
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ba4Ru3O10.2(OH)1.8: a new member of the layered hexagonal perovskite family crystallised from water†
Craig I.
Hiley
a,
Martin R.
Lees
b,
David L.
Hammond
b,
Reza J.
Kashtiban
b,
Jeremy
Sloan
b,
Ronald I.
Smith
c and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
cISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire, OX11 0QX, UK
First published on 7th April 2016
Abstract
A new barium ruthenium oxyhydroxide Ba4Ru3O10.2(OH)1.8 crystallises under hydrothermal conditions at 200 °C: powder neutron diffraction data show it adopts an 8H hexagonal perovskite structure with a new stacking sequence, while high resolution electron microscopy reveals regions of ordered layers of vacant Ru sites, and magnetometry shows antiferromagnetism with TN = 200(5) K.
Oxides of the second- and third-row transition elements are of topical interest for their exotic magnetic properties, which have been rather less explored compared to their first-row analogues. The range of striking magnetic and electronic properties is exemplified by ruthenates, such as superconductivity in Sr2RuO4,1 ferromagnetism in SrRuO3,2 antiferromagnetism persisting to 560 K in SrRu2O63 and non-Fermi-liquid behaviour in La4Ru6O19.4 Complex ruthenium oxides are also of interest in heterogeneous catalysis,5,6 and for electrocatalytic splitting of water.7 The structural chemistry of barium ruthenates is known already to be particularly extensive, and in this family there is a notable propensity to adopt hexagonal perovskite structures. These consist of layers of hexagonal close-packed [BaO3] units with Ru atoms occupying a fraction of the octahedral interstices.8,9 Layers are stacked along the c-axis such that octahedra are linked in either a corner-sharing manner (as in the cubic perovskite, denoted c) or in a face-sharing manner (hexagonal close packing, h), which may be stabilised by metal–metal bonding. Whilst some of these phases are stoichiometric, namely the 4H-, 9R- and 10H-BaRuO3 variants,10–12 hexagonal perovskites with compositional variation have also been discovered. Deviation from the ideal ABO3 composition has been observed in the form of both Ru deficiency (as observed in Ba5Ru2O10 and Ba5Ru2O9(O2)13,14) and substitution of Ru for other metals (Li, Na, Mg, Zn;15 Ni, Co, Zn16).
When preparing new complex ruthenates, highly oxidising environments are often required to access oxidation states of Ru higher than +4, and, indeed, to prevent reduction to Ru metal, however high oxygen pressure at elevated temperatures can lead to formation and volatilisation of considerable quantities of RuVIIIO4 making control of composition challenging.17 Low temperature synthesis techniques that can stabilise highly oxidised Ru, such as molten salt flux crystal growth18 and hydrothermal crystallisation,19 are therefore attractive propositions for the isolation of complex ruthenium oxides. Recently we reported the unique structure of Ba2Ru3O9(OH), an oxyhydroxide prepared by a hydrothermal reaction.20 We demonstrate herein that by simply altering the ratio of the two reagents, this synthetic procedure yields a structurally unrelated, 8H-hexagonal perovskite phase Ba4Ru3O10.2(OH)1.8, which contains a new stacking sequence, and regions of vacancy ordering.
Phase-pure Ba4Ru3O10.2(OH)1.8 was prepared by the hydrothermal reaction of BaO2 and KRuO4 at 200 °C for 24 hours in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (i.e. an excess of Ba). A silver-grey powder was recovered by suction filtration and washed with 0.1 M HCl to remove any Ba(OH)2 or BaCO3 byproducts, as we found necessary in earlier work on other oxides.20 The powder X-ray diffraction pattern of the new phase was indexed to a hexagonal unit cell with lattice parameters a = 5.79905(7) Å, c = 18.7562(5) Å, similar to those of the barium ruthenate 8H-perovskites Ba4Ru3NaO1215 and Ba2Ru5O10.21 ICP measurements give a Ba:Ru 1.33(1):1 ratio, and no K was detected (detection limit 10 ppm). A structural solution was initially found by a Monte Carlo optimisation in direct space using the software FOX,22 and a Rietveld refinement of this structure against time-of-flight powder neutron diffraction data (PND, GEM, ISIS; Fig. 1(a)) was carried out in GSAS23 implemented using EXPGUI.24 The best fit was obtained in the space group P63/mmc.
:1 ratio (i.e. an excess of Ba). A silver-grey powder was recovered by suction filtration and washed with 0.1 M HCl to remove any Ba(OH)2 or BaCO3 byproducts, as we found necessary in earlier work on other oxides.20 The powder X-ray diffraction pattern of the new phase was indexed to a hexagonal unit cell with lattice parameters a = 5.79905(7) Å, c = 18.7562(5) Å, similar to those of the barium ruthenate 8H-perovskites Ba4Ru3NaO1215 and Ba2Ru5O10.21 ICP measurements give a Ba:Ru 1.33(1):1 ratio, and no K was detected (detection limit 10 ppm). A structural solution was initially found by a Monte Carlo optimisation in direct space using the software FOX,22 and a Rietveld refinement of this structure against time-of-flight powder neutron diffraction data (PND, GEM, ISIS; Fig. 1(a)) was carried out in GSAS23 implemented using EXPGUI.24 The best fit was obtained in the space group P63/mmc.
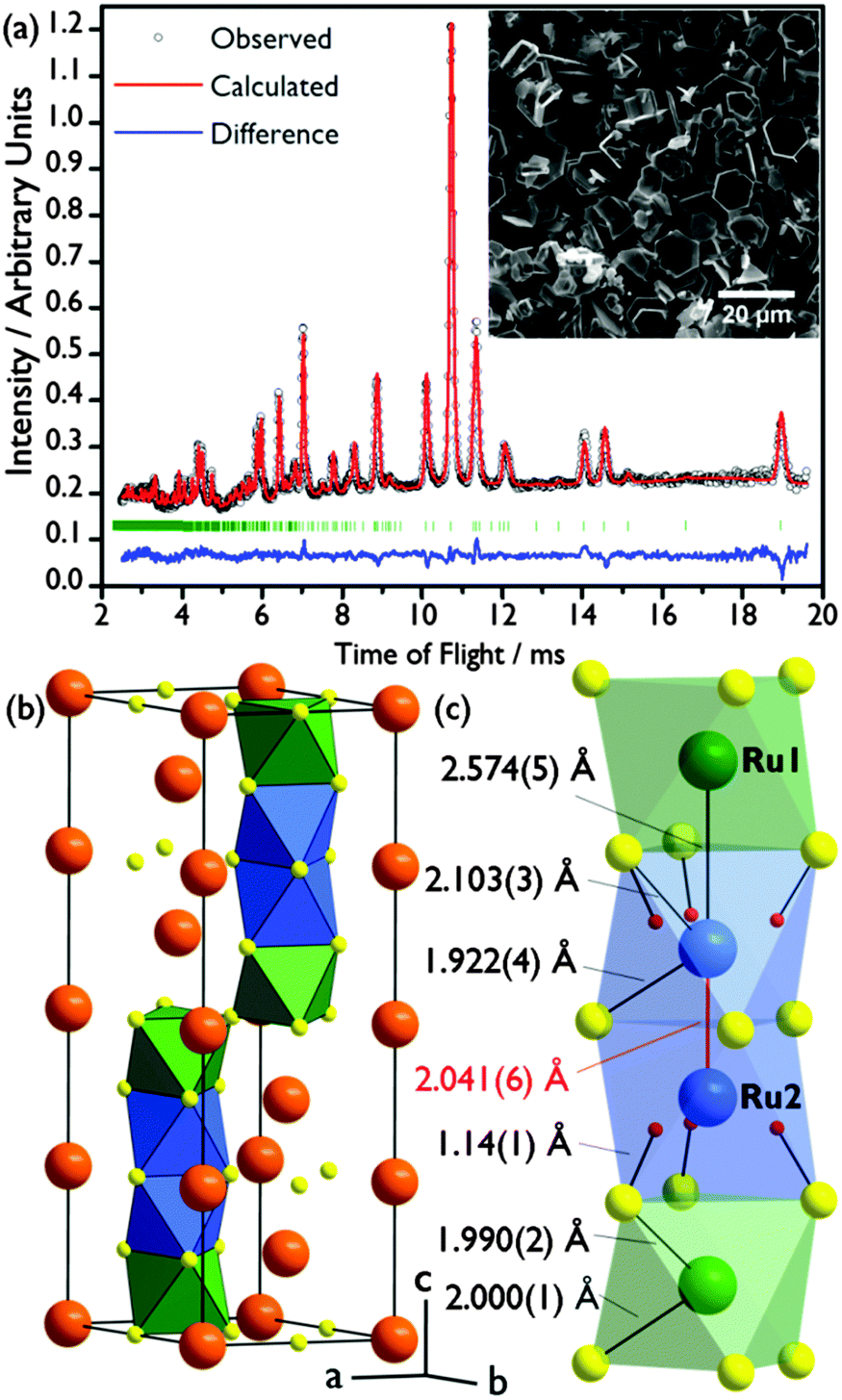 | ||
| Fig. 1 (a) Rietveld fit to powder neutron diffraction (GEM Bank 4) with scanning electron micrograph inset. (b) Representation of the unit cell of Ba4Ru3O10.2(OH)1.8 with Ba atoms in orange, oxygen atoms in yellow, fully occupied octahedral sites in green and half-occupied octahedral sites in blue. (c) One chain of four octahedral Ru sites with refined bond lengths labelled. H sites in red. | ||
The structure of Ba4Ru3O10.2(OH)1.8 consists of an eight-layer stacking sequence, (chhh)2 (Fig. 2(a)), with chains of four face-sharing octahedral sites (Table 1 and Fig. 1(b)). However, the Ru2 site, which lies within the two central octahedral sites of the quasi-tetrameric chains (Fig. 1(c)), is only half-occupied. Examination of high angle annular dark field images obtained in scanning transmission electron microscopy (HAADF STEM) shows that regions of the sample show extensive vacancy ordering (Fig. 2(b)). A refinement to the powder neutron diffraction using a model in the lower symmetry space group P63mc with ordered vacant sites yields a poorer fit to the data (see ESI†), suggesting that although vacancies may order in domains on the nanoscale, on average the Ru2 site is randomly occupied. Whilst the Ru1 site sits on the highly symmetrical octahedral site (Ru–O bond distances in the range 1.990(2)–2.000(1) Å), the Ru2 sits off-centre in an octahedral site, generating three long (2.103(3) Å) and three short (1.922(4) Å) Ru–O distances (Fig. 1(c)). This gives a Ru1–Ru2 bond distance of 2.574(5) Å, a value consistent with an average Ru oxidation state of +4.5–4.75 in barium ruthenates, using the correlation established by Kimber et al. in their study of 6H perovskites Ba3ARu2O9.26 A second consequence of the Ru2 site asymmetry is the unphysically short distance between neighbouring Ru2 sites of 2.041(6) Å, indicating that whilst there is no preferential ordering within the quasi-tetrameric chains, there must be exactly one octahedral vacancy per chain. The structure can be compared to the structure adopted in another ruthenate 8H-perovskite, Sr4Ru3.05O12, which also contains partially occupied face-sharing Ru octahedra and, due to the (ccch)2 stacking sequence, also has no Ru–Ru bonding (see ESI†).25
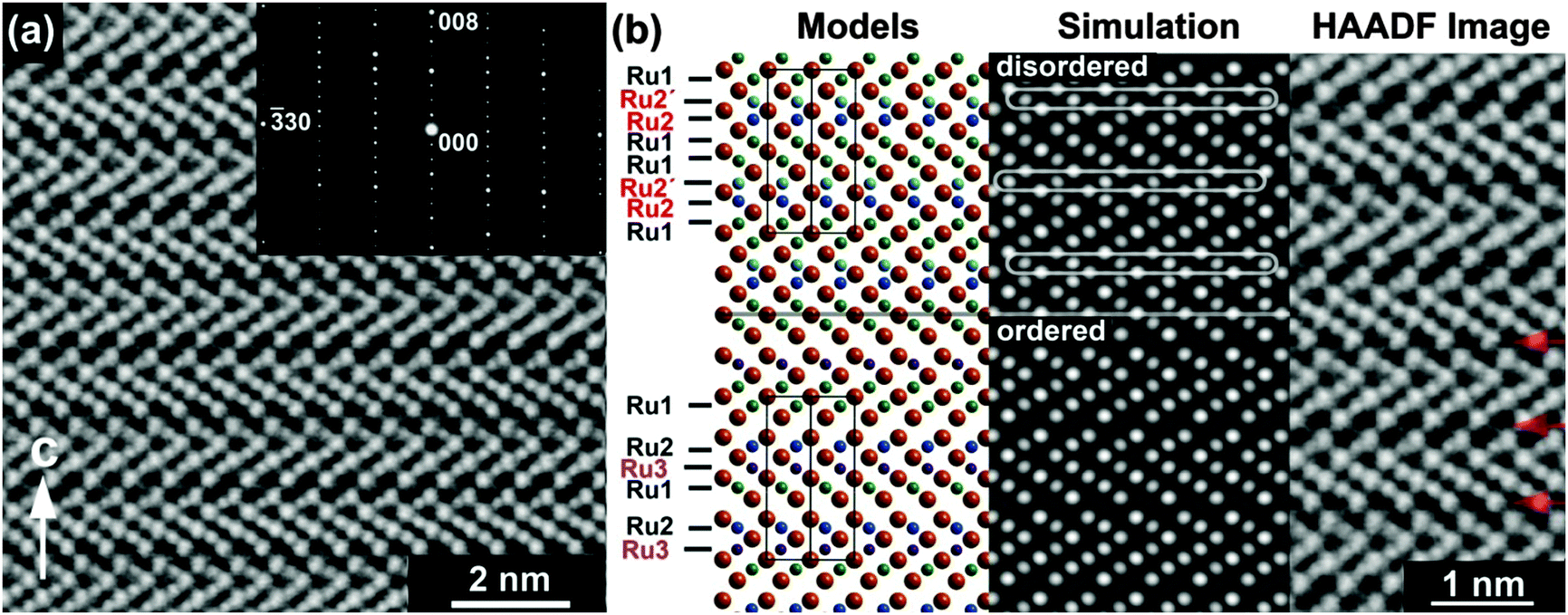 | ||
| Fig. 2 (a) HAADF STEM image of Ba4Ru3O10.2(OH)1.8 with inset electron diffraction pattern. (b) Comparison of simulated HAADF images from disordered and ordered Ru vacancies to an experimental HAADF image obtained from a [110] projection. In the disordered simulation partially occupied Ru positions (Ru2′) are indicated by a loop in the simulated image. The diffuse red arrows in the experimental HAADF indicate that these are absent observable by comparison with the ‘ordered’ simulation. | ||
| Atom | x/a | y/b | z/c | Occupancy | ADP/Å2 | BVS |
|---|---|---|---|---|---|---|
| Ba1 | 0 | 0 | 0 | 1 | 0.010(1) | 2.34 |
| Ba2 | 0 | 0 | 1/4 | 1 | 0.015(1) | 2.10 |
| Ba3 | 1/3 | 2/3 | 0.1280(2) | 1 | 0.0070(5) | 2.32 |
| Ru1 | 1/3 | 2/3 | 0.5584(1) | 1 | 0.0159(4) | 4.39 |
| Ru2 | 1/3 | 2/3 | 0.8044(2) | 0.5 | 0.0080(6) | 4.41 |
| O1 | 0.5045(3) | 0.0090(6) | 1/4 | 1 | 0.0151(4) | 1.72 |
| O2 | 1/2 | 0 | 0 | 1 | 0.0046(2) | 2.22 |
| O3 | 0.1751(2) | 0.3502(4) | 0.8778(1) | 1 | 0.0129(2) | 1.50/2.06/2.09* |
| H1 | 0.2349(8) | 0.470(2) | 0.6741(6) | 0.294(6) | 0.056(5) | 0.59 |
Examination of difference Fourier maps revealed a site containing negative scattering density within these partially occupied octahedra, approximately 1 Å from the O3 site (which is otherwise under-coordinated due to Ru vacancies, Table 1), strongly suggesting the presence of H. This 12-fold site has a refined occupancy of 0.294(6), equivalent to 1.76(4) H per formula unit, giving the refined empirical formula Ba4Ru3O10.24(4)(OH)1.76(4) and an O–H bond length of 1.14(1) Å (Fig. 1(c)). The O3 bond valence sum has a range of values, depending on its local environment. When it is bonded to either a Ru2 or H atom the value is close to the expected value of 2 (2.06 and 2.09 respectively). However, in the refined model approximately one in six oxygen atoms is under-coordinated, giving a bond valence sum of 1.50. The high correlation of the H1 and Ru2 atomic parameters led us to consider the possibility that the H1 occupancy is truly 0.5, which would result in O3 BVS much closer to 2. However, refinements carried out with fixed H1 occupancies show a poorer fit to the data, with unphysical refined values for Ru2 and H1 atomic displacement parameters (see ESI†).
Thermogravimetric analysis (TGA) under a N2 gas flow combined with mass spectrometry (MS) of the evolved gas confirms the presence of structural hydroxide: after a small initial loss of surface adsorbed water from the ∼10 μm crystallites (Fig. 1(a)), a mass loss of 1.55% occurs at ∼300 °C (Fig. 3(a)), with MS allowing this to be attributed to the loss of H2O due to the loss of the 1.80 hydroxide groups per formula unit, consistent with the structural model refined from powder neutron diffraction. This also correlates with in situ X-ray thermodiffraction data, which show a structural transition at around 300 °C, which must be brought about by dehydroxylation (see ESI†).
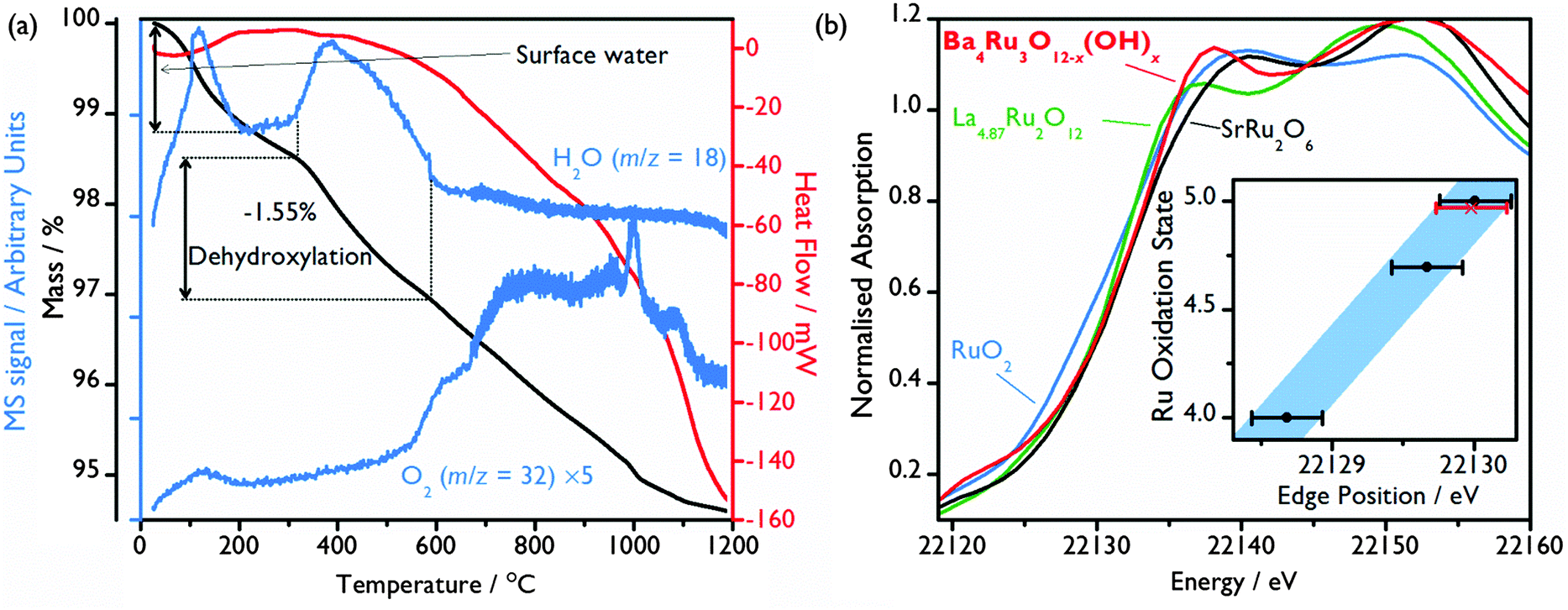 | ||
| Fig. 3 (a) TGA-DSC under N2 flow (heating rate 10 °C min−1) with in situ MS signals associated with H2O (m/z = 18) and O2 (m/z = 32, multiplied by 5 for clarity). (b) Normalised Ru K-edge XANES spectrum of Ba4Ru3O10.2(OH)1.8 compared to reference spectra of Ru(IV)O2, La4.87Ru(4.70)2O12 and SrRu(V)2O6. Inset shows a plot of Ru average oxidation state vs. edge position (measured where normalised absorption = 0.5). Black points correspond to reference samples, with a linear fit (blue, 0.5 eV wide, see text) which was then used to infer an average oxidation state from the Ba4Ru3O10.2(OH)1.8 (red point). Error bars were estimated from the core-hole broadening. | ||
Structural hydroxide has been previously observed in metal-deficient perovskites,27,28 although previously reported examples are typically ReO3-type structures where the H atoms project into the voids left by unoccupied A-sites. These are significantly larger than the B-site voids observed in Ba4Ru3O10.2(OH)1.8, explaining the partial occupancy of the H-site (1.8 H per vacant octahedron, compared with 3 in ReO3-type hydroxides).27,28 Hydroxylation may be driven by both the highly basic, oxidising synthesis conditions and the unique packing sequence observed in this hexagonal perovskite which necessitates Ru deficiency for steric reasons, leading to hydroxylation of otherwise under-coordinated oxide.
From the refined chemical formula, an average Ru oxidation state of +4.75 is deduced. Ru bond valence sums suggest a similar oxidation state of +4.40, with little difference between the two crystallographically distinct sites despite the large asymmetry seen in Ru2 (Table 1). X-ray absorption near edge structure (XANES) spectroscopy experiments were undertaken for further evaluation of the Ru oxidation state, with the Ru K-edge XANES spectrum of the Ba4Ru3O10.2(OH)1.8 compared to spectra of oxides of known oxidation state, Fig. 3(b) (see ESI† for full spectra). Although Ru K-edge XANES has previously been shown to be sensitive to the Ru average oxidation state,19,29,30 significant core-hole broadening at the Ru K-edge31 introduces large uncertainty in the measured edge position, and differences in local environment are also known to affect edge position. Despite this, the Ru K-edge position of the standards was found to have a linear relationship with oxidation state, with the Ba4Ru3O10.2(OH)1.8 K-edge position consistent with Ru oxidation state between 4.70 and 5 (Fig. 3(b)).
A Curie–Weiss law fit to the magnetic susceptibility versus temperature data in the paramagnetic region (250 < T < 400 K), yields an effective Ru moment, μeff, of 3.13 μB and a Weiss temperature, θ, of −629.2(8) K, indicative of significant antiferromagnetic correlations, Fig. 4. The Ru average valence state of +4.75 suggests a mixture of Ru4+ (t2g4) and Ru5+ (t2g3) ions with spin-only moments of 2.83 μB and 3.87 μB respectively, which would give an average spin-only moment of 3.64 μB in Ba4Ru3O10.2(OH)1.8. The difference between the spin-only value and μeff can be explained by a combination of crystalline field and spin–orbit coupling effects, commonly observed in Ru(V) oxides.20,32,33
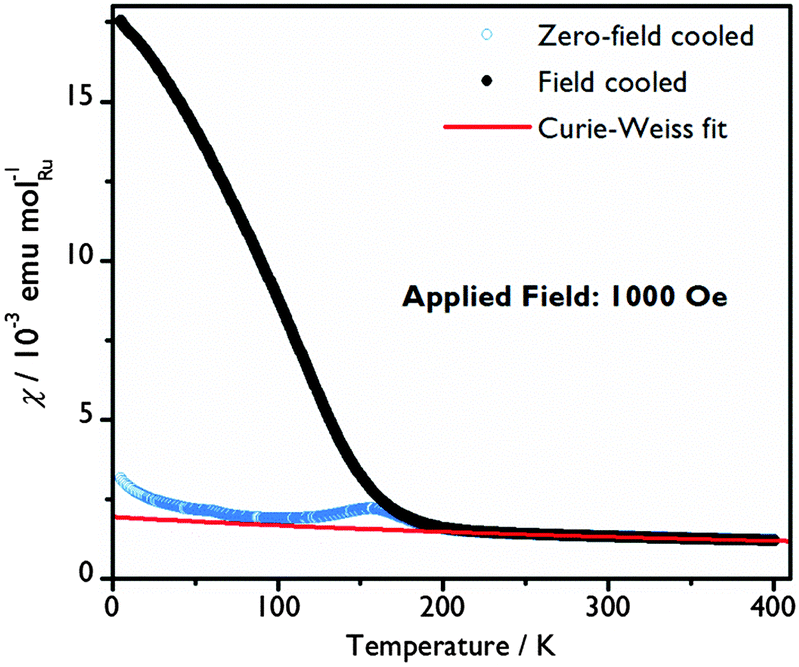 | ||
| Fig. 4 Molar susceptibility as a function of temperature with a Curie–Weiss fit to the paramagnetic region of the field cooled data. | ||
The system orders magnetically at TN = 200(5) K. A clear change in dχFC/dT that coincides with the minimum in the zero-field-cooled-warming χ(T) data may indicate a spin reorientation. At 5 K, the magnetisation increases almost linearly with applied field curve, with a moment of just 0.082(1) μB per Ru atom at 70 kOe indicating that the moments are principally aligned antiferromagnetically. A hysteresis in the χ(T) and M(H) curves only seen below TN reflects a small net magnetic moment and suggests that there may be a slight canting or a ferrimagnetic component to the spin arrangement at low temperature. A |θ|/TN ratio of over 3, although still smaller than for some other Ru oxides,20,32,33 reveals that in Ba4Ru3O10.2(OH)1.8 there is not a simple relationship between θ and TN. Similar behaviour in other Ru oxides has been rationalised by considering the variation in the strength of the transfer energy and a crossover from localised to itinerant electrons.34
In summary, we have reported the hydrothermal preparation of a complex barium ruthenium oxyhydroxide using just two reagents and avoiding the presence of a second, competing phase. This illustrates how solution chemistry can be utilised to exert a high degree of control of both crystal structure and oxidation state in the solid state for 4d elements that are easily reduced to the elemental state under other synthesis conditions typically used. The use of a metal peroxide as a reagent provides a reactive metal source, and has potential in the discovery of other new phases under mild reaction conditions. Ba4Ru3O10.2(OH)1.8 is shown to be thermally unstable, with a structural transition and loss of hydroxide occurring at temperatures as low as 300 °C, suggesting it would be impossible to prepare by conventional solid-state synthesis. The hexagonal perovskite-type structure with hydroxide ions found close to vacant octahedral B-sites is a highly unusual (if not unique) H-environment in oxide chemistry. It may be noted that another hexagonal perovskite, Sr4Ru3O12, previously reported to be synthesised under hydrothermal conditions (at 500 °C), contains partially occupied B-site octahedral sites but was characterised only by X-ray diffraction;25 the presence of H would be undetectable by this method, and that phase could potentially be another example of a ruthenium oxyhydroxide. Our work suggests that the oxyhydroxide chemistry of 4d transition metal could be a fruitful place to discover materials with new structures and interesting magnetic and electronic properties, especially when mild solution synthesis allows access to phases not accessible at high temperatures.
We thank Johnson Matthey plc for their contribution to an EPSRC industrial CASE award to CIH, STFC for provision of neutron beamtime at ISIS via the GEM Xpress Access route, and Diamond Light Source for access to beamline B18.
References
- K. Ishida, H. Mukuda, Y. Kitaoka, K. Asayama, Z. Q. Mao, Y. Mori and Y. Maeno, Nature, 1998, 396, 658 CrossRef CAS.
- R. J. Bouchard and J. L. Gillson, Mater. Res. Bull., 1972, 7, 873–878 CrossRef CAS.
- C. I. Hiley, D. O. Scanlon, A. A. Sokol, S. M. Woodley, A. M. Ganose, S. Sangiao, J. M. De Teresa, P. Manuel, D. D. Khalyavin, M. Walker, M. R. Lees and R. I. Walton, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 104413 CrossRef.
- P. Khalifah, K. D. Nelson, R. Jin, Z. Q. Mao, Y. Liu, Q. Huang, X. P. A. Gao, A. P. Ramirez and R. J. Cava, Nature, 2001, 411, 669–671 CrossRef CAS.
- J. A. Kurzman, L. M. Misch and R. Seshadri, Dalton Trans., 2013, 42, 14653–14667 RSC.
- A. T. Ashcroft, A. K. Cheetham, J. S. Foord, M. L. H. Green, C. P. Grey, A. J. Murrell and P. D. F. Vernon, Nature, 1990, 344, 319–321 CrossRef CAS.
- N. F. Atta, A. Galal and S. M. Ali, Int. J. Electrochem. Sci., 2012, 7, 725–746 CAS.
- J. Darriet and M. A. Subramanian, J. Mater. Chem., 1995, 5, 543–552 RSC.
- R. H. Mitchell, Perovskites: Modern and Ancient, Almaz Press, 2002 Search PubMed.
- S.-T. Hong and A. W. Sleight, J. Solid State Chem., 1997, 128, 251–255 CrossRef CAS.
- P. C. Donohue, L. Katz and R. Ward, Inorg. Chem., 1965, 4, 306–310 CrossRef CAS.
- T. Ogawa and H. Sato, J. Alloys Compd., 2004, 383, 313–318 CrossRef CAS.
- F. Grasset, C. Dussarrat and J. Darriet, J. Mater. Chem., 1997, 7, 1911–1915 RSC.
- F. Grasset, M. Zakhour and J. Darriet, J. Alloys Compd., 1999, 287, 25–31 CrossRef CAS.
- P. D. Battle, S. H. Kim and A. V. Powell, J. Solid State Chem., 1992, 101, 161–172 CrossRef CAS.
- P. Lightfoot and P. D. Battle, J. Solid State Chem., 1990, 89, 174–183 CrossRef CAS.
- C. Mallika and O. M. Sreedharan, J. Alloys Compd., 1993, 191, 219–222 CrossRef CAS.
- D. E. Bugaris and H.-C. zur Loye, Angew. Chem., Int. Ed., 2012, 51, 3780–3811 CrossRef CAS.
- R. J. Darton, S. S. Turner, J. Sloan, M. R. Lees and R. I. Walton, Cryst. Growth Des., 2010, 10, 3819–3823 CAS.
- C. I. Hiley, M. R. Lees, J. M. Fisher, D. Thompsett, S. Agrestini, R. I. Smith and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 4423–4427 CrossRef CAS PubMed.
- C. Dussarrat, J. Fompeyrine and J. Darriet, Eur. J. Solid State Inorg. Chem., 1994, 31, 289–300 CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- A. C. Larson and R. B. Van Dreele, Los Alamos National Laboratory Report LAUR, 1994, 86–748.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- C. Renard, S. Daviero-Minaud, M. Huve and F. Abraham, J. Solid State Chem., 1999, 144, 125–135 CrossRef CAS.
- S. A. J. Kimber, M. S. Senn, S. Fratini, H. Wu, A. H. Hill, P. Manuel, J. P. Attfield, D. N. Argyriou and P. F. Henry, Phys. Rev. Lett., 2012, 108, 217205 CrossRef PubMed.
- J. R. Neilson, J. A. Kurzman, R. Seshadri and D. E. Morse, Inorg. Chem., 2011, 50, 3003–3009 CrossRef CAS PubMed.
- W. D. Birch, A. Pring, A. Reller and H. Schmalle, Naturwissenschaften, 1992, 79, 509–511 CrossRef CAS.
- I. Arčon, A. Benčan, A. Kodre and M. Kosec, X-Ray Spectrom., 2007, 36, 301–304 CrossRef.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. B. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 10960–10964 CrossRef CAS PubMed.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, Cambridge University Press, 2010 Search PubMed.
- P. D. Battle and W. J. Macklin, J. Solid State Chem., 1984, 52, 138–145 CrossRef CAS.
- J. Darriet, F. Grasset and P. D. Battle, Mater. Res. Bull., 1997, 32, 139–150 CrossRef CAS.
- P. D. Battle, J. B. Goodenough and R. Price, J. Solid State Chem., 1983, 46, 234–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details, characterisation data and supplementary materials. See DOI: 10.1039/c6cc02121b |
| This journal is © The Royal Society of Chemistry 2016 |