
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon nanotube anions for the preparation of gold nanoparticle–nanocarbon hybrids†
Mustafa K.
Bayazit‡
*,
Stephen A.
Hodge§
,
Adam J.
Clancy
,
Robert
Menzel¶
,
Shu
Chen
and
Milo S. P.
Shaffer
*
Department of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk; m.bayazit@ucl.ac.uk
First published on 10th December 2015
Abstract
Gold nanoparticles (AuNPs) can be evenly deposited on single-walled carbon nanotubes (SWCNTs) via the reduction of the highly stable complex, chloro(triphenylphosphine) gold(I), with SWCNT anions (‘nanotubides’). This methodology highlights the unusual chemistry of nanotubides and provides a blueprint for the generation of many other hybrid nanomaterials.
The surface modification of single-walled carbon nanotubes (SWCNTs) has been explored extensively for more than a decade.1 The properties of SWCNTs can be adjusted to afford greater solubility, improved interactions with other materials, or specific functions.2,3 Hybrid materials, in particular the combination of SWCNTs with metal nanoparticles (NPs), is an area of intense interest, including for applications such as catalysis and sensing; in this context, the SWCNTs act as a high surface area support and/or electrically conductive electrode.
The chemical reduction of metal salts is a well-studied solution phase method to produce a wide variety of metal NPs.4–6 Interestingly, pristine neutral SWCNT bundles grown on a SiO2 substrate have been shown to behave as a reducing agent towards aqueous solutions of the metal salts HAuCl4 and Na2PtCl4, spontaneously generating the respective Au0/Pt0 NPs via an electroless plating mechanism.7 In principle, this approach is limited: firstly, by the choice of metal precursor. The electroless plating mechanism relies on the position of the SWCNT reduction potential (i.e. the Fermi level) in relation to the metal complex reduction potential.7,8 Neutral SWCNTs were estimated to lie between hydrogen and copper in the electrochemical series.9 Secondly, to maximize the surface area of the SWCNTs as a support, they must be individualised, either grown dilutely on substrates by CVD,7 or dispersed in non-aqueous solutions via prolonged ultrasonication.10
Here the use of SWCNT anions,9,11–13 known as nanotubides, is proposed as a solution to these issues. Nanotubides have an increased reduction potential, offering the ability to reduce a wider range of metal NP precursors, and can also spontaneously dissolve without the use of damaging ultrasonication, potentially leading to a versatile and scalable process. Metal reduction and NP formation on fully individualized SWCNTs should result in a significantly more homogeneous distribution of the NPs on the nanostructured carbon support, an important requisite for practical applications. Nanotubides are expected to enable much greater control over nanoparticle size and size distribution by manipulating the initial SWCNT charge density or metal precursor:charge stoichiometry.10
Herein, SWCNT/AuNP hybrid materials are produced in a two-step process. (i) SWCNTs are reduced chemically to generate solutions of highly reactive nanotubides in anhydrous, aprotic media; (ii) a gold complex is introduced and is electrolessly plated onto the reducing SWCNTs. In this case, the highly stable gold complex chloro(triphenylphosphine)gold(I), ClAu(PPh3) is compared to auric acid, to probe the effect of its more negative reduction potential, which should prevent its reaction with pristine SWCNTs. Specifically, the reaction scheme (Fig. 1a) involved the lithiation of HiPco SWCNTs by n-BuLi (−2.04 V vs. SHE) in THF to form [(nBu–SWCNT)x−·Lix+]; this reaction was previously demonstrated to yield ∼1 butyl group/25 C atoms (corresponding to 1 e− charged per 25 C atoms).12 In order to investigate the effect of the increased reduction potential of nanotubides, the stable gold complex chloro(triphenylphosphine)gold(I), ClAu(PPh3) was reacted with [(nBu–SWCNT)x−·Lix+]. Although the reported reduction potential of ClAu(PPh3) appears to show significant solvent dependency (−1.76 V in dichloromethane,14 −1.59 V in acetonitrile,15 −0.39 V in dimethylformamide,15 and −0.31 V in THF (ESI,† Fig. S5), all adjusted to SHE (standard hydrogen electrode)), the formation of AuNPs should not occur spontaneously with neutral SWCNTs (∼+0.30 V vs. SHE), unlike previous reports using HAuCl4 (+1.002 V vs. SHE) as the gold source. Subsequent addition of ClAu(PPh3) to [(nBu–SWCNT)x−·Lix+] under inert conditions generated nBu–SWCNTs/AuNPs, (1). For control experiments, neutral HiPco SWCNTs were dispersed in THF and mixed with ClAu(PPh3) to give (2), and HAuCl4 to give (3) under the same conditions. In all cases, the atomic ratio of carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :gold was kept constant at 5:1 (see ESI†).
:gold was kept constant at 5:1 (see ESI†).
 | ||
| Fig. 1 (a) Scheme showing the generation of [(nBu–SWCNT)x−·Lix+] and proposed formation of AuNPs following ClAu(PPh3) addition to afford nBu–SWCNTs/AuNPs, (1). Electron microscopy characterisation of (1): (b) SEM characterisation. Inset shows the particle size distribution of AuNPs. (c) Low resolution TEM image showing AuNPs supported on SWCNT bundles (note the presence of a holey amorphous carbon film support). Inset shows the particle size distribution of AuNPs. (d) HRTEM image of the selected region in (c) (blue box) showing the presence of SWCNT bundles and individualized SWCNTs. (e) High magnification HR-TEM image showing two NPs with lattice spacing consistent with Au0 and Fe0. Iron catalyst nanoparticles are present, remaining from the HiPco SWCNT synthesis. | ||
Both scanning (SEM) and transmission electron microscopy (TEM) of (1) consistently demonstrated the formation of evenly distributed AuNPs on the SWCNT networks (Fig. 1b and c). High-resolution (HR) TEM images showed the presence of both individual SWCNTs and bundles (Fig. 1d). The presence of Au was confirmed by scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (STEM-EDX) (ESI,† Fig. S1). HRTEM images of the NP lattice spacings (Fig. 1e) demonstrate that the AuNPs can be distinguished from the smaller iron catalyst nanoparticles, present from the HiPco SWCNT synthesis. One of the advantages of the nanotubide route is that pristine, unpurified SWCNTs can be processed directly; however, the typical amorphous carbon content associated with the original growth remains in the sample (visible in Fig. 1e); the presence of a large fraction of SWCNTs is, nevertheless, confirmed by microscopy and Raman spectroscopy (ESI,† Fig. S1 & S3). Image analysis using TEM and SEM showed a relatively narrow diameter distribution for the new particles, with means 12.4 ± 4.2 nm and 14.6 ± 3.8 nm, respectively. SEM did not identify any AuNPs in control sample (2), while control sample (3) was very heterogeneous with large aggregates of metallic gold (Fig. 2).
 | ||
| Fig. 2 SEM characterisation following control reactions between neutral SWCNTs and (a) ClAu(PPh3), and (b) HAuCl4, to afford samples (2) and (3), respectively. | ||
X-ray photoelectron spectroscopy (XPS) survey spectra of (1) (Fig. 3a) clearly showed peaks related to metallic gold at ca. 82.5–88.5 eV (4f, 2 peaks), and 335.2–353.2 eV (4d, 2 peaks). Relatively small nitrogen (N 1s; ∼400.5 eV) and iron (Fe 3p and 2p; ∼56.5 and 710.5 eV, respectively) signals were also detected, attributed to adsorbed DMF used for washing and the original iron catalyst nanoparticles, respectively. Interestingly, no measurable phosphorous related EDX or XPS features (P 2s ∼190.0 eV or P 2p at ∼132.0 eV) were detected for (1), suggesting excess PPh3 ligand removal, following reduction. Further inspection of the enlarged XPS region for P 2p, as inset in Fig. 3a, showed only very low intensity XPS peaks positioned at ca. 197.5 and 152.5 eV, attributed to Cl 2p and Si 2s, probably due to trace ClAu(PPh3) and silicone grease, respectively. The lack of phosphorus signal is consistent with the reduction of the Au complex; any remaining PPh3, present only on the surface of the Au NPs, would be too low concentration to be detected.
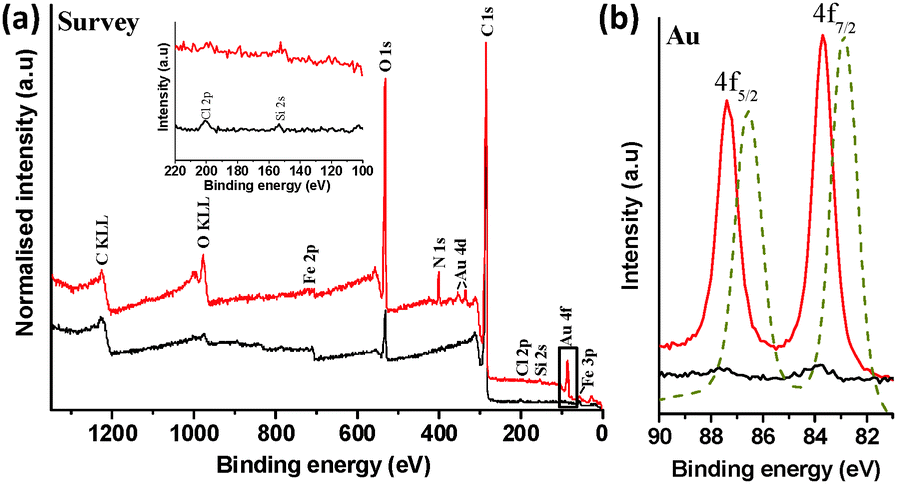 | ||
| Fig. 3 (a) XPS survey spectrum of (1) (red) and control sample (2) (black). Survey spectrum of (1) shows the presence of Au, O, C, Si, Cl, Fe and N elements in the analysed sample. The highlighted box shows the Au 4f region. Intensities are normalised to the C 1s peak. Inset shows the enlarged XPS region for P 2p. (b) High-resolution scan of the Au 4f region of (1), (2) and gold foil reference (dashed gold). The gold reference spectrum intensity is normalised the 4f7/2 peak of (1). | ||
A high resolution scan of the Au 4f region revealed Au signals at 83.7 and 87.4 eV (Fig. 2b) while Au peak positions in the gold reference appeared at 82.9 and 86.6 eV, confirming the formation of metallic Au on SWCNT surface.16 This binding energy shift of ∼0.8 eV can reasonably be attributed to a strong charge transfer between the SWCNTs and AuNPs.17 XPS elemental composition analysis indicated ∼0.5 at% Au in (1). AuNPs are well known to be stabilised on a range of solid supports, including carbon nanotubes and SiO2, which can provide active sites at the metal-support boundary, potentially influencing the average oxidation state of gold.7,18–20 The shift in XPS binding energy of Au 4f electrons is consistent with such an interaction, whilst the stability of the samples during extensive washing steps, including sonication procedures to disperse samples for microscopy, indicates relatively strong electrostatic and dispersive interactions between AuNPs and SWCNTs, as reported in previous literature.20
Since XPS measurements are highly surface sensitive, bulk quantification was performed by thermogravimetric analysis-mass spectroscopy (TGA MS) and inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (ESI,† Fig. S2 for TGA MS). Under a nitrogen atmosphere, (1) underwent significant weight loss (12.2 wt%) between 200–600 °C, corresponding to m/z 43 amu (–CH2CH2CH3), confirming the n-butyl group addition to the SWCNT surface (∼1 butyl group/33 C atoms, based on this TGA data).11,21 This fragment was not observed for as-received HiPco SWCNTs, or control samples (2) and (3). The Raman spectra (excited at 532 nm and normalized to the G-band intensity) of (1) showed an enhanced D-band at ∼1350 cm−1 when compared with (2), with an IG/ID ratio of 11.3 and 15.8, respectively, indicative of groups attached to the surface of the nanotubes (ESI,† Fig. S3), but with a low level of sp2 framework disruption.2,3,11,21 Complementary FTIR spectroscopy also showed bands characteristic of the C–H bonds in the butyl group (ν(CH) 2914 and 2840 cm−1) present in (1) but not in (2) (ESI,† Fig. S4).11,21 Residues following oxidative TGA were analysed by ICP-AES for accurate quantification of the Au content. Sample (1) contained 1 Au/46 C atoms (71.7% charge utilisation yield, based on the 1 charge/33 C atom stoichiometry), whilst the reaction of neutral SWCNTs with ClAuPPh3 (2) and HAuCl4 (3) generated ∼1 Au/7000 C atoms and 1 Au/25 C atoms, respectively. As reported recently,8 the charge utilisation yield of nanotubides reactions can be related semi-quantitatively to the SWCNT density of states (DOS).
The hypothesis is that the introduction of charge into the nanotubes gradually fills the available DOS, raising the reduction potential (and hence reactivity) of the resulting nanotubide. The extent of reaction with a given reagent should then depend on its redox potential. The reduction peak for ClAu(PPh3) in THF was determined to be −0.31 V vs. SHE using cyclic voltammetry (ESI,† Fig. S5). Based on this value, the estimated DOS of the combined SWCNTs (Fig. 4), and the extent of metallic gold deposition, it is possible to deduce the reduction potential of the lithiated SWCNTs, [(nBu–SWCNT)x−·Lix+], where x is 33, as −0.82 V vs. SHE. This value is consistent with those obtained for potassium graphenides derived previously8 (e.g. KC24, −1.21 V vs. SHE). Since the reduction potential of ClAuPPh3 lies above that of the neutral SWCNTs, negligible metallic gold is observed in control (2). Conversely, the reduction potential of HAuCl4 lies below even the neutral SWCNTs leading to spontaneous gold deposition in control (3), in line with previous reports.7 It is worth noting, however, that this reaction self-limits, even in excess HAuCl4, as the depletion of electrons from the SWCNTs decreases their reducing power. Fig. 4 summarises, approximately, the positions of the AuCl(PPh3) and HAuCl4 reduction potentials relative to the Fermi levels (EF) of undoped and lithiated SWCNTs. Although these potentials may vary, for reasons discussed previously,10 notably including electrostatic capacitive effects, it is apparent that the relative reduction potentials are critical factors in driving the electroless plating mechanism.
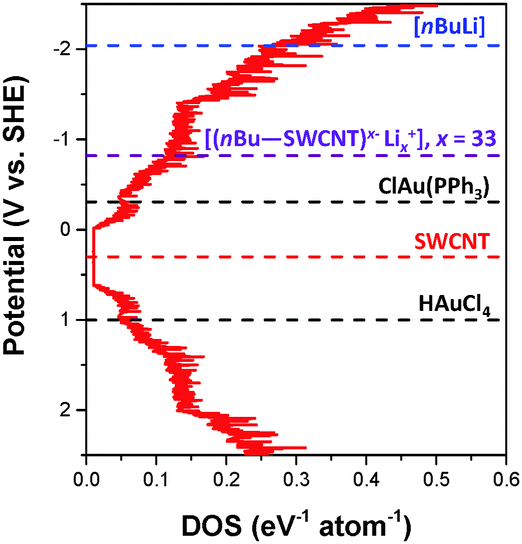 | ||
| Fig. 4 The averaged DOS of the (n,m) distribution in a HiPco sample was derived from ref. 13 and 22. The averaged DOS comprises 28 semi-conducting and 18 metallic SWCNTs as identified from resonance Raman spectroscopy.23 For simplicity, all (n,m)-SWCNTs equally contribute to the DOS, although, as in a typical SWCNT sample the proportion of semi-conducting:metallic SWCNTs is adjusted to 2:1. The position of the Fermi level, EF, of undoped HiPco SWCNTs was determined from cyclic voltammetry (∼+0.30 V vs. SHE), although in principle, the EF will vary for each (n,m)-SWCNT.1,24 | ||
In conclusion, the nanotubide, [(nBu–SWCNT)x−·Lix+], has been shown to be an efficient reducing agent, even towards a stable Au(I) complex, as a route to generate supported AuNPs. These SWCNT–AuNP hybrids in principle may be useful candidates for sensor25 and catalysis26 applications, although the process should ideally be adjusted towards smaller nanoparticles, by for example varying the charge density on SWCNTs, the metal precursor redox potential, and/or the metal precursor/CNT stoichiometry. The mechanism of electroless plating is driven by the relative difference between the SWCNT and metal complex reduction potentials. The continuum density of states in the conductive SWCNTs generates an unusual reactivity for these discrete, molecular species. Their reducing power is related to the charge density on the backbone, and changes dynamically with the extent of the reaction, due to electron transfer. This behaviour is worthy of further investigation from a fundamental perspective, since, for example, multiple electron affinities/ionisation potentials have not yet been calculated for SWCNTs; these values will be also influenced by the local environment, solvent, and counterions. Whilst not yet attained in this study, the possible formation of discrete C–Au or C–metal covalent bonds on the nanotube framework as demonstrated recently for fulleride anions,27 is an exciting avenue for exploration. Nanotubides may also be relevant as general reducing agents for in the manner of for example KC8, but with more flexible control of reducing power.8,28
Practically, the use of nanotubides as reducing agents offers a range of important advantages compared to the use of the neutral nanocarbon; firstly, there is a greater reduction potential which can also be tuned via the charge stoichiometry, greatly expanding the useful range of metal complex precursors for novel hybrid material synthesis. By controlling the redox potentials of the reagents, sequential functionalisation reactions can be envisioned. It is worth mentioning that the band structure of SWCNTs will be altered with increasing covalent functionalisation. Thus, modest level of functionalization, as reported here, is preferred for minimal electronic disruption to the reduction chemistry or subsequent application. Whilst alkylation offers potential benefits in solubility, alternative nanotubide chemistries without any accompanying functionalisation reaction are available.9,29 Secondly, the excellent solubility of nanotubide provides a greater accessible surface area for nanoparticle support, and numerous further processing options to form useful constructs.30,31 Generally speaking, the use of charged nanoions as a reducing agent is anticipated to be applicable to other systems including graphene and its vast two-dimensional layered analogues in organic phase.
The authors would like to thank EPSRC EP/G007314/1 and EP/L001896/1 for the funding of this project and Hui Huang Tay for assistance. M. K. B. thanks the Scientific and Technological Research Council of Turkey (TÜBTAK).
Notes and references
- S. A. Hodge, M. K. Bayazit, K. S. Coleman and M. S. P. Shaffer, Chem. Soc. Rev., 2012, 41, 4409–4429 RSC.
- M. K. Bayazit, L. S. Clarke, K. S. Coleman and N. Clarke, J. Am. Chem. Soc., 2010, 132, 15814–15819 CrossRef CAS PubMed.
- M. K. Bayazit and K. S. Coleman, J. Am. Chem. Soc., 2009, 131, 10670–10676 CrossRef CAS PubMed.
- Y. Vasquez, Z. Luo and R. E. Schaak, J. Am. Chem. Soc., 2008, 130, 11866–11867 CrossRef CAS PubMed.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem. Soc. Rev., 2000, 29, 27–35 RSC.
- M. Hu, L. Qian, R. P. Brinas, E. S. Lymar and J. F. Hainfeld, Angew. Chem., Int. Ed., 2007, 46, 5111–5114 CrossRef CAS PubMed.
- H. C. Choi, M. Shim, S. Bangsaruntip and H. J. Dai, J. Am. Chem. Soc., 2002, 124, 9058–9059 CrossRef CAS PubMed.
- S. A. Hodge, H. H. Tay, D. B. Anthony, R. Menzel, D. J. Buckley, P. L. Cullen, N. T. Skipper, C. A. Howard and M. S. P. Shaffer, Faraday Discuss., 2014, 172, 311–325 CAS.
- S. A. Hodge, S. Fogden, C. A. Howard, N. T. Skipper and M. S. P. Shaffer, ACS Nano, 2013, 7, 1769–1778 CrossRef CAS PubMed.
- S. Giordani, S. D. Bergin, V. Nicolosi, S. Lebedkin, M. M. Kappes, W. J. Blau and J. N. Coleman, J. Phys. Chem. B, 2006, 110, 15708–15718 CrossRef CAS PubMed.
- M. K. Bayazit, A. Suri and K. S. Coleman, Carbon, 2010, 48, 3412–3419 CrossRef CAS.
- Y. Maeda, K. Saito, N. Akamatsu, Y. Chiba, S. Ohno, Y. Okui, M. Yamada, T. Hasegawa, M. Kako and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 18101–18108 CrossRef CAS PubMed.
- S. Fogden, C. A. Howard, R. K. Heenan, N. T. Skipper and M. S. P. Shaffer, ACS Nano, 2012, 6, 54–62 CrossRef CAS PubMed.
- U. Koelle and A. Laguna, Inorg. Chim. Acta, 1999, 290, 44–50 CrossRef CAS.
- K. N. Kouroulis, S. K. Hadjikakou, N. Kourkoumelis, M. Kubicki, L. Male, M. Hursthouse, S. Skoulika, A. K. Metsios, V. Y. Tyurin, A. V. Dolganov, E. R. Milaeva and N. Hadjiliadis, Dalton Trans., 2009, 10446–10456 RSC.
- P. Gobbo, M. C. Biesinger and M. S. Workentin, Chem. Commun., 2013, 49, 2831–2833 RSC.
- H. Chu, J. Wang, L. Ding, D. Yuan, Y. Zhang, J. Liu and Y. Li, J. Am. Chem. Soc., 2009, 131, 14310–14316 CrossRef CAS PubMed.
- Z. Ma and S. Dai, Nano Res., 2011, 4, 3–32 CrossRef CAS.
- P. V. Dudin, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2010, 114, 13241–13248 CAS.
- G. A. Rance, D. H. Marsh, S. J. Bourne, T. J. Reade and A. N. Khlobystov, ACS Nano, 2010, 4, 4920–4928 CrossRef CAS PubMed.
- M. K. Bayazit and K. S. Coleman, J. Mater. Sci., 2014, 49, 5190–5198 CrossRef CAS.
- S. Maruyama, 2002, http://www.photon.t.u-tokyo.ac.jp/%E2%88%BCmaruyama/kataura/kataura.html#DOS.
- C. Fantini, A. Jorio, M. Souza, M. S. Strano, M. S. Dresselhaus and M. A. Pimenta, Phys. Rev. Lett., 2004, 93, 147406 CrossRef CAS PubMed.
- Y. Tanaka, Y. Hirana, Y. Niidome, K. Kato, S. Saito and N. Nakashima, Angew. Chem., Int. Ed., 2009, 48, 7655–7659 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- M. Halim, R. D. Kennedy, M. Suzuki, S. I. Khan, P. L. Diaconescu and Y. Rubin, J. Am. Chem. Soc., 2011, 133, 6841–6851 CrossRef CAS PubMed.
- L. B. Ebert, Carbon, 1985, 23, 585–587 CrossRef CAS.
- A. J. Clancy, J. Melbourne and M. S. P. Shaffer, J. Mater. Chem. A, 2015, 3, 16708–16715 CAS.
- V. A. Davis, A. N. G. Parra-Vasquez, M. J. Green, P. K. Rai, N. Behabtu, V. Prieto, R. D. Booker, J. Schmidt, E. Kesselman, W. Zhou, H. Fan, W. W. Adams, R. H. Hauge, J. E. Fischer, Y. Cohen, Y. Talmon, R. E. Smalley and M. Pasquali, Nat. Nanotechnol., 2009, 4, 830–834 CrossRef CAS PubMed.
- N. Behabtu, C. C. Young, D. E. Tsentalovich, O. Kleinerman, X. Wang, A. W. K. Ma, E. A. Bengio, R. F. ter Waarbeek, J. J. de Jong, R. E. Hoogerwerf, S. B. Fairchild, J. B. Ferguson, B. Maruyama, J. Kono, Y. Talmon, Y. Cohen, M. J. Otto and M. Pasquali, Science, 2013, 339, 182–186 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRTEM, HAADF STEM and STEM-EDX of (1), TGA-MS of (1), (2) and as-received SWCNTs, FTIR and Raman spectra of (1) and (2), and cyclic voltammetry data for ClAu(PPh3). See DOI: 10.1039/c5cc08726k |
| ‡ Current Address: Department of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. |
| § Current Address: Cambridge Graphene Centre, Engineering Department, University of Cambridge, 9, JJ Thomson Avenue, Cambridge, CB3 0FA, UK. |
| ¶ Current Address: School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. |
| This journal is © The Royal Society of Chemistry 2016 |