
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium-catalyzed asymmetric [3+2] annulation of aromatic ketimines with alkynes via C–H activation: unexpected inversion of the enantioselectivity induced by protic acids†
Midori
Nagamoto
,
Daisuke
Yamauchi
and
Takahiro
Nishimura
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: tnishi@kuchem.kyoto-u.ac.jp
First published on 29th March 2016
Abstract
A cationic iridium/binap catalyst enabled the asymmetric [3+2] annulation of cyclic N-acyl ketimines with internal alkynes via C–H activation to give spiroaminoindene derivatives with high enantioselectivity. The stereochemical course of this annulation was switchable by acid additives.
Transition metal-catalyzed direct functionalization of aromatic C–H bonds has enabled a short-step synthesis of various useful compounds.1 The regioselective functionalization has been achieved by use of directing groups, which are often involved in the reactions leading to cyclic compounds through the catalytic process.2 In this regard, there have been several successful reports on the formal [3+2] annulation reactions of aromatic imines or ketones with C–C multiple bonds giving indene or indane derivatives in the presence of Re,3 Ru,4 Rh,5 and Ir catalysts.6 The asymmetric annulation reaction has also been reported by means of Rh5c,d and Ir6c catalysis giving chiral 1-aminoindane derivatives with high enantioselectivity.
Recently, we reported that a cationic iridium(cod) complex catalyzes a [3+2] annulation of aromatic ketimines with alkynes via C–H activation to give spiroaminoindenes (Scheme 1a).7 Mechanistic studies revealed that the reaction proceeds via sequential ortho-alkenylation and intramolecular cyclization. For the development of the asymmetric reaction, we envisioned two possible asymmetric induction methods in light of the tandem reaction mechanism, which involves the intramolecular cyclization of the alkenylated intermediate C as the enantio-determining step. One is the transfer of a transient axial chirality of intermediate C to the chiral annulation product 3,8 and the other is the use of the cationic iridium species as a chiral Lewis acid to invoke the enantioselective cyclization of intermediate C.9 We tested several chiral ligands for the Ir-catalyzed annulation of 3-hydroxy-3-phenylisoindolin-1-one (1a), which in situ generates an N-acyl ketimine by dehydration,7 but only low levels of the asymmetric induction were obtained (e.g., 25% ee with (R)-binap, Scheme 1b). In contrast, it was found that the reaction of ketimines having substituted phenyl groups gave the annulation products in high yields and enantioselectivity. We also found that the stereochemical course of this annulation was switchable by acid additives. Here we report that the asymmetric [3+2] annulation of cyclic N-acyl ketimines with internal alkynes via C–H activation gives chiral spiroaminoindene derivatives.
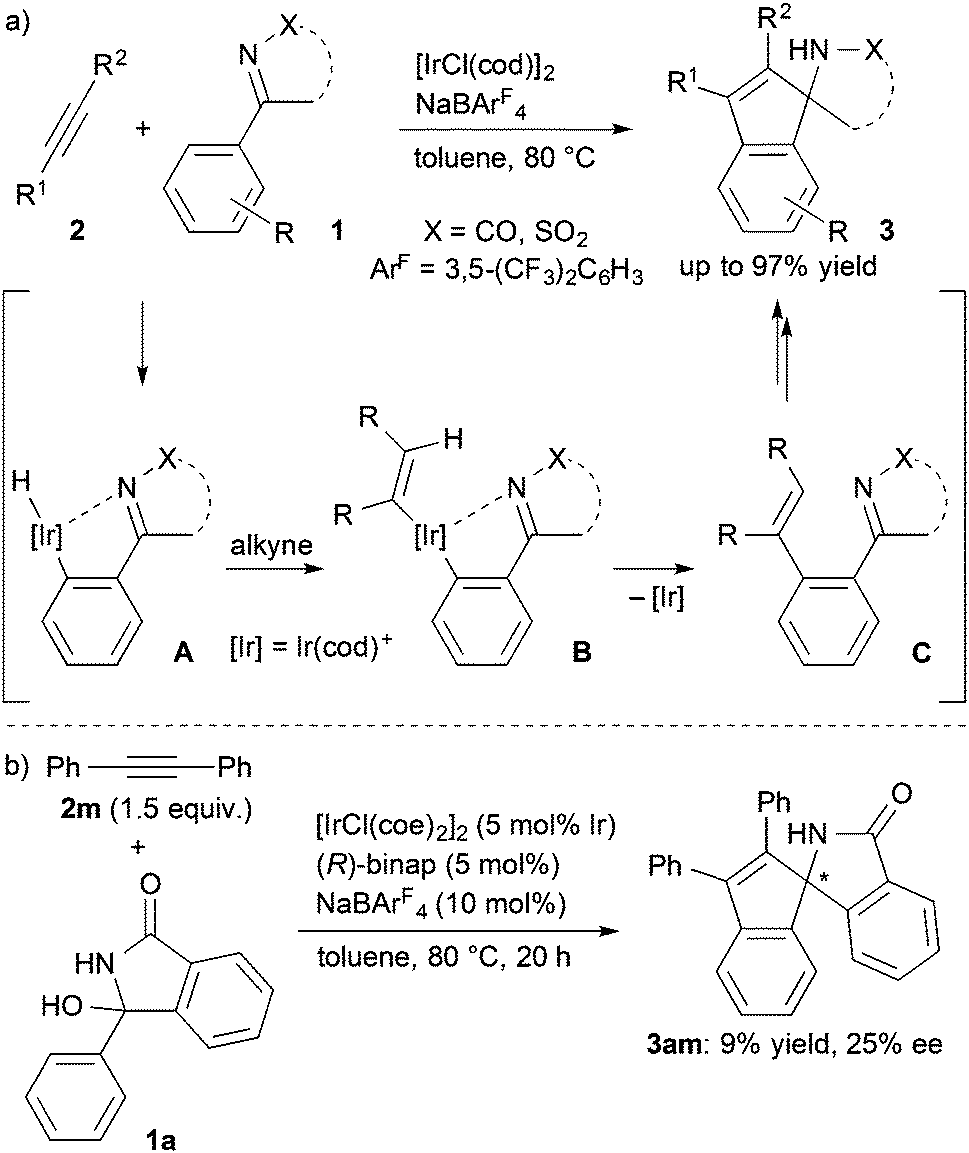 | ||
| Scheme 1 Ir-catalyzed annulation of aromatic ketimines 1 with alkynes 2. | ||
An enantiodivergent synthesis of both enantiomers has drawn a lot of attention, particularly in the field of pharmaceutical chemistry because each enantiomer often exhibits different bioactivity.10 It is an attractive and important challenge to induce both enantioselectivities using a single chiral catalyst by changing the reaction conditions such as the reaction temperature,11 solvents,12 and additives.13 We found that the enantiodivergent synthesis of the annulation products formed from ketimines and alkynes under Ir catalysis can be achieved by means of just a slight modification of the reaction conditions. Treatment of 3-hydroxy-3-(3,5-dimethoxyphenyl)isoindolin-1-one (1b) with diphenylacetylene (2m) in chlorobenzene in the presence of [Ir(cod)2]BF4 (5 mol%, cod = 1,5-cyclooctadiene) and (R)-binap (7.5 mol%) at 80 °C for 20 h gave the annulation product (+)-3bm in 79% yield with 85% ee (Scheme 2). To our surprise, the presence of a carboxylic acid inverted the enantioselectivity giving (−)-3bm under otherwise the same reaction conditions; the reaction in the presence of benzoic acid (10 mol%) gave (−)-3bm in 81% yield with 84% ee. The same inverse effect of the enantioselectivity was also observed in the presence of acetic acid (83% ee) and pivalic acid (50% ee).
 | ||
| Scheme 2 Ir-catalyzed asymmetric annulation of ketimine 1b with alkyne 2m. | ||
The enantiodivergent synthesis of the annulation products can be achieved in the reaction of 1b with several internal alkynes 2 (Table 1). The reactions of symmetrically substituted diaryl acetylenes 2m–2s gave the corresponding annulation products (+)-3bm–3bs with 83–93% ee in the absence of benzoic acid (Condition A, odd entries of 1–14). In contrast, the reactions in the presence of benzoic acid (Condition B) gave (−)-3bm–3bs with 59–85% ee (even entries of 1–14). The reaction of 1-phenyl-1-hexyne (2t) gave 3bt with high regioselectivity and good enantioselectivity under both Conditions A and B (entries 15 and 16). In contrast, the switch of the enantioselectivity was not observed in the reaction of 1-octyne (2u); the reaction gave (+)-3bu with moderate ee under both Conditions A and B (entries 17 and 18). The absolute configuration of (+)-3br obtained under Condition A was determined to be S by X-ray crystallographic analysis.14
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne | Condition | Yield (%) | ee (%) |
| a Condition A: 1b (0.20 mmol), alkyne 2 (0.30 mmol), [Ir(cod)2]BF4 (5 mol%), and (R)-binap (7.5 mol%) in PhCl (0.80 mL) at 80 °C for 48 h. Condition B: with PhCO2H (10 mol%). Isolated yields are shown and the ee was determined by chiral HPLC analysis. b [Ir(cod)2]BF4 (10 mol%) and (R)-binap (15 mol%). c Combined yield of two regioisomers (97/3). d Combined yield of two regioisomers (99/1). | ||||
| 1 | R1 = R2 = Ph (2m) | A | 81 (3bm) | 88 (+) |
| 2 | 2m | B | 94 (3bm) | 83 (−) |
| 3b | R1 = R2 = 4-MeC6H4 (2n) | A | 68 (3bn) | 92 (+) |
| 4 | 2n | B | 89 (3bn) | 76 (−) |
| 5 | R1 = R2 = 4-MeOC6H4 (2o) | A | 73 (3bo) | 93 (+) |
| 6 | 2o | B | 97 (3bo) | 59 (−) |
| 7b | R1 = R2 = 4-CF3C6H4 (2p) | A | 53 (3bp) | 83 (+) |
| 8 | 2p | B | 50 (3bp) | 87 (−) |
| 9 | R1 = R2 = 4-FC6H4 (2q) | A | 72 (3bq) | 92 (+) |
| 10 | 2q | B | 98 (3bq) | 79 (−) |
| 11 | R1 = R2 = 4-ClC6H4 (2r) | A | 89 (3br) | 87 (+) |
| 12 | 2r | B | 93 (3br) | 82 (−) |
| 13 | R1 = R2 = 3-ClC6H4 (2s) | A | 52 (3bs) | 84 (+) |
| 14 | 2s | B | 71 (3bs) | 85 (−) |
| 15 | R1 = Ph, R2 = n-Bu (2t) | A | 56c (3bt) | 73 (−) |
| 16 | 2t | B | 50d (3bt) | 92 (+) |
| 17 | R1 = R2 = n-Pr (2u) | A | 34 (3bu) | 37 (+) |
| 18 | 2u | B | 23 (3bu) | 57 (+) |

Scheme 3 summarizes the results obtained for the reaction of several hemiaminals 1 with diphenylacetylene 2m. The reaction of 1c, having a 3,5-dibenzyloxyphenyl group, gave (+)-3cm with 93% ee under Condition A, and (−)-3cm with 42% ee under Condition B. The inversion of enantioselectivity was also observed in the reaction of 1d having a 3,4,5-trimethoxyphenyl group (86% ee (+) under Condition A and 33% ee (−) under Condition B). In contrast, in the reaction of 1e substituted with a 2,5-dimethoxypheny group and 1f having a 3-methoxy-5-methyl group, the inversion of the enantioselectivity did not take place.
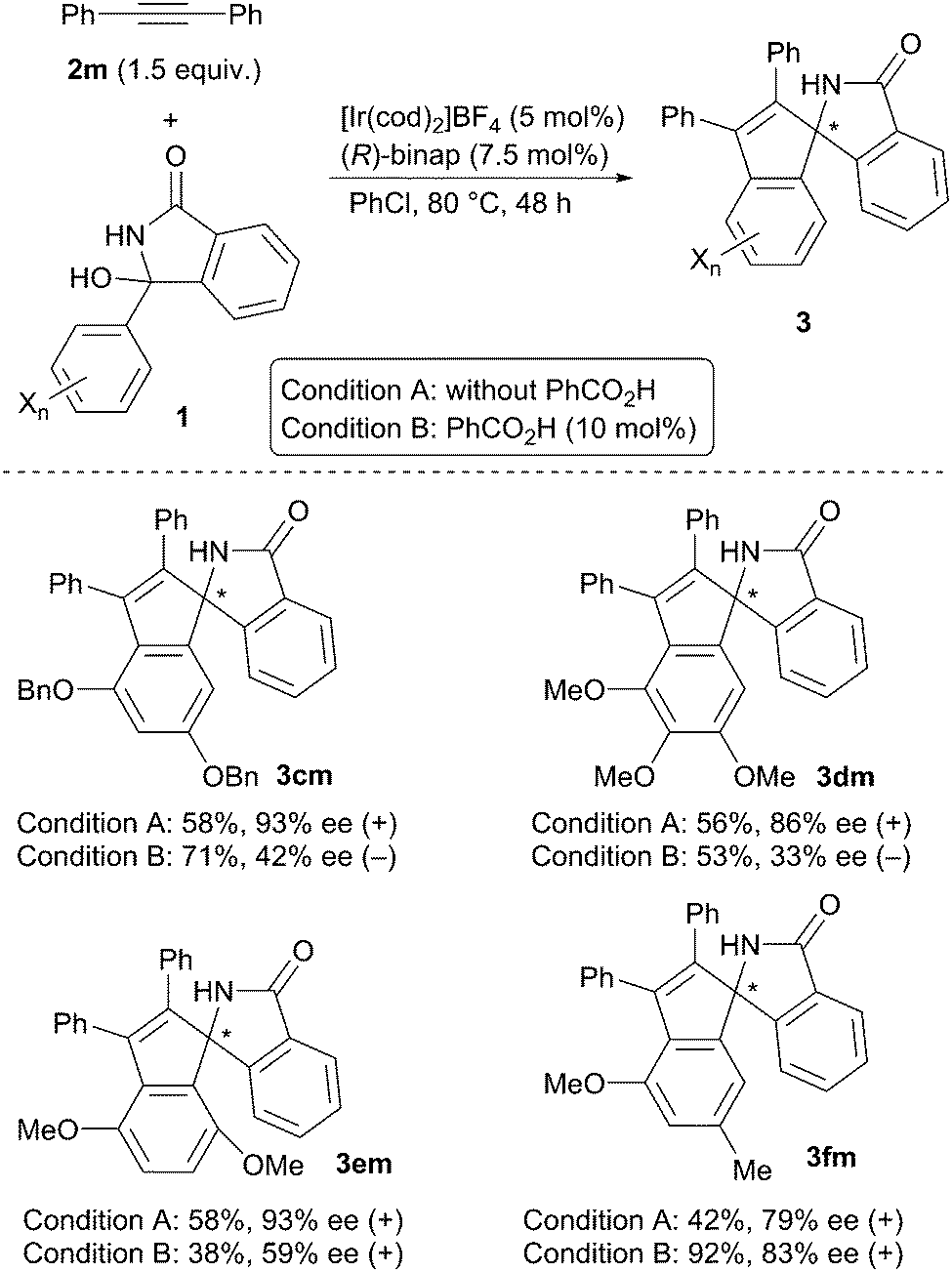 | ||
| Scheme 3 Asymmetric annulation of hemiaminals 1 with 2m. | ||
The reaction of hemiaminals bearing monosubstituted aryl groups was also conducted in order to gain some insights into the origin of the enantioselectivity (Scheme 4). The reaction of 1g having a meta-methoxyphenyl group gave an isomeric mixture of 3gm and 3gm′ with good regioselectivity (Scheme 4a). The enantioselectivity of the major regioisomer 3gm, which was formed via C–H activation at the less sterically hindered ortho-position, was much higher than that of 3gm′ under both Conditions A and B. The inversion of enantioselectivity was only observed for the major regioisomer 3gm. It should be noted that the ee of 3gm′ was much lower than that of 3gm, although it was expected that the methoxy group can restrict the axial rotation of the alkenylated intermediate formed via C–H activation at the more sterically hindered ortho-position. These results are consistent with the chiral Lewis acid-induced cyclization rather than the chirality transfer as the enantio-determining step. The reactions of 1h and 1i, having a methyl and a chloro group at the meta-position, respectively, gave the corresponding annulation products. The inversion of the enantioselectivity was observed in both cases, albeit with a low ee (Scheme 4b). In contrast, the inversion was not observed in the reaction of 1j having an ortho-methoxyphenyl group, while a decrease of the ee was observed in the presence of the acid (Scheme 4c). The para-methoxy group on 1k had no effect on the asymmetric induction in the absence of acid (Scheme 4d). The reaction of 1k in the presence of the acid gave 3km with 20% ee. Hemiaminal 1a having a non-substituted phenyl group exhibited a similar reactivity and selectivity to 1k. These results indicate that the meta-substituent of the aromatic rings, which undergo the C–H activation, plays some roles in the inversion of the enantioselectivity in the presence of the acid, regardless of their electronic character. Although we assume that the asymmetric induction is due to the chiral Lewis acid catalyst shown in Scheme 1, the acid additive might alter the reaction mechanism, which may result in the different stereochemical outcome.15,16 At this stage, the exact roles of the acid additives and the substituents on aryl group remain unclear.
 | ||
| Scheme 4 Annulation of monosubstituted hemiaminals 1 with 2m. | ||
In summary, we have developed an asymmetric [3+2] annulation of cyclic N-acyl ketimines with alkynes using a cationic iridium/binap catalyst. The enantioselectivity was found to be switchable by acid additives.
This work was supported by JSPS KAKENHI Grant No. 15H03810. M. N. thanks the JSPS for a Research Fellowship for Young Scientists.
Notes and references
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS . For selected recent reviews, see: ; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed; (e) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (f) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764 CrossRef CAS PubMed; (b) V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416 CrossRef CAS PubMed; (c) L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176 CrossRef CAS PubMed; (d) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (e) M. P. Huestis, L. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., Int. Ed., 2011, 50, 1338 CrossRef CAS PubMed; (f) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed; (g) Z. Shi, C. Grohmann and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5393 CrossRef CAS PubMed; (h) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed; (i) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (j) Y. Su, M. Zhao, K. Han, G. Song and X. Li, Org. Lett., 2010, 12, 5462 CrossRef CAS PubMed; (k) Y. Unoh, Y. Hashimoto, D. Takeda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3258 CrossRef CAS PubMed; (l) K. Morimoto, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 9548 CrossRef CAS PubMed; (m) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed.
- (a) Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed; (b) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (c) Y. Kuninobu, Y. Nishina, M. Shouho and K. Takai, Angew. Chem., Int. Ed., 2006, 45, 2766 CrossRef CAS PubMed; (d) Y. Kuninobu, P. Yu and K. Takai, Org. Lett., 2010, 12, 4274 CrossRef CAS PubMed.
- (a) P. W. R. Harris, C. E. F. Rickard and P. D. Woodgate, J. Organomet. Chem., 1999, 589, 168 CrossRef CAS; (b) R. K. Chinnagolla and M. Jeganmohan, Eur. J. Org. Chem., 2012, 417 CrossRef CAS; (c) P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14, 5506 CrossRef CAS PubMed; (d) J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681 CrossRef CAS PubMed; (e) C.-H. Hung, P. Gandeepan and C.-H. Cheng, ChemCatChem, 2014, 6, 2692 CrossRef CAS.
- Rh(I): (a) Z.-M. Sun, S.-P. Chen and P. Zhao, Chem. – Eur. J., 2010, 16, 2619 CrossRef CAS PubMed; (b) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181 CrossRef CAS PubMed; (c) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098 CrossRef CAS PubMed; (d) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630 CrossRef CAS PubMed ; Rh(III): ; (e) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS PubMed; (f) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 4169 CrossRef CAS PubMed; (g) Y. Chen, F. Wang, W. Zhen and X. Li, Adv. Synth. Catal., 2013, 355, 353 CAS; (h) L. Dong, C.-H. Qu, J.-R. Huang, W. Zhang, Q.-R. Zhang and J.-G. Deng, Chem. – Eur. J., 2013, 19, 16537 CrossRef CAS PubMed; (i) M. V. Pham and N. Cramer, Chem. – Eur. J., 2016, 2270 CrossRef CAS PubMed.
- (a) K. Tsuchikama, M. Kasagawa, K. Endo and T. Shibata, Synlett, 2010, 97 CAS; (b) T. Nishimura, Y. Ebe and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 2092 CrossRef CAS PubMed; (c) T. Nishimura, M. Nagamoto, Y. Ebe and T. Hayashi, Chem. Sci., 2013, 4, 4499 RSC; (d) Y. Ebe, M. Hatano and T. Nishimura, Adv. Synth. Catal., 2015, 357, 1425 CrossRef CAS.
- M. Nagamoto and T. Nishimura, Chem. Commun., 2014, 50, 6274 RSC.
- For a review, see: (a) D. Campolo, S. Gastaldi, C. Roussel, M. P. Bertrand and M. Nechab, Chem. Soc. Rev., 2013, 42, 8434 RSC ; for a recent example, see: ; (b) L. Yang, H. Zheng, L. Luo, J. Nan, J. Liu, Y. Wang and X. Luan, J. Am. Chem. Soc., 2015, 137, 4876 CrossRef CAS PubMed.
- The ortho-alkenylated N-sulfonylketimine was isolated in the annulation of N-sulfonylketimine with an alkyne, and the cyclization of it was found to proceed on a slightly acidic silica gel without the iridium catalyst; see ref. 7 for details.
- For reviews, see: (a) J. Escorihuela, M. I. Burguete and S. V. Luis, Chem. Soc. Rev., 2013, 42, 5595 RSC; (b) M. Bartók, Chem. Rev., 2010, 110, 1663 CrossRef PubMed; (c) T. Tanaka and M. Hayashi, Synthesis, 2008, 3361 CAS; (d) G. Zanoni, F. Castronovo, M. Franzini, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115 RSC; (e) Y. H. Kim, Acc. Chem. Res., 2001, 34, 955 CrossRef CAS PubMed.
- (a) V. S. Chen, M. Chiu, R. D. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef PubMed; (b) K. Manna, N. Eedugurala and A. D. Sadow, J. Am. Chem. Soc., 2015, 137, 425 CrossRef CAS PubMed.
- (a) J. C. Anderson, G. J. Stepney, M. R. Mills, L. R. Horsfall, A. J. Blake and W. Lewis, J. Org. Chem., 2011, 76, 1961 CrossRef CAS PubMed; (b) N. Haddad, B. Qu, S. Rodriguez, L. van der Veen, D. C. Reeves, N. C. Gonnella, H. Lee, N. Grinberg, S. Ma, D. Krishnamurthy, T. Wunberg and C. H. Senanayake, Tetrahedron Lett., 2011, 52, 3718 CrossRef CAS; (c) H. Yu, F. Xie, Z. Ma, Y. Liu and W. Zhang, Org. Biomol. Chem., 2012, 10, 5137 RSC; (d) R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem. – Eur. J., 2015, 21, 4800 CrossRef CAS PubMed.
- (a) T. Nishimura, X.-X. Guo and T. Hayashi, Chem. – Asian J., 2008, 3, 1505 CrossRef CAS PubMed; (b) T. Inagaki, A. Ito, J. Ito and H. Nishiyama, Angew. Chem., Int. Ed., 2010, 49, 9384 CrossRef CAS PubMed.
- See the ESI† for the details.
- For an example of the formation of hydridoiridium(III) complexes by the reaction of iridium/bisphosphine complexes with carboxylic acids, see: T. Yamamoto, H. Tadaoka, M. Nagata, T. Hirao, Y. Kataoka, V. Ratovelomanana-Vidal, J. P. Genet and K. Mashima, Organometallics, 2006, 25, 2505 CrossRef.
- The carbometalation of the aryliridium(III) species to an alkyne and a subsequent intramolecular cyclization are alternative reaction pathways leading to the annulation product. For an example of Rh(III)-catalyzed asymmetric annulation of N-sufonylketimines with alkynes involving the carbometalation step, see ref. 5i.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and X-ray crystallographic data of compound 3br. CCDC 1449863. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01398h |
| This journal is © The Royal Society of Chemistry 2016 |