
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of benzofurans via an acid catalysed transacetalisation/Fries-type O → C rearrangement/Michael addition/ring-opening aromatisation cascade of β-pyrones†
Siddheshwar K.
Bankar
,
Jopaul
Mathew
and
S. S. V.
Ramasastry
*
Organic Synthesis and Catalysis Lab, Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Knowledge City, Sector 81, S. A. S. Nagar, Manuali PO, Punjab 140306, India. E-mail: ramsastry@iisermohali.ac.in; ramsastrys@gmail.com; Web: http://14.139.227.202/faculty/sastry/
First published on 17th March 2016
Abstract
An unusual and facile approach for the synthesis of 2-benzofuranyl-3-hydroxyacetones from 6-acetoxy-β-pyrones and phenols is presented. The synthetic sequence involves a cascade transacetalisation, Fries-type O → C rearrangement followed by Michael addition and ring-opening aromatisation. The versatility of this method was further demonstrated via the synthesis of 4,4a-dihydropyrano[3,2-b]benzofuran-3-ones, furo[3,2-c]coumarins, and spiro[benzofuran-2,2′-furan]-4′-ones. The unexpected cascade event would also provide new possible considerations in the β-pyrone-involved organic synthesis.
Benzofurans are ubiquitous building blocks in many bioactive natural products and primary structural motifs in several pharmaceuticals and molecular electronics.1 Furthermore, benzofurans are acclaimed privileged scaffolds in drug discovery.2 These distinguished features stimulated the development of several efficient and concise strategies for the synthesis of diverse benzofuran derivatives.3 However, owing to the limitations of the conventional approaches, such as the harsh reaction conditions, broad substrate scope and limited functional-group tolerability, there still exists ample scope for exploring new approaches for the synthesis of benzofurans. Herein, we report a new access to benzofurans via a Lewis acid catalysed one-pot cascade process.
Development of novel cascade processes has received great attention owing to their exceptional ability to rapidly assemble intricate molecular scaffolds.4 As part of our recent efforts to develop new cascade approaches for the O,S-containing heterocycles, it necessitated us to have rapid and efficient access to 6-aryloxy-β-pyrones of the type 1a.5 For this purpose, the Lewis acid catalysed protocols of Grynkiewicz6a and Feringa6b were tried with 6-acetoxy-β-pyrone 2a and phenol 3a, but the required product 1a was isolated in low yields, Scheme 1.7 A detailed investigation revealed that the formation of a polar compound (on TLC) was responsible for the yield loss. Further study of the reaction under Feringa's conditions revealed that the concentration of the initially formed phenyl ether 1a started diminishing and simultaneous build-up of the unanticipated product 4a was observed. Thus, we have drawn the conclusion at this stage that the unexpected product 4a formed via the intermediacy of 1a. The structure of the unexpected product 4a was deduced from 1H and 13C NMR data and was further confirmed by single-crystal X-ray diffraction analysis (vide infra).8 Since the 6-acetoxy-β-pyrone 2a can be accessed from furyl carbinol 5a in two straightforward steps,5 this protocol thus represents an unique three-step conversion of furans (of the type 5a) to benzofurans (such as 4a), Scheme 1.9
 | ||
| Scheme 1 Unprecedented cascade reaction of 6-acetoxy-β-pyrones and phenols leading to the synthesis of 1-(2-benzofuranyl)-3-hydroxyacetones. | ||
Having realised the significance of benzofurans especially generated under mild Lewis acidic conditions from readily accessible starting compounds, and considering the potential implications of this rearrangement in organic synthesis, we turned to optimising the reaction conditions. Towards this, various Lewis acid and solvent combinations were investigated, and few important results are shown in Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | Acid (10 mol%) | Solvent | Time (h) | Yieldb (%) |
| a A 5 mL glass vial was filled with 2a (0.2 mmol), 3a (0.22 mmol), and a solvent (1 mL). A catalyst (0.02 mmol) was then added at 0–5 °C. After stirring at the same temperature for about 30 min, the reaction continued at room temperature until 1a and 2a disappeared (by TLC). b Isolated yield after column chromatography. c 1a exclusively formed. d 20 mol% TMSOTf was employed. e 5 mol% TMSOTf was employed. f 1a and 2a were also recovered. g In the presence of 2,6-di-tert-butyl-4-methylpyridine (1 equiv.). | ||||
| 1 | La(OTf)3 | DCE | 48 | 74c |
| 2 | SnCl4 | DCE | 3 | 50 |
| 3 | BF3OEt2 | DCE | 18 | 55 |
| 4 | FeCl3 | DCE | 18 | 45 |
| 5 | In(OTf)3 | DCE | 20 | 48 |
| 6 | Zn(OTf)3 | DCE | 20 | 45 |
| 7 | Bi(OTf)3 | DCE | 20 | 61 |
| 8 | AgOTf | DCE | 21 | 40 |
| 9 | TMSOTf | DCE | 20 | 74 |
| 10d | TMSOTf | DCE | 20 | 47 |
| 11e,f | TMSOTf | DCE | 30 | 45 |
| 12g | TMSOTf | DCE | 72 | — |
| 13 | TfOH | DCE | 20 | 63 |
| 14 | PTSA | DCE | 20 | 51 |
| 15 | TMSOTf | CH3CN | 21 | 48 |
| 16 | TMSOTf | Toluene | 72 | 25 |
| 17 | TMSOTf | THF | 72 | — |
The reaction catalysed by La(OTf)3 generated exclusively the 6-phenoxy-β-pyrone 1a even after extended reaction times, thereby establishing a high-yielding method for its selective synthesis (Table 1, entry 1). Most of the Lewis acids employed during the screening otherwise furnished the desired product 4a in varied yields, with TMSOTf giving the best result (entries 2–9). Reaction with higher TMSOTf loading (20 mol%) gave a poor result due to the formation of undesired side products (entry 10). On the other hand, reaction in the presence of 5 mol% TMSOTf was found to be sluggish (entry 11). So, 10 mol% TMSOTf loading was realised to be optimal for this transformation.
Interestingly, the reaction in the presence of a proton sponge such as 2,6-di-tert-butyl-4-methylpyridine completely inhibited the product formation, indicating most likely that catalytic amounts of TfOH generated in situ might be promoting this process (entry 12). However, despite repeated attempts, TfOH furnished the required product in lower yields when compared to TMSOTf (entry 13). Among few other Brønsted acids employed, PTSA generated 4a in satisfactory yield (entry 14). So, TMSOTf was identified as the catalyst of choice for this study considering its mild nature and ease of handling. Brief solvent screening with TMSOTf offered no further improvement in the yield (Table 1, entries 15–17).
With the optimised reaction conditions in hand, the scope of the reaction was subsequently investigated, and the representative results are presented in Table 2. Since the 6-benzoyloxy-β-pyrones (2b and 2g) afforded the respective products (4b, 4d, 4l, and 4m) consistently in low yields under the optimised conditions, acetates of β-pyrones were preferred over benzoates during this study.
| a A 5 mL glass vial was filled with 2 (0.2 mmol), 3 (0.22 mmol), and DCE (1 mL). TMSOTf (0.02 mmol) was then introduced at 0–5 °C. After stirring at the same temperature for 30 min, the reaction continued at room temperature until 1 and 2 disappeared (by TLC). b Isolated yield after column chromatography. c Structure confirmed by single crystal X-ray diffraction analysis, see the ESI for details.10 |
|---|

|
A variety of 6-acetoxy-β-pyrones (2c–2f, 2h–2j) and phenols (3b–3e) conveniently generated the respective benzofurans 4b–4x in good to excellent yields.11 A range of possible substitution patterns on the pyrones were considered that provided 2-benzofuranyl propanones possessing 1°, 2° and 3°-alcoholic centres. Notably, chiral hydroxyacetones such as 4g–4j and 4t can be easily assembled by employing this strategy. In particular, isolation of alcohols 4j and 4t in 98% and 93% ee, respectively, indicates the involvement of a non-racemising process during the transformation which in turn signifies the mildness of the reaction conditions.
Interestingly, the reaction of the pyrone 2a with halogenated phenols 3f–3h generated only the 4,4a-dihydropyrano[3,2-b]benzofurans 4y, 4z, and 4aa. Even prolonged reaction times did not yield the expected 2-benzofuranyl-3-hydroxyacetones. This result has two-fold significance; it not only provided mechanistic insights into the conversion of 2 to 4, but also provided a new entry for the synthesis of pyrano[3,2-b]benzofuran-3-ones.12
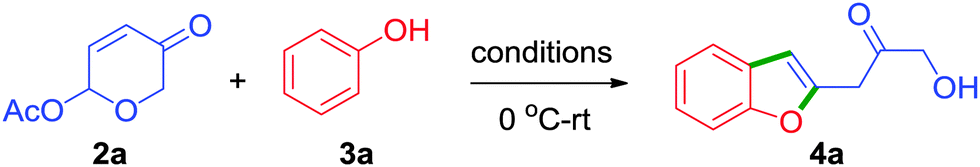
Apart from phenols, strikingly, enol such as 4-hydroxycoumarin 3i also proved to be a distinctive reactive partner in producing furocoumarins 4ab–4ad in one simple step from 6-acetoxy-β-pyrones, Scheme 2. Furocoumarins are part of several bioactive natural products and medicinally interesting compounds.13 Most of the synthetic approaches have focused on the construction of coumestans. Only a few methods have been described for the synthesis of furo[3,2-c]coumarins.14 In this regard, our approach depicted herein provides an unprecedented access for the synthesis of 2-alkylated furo[3,2-c]coumarins.
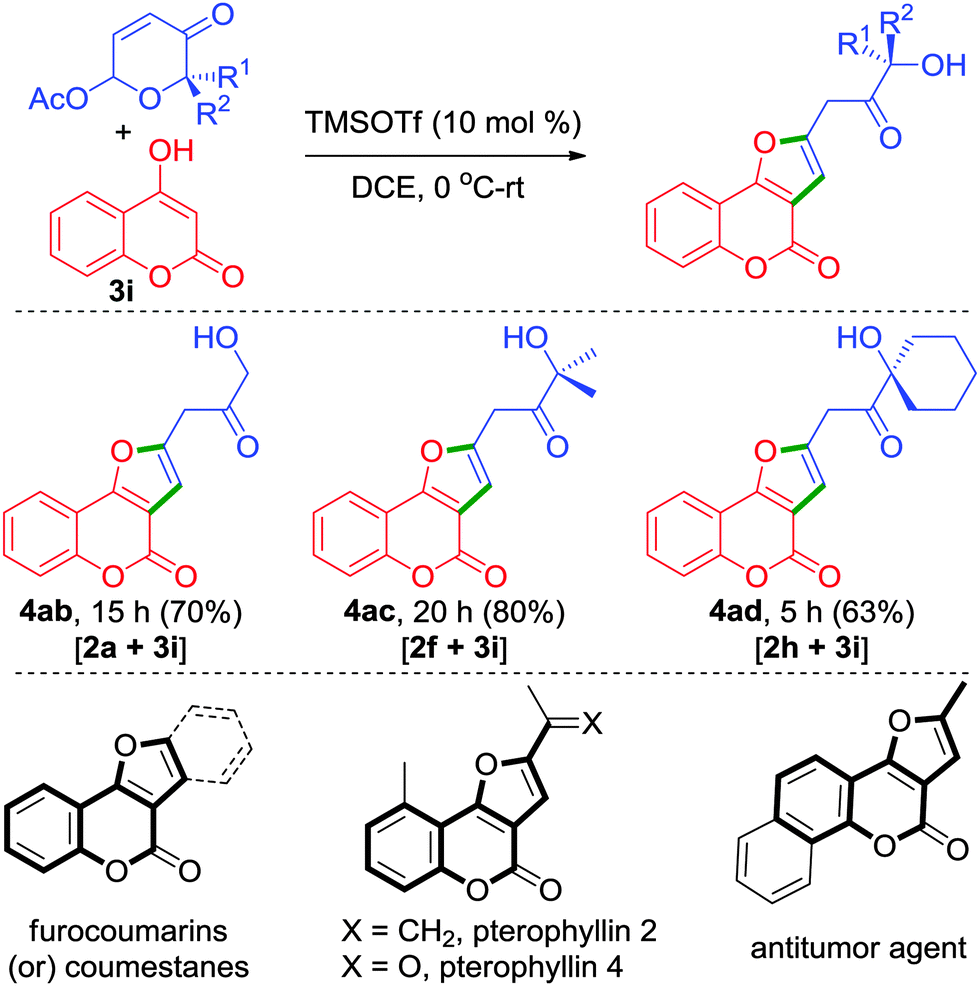 | ||
| Scheme 2 Unprecedented approach for the synthesis of furo[3,2-c]coumarins and a few representative bioactive furocoumarins. | ||
Based on the experimental observations, a plausible mechanism has been proposed in Scheme 3.15 The cascade process begins with an acid catalysed transacetalisation6 followed by an unusual Fries-type O → C rearrangement16 which leads to the formation of a neutral but unstable intermediate 6a in a highly regio- and chemoselective manner.17 Subsequently, 6a undergoes aromatisation and concomitant oxa-Michael addition to form intermediate 4a′. Furthermore, acid-induced ring-opening aromatisation of 4a′ affords the 2-benzofuranyl-3-hydroxyacetone 4a.
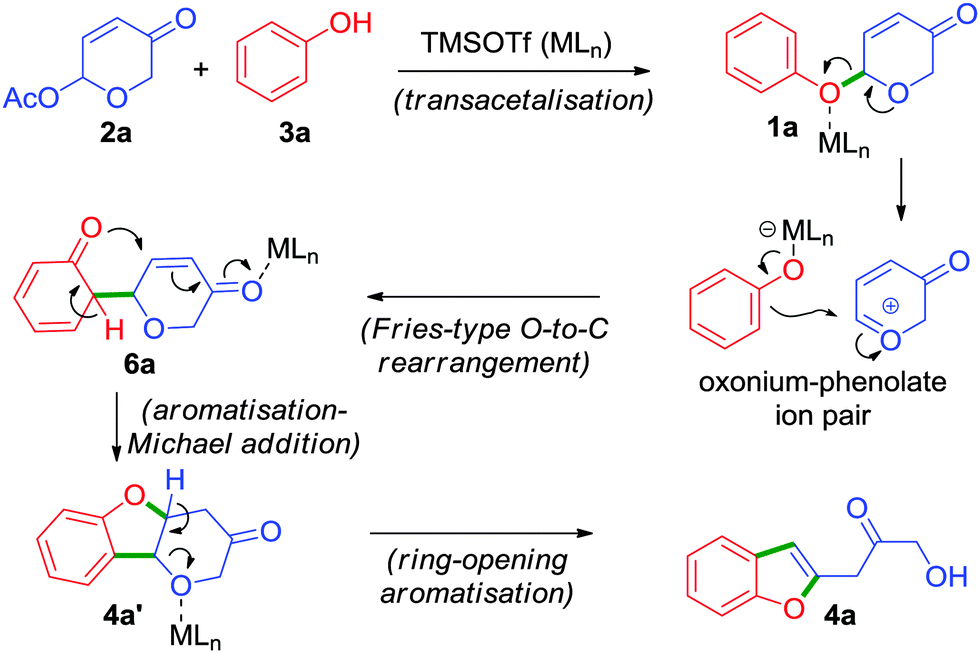 | ||
| Scheme 3 Plausible mechanism. | ||
To further illustrate the generality and synthetic utility of this methodology, we considered an elaboration, Scheme 4. We intended to exploit the presence of alcohol functionality in the side chain in an intramolecular haloetherification reaction which would potentially generate spiro[benzofuran-2,2′-furan]-4′-ones.18 Accordingly, reaction of the keto-alcohols 4a, 4b and 4l with NBS at room temperature conveniently furnished the respective 5,5-spiroketals 4ae–4ag in excellent yields, thereby establishing a mere two-step unprecedented access from readily accessible 6-acetoxy-β-pyrones. The relative stereochemistry of 4ae–4ag was assigned based on the X-ray crystal analysis of 4ag.19 Prevalence of several bioactive natural products possessing the 5,5-spiroketal scaffold renders this an attractive strategy for their easy synthesis, Scheme 4.20
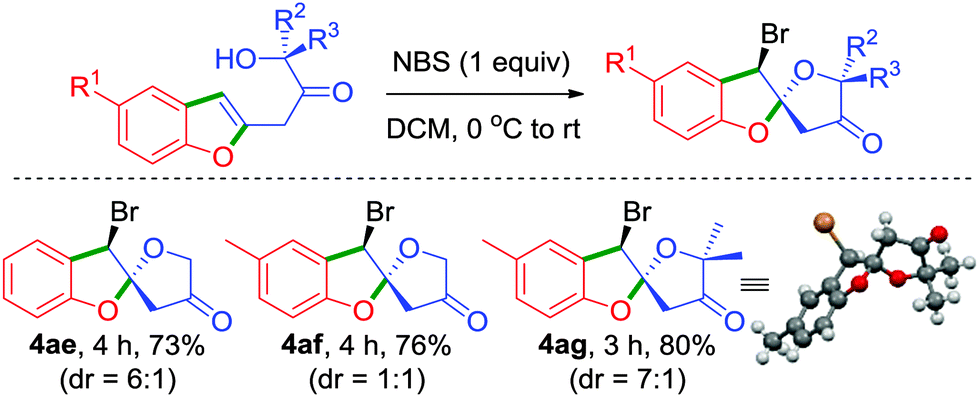 | ||
| Scheme 4 An unusual two-step synthetic approach for spiro[benzofuran-2,2′-furan]-4′-ones from β-pyrones. | ||
Finally, scalability and practicality of the cascade process were verified by conducting gram scale reactions of 2a.8
In conclusion, we have described a cascade event of β-pyrones and phenols, originating out of serendipity, leading to the synthesis of 2-benzofuranyl-3-hydroxyacetones. The versatility of this strategy lies in its ability to establish unprecedented access for medicinally significant scaffolds such as 4,4a-dihydropyrano[3,2-b]benzofuran-3-ones, furo[3,2-c]coumarins, and spiro[benzofuran-2,2′-furan]-4′-ones in a short and efficient manner. Efforts to extend these methods for the total synthesis of natural products are in progress and will be communicated in due course.
We thank the DST (SR/FT/CS-156/2011) and IISER Mohali for funding. We also thank NMR, mass, and departmental X-ray facilities at IISER Mohali. S. K. B. thanks IISER Mohali for a research fellowship and J. M. thanks DST, Govt. of India, for the INSPIRE fellowship.
Notes and references
- (a) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS; (b) T. Nagahara, Y. Yokoyama, K. Inamura, S. Katakura, S. Komoriya, H. Yamaguchi, T. Hara and M. Iwamoto, J. Med. Chem., 1994, 37, 1200 CrossRef CAS PubMed; (c) J. R. Hanson, Nat. Prod. Rep., 1995, 12, 381 RSC; (d) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS; (e) T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 2001, 2491 RSC; (f) M. Watanabe, W. T. Su, Y. J. Chang, T. H. Chao, Y. S. Wen and T. J. Chow, Chem. – Asian J., 2012, 8, 60 CrossRef PubMed; (g) S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Nat. Prod. Rep., 2013, 30, 941 RSC; (h) P. Han, X. Gong, B. Lin, Z. Jia, S. Ye, Y. Suna and H. Yanga, RSC Adv., 2015, 5, 50098 RSC.
- (a) B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B. Chen, P. J. Kling, K. A. Kunkel, J. P. Springer and J. Hirshfield, J. Med. Chem., 1988, 31, 2235 CrossRef CAS PubMed; (b) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (c) A. K. Ghosh, P. R. Sridhar, N. Kumaragurubaran, Y. Koh, I. T. Weber and H. Mitsuya, ChemMedChem, 2006, 1, 939 CrossRef CAS PubMed; (d) H. Sunden and R. Olsson, Org. Biomol. Chem., 2010, 8, 4831 RSC.
- (a) T. Sakamoto, Y. Kondo and H. Yamanaka, Heterocycles, 1988, 27, 2225 CrossRef CAS; (b) D. Nematollahi, D. Habibi, M. Rahmati and M. Rafiee, J. Org. Chem., 2004, 69, 2637 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi and A. Goggiamani, Curr. Org. Chem., 2006, 10, 1423 CrossRef CAS; (d) M. Nagamochi, Y. Q. Fang and M. Lautens, Org. Lett., 2007, 9, 2955 CrossRef CAS PubMed; (e) Y. Liu, M. Wang, H. Yuan and Q. Liu, Adv. Synth. Catal., 2010, 352, 884 CrossRef CAS; (f) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC; (g) M. R. Kuram, M. Bhanuchandra and A. K. Sahoo, Angew. Chem., Int. Ed., 2013, 52, 4607 CrossRef CAS PubMed; (h) U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669 CrossRef CAS PubMed; (i) Y. Gao, W. Xiong, H. Chen, W. Wu, J. Peng, Y. Gao and H. Jiang, J. Org. Chem., 2015, 80, 7456 CrossRef CAS PubMed.
- Few selected articles: (a) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131 CrossRef; (b) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed; (c) S. Sato, M. Miura, T. Sekito and T. Kumazawa, J. Carbohydr. Chem., 2008, 27, 86 CrossRef CAS; (d) D. B. Ramachary, C. Venkaiah and P. M. Krishna, Chem. Commun., 2012, 48, 2252 RSC; (e) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC; (f) L. Q. Lu, J. R. Chen and W. J. Xiao, Acc. Chem. Res., 2012, 45, 1278 CrossRef CAS PubMed; (g) S. Dhiman and S. S. V. Ramasastry, J. Org. Chem., 2013, 78, 10427 CrossRef CAS PubMed; (h) B. Satpathi, S. Dhiman and S. S. V. Ramasastry, Eur. J. Org. Chem., 2014, 2022 CrossRef CAS; (i) L. Zhang, Y. Xia and D. G. Peterson, J. Agric. Food Chem., 2014, 62, 8470 CrossRef CAS PubMed; (j) S. Dhiman and S. S. V. Ramasastry, Chem. Commun., 2015, 51, 557 RSC; (k) S. Dhiman and S. S. V. Ramasastry, Org. Lett., 2015, 17, 5116 CrossRef CAS PubMed; (l) D. H. Dethe and G. M. Murhade, Chem. Commun., 2015, 51, 10891 RSC.
- (a) S. Kasare, S. K. Bankar and S. S. V. Ramasastry, Org. Lett., 2014, 16, 4284 CrossRef CAS PubMed; (b) S. K. Bankar, R. P. Shirke and S. S. V. Ramasastry, Adv. Synth. Catal., 2015, 357, 3284 CrossRef CAS.
- (a) G. Grynkiewicz, B. Barszczak and A. Zamojski, Synthesis, 1979, 364 CrossRef CAS; (b) J. Knol, J. F. G. A. Jansen, F. van Bolhuis and B. L. Feringa, Tetrahedron Lett., 1991, 32, 7465 CrossRef CAS . For other outstanding contributions on this subject, see: ; (c) G. Grynkiewicz and A. Zamojski, Z. Naturforsch., 1980, 35B, 1024 CAS; (d) M. T. Reetz and H. Muller-Starke, Liebigs Ann. Chem., 1983, 1726 CrossRef CAS; (e) O. R. Martin, Tetrahedron Lett., 1985, 26, 2055 CrossRef CAS; (f) T. Matsumoto, M. Katsuki, H. Jona and K. Suzuki, Tetrahedron Lett., 1989, 30, 6185 CrossRef CAS; (g) K. Toshima, G. Matsuo, T. Ishizuka, M. Nakata and M. Kinoshita, J. Chem. Soc., Chem. Commun., 1992, 1641 RSC; (h) K. Suzuki, Pure Appl. Chem., 1994, 66, 2175 CAS; (i) R. S. Babu and G. A. O'Doherty, J. Am. Chem. Soc., 2003, 125, 12406 CrossRef CAS PubMed.
- 1a was obtained in <10% yield when the reaction was performed at room temperature as suggested in the original protocols of Grynkiewicz and Feringa. Reaction at reduced temperature (0–5 °C) afforded 1a in reasonable yields.
- See the ESI† for details.
- For our recent work on the conversion of furans to benzofurans, see: R. P. Shirke, V. Reddy, R. V. Anand and S. S. V. Ramasastry, Synthesis, 2016 DOI:10.1055/s-0035-1560432 . For our work on an interesting transformation of benzofurans to furans, see ref. 4g.
- X-ray crystallographic data of compound 4l are provided in the ESI.†4l, CCDC 1437901.
- The observed regioselectivity (C-1 vs. C-3) during the Fries-type rearrangement of 2-naphthyl derivatives (4s–4x) is in agreement with literature reports, see: C. Cui, X. Wang and R. G. Weiss, J. Org. Chem., 1996, 61, 1962 CrossRef CAS.
- (a) S. J. Gharpure and V. Prasath, Org. Biomol. Chem., 2014, 12, 7397 RSC; (b) F. Wang, C. Luo, Y. Y. Shen, Z. D. Wang, X. Li and J. P. Cheng, Org. Lett., 2015, 17, 338 CrossRef CAS PubMed.
- For pterophyllins, see: (a) D. A. Mulholland, S. E. Iourine, D. A. H. Taylor and F. M. Dean, Phytochemistry, 1998, 47, 1641 CrossRef CAS . For medicinal significance of furocoumarins, see: ; (b) L. Santana, E. Uriarte, F. Roleira, N. Milhazes and F. Borges, Curr. Med. Chem., 2004, 11, 3239 CrossRef CAS PubMed; (c) Y. Dong, Q. Shi, H. C. Pai, C. Y. Peng, S. L. Pan, C. M. Teng, K. Nakagawa-Goto, D. Yu, Y. N. Liu, P. C. Wu, K. F. Bastow, S. L. Morris-Natschke, A. Brossi, J. Y. Lang, J. L. Hsu, M. C. Hung, E. Y. H. P. Lee and K. H. Lee, J. Med. Chem., 2010, 53, 2299 CrossRef CAS PubMed.
- (a) G. Cheng and Y. Hu, Chem. Commun., 2007, 3285 RSC; (b) G. Raffa, M. Rusch, G. Balme and N. Monteiro, Org. Lett., 2009, 11, 5254 CrossRef CAS PubMed; (c) R. G. dos Santos, A. R. Jesus, J. M. Caio and A. P. Rauter, Curr. Org. Chem., 2011, 15, 128 CrossRef CAS; (d) C.-J. Lee, C.-C. Tsai, S.-H. Hong, G.-H. Chang, M.-C. Yang, L. Möhlmann and W. Lin, Angew. Chem., Int. Ed., 2015, 54, 8502 CrossRef CAS PubMed; (e) W. L. Zhang, S. N. Yue, Y. M. Shen, H. Y. Hu, Q.-H. Meng, H. Wu and Y. Liu, Org. Biomol. Chem., 2015, 13, 3602 RSC; (f) X. Y. Zhang, L. L. Hu, Z. Shen, Z. Z. Chen, Z. G. Xu, S. Q. Li, J. W. Xie and H. L. Cui, Synlett, 2015, 2821 CAS.
- See the ESI† for additional experiments carried out to get insights into the mechanism.
- (a) T. Kometani, H. Kondo and Y. Fujimori, Synthesis, 1988, 1005 CrossRef CAS; (b) T. Matsumoto, M. Katsuki and K. Suzuki, Tetrahedron Lett., 1988, 29, 6935 CrossRef CAS; (c) T. Matsumoto, T. Katsuki, H. Jona and K. Suzuki, J. Am. Chem. Soc., 1991, 113, 6982 CrossRef CAS; (d) J. A. Mahling and R. R. Scmidt, Synthesis, 1993, 325 CrossRef CAS; (e) C. Booma and K. K. Balasubramanian, Tetrahedron Lett., 1995, 36, 5807 CAS; (f) E. R. Palmacci and P. H. Seeberger, Org. Lett., 2001, 3, 1547 CrossRef CAS PubMed; (g) E. R. Palmacci, O. J. Plante and P. H. Seeberger, Eur. J. Org. Chem., 2002, 595 CrossRef CAS; (h) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240 RSC; (i) C. Wiebe, C. Schlemmer, S. Weck and T. Opatz, Chem. Commun., 2011, 47, 9212 RSC; (j) X. H. Xu, M. Taniguchi, X. Wang, E. Tokunaga, T. Ozawa, H. Masuda and N. Norio Shibata, Angew. Chem., Int. Ed., 2013, 52, 12628 CrossRef CAS PubMed; (k) C. N. Kona and C. V. Ramana, Chem. Commun., 2014, 50, 2152 RSC.
- Intermediacy of an oxidopyrylium intermediate at this stage can be ruled out based on the fact that alcohols 4j and 4t were isolated in 98% and 93% ee, respectively.
- (a) J. Sperry, Z. E. Wilson, D. C. K. Rathwell and M. A. Brimble, Nat. Prod. Rep., 2010, 27, 1117 RSC; (b) I. Sharma, J. M. Wurstand and D. S. Tan, Org. Lett., 2014, 16, 2474 CrossRef CAS PubMed; (c) Z. Li and Y. Shi, Org. Lett., 2015, 17, 5752 CrossRef CAS PubMed.
- X-ray crystallographic data of compound 4ag are provided in the ESI.†4ag, CCDC 1441109.
- For aquilarinoside A, see: (a) J. Qi, J. J. Lu, J. H. Liu and B. Y. Yu, Chem. Pharm. Bull., 2009, 57, 134 CrossRef CAS PubMed . For pinnatifinoside A, see: ; (b) P. C. Zhang and S. X. Xu, Phytochemistry, 2001, 57, 1249 CrossRef CAS PubMed . For other 5,5-spiroketal natural products, see: ; (c) Y. C. Hu, X. F. Wu, S. Gao, S. S. Yu, Y. Liu, J. Qu, J. Liu and Y. B. Liu, Org. Lett., 2006, 8, 2269 CrossRef CAS PubMed; (d) R. Raju, O. Gromyko, V. Fedorenko, A. Luzhetskyy and R. Muller, Org. Lett., 2013, 15, 3487 CrossRef CAS PubMed; (e) H. Wang, J. Hong, J. Yin, H. R. Moon, Y. Liu, X. Wei, D. C. Oh and J. H. Jung, J. Nat. Prod., 2015, 78, 2832 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1437901 and 1441109. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01016d |
| This journal is © The Royal Society of Chemistry 2016 |