gel construct will retain all the desirable gel physical properties of MAX8.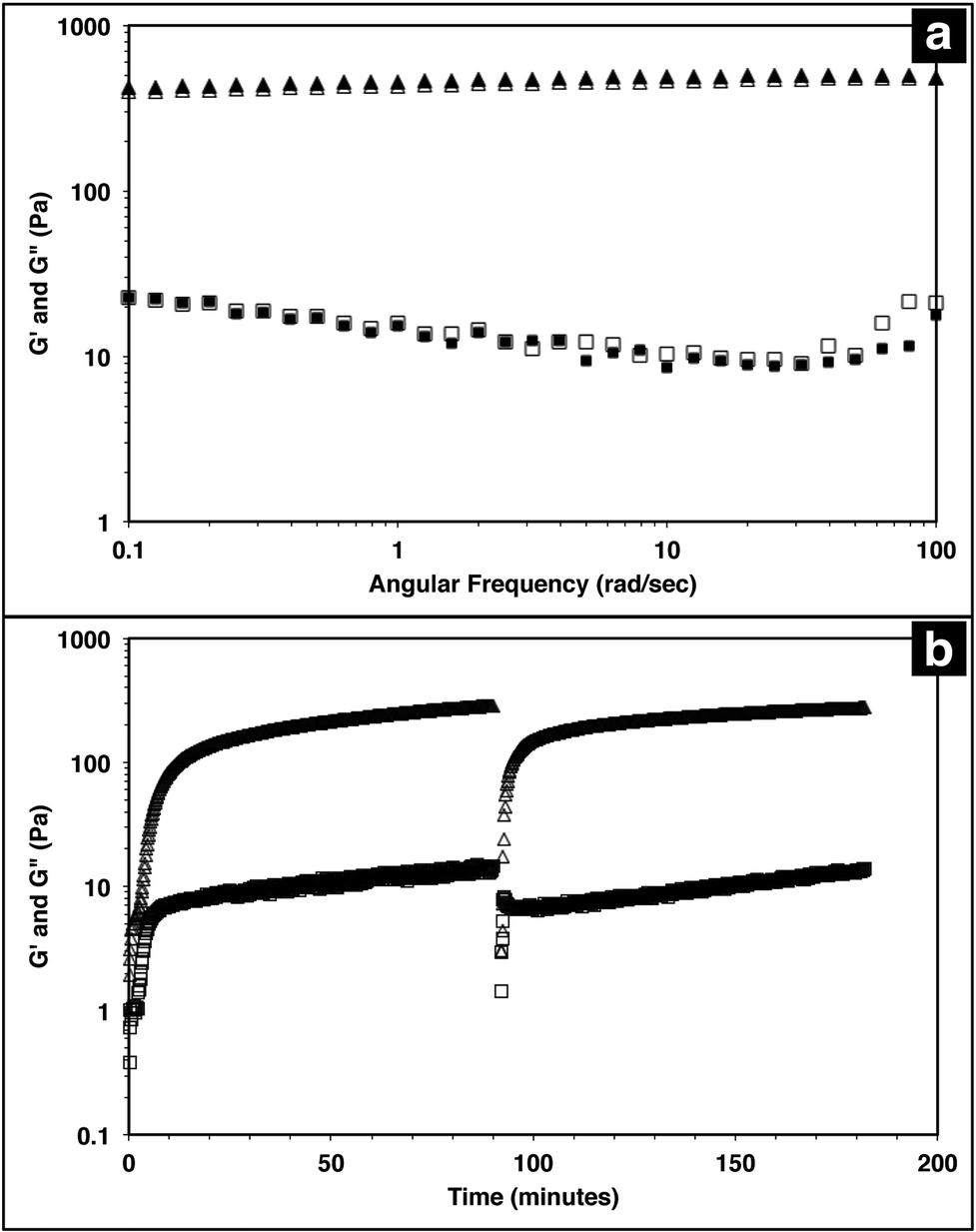 | ||
| Fig. 1 Triangles correspond to G′ (storage modulus), and squares correspond to G′′ (loss modulus). (a) A frequency sweep from 0.1–100 rad s−1 with 0.2% strain was run for 0.5 wt% MAX8 hydrogels with 500 μM vincristine (filled symbols) and without vincristine (open symbols). No difference is observed in the viscoelastic properties of the hydrogel with and without vincristine encapsulated. (b) A time sweep at a frequency of 6 rad s−1 with a 0.2% strain was run on a 0.5 wt% MAX8 hydrogel with 500 μM vincristine encapsulated. Early time shows the initial gelation within 10 minutes. A constant shear at a steady-state shear of 1000 s−1 is applied for 30 seconds at 90 minutes. As soon as the large shear ceases, the time sweep data shows the hydrogel immediately as a solid material and quickly recovering original gel properties. | ||
Previous studies have shown that when a constant shear force is applied on the hydrogel, the material flows with properties of a low viscosity material.23,62 Once shear forces cease, the hydrogel has been shown to immediately recover solid gel properties, reaching pre-shear peak G′ and G′′ values quickly after shearing. Fig. 1b demonstrates the same shear-thinning and re-healing properties of MAX8 with 500 μM of vincristine encapsulated. Thus, after gelation the drug-loaded hydrogel flows easily when sheared and recovers original properties of the presheared gel after shear cessation. This ability is critical for delivery applications, allowing the hydrogel to be injected into a specific site and trusted to recover to a gel state with known properties and to stay in place at the injection site.
In order to better understand all of the drug-hydrogel construct properties, it is key to characterize where the drug molecule sits within the network. The rheology seen in Fig. 1 shows no change in hydrogel behavior with or without drug loading, indicating that the drug is not affecting the overall hydrogel network itself. However, the rheology data does not help determine specifically the location of the vincristine within the network. Small-angle neutron scattering (SANS) was performed to determine whether vincristine alters the structure of the fibrillar nanostructure and where the drug is located within the nanostructure of the network. Fig. 2 shows the scattering profile of 0.5 wt% MAX8 hydrogel with 500 μM vincristine (shaded squares) and without (open squares). The I(q) versus q measurement determines sample structure, giving information in the length scale of nanometers to hundreds of nanometers. The presence of the vincristine does not alter significantly the overall shape and intensity, implying that the hydrogel structure is practically identical in both cases. The SANS results reveal that when encapsulated, there are only two ways the drug could be incorporated into the overall hydrogel network: (A) either in aggregated vincristine clusters with as little exposure to the surrounding aqueous environment or (B) intimately associated along the fibrils throughout the network. Vincristine domains would both scatter as individual particles of polydisperse size and shape due to the large hydrogen content within the drug molecules as well as most likely displaying interparticle correlations due to their presence throughout the gel network. Both of these effects would increase significantly intensity at both low and mid-q. The lack of a significant difference in overall curve shape for low and mid q scattering, in both intensity and slope, confirms that there are no size differences in the morphology of the hydrogel networks with or without vincristine. While not significant enough to change the curve shape, there is a definite, albeit slight, increase in intensity within the mid-q range, associated with the nanofibrillar characteristics of the overall hydrogel, most likely comes from the increase in contrast between the hydrogel nanofibrils and the deuterated buffer solvent. This difference in intensity due to a higher contrast suggests, like the cartoon, that the vincristine is organized along the fibrils of the network. If the vincristine were able to incorporate significantly into the core of the nanofibrils, there would most likely be a distortion in fibril width and an increase in fibril branching leading to a large difference in gel stiffness as well as fibril nanostructure. The rheology shown in Fig. 1 shows that the storage moduli are the same with or without drug, indicating no fibrillar disruption or gel network differences, suggesting that the vincristine is not within the fibrils. Another way of confirming the presence of fibrils, is to measure the slope in the mid-q range of a SANS scattering measurement. A slope around −1 in this range is indicative of nanofibrillar structure. In this case, as seen in Fig. 2, the slope was measured in the q-range of 0.015 to 0.05. For hydrogels with and without 500 μM of vincristine the slope was 0.922 and 0.948 respectively, both close to one, indicating preservation of nanofibrillar structure. TEM images included in the ESI† do not clearly show the location or presence of vincristine within the MAX8 fibrils, but do show the fibril width for the samples with and without vincristine are not different. Showing that the presence of vincristine does not interrupt or alter the structure of vincristine itself, important for the preservation of MAX8's shear-thinning properties.
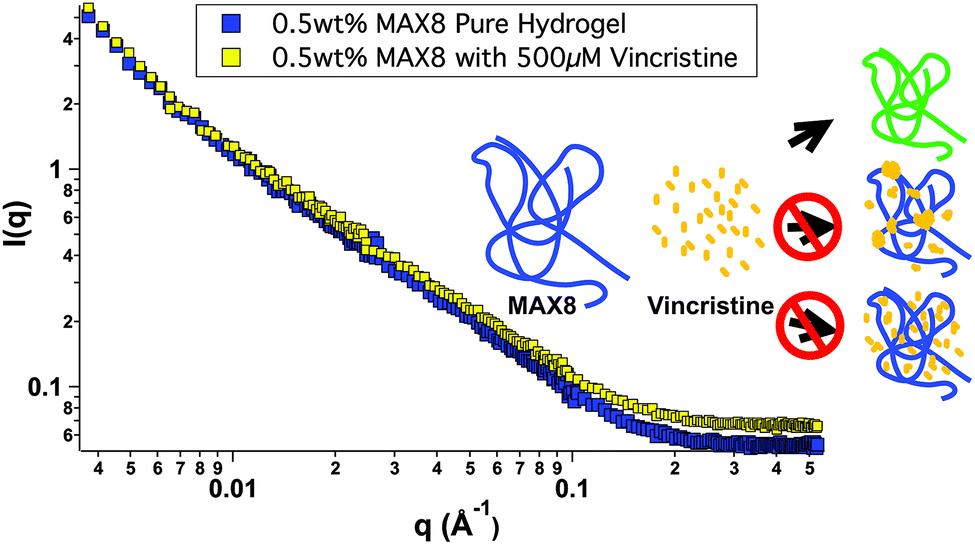 | ||
| Fig. 2 Small-angle neutron scattering from 0.5 wt% MAX8 hydrogels with 500 μM vincristine (yellow) and without (blue) as a function of scattering variable q. Both lines have a similar overall shape and slope throughout the measured q range, implying that the presence of vincristine does not alter the structure of the MAX8 gel or the intramolecular folding of individual MAX8 chains. The cartoon inset shows the possible drug-gel configurations, (a) green fibrils indicating the yellow vincristine bound to the blue MAX8 fibrils, (b) domains of yellow vincristine mostly at the branch and entanglement points, or (c) yellow vincristine evenly scattered throughout the MAX8 network. | ||
Most likely, the vincristine evenly incorporates itself around the outside of the fibrils, perhaps buried within the hydrophobic lysine side chains.47 A third possibility of vincristine freely moving throughout the entire network is discounted because of the lack of change in intensity of the scattering. Were the vincristine unassociated with the fibrillar network and freely soluble in the buffer background, the intensity of the hydrogel-drug sample curve would be less than the pure hydrogel at low and mid q due to the presence of the hydrogenated drug compounds floating freely in solution and lowering the contrast between the peptide fibrils and the deuterated solvent. The fact that the intensity goes up slightly in the drug-containing hydrogel signifies a slight increase in contrast due to the association of the drug compound along the length of the hydrogel fibril nanostructure. A more in-depth SANS experiment is needed for a longer period of release to better understand the nanostructure of the network with drug release for a prolonged period of time.
In vitro study
In order to show MAX8 would be an effective delivery vehicle, releasing vincristine to induce cell death, a series of in vitro studies were performed. The immortalized DAOY cell line was chosen as an acceptable model for medullablastoma. In order to show the IC50 value, the concentration of vincristine directly applied for treatment was in the picomolar range. These picomolar concentrations agreed with previous in vitro studies, consistent with the potency of vincristine.53 In order to measure the IC50 for cells being treated either directly with vincristine, or by vincristine released from a MAX8 gel, a series of decreasing concentrations for both directly applied and hydrogel-released vincristine were prepared. An LDH assay was performed for both models to find the IC50 as presented in Fig. 3. Fig. 3a and b both show that cell death increases as the concentration of vincristine increases. For direct treatment, the IC50 was determined to be between 5 nM and 25 nM, after showing a clear trend of cell death with increasing drug concentration of treatment. For the encapsulated vincristine the IC50 is reached when 8 nM of vincristine is encapsulated into a hydrogel and then exposed to cells. It should be noted that the released drug concentration for the direct applied treatment are extremely low.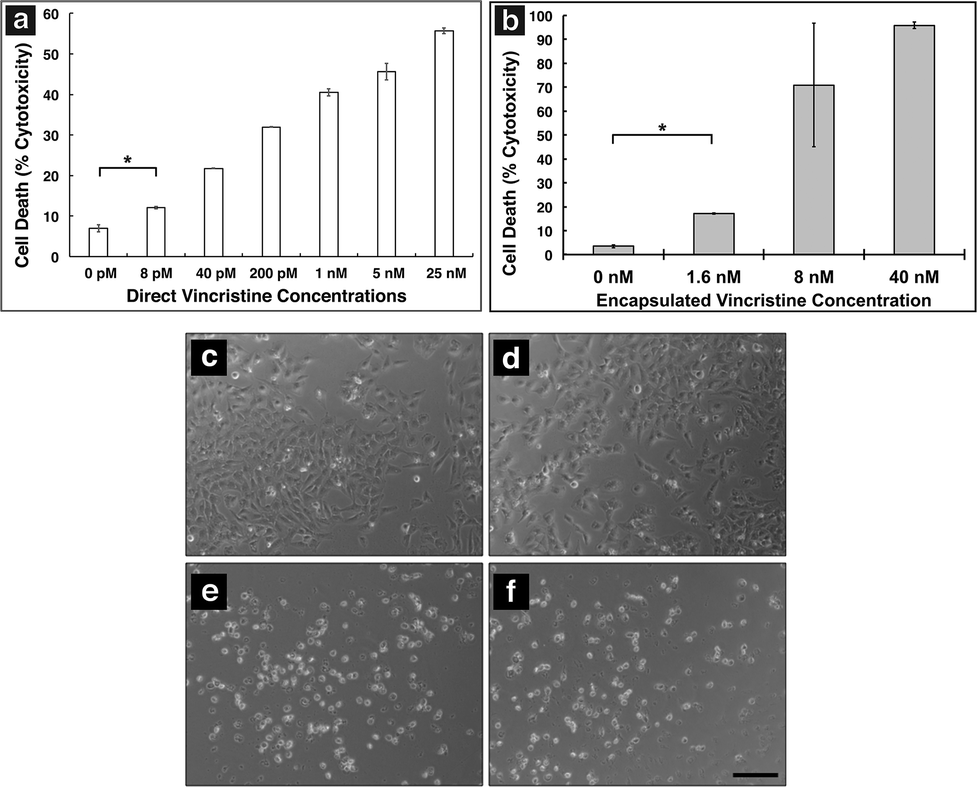 | ||
| Fig. 3 LDH assays showing percent cell death of DAOY cells (a) after direct treatment with vincristine and (b) vincristine encapsulated and released from a 0.5 wt% MAX8 hydrogel. All measurements are taken after 2 days of cell incubation with each listed concentration. * indicates significance (p < 0.05). Optical micrographs of DAOY cells treated with (c) 0 nM (d) 1.6 nM (e) 8 nM and (f) 40 nM of vincristine released from a MAX8 hydrogel. Live cells appear elongated and transparent whereas dead cells are rounded and opaque. The scale bar is 200 μm. | ||
Determining the IC50 concentrations was important in ensuring that vincristine encapsulated in MAX8 would still induce cell death, and drug concentration affected cell death percentage. When beginning the in vitro experiments, 500 μM was first attempted. This first concentration was chosen since it is the highest concentration that could be encapsulated due to the limited solubility in aqueous solution of hydrophobic vincristine. But the potency of vincristine quickly showed that micromolar was too high of a concentration, killing cell populations completely. But the result clearly shows that the concentrations of drug required for original encapsulation prior to release can be very low and still effective/useful for local delivery. These low values demonstrate that lower vincristine doses are still effective and would minimize the amount of undesirable side effects and healthy cell death during local delivery. Fig. 3c–f shows light microscope images of the cells treated with the corresponding concentrations of vincristine encapsulated in the hydrogel to confirm the presence of the drug is responsible for cell death. The 0 nM sample consisted of pure MAX8 hydrogel without any vincristine. The presence of the MAX8 does not result in significant cell death, indicating any cell death with vincristine is a result of the drug, while at 40 nM, the cells are almost all round and opaque, showing clear signs of cell death.
Vincristine release and sustained drug potency
A month long time release of vincristine from a 0.5 wt% MAX8 hydrogel containing 10 μM tritiated vincristine encapsulated in 0.5 wt% is shown in Fig. 4. The time points are of concentrations measured at days 1 (accumulated from measurements between hours 1 through 6, and 24 hours), 3, 7, 10, 14, 17, 21, 24, and 28 in the release. The inset of Fig. 4 highlights days 14, 17, 21, 24, and 28 to show that the released concentrations are non-zero at these long time points of release. In particular, note that after 28 days, approximately 2 nM concentration vincristine is still released from the gel.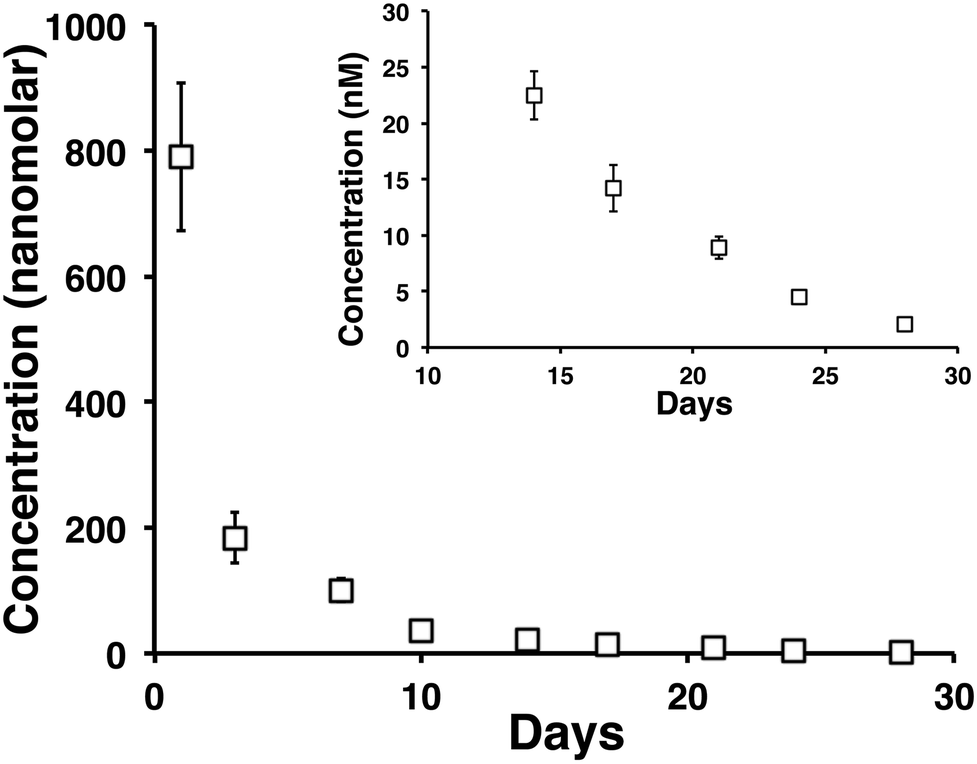 | ||
| Fig. 4 The 28 day release profile of tritiated vincristine from a 0.5 wt% MAX8 peptide hydrogel that initially encapsulated 10 μM vincristine. After 28 days of release, the amount of drug being released was still in nanomolar quantities, as seen in the inset which highlights days 14 through 28 of the release study. | ||
In order to ensure the vincristine released from the hydrogel is still biologically active after prolonged hydrogel encapsulation we determined the efficacy of vincristine to induce cell death in DAOY cells at extended time points The experimental set up mimicked the release study but with an additional interaction step with fresh DAOY cells after long time points of drug release. A negative control of 0.5 wt% MAX8 hydrogel without vincristine was run at the same time to establish that the cells were dying from the presence of the drug and not the hydrogel or environment.
Two encapsulated drug concentrations were used to test the sustained drug potency. The first set up was for 10 μM concentration of encapsulated vincristine. This concentration was chosen to match the concentration that was used for the release study in Fig. 4. The second set up considered a higher concentration of 500 μM to show a difference in release amounts at the highest possible initial drug concentration due to the limited solubility of vincristine. Fig. 5 shows a clear increase in cell death for the higher vincristine concentration, confirming that the cell death is a result of the encapsulated vincristine. At first glance, it may seem contrary that the encapsulated vincristine experiment in Fig. 3b showed higher percentage cell death at 8 nM and 40 nM, both lower than 10 μM, than in the efficacy study in Fig. 5. However, the experiment setups for the two are greatly different. The cell death measured for time 0 in Fig. 5 is after only an hour of cell exposure to the drug-gel construct, as opposed to Fig. 3, where the cells were exposed for two days, until the LDH assay was performed. These two days meant that there was an accumulation of released drug within the wells. The later release and efficacy studies were modified to simulate a more realistic environment, closer to an infinite sink.
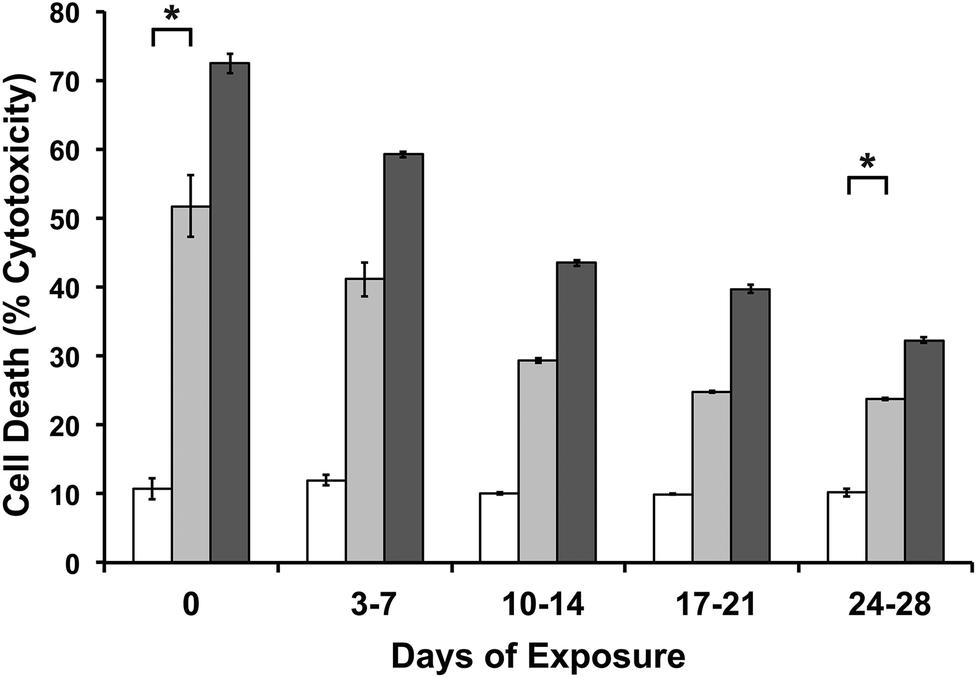 | ||
| Fig. 5 To ensure efficacy of the drug released from they hydrogel, 0 μM (empty bars), 10 μM (lighter gray bars) and 500 μM (darker gray bars) of vincristine were encapsulated in 0.5 wt% MAX8 hydrogels. There is a noticeable difference in the effectiveness of the hydrophobic drug on cells despite encapsulation in an aqueous. DAOY cell death was measured using LDH assays after cells were exposed to the drug-gel constructs in the listed days of release. Day 0 cells were exposed for an hour to drug-gel transwells, then measured two days later. * indicates noted concentrations are significantly different (p < 0.05). | ||
In Fig. 5, for both concentrations, cell death is significantly greater in vincristine encapsulated MAX8 than hydrogels without vincristine, even after a month of continuous release in an aqueous environment. The concentration of 2.04 nM ± 0.31 nM released after 28 previous days of release for the 10 μM vincristine encapsulated hydrogel should be sufficient to kill almost half the population of cells according to Fig. 3a containing direct treatment data. However, as seen in Fig. 5, the cells dying due to the presence of vincristine is to a lower extent than predicted by Fig. 3a, implying the vincristine is slightly less effective after 28 days of being encapsulated inside the hydrogel. This indicates that there is a percentage of vincristine that deteriorates in the aqueous environment, but, more importantly, that there is also a significant percentage of vincristine that remains effective after 28 days and significant previous release. Fig. 5 shows that the percentage of effective vincristine also increases with increased initial encapsulated drug concentration.
Previous studies of vincristine have shown that very low amounts of vincristine are extremely effective. Tsuruo et al. showed IC50 values of less than 2 nM for direct treatment of leukemia cells.53 However, much higher concentrations, ranging from 1 μM to 100 μM, are used for intravenous treatments because of the poor target specificity of the drug.52,63,64 Vincristine has a bulk solution half life range of 164 minutes to 32 hours65,66 within the body due to its hydrophobicity and functionality. While in aqueous solution, vincristine has a half-life of 136 hours, this is at its most stable in a pH range of 3.5 to 5.6, much lower than physiological pH.67 These studies have shown these cytotoxic effectiveness of released drug has been protected by the MAX8 hydrogel for longer than what has been measured in the body. In usage, once injected, the vincristine-loaded hydrogel can be relied on to continuously release low but effective concentrations of vincristine to the intended site to treat cancers and other diseases. The SANS data in Fig. 2 looks at overall structure and vincristine location to help in understanding the mechanics of the encapsulation and ultimately, the release from the hydrogel. The efficacy study suggests the vincristine's location within the fibrils as suggested by Fig. 2 is important to its protection from an aqueous environment. There is clearly some delay of drug exposure to a degrading environment, shielding the vincristine from its surroundings to achieve the high half-life, similar to pro-drugs or time-release drugs.
Attempts to prolong hydrophobic drug half-life in aqueous environments do so by isolating the drug from the environment in a separate hydrophobic area through encapsulation.68,69 The difference with the MAX8 hydrogel is that there is no distinctly hydrophobic cavity that would offer overall obvious protection. The SANS data of Fig. 2 shows that with or without vincristine there are no major differences in nanofibril or overall network characteristics. As mentioned earlier, the vincristine is mostly likely shielded by the lysine side chains, providing long-time drug stability. This protection coupled with the continued release of vincristine from the 0.5 wt% MAX8 hydrogel further support the use of the drug-hydrogel construct for local and targeted drug delivery to a tumor environment while decreasing the exposure and effects on healthy tissue.
Experimental
Materials and methods
500 μM vincristine encapsulated in 0.5 wt% MAX8 was prepared for the shear-thinning experiment. The shear-thinning experiment was subjected to a time sweep at a frequency of 6 rad s−1 with 0.2% strain as the hydrogel assembled after mixing. Next, the hydrogel was subjected to a steady-state shear at 1000 s−1 for 30 seconds. After 30 seconds, the rheometer returned to a dynamic sweep oscillatory measurement, and the hydrogel was monitored for 90 minutes.
MAX8 hydrogels (0.5 wt%) were prepared with a final concentration of 1.6 nM, 8 nM, and 40 nM vincristine. Additionally, a hydrogel without any vincristine was prepared as a control. For the in vitro studies, vincristine applied directly to cells in culture was compared to the vincristine that was released into the culture medium after encapsulation in the hydrogel. DAOY cells were plated in a 24-well plate and incubated overnight in DMEM. For hydrogel drug delivery, 100 μL of MAX8-vincristine gel-drug construct was pipetted into a transwell polyester membrane insert and allowed an additional 20 minutes to complete assembly/rehealing after injection. After the initial wait, each transwell was inserted into a well of 2 mL of DMEM to remove unencapsulated vincristine. The transwell inserts were left in the wash for 20 minutes before being added subsequently to the DAOY cell plates. For experiments with direct treatment of vincristine, 100 μL of vincristine at the desired concentration was added directly into wells with 2 mL of DMEM and plated DAOY cells. Each measurement was measured three times and averaged. The direct treatment cell wells had 8 pM, 40 pM, and 200 pM vincristine concentrations directly in contact with the cultured cells.
To measure cell death, released LDH from dead cells was isolated through centrifugation from the supernatant medium of the cells at desired time points. To measure LDH within live cells, the cells were lysed and crushed after freeze–thawing. Cytotoxicity was then determined on the basis of the ratio of LDH released into the medium to the sum of medium LDH and viable cell LDH.
To correlate scintillation counts with vincristine concentration, a calibration curve was created for each day of measurements at five known concentrations of 10 pM, 100 pM, 1 nM, 10 nM, and 100 nM. 100 μL of each known concentrations was added to 3 mL of scintillation fluid and measured for 5 minutes on the LSC. The calibration was performed separately for each day of measurement in order to account for fluctuations in sample radioactivity and background radiation.
Conclusions
Vincristine, a hydrophobic chemotherapeutic, was successfully encapsulated and subsequently released from the shear-thinning, re-healing peptidic hydrogel MAX8. The release of vincristine is shown to be continuous over the course of a month, with the released drug remaining effective at kill cancer cell populations. SANS and rheology were used to characterize vincristine's interactions with MAX8, and to better understand where the vincristine is positioned in relation to the hydrogel. The vincristine does not disrupt the fibrillar nature of MAX8 or its physically cross-linked properties, insuring the drug-gel construct is an ideal injectable, delivery vehicle.While direct treatment of cells is prudent in an in vitro setting, during actual cancer treatment other non-cancer cells present in the environment should not be exposed to chemotherapy compounds such as vincristine. Current methods of treating cancers with vincristine lead to negative side effects due to the large, systemic dosages required and healthy tissue exposed. These large dosages are needed because of the lack of specific drug targeting. In order to better treat specific regions of the body, such as the site of a newly resected tumor, a specific, local delivery with an injectable solid delivery system using a shear-thinning hydrogel is a viable strategy. This deposition of chemotherapeutic would minimize the need for repeated treatments or intrusions.
In practice, the sustained release will allow a targeted area to receive treatment continuously over long time periods that will alleviate problems seen in multiple, frequent chemotherapy treatments that are used for systemic treatment today. These multiple treatments expose healthy tissue to vincristine, leading to negative side effects. The shear thinning and immediate re-healing properties of MAX8 hydrogel allows the deposition of the drug-loaded, solid hydrogel directly to a desired injection site. Additionally, the injection would be ideal for post-operative treatment after tumor removal surgeries by depositing the drug-gel construct into the cancer's previous location. The low dosage and continuous release of the vincristine can target any cancerous cells that may not have been resected as well as preventing the return of any cancer in that area.
Acknowledgements
This work was funded by a seed grant from the University of Delaware's NIH Center of Biomedical Research Excellence, entitled Molecular Design of Advanced Biomaterials (P20-RR017716) and the Nemours Foundation. Research facilities were supported in part by the current Delaware COBRE program, supported by a grant from the National Institute of General Medical Sciences – NIGMS (1 P30 GM110758-01) from the National Institutes of Health. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. This work utilized facilities supported in part by the National Science Foundation under Agreement no. DMR-0944772. This manuscript was prepared under cooperative agreement 70NANB12H239 from NIST, U.S. Department of Commerce. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the view of NIST or the U.S. Department of Commerce.Notes and references
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853–862 CrossRef CAS PubMed.
- A. Altunbas, N. Sharma, M. S. Lamm, C. Yan, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, ACS Nano, 2010, 4, 181–188 CrossRef CAS PubMed.
- M. Boustta, P.-E. Colombo, S. Lenglet, S. Poujol and M. Vert, J Control Release, 2014, 174, 1–6 CrossRef CAS PubMed.
- D. Zhang, P. Sun, P. Li, A. Xue, X. Zhang, H. Zhang and X. Jin, Biomaterials, 2013, 34, 10258–10266 CrossRef CAS PubMed.
- J.-K. Cho, K.-Y. Hong, J. W. Park, H.-K. Yang and S.-C. Song, J. Drug Targeting, 2011, 19, 270–280 CrossRef CAS PubMed.
- F. P. Seib and D. L. Kaplan, Biomaterials, 2012, 33, 8442–8450 CrossRef CAS PubMed.
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 CrossRef CAS PubMed.
- R. Mooney, Y. Weng, E. Garcia, S. Bhojane, L. Smith-Powell, S. U. Kim, A. J. Annala, K. S. Aboody and J. M. Berlin, J. Controlled Release, 2014, 191, 82–89 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2012, 42, 202–224 RSC.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- E. S. Glazer, C. Zhu, A. N. Hamir, A. Borne, C. S. Thompson and S. A. Curley, Nanotoxicology, 2011, 5, 459–468 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS PubMed.
- E. Fournier, C. Passirani, C. N. Montero-Menei and J. P. Benoit, Biomaterials, 2003, 24, 3311–3331 CrossRef CAS PubMed.
- W. H. Blackburn, E. B. Dickerson, M. H. Smith, J. F. McDonald and L. A. Lyon, Bioconjugate Chem., 2009, 20, 960–968 CrossRef CAS PubMed.
- S. Lindsey, J. H. Piatt, P. Worthington, C. Sönmez, S. Satheye, J. P. Schneider, D. J. Pochan and S. A. Langhans, Biomacromolecules, 2015, 16, 2672–2683 CrossRef CAS PubMed.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2012, 18, 806–815 CrossRef CAS PubMed.
- K. J. Lampe and S. C. Heilshorn, Neurosci. Lett., 2012, 519, 138–146 CrossRef CAS PubMed.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315–4323 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2011, 8, 260–272 RSC.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS PubMed.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. A. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 Search PubMed.
- D. Pochan, J. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS PubMed.
- J. Schneider, D. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- J. H. Collier, B. H. Hu, J. W. Ruberti, J. Zhang, P. Shum, D. H. Thompson and P. B. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS PubMed.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- J. Kopecek and J. Yang, Acta Biomater., 2009, 5, 805–816 CrossRef CAS PubMed.
- D. N. Woolfson, Biopolymers, 2010, 94, 118–127 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, Biopolymers, 2012, 98, 169–184 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2010, 132, 9526–9527 CrossRef CAS PubMed.
- J. H. Collier and T. Segura, Biomaterials, 2011, 32, 4198–4204 CrossRef CAS PubMed.
- J. Kopecek and J. Yang, Angew. Chem., Int. Ed., 2012, 51, 7396–7417 CrossRef CAS PubMed.
- T. Nicolai and D. Durand, Curr. Opin. Colloid Interface Sci., 2013, 18, 249–256 CrossRef CAS.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2011, 3, 18–33 RSC.
- A. K. Vellimana, V. R. Recinos, L. Hwang, K. D. Fowers, K. W. Li, Y. Zhang, S. Okonma, C. G. Eberhart, H. Brem and B. M. Tyler, J. Neurooncol., 2012, 111, 229–236 CrossRef PubMed.
- N. B. Varukattu and S. Kannan, Int. J. Biol. Macromol., 2012, 51, 1103–1108 CrossRef PubMed.
- N. L. Elstad and K. D. Fowers, Adv. Drug Delivery Rev., 2009, 61, 785–794 CrossRef CAS PubMed.
- L. Tavano, M. Vivacqua, V. Carito, R. Muzzalupo, M. C. Caroleo and F. Nicoletta, Colloids Surf., B, 2013, 102, 803–807 CrossRef CAS PubMed.
- C. Lorenzato, A. Cernicanu, M. E. Meyre, M. Germain, A. Pottier, L. Levy, B. D. Senneville, C. Bos, C. Moonen and P. Smirnov, Contrast Media Mol. Imaging, 2013, 8, 185–192 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- J. K. Kretsinger, L. A. Haines, B. Ozbas and D. J. Pochan, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS PubMed.
- C. Yan, M. E. Mackay, K. Czymmek, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Langmuir, 2012, 28, 6076–6087 CrossRef CAS PubMed.
- L. A. Haines-Butterick, D. A. Salick, D. J. Pochan and J. P. Schneider, Biomaterials, 2008, 29, 4164–4169 CrossRef CAS PubMed.
- V. Basile, E. Ferrari, S. Lazzari, S. Belluti, F. Pignedoli and C. Imbriano, Biochem. Pharmacol., 2009, 78, 1305–1315 CrossRef CAS PubMed.
- T. J. Smith, J. Khatcheressian, G. H. Lyman, H. Ozer, J. O. Armitage, L. Balducci, C. L. Bennett, S. B. Cantor, J. Crawford and S. J. Cross, J. Clin. Oncol., 2006, 24, 3187–3205 CrossRef CAS PubMed.
- P. P. Carbone, V. Bono, E. Frei and B. C. O, Blood, 1963, 21, 640–647 CAS.
- B. Coiffier, E. Lepage, J. Brière, R. Herbrecht, H. Tilly, R. Bouabdallah, P. Morel, E. Van Den Neste, G. Salles, P. Gaulard, F. Reyes, P. Lederlin and C. Gisselbrecht, N. Engl. J. Med., 2002, 346, 235–242 CrossRef CAS PubMed.
- M. van Oers, R. Klasa, R. E. Marcus, M. Wolf, E. Kimby, R. D. Gascoyone, A. Jack, M. V. Veer, A. Vranovsky, H. Holte, M. von Glabbeke, I. Teodorovic, C. Rozewicz and A. Hagenbeek, Blood, 2006, 108, 3295–3301 CrossRef CAS PubMed.
- T. Tsuruo, H. Iida, S. Tsukagoshi and Y. Sakurai, Cancer Res., 1981, 41, 1967–1972 Search PubMed.
- J. P. Fermand, P. Ravaud, S. Chevret, M. Divine, V. Leblond, C. Belanger, M. Macro, E. Pertuiset, F. Dreyfus, X. Mariette, C. Boccacio and J. C. Brouet, Blood, 1998, 92, 3131–3136 CAS.
- B. Shofty, M. Mauda Havakuk, L. Weizman, S. Constantini, D. Ben Bashat, R. Dvir, L. T. Pratt, L. Joskowicz, A. Kesler, M. Yalon, L. Ravid and L. Ben Sira, Pediatr. Blood Cancer, 2015, 62, 1353–1359 CAS.
- M. Chintagumpala, S. P. Eckel, M. Krailo, M. Morris, A. Adesina, R. Packer, C. Lau and A. Gajjar, Neuro – Oncology, 2015, 17, 1132–1138 CrossRef PubMed.
- G. J. D'Angio, N. Breslow, J. B. Beckwith, A. Evans, E. Baum, A. deLorimier, D. Fernbach, E. Hrabovsky, B. Jones, P. Kelalis, H. B. Otherson, M. Tefft and P. R. M. Thomas, Cancer, 1989, 64, 349–360 CrossRef.
- A. Spira and D. S. Ettinger, N. Engl. J. Med., 2004, 350, 379–392 CrossRef CAS PubMed.
- E. Aboutaleb, F. Atyabi, M. R. Khoshayand, A. R. Vatanara, S. N. Ostad, F. Kobarfard and R. Dinarvand, J. Biomed. Mater. Res., Part A, 2013, 102, 2126–2136 Search PubMed.
- A. Jordan, J. A. Hadfield, N. J. Lawrence and A. T. McGown, Med. Res. Rev., 1998, 18, 259–296 CrossRef CAS PubMed.
- B. Gigant, C. Wang, R. B. G. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Nature, 2005, 435, 519–522 CrossRef CAS PubMed.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- R. I. Fisher, E. R. Gaynor, S. Dahlberg, M. M. Oken, T. M. Grogan, E. M. Mize, J. H. Glick, C. A. Coltman Jr. and T. P. Miller, N. Engl. J. Med., 1993, 328, 1002–1006 CrossRef CAS PubMed.
- D. L. Longo, V. T. DeVita Jr., P. L. Duffey, M. N. Wesley, D. C. Ihde, S. M. Hubbard, M. Gilliom, E. S. Jaffe, J. Cossman and R. I. Fisher, J. Clin. Oncol., 1991, 9, 25–38 CAS.
- V. S. Sethi, D. V. Jackson, D. R. White, F. Richards, J. J. Stuart, H. B. Muss, M. R. Cooper and C. L. Spurr, Cancer Res., 1981, 41, 3551–3555 CAS.
- V. S. Sethi and K. N. Thimmaiah, Cancer Res., 1985, 45, 5386–5389 CAS.
- D. Vendrig, J. H. Beijnen and O. van der Houwen, Int. J. Pharm., 1989, 50, 189–196 CrossRef CAS.
- M. Coimbra, B. Isacchi, L. van Bloois, J. S. Torano, A. Ket, X. Wu, F. Broere, J. M. Metselaar, C. J. F. Rijcken, G. Storm, R. Bilia and R. M. Schiffelers, Int. J. Pharm., 2011, 416, 433–442 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- R. A. Hule, R. P. Nagarkar, A. Altunbas, H. R. Ramay, M. C. Branco, J. P. Schneider and D. J. Pochan, Faraday Discuss., 2008, 139, 251–264 RSC.
- M. C. Branco, F. Nettesheim, D. J. Pochan, J. P. Schneider and N. J. Wagner, Biomacromolecules, 2009, 10, 1374–1380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5bm00538h |
| This journal is © The Royal Society of Chemistry 2016 |