
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective OFF ↔ ON activation processes initiated by hydrogen transfer: concepts and challenges
Adrien
Quintard
* and
Jean
Rodriguez
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: adrien.quintard-inv@univ-amu.fr; jean.rodriguez@univ-amu.fr
First published on 22nd June 2016
Abstract
Hydrogen transfer initiated processes are eco-compatible transformations allowing the reversible OFF ↔ ON activation of otherwise unreactive substrates. The minimization of stoichiometric waste as well as the unique activation modes provided by these transformations make them key players for a greener future for organic synthesis. Long limited to catalytic reactions that form racemic products, considerable progress on the development of strategies for controlling diastereo- and enantioselectivity has been made in the last decade. The aim of this review is to present the different strategies that enable enantioselective transformations of this type and to highlight how they can be used to construct key synthetic building blocks in fewer operations with less waste generation.
 Adrien Quintard | Adrien Quintard studied chemistry at the University of Toulouse and then at the CPE/University of Lyon. He completed his PhD in 2011 under the supervision of Prof. A. Alexakis at the University of Geneva. He subsequently moved to the University of Stanford for a one year post-doctoral stay working with Prof. B. M. Trost. He was appointed as an associate researcher at Aix-Marseille University in 2012 and has been a CNRS French researcher since 2014. His research interests include the development of organometallic/organocatalytic enantioselective methodologies and their application to natural product synthesis. |
 Jean Rodriguez | Jean Rodriguez was born in Cieza (Spain) in 1958 and in 1959 his family immigrated to France. After studying chemistry at the University of Aix-Marseille (France), he completed his PhD as a CNRS researcher with Prof. B. Waegell and Prof. P. Brun in 1987. He completed his Habilitation in 1992, also at Marseille, where he is currently Professor and Director of the UMR-CNRS-7313-iSm2. His research interests include the development of domino and multicomponent reactions, and their application in stereoselective organocatalyzed synthesis. In 1998 he was awarded the ACROS prize in Organic Chemistry, in 2009 he was awarded the prize of the Division of Organic Chemistry from the French Chemical Society and in 2013 he became a “Distinguished member” of the French Chemical Society. |
1. Introduction
(a) OFF ↔ ON activation by hydrogen transfer
From fundamental to applied research, most efforts from synthetic organic chemists are focused on the same goal: how can we obtain valuable molecular objects (materials, bioactive molecules…) in the most practical, straightforward and economically reliable approach? To conceptualize the efforts towards more eco-compatible transformations, several principles such as atom-,1 step-2 or redox-3 economies have been formulated. Consequently, and in agreement with the concept of green chemistry, the chemical synthetic route should be the shortest with minimum waste production and energy consumption.4 This is a daunting challenge when relatively inert and simple substrates need to be selectively functionalized to construct complex enantioenriched products. Such a situation often results in the use of tedious and non-user friendly multiple operations such as separate stoichiometric oxidation and reduction steps.In the quest for new eco-compatible reactions, hydrogen transfer based reactions have recently changed the way people conceive chemical transformations. They provide genuine solutions for the transient activation of otherwise unreactive species triggering carbon–carbon or carbon-heteroatom bond-formation. As a result, by using the smallest possible atom (hydrogen) as an activating agent, considerable step, waste and atom economies have been achieved in reactions of fundamental importance.
The general principle behind these hydrogen based transformations lies in the ability of a metal complex to catalytically promote reversible hydrogen transfer reactions through OFF ↔ ON activation processes (Scheme 1).5
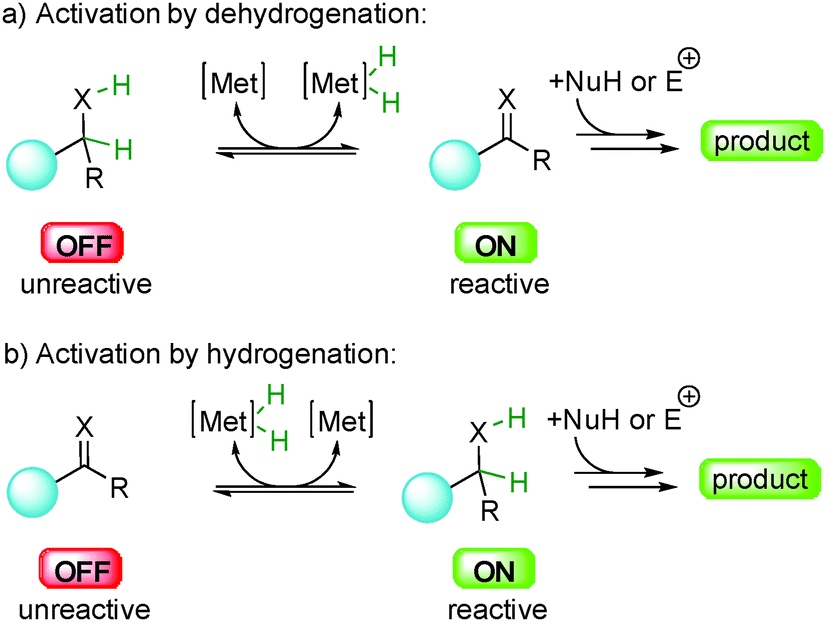 | ||
| Scheme 1 General concept of OFF ↔ ON activation by hydrogen transfer. | ||
For example, dehydrogenation of an unreactive saturated OFF compound can form a reactive unsaturated ON transient derivative ready for subsequent in situ bond-forming events (Scheme 1a).6 On the contrary, an unreactive unsaturated OFF compound can be hydrogenated to form a reactive saturated ON substrate involved in the formation of the product of the reaction (Scheme 1b). Overall this allows the combination of catalytic dehydrogenation and hydrogenation that simultaneously activates and merges together several unsaturated and saturated compounds allowing for unique reactivities. The main advantage of this strategy is to associate distinct redox operations that minimize the utilization of stoichiometric activating agents such as oxygen, quinones or TEMPO. The aim of this paper is to present an overview of the development of enantioselective transformations based on this OFF ↔ ON activation approach initiated by hydrogen transfer.7 The concepts behind this general reactivity, the advantages, the challenges and future research directions will be discussed in detail. Notably, while the racemic version of this approach has witnessed an impressive number of publications, the development of enantioselective counterparts is still in its infancy. This is mainly due to the difficulty in discovering efficient enantioselective catalytic systems compatible with hydrogen transfer conditions.8 Indeed, the crucial question in all the presented systems is how to insert a point of stereocontrol? As will be discussed later, each fundamental step of these processes (dehydrogenation, hydrogenation and subsequent carbon–carbon or carbon-heteroatom bond-formation) can become a strategy to control the stereochemistry of the final product. This challenge has pushed chemists to find innovative solutions to introduce chirality control notably using multiple catalysis as discussed here in detail.9
(b) Different approaches for hydrogen activation
The general concept highlighted in Scheme 1 shows several ways to initiate a process by hydrogen transfer. The first one concerns the rapid racemization of a stereogenic center, which has found numerous applications in the resolution of racemic alcohols (vide infra). The other famous approach is the so-called borrowing hydrogen or hydrogen auto-transfer resulting in an overall redox-free sequence (Scheme 2).10 From an initial saturated unreactive OFF substrate (alcohol for example), and in the presence of a nucleophilic species (primary amine for example), a metal complex [Met] abstracts H2 forming a [Met]–H2 complex and an unsaturated reactive ON intermediate prone to react with the nucleophile. After dehydration, the resulting new unsaturated ON derivative (imine for example) suffers hydrogen back-transfer delivering the final OFF product. Without the hydrogen transfer, the direct reaction from the unreactive OFF starting material to the OFF product would be impossible. The metal makes this transformation possible by an efficient transient catalytic activation avoiding the use of any stoichiometric redox reagents.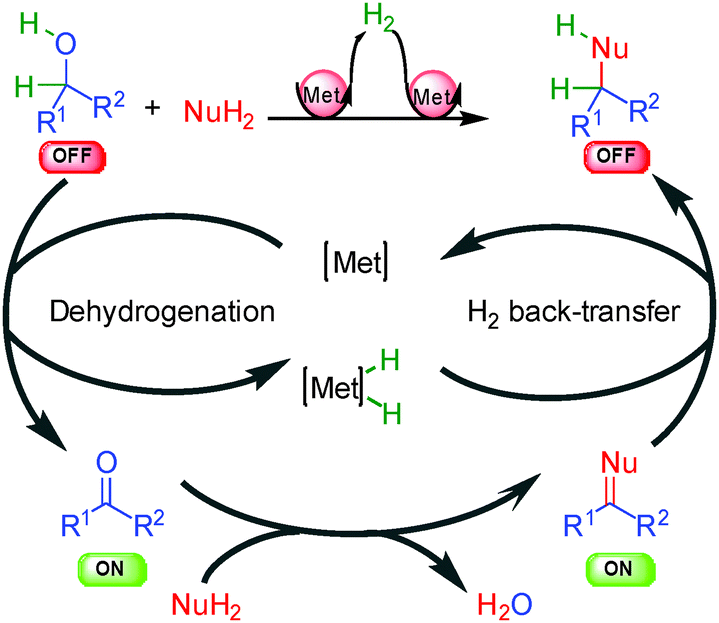 | ||
| Scheme 2 General concept of borrowing hydrogen. | ||
The main challenge in performing this type of process in an enantioselective manner is to find a way to displace the overall hydrogen transfer equilibrium towards product formation at temperatures where good enantiodiscrimination can be achieved. Moreover, as discussed in the following examples, the chirality of the product can be created at each fundamental step of the process and there is no general solution to determine the catalytic enantiodetermining event.
In some specific cases, the borrowing hydrogen process can be truncated and oxidation of the starting material occurs by hydrogen removal from the mixture either as a gas or using a hydrogen acceptor (Scheme 3). As compared to borrowing hydrogen, the result of the process is a saturated reactive ON final product that might be ready for additional cascade transformations. Interestingly, since the metal catalyst is still present in the mixture at the end of the process, it can also be used in one-pot for further transformations (like hydrogenation) by simply modulating the reaction conditions.
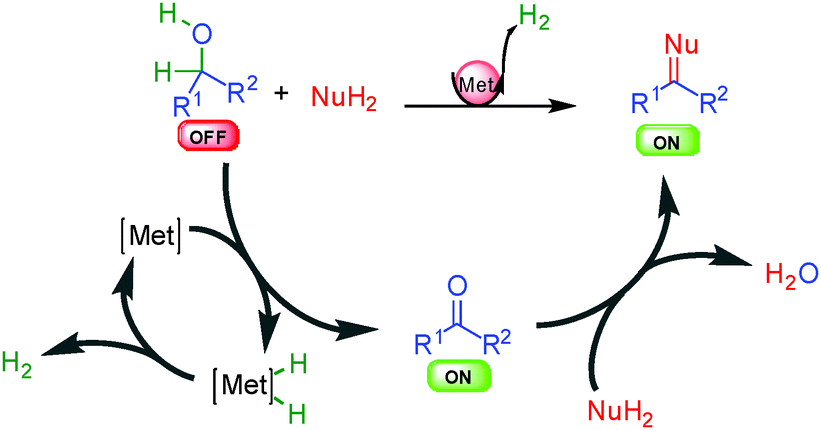 | ||
| Scheme 3 General concept of dehydrogenative activation. | ||
The last general approach for the hydrogen transfer activation involves metal hydride species and has found numerous applications in enantioselective carbon–carbon bond formation (Scheme 4). In these types of transformations, two distinct unreactive OFF substrates can be activated synergistically by two concomitant catalytic cycles using the same metallic center. The sequence is initiated by dehydrogenation of the first OFF substrate (alcohol for example) leading to the corresponding reactive ON oxidized intermediate (carbonyl compound) and a crucial reactive metal hydride able to activate the second OFF substrate (unsaturated double bond) to an ON metal alkyl or alkenyl complex. A final addition liberates the OFF product and turns back the metal complex. The chirality can arise from two distinct steps, either from the metal hydride insertion on the unsaturated substrate or during the last organometallic addition to the in situ generated ON electrophile. Alternative pathways to this general scheme also exist such as insertion into alkyl halide or metal promoted cyclometallation and will be discussed in more detail in part 6.
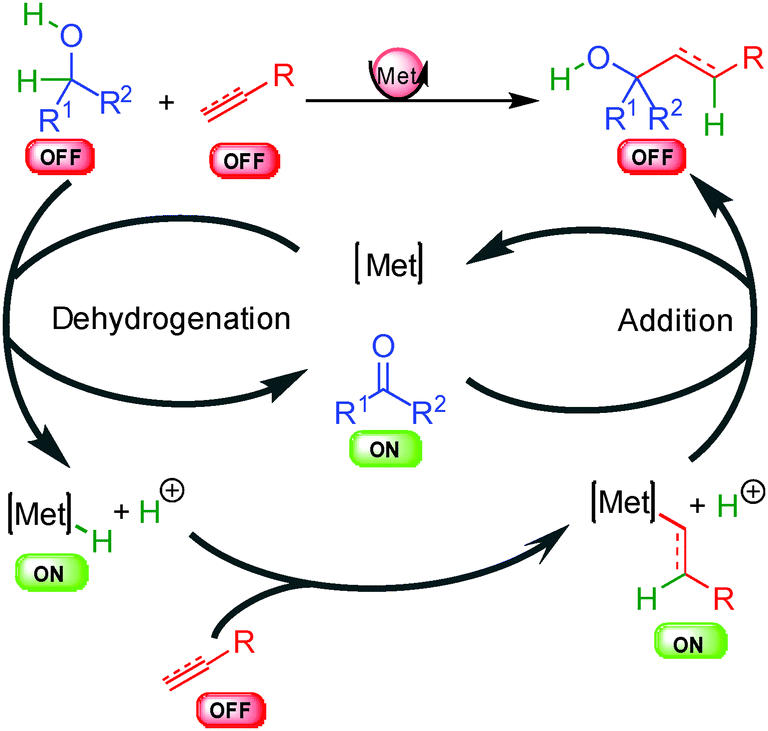 | ||
| Scheme 4 General concept of the hydrogenative metal hydride activation. | ||
2. Alcohols and amines as ON substrates
(a) Dynamic kinetic resolution of alcohols
As stated in the introduction, alcohols can be used either as OFF or ON reactants depending on the reaction. The dynamic kinetic resolution (DKR) of alcohols by enantioselective acylation is an example of hydrogen transfer process where an alcohol is the actual ON reactant in equilibrium with its corresponding OFF carbonyl derivative (Scheme 5). This property was advantageously used at the end of the 90s in pioneering studies by the group of Bäckvall in Sweden and Williams from United Kingdom on the DKR of racemic alcohols.11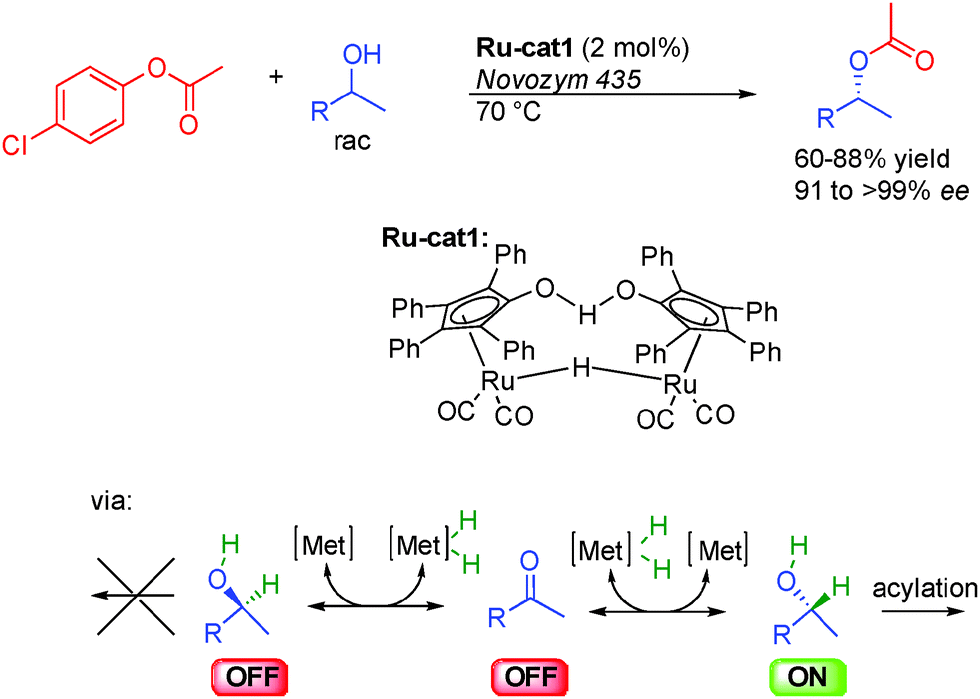 | ||
| Scheme 5 Dynamic kinetic resolution of alcohols by hydrogen transfer racemization. | ||
Notably, to efficiently develop this concept, Bäckvall took advantage of dual catalysis based on a ruthenium complex12 for the rapid isomerization of alcohols, and enzymes such as Novozym-435 for enantioselective acylation.13 Only one enantiomer of the chiral alcohol is acylated by the chiral enzyme while in the presence of the Shvo ruthenium complex Ru-cat1, the other less reactive one is rapidly inter-converted by reversible hydrogen transfer through the prochiral ketone.14 As a result, both enantiomers of the starting racemic alcohol converge to the final acetylated product demonstrating the potential of this method for the preparation of enantioenriched alcohols in 91 to >99% ee.15
The challenge in the development of these types of dynamic processes is to find a metal complex able to promote racemization at a rate considerably higher than the rate of acylation of the undesired enantiomer. In order to obtain systems able to promote the fastest isomerization rates at low temperatures and potentially improve the reaction efficiency and its scope, a great deal of work has been done on the design of the best possible isomerization catalysts. Most notably, in addition to the classical Shvo complex Ru-cat1, complexes based on either Ruthenium or Iridium presented in Scheme 6 perform efficiently in the DKR of alcohols. Complexes Ru-cat2 and Ru-cat3 are easily synthesized alternatives to the Shvo complex, efficient for a wide range of alcohols even at room temperature.16 However, a base is needed to convert these pre-complexes to active catalysts possessing a vacant site on the ruthenium.17 On the contrary, iridium complexes Ir-cat1 and Ir-cat2 are efficient for a broad range of alcohols in the absence of any basic additive.18
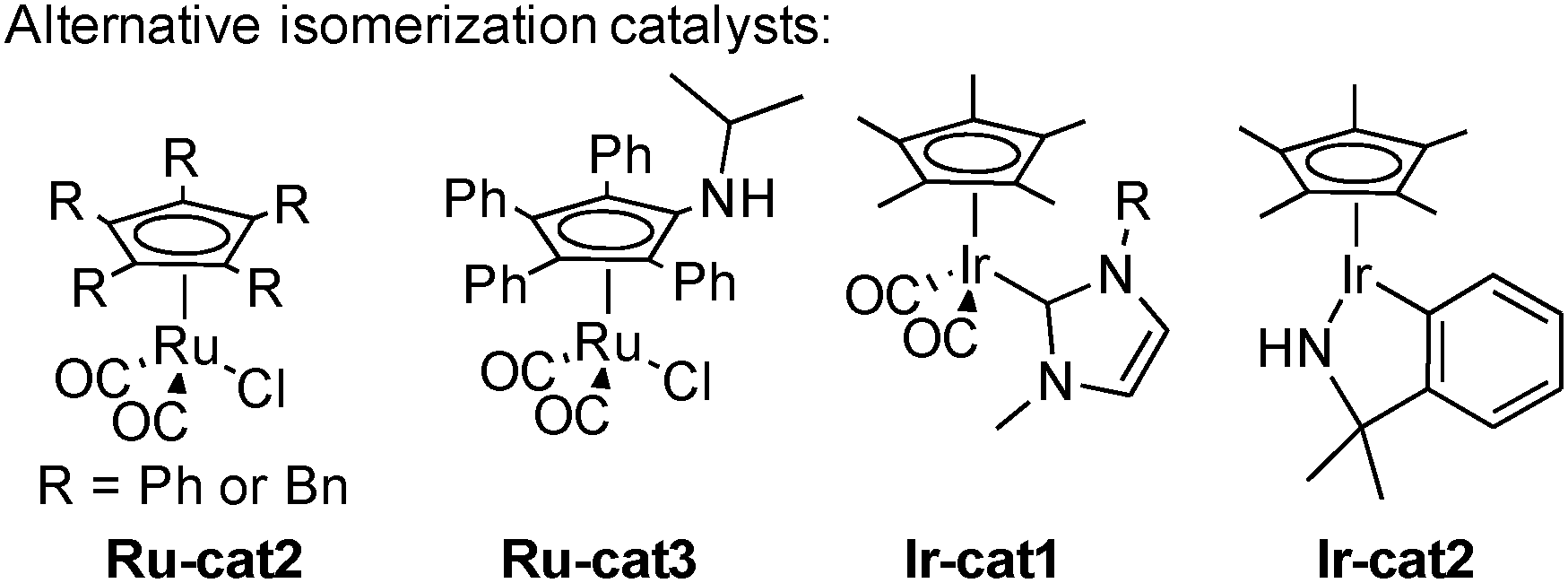 | ||
| Scheme 6 Efficient metal catalysts for the DKR of alcohols. | ||
Since the original developments of this efficient approach, a large number of papers on the topic have appeared and this chemistry has been applied to the resolution of a broad range of racemic alcohols (Scheme 7).19 For example, beside classical aromatic or aliphatic substituted alcohols 1a, optically active 1,2- to 1,5-diols can be prepared with perfect stereocontrol in some cases. It is interesting to note that starting from a mixture of 4 stereoisomers of racemic diol 1b, the system converges to a single major diastereomer with high enantioselectivity.20 Functionalized alcohols such as 1c or 1d are also tolerated in the ruthenium-enzyme catalytic system providing synthetic entries to amino-alcohols or epoxides for example.21 In addition, structurally diversified allylic alcohols 1e can also be resolved by this method introducing a versatile adjacent double-bond to the alcohols.22 Finally, primary alcohols 1f with a vicinal stereogenic center can also be prepared with enantioselectivities up to 99%.23 The broad scope and the high efficiency to create enantiopure building blocks have largely been exploited for the synthesis of various natural products, highlighting the robustness of the processes.24
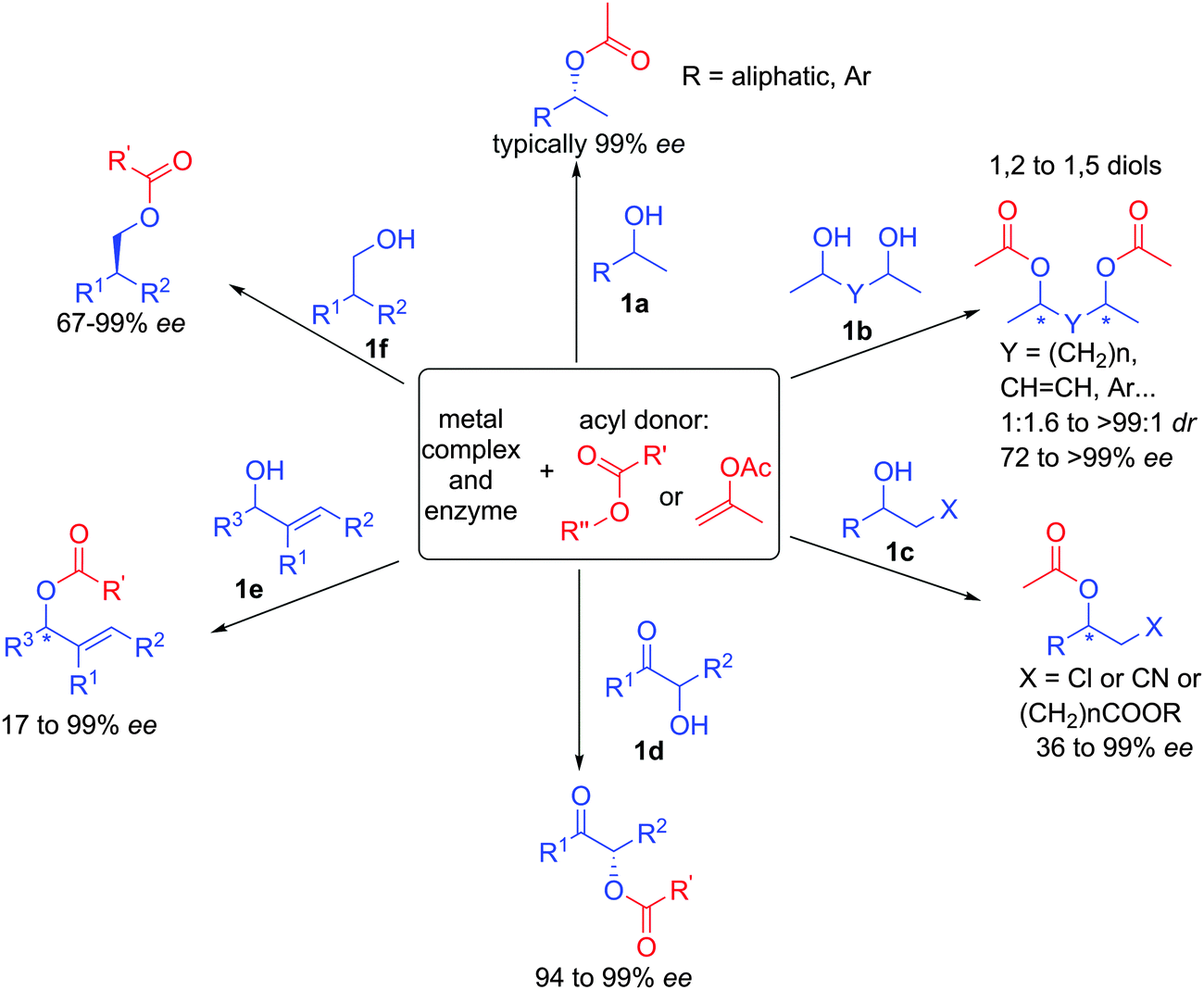 | ||
| Scheme 7 Synthetic potential of the dynamic kinetic resolution of alcohols. | ||
In an interesting alternative study, the groups of Janssen, Feringua and de Vries from Netherlands reported that racemic β-haloalcohols could be resolved in the presence of enzymes in a buffered solution to provide directly enantioenriched epoxides (Scheme 8).25 They found that iridacycle Ir-cat3 in the presence of a catalytic amount of base was the catalyst of choice to promote the fast alcohol racemization at room temperature, providing an efficient DKR route for these types of alcohols.
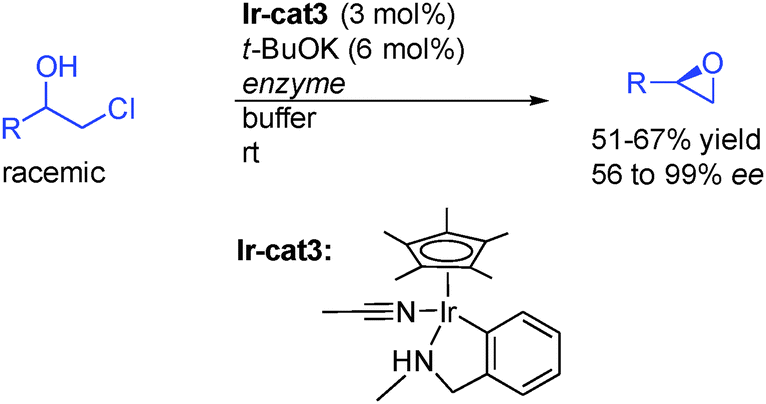 | ||
| Scheme 8 Direct enantioselective epoxide synthesis. | ||
Beside enzymes, there is a unique example of DKR of alcohols employing an organocatalyst proposed by the group of Fu with a chiral aminopyridine DMAP* (Scheme 9).26 This is of particular interest because of the potential complementary scope achievable using this catalytic system. For example, a wider variety of R2 groups (longer alkyl chain for example) can be used in the present transformation forming enantioenriched products in 82 to 93% ee.
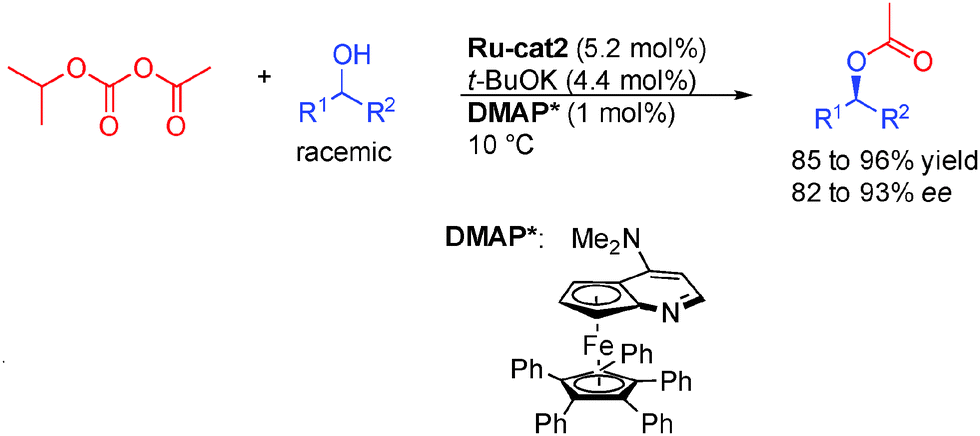 | ||
| Scheme 9 Dual organocatalytic/ruthenium catalysis for DKR of alcohols. | ||
(b) Dynamic kinetic resolution of amines
In addition to alcohols, enantiopure amines can also be efficiently prepared through the isomerization mechanism by the combination between metals and enzymes. In these cases, amines act as the reactive ON reagents while the corresponding imines represent the unreactive OFF substrates. The first report on hydrogen initiated preparation of enantiopure amines came in 2001 from the group of Kim in Korea. Starting from a ketoxime, under a positive hydrogen pressure, Pd on charcoal promoted both the reduction of the ketoxime to the racemic amine as well as its racemization through imine formation. In combination with an appropriate enzyme and an acyl transfer agent, the DKR is efficiently promoted allowing the formation of chiral amines with excellent stereocontrol (94 to 99% ee, Scheme 10).27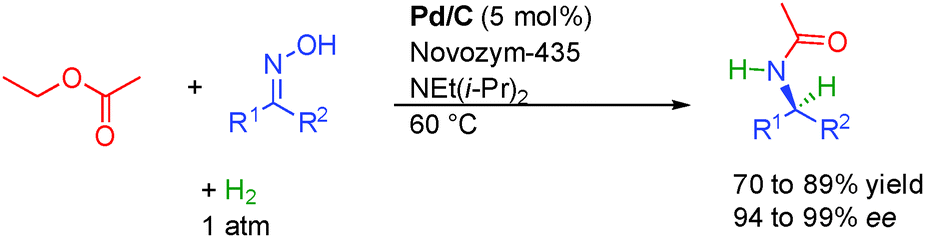 | ||
| Scheme 10 Preparation of chiral amines from ketoximes. | ||
Subsequently, several groups showed that amines could efficiently be resolved through in situ imine formation, by various heterogeneous catalysts or by the Shvo-type complex Ru-cat4 (Scheme 11). In combination with enzymes notably Novozym-435, clean convergence to the formation of one single acetylated enantioenriched amine (93 to 98% ee) could be observed. Using Pd/BaSO4, RANEY®-Ni or RANEY®-Co, low hydrogen pressure is also necessary to form the active heterogeneous catalyst. Even-though it has been less studied, this process interestingly complements the chemistry of alcohol DKR providing access to various substituted amines.
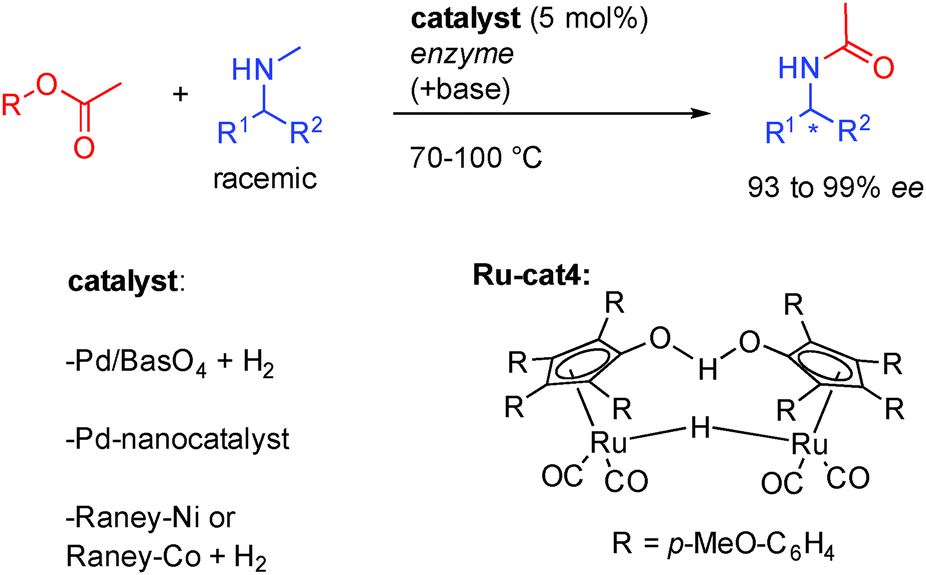 | ||
| Scheme 11 Amines DKR in the presence of various metal complexes. | ||
(c) Enantioselective lactone formation
Pioneered by early work in 1990 from the group of Noyori in Japan, alcohols were also used as ON reactants in lactone synthesis. Using a chiral ruthenium complex based on BINAP, under a high hydrogen pressure (100 atm), several keto-esters could be efficiently hydrogenated providing directly or after subsequent acidic treatment six-membered ring lactones in excellent 96 to 99% ee (Scheme 12).28 This approach constitutes an attractive enantioselective eco-compatible access to this heterocyclic framework that prevails in many important natural products and fragrances.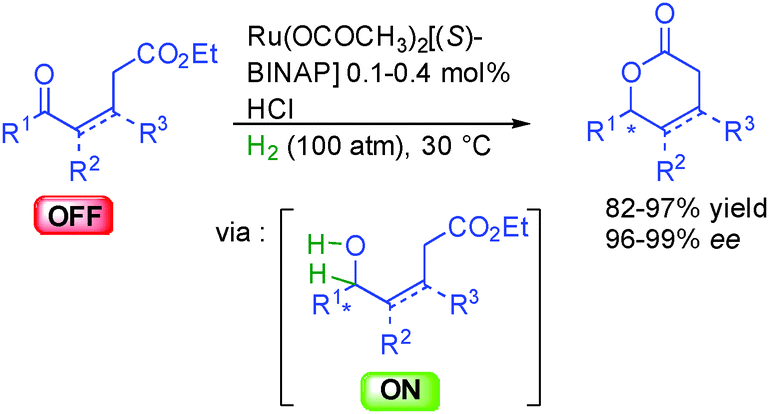 | ||
| Scheme 12 Enantioselective lactone formation from keto-esters. | ||
The process was subsequently extended to transfer hydrogen using isopropanol as a hydrogen source notably by the group of Carpentier in France.29 By employing a chiral diamine ruthenium complex, Ru-cat5, they could avoid the use of high hydrogen pressure from the original Noyori's report (Scheme 13). Unfortunately, the transformation was less general providing enantiocontrol on the final lactone from low 10 to excellent 97% ee.
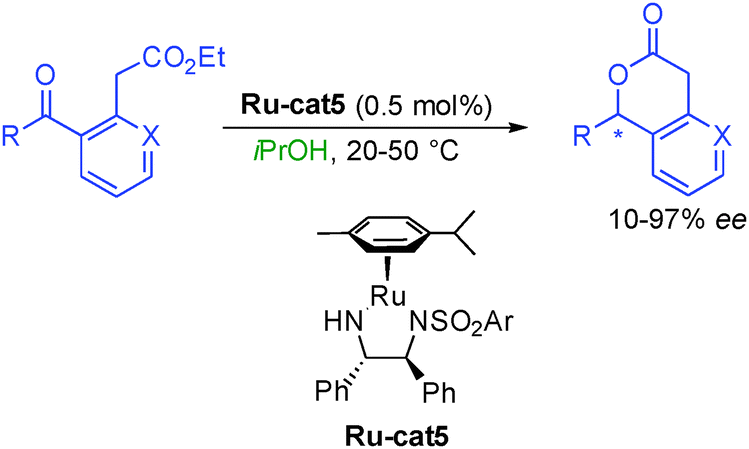 | ||
| Scheme 13 Enantioselective lactone synthesis from keto-esters using transfer hydrogenation. | ||
A related lactonization process was proposed in 2003 by the group of Hiroi in Japan showing that meso-diols could be oxidatively desymmetrized by the use of different amino-alcohols as chiral ligands of [Cp*IrCl2]2 (Scheme 14).30 The key enantiodiscriminating step of the reaction is the dehydrogenation of one of the two prochiral alcohols to form an ON hydroxyaldehyde that evolves by hemiacetalization. The resulting lactol suffers a final dehydrogenation giving the corresponding optically active lactone. The results in terms of enantiocontrol remained moderate (42 to 81% ee) and the reaction required a stoichiometric amount of acetone to perform the net-oxidation of the substrate. Given the recent progress in acceptorless dehydrogenation, a greener and more efficient version of this transformation might be envisaged.
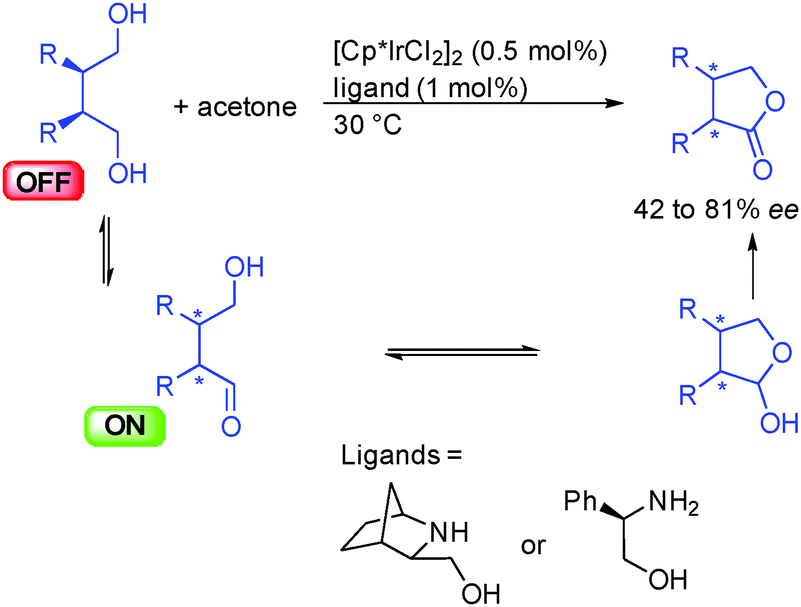 | ||
| Scheme 14 Enantioselective lactone formation from meso-diols. | ||
The group of Johnson in the United States reported the dynamic kinetic asymmetric reduction of racemic α-keto-esters by hydrogenation using a HCOOH/Et3N mixture as the hydrogen source (Scheme 15).31 Thanks to the appropriate ruthenium complex in the presence of a chiral 1,2-aminosulfonamide ligand, one single enantiomer of the starting material is selectively reduced, providing two contiguous stereocenters with excellent diastereocontrol (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and 78 to 92% ee. The other non-reactive enantiomer is racemized through reversible enolization, providing a pathway for the dynamic resolution. Finally, the generated secondary ON hydroxyester spontaneously evolved by diastereoselective lactonization leading to synthetically attractive γ-butyrolactones bearing three contiguous stereocenters.
:1 dr) and 78 to 92% ee. The other non-reactive enantiomer is racemized through reversible enolization, providing a pathway for the dynamic resolution. Finally, the generated secondary ON hydroxyester spontaneously evolved by diastereoselective lactonization leading to synthetically attractive γ-butyrolactones bearing three contiguous stereocenters.
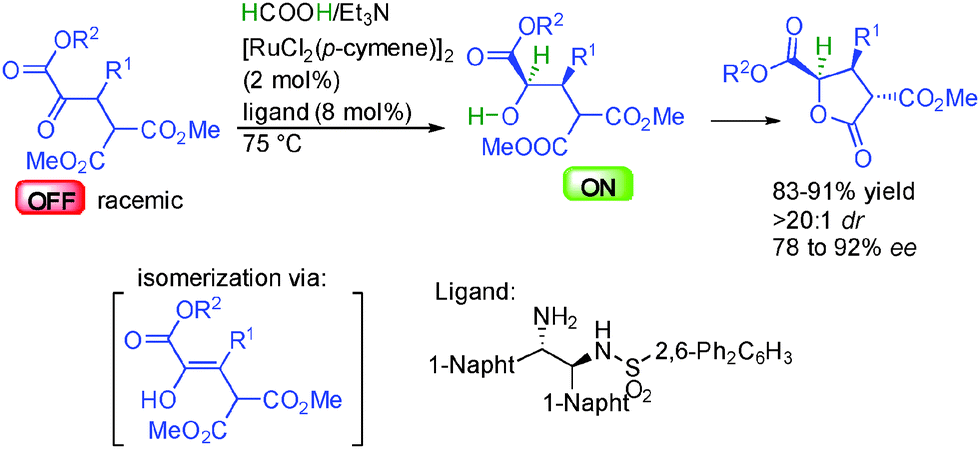 | ||
| Scheme 15 Lactone synthesis by enantioselective hydrogenative DKR. | ||
Finally, the group of Dong in the United States showed in 2013 that starting from 1,5-hydroxyketones, the chiral Noyori's ruthenium complex Ru-cat6 was able to catalyze successive hydrogen transfers resulting in the formation of optically active δ-valerolactones (Scheme 16).32 Initially, the enantioselective reduction of the ketone to 1,5-diol is followed by dehydrogenation of the primary alcohol providing an enantioenriched ON secondary hydroxyaldehyde able to form a lactol intermediate by hemiacetalization from which final dehydrogenation gave the enantioenriched δ-valerolactones in 86 to 96% ee. The transformation is significantly accelerated by using a mixture of acetone and isopropanol to facilitate both successive reduction and oxidation reactions, and the stability of the final lactone drives the thermodynamic equilibrium. As a result of this strong thermodynamic gain, only 1.2 equivalent of acetone is required for the net oxidation of the product and 3 equivalents of isopropanol are used as an autocatalyst to increase the transfer hydrogenation rate. This reaction highlights the power of hydrogen transfer initiated-transformations where both reduction and oxidation occur under the same conditions and use one single catalyst to act simultaneously in apparently opposite reactions (oxidation and reduction).
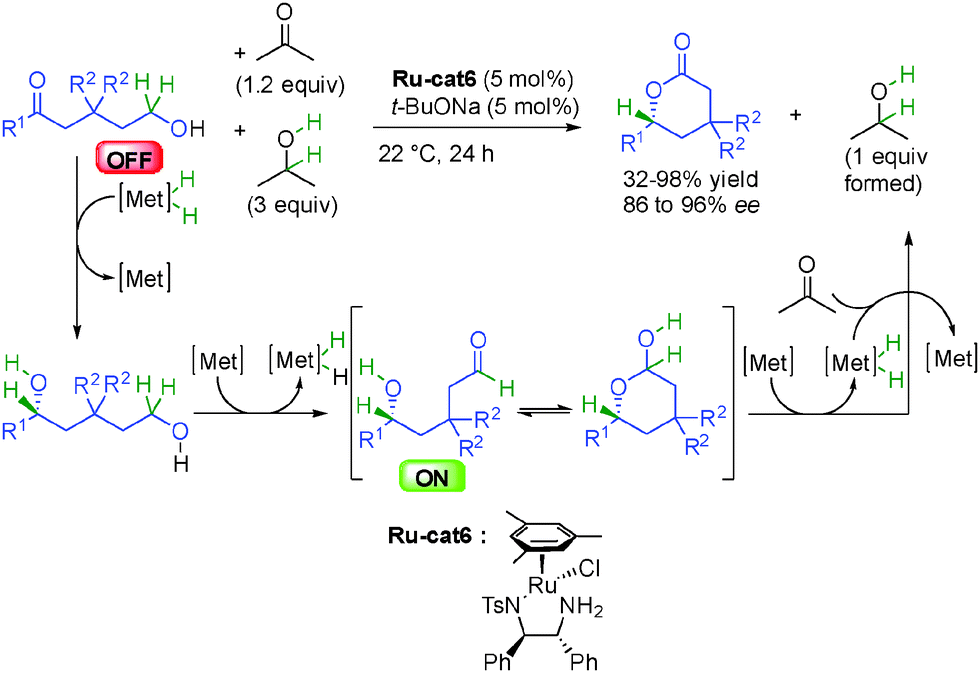 | ||
| Scheme 16 Enantioselective lactone synthesis by multiple hydrogen transfers. | ||
From all these transformations using alcohols and amines as ON or OFF substrates, it appears that the DKR of alcohols and amines by acylation is already a mature field allowing excellent results on a wide array of substrates. On the contrary, other transformations such as the formation of lactones, even-though they seem attractive in terms of eco-compatibility have only remained limited examples for now. Given the value of these processes it would not be surprising that these studies should inspire other research on the topic.
3. Carbonyl alkylation with alcohols as OFF substrates
(a) Hydrogenation as the enantioselective step
The use of alcohols as initial OFF reagents for the catalytic dehydrogenative alkylation of ketones is a modern eco-compatible approach to rapidly combine products without the need to pre-functionalize the substrates or the requirement of any stoichiometric redox agents.33 Notably, when used in a borrowing hydrogen process, alcohols act both as an alkyl source and also as a hydrogen source increasing the eco-compatibility of the transformations.Despite the interest in borrowing hydrogen for ketone alkylation and an intensive research in the discovery of more and more efficient catalysts for the racemic version, the enantioselective variant is particularly over-looked. This is rather surprising given the possibility of inserting a point of stereocontrol during the hydrogenation event for which a huge number of metal catalysts should potentially be highly efficient.
As highlighted in the precedent chapters, enzymes have been known for a long time to promote enantioselective redox-neutral transformations. Therefore, the first successful examples of efficient borrowing hydrogen with alcohols as alkylating reagents were disclosed using different enzymes as catalysts.34 In this area, Baker's yeast or C.lunata notably allowed the dehydrative condensation of α-cyano-ketones with different alcohols without the requirement of any extra stoichiometric reagent (Scheme 17). Interestingly, the alcohol used as a solvent not only acts as an alkyl donor by in situ forming the required ON aldehyde but also as an extra hydrogen source triggering the successive reduction of the transient Knoevenagel-type unsaturated adduct. The resulting ON cyanoketone is directly reduced to the expected β-cyanoalcohol with good to high diastereoselectivity and 86 to 99% ee. It must be pointed out that the enzymes used in these studies are able to promote both reversible hydrogen transfers but also the C–C bond-forming event highlighting the versatility of this bioconversion.
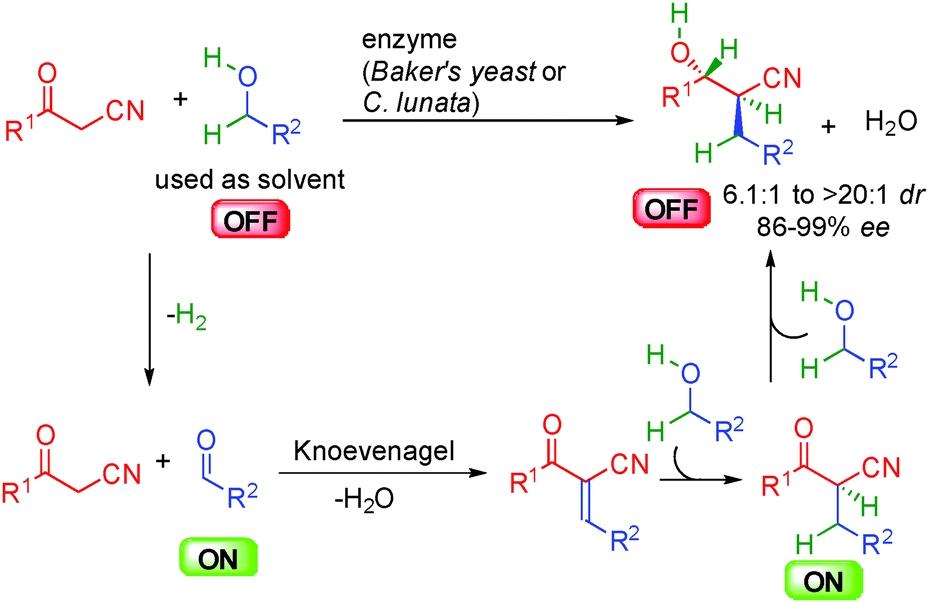 | ||
| Scheme 17 Enzyme catatalyzed α-cyano-ketone alkylation-reduction sequence. | ||
The difficulty in finding an appropriate catalytic system able to promote enantioselectively the several distinct reactions explains why besides enzymes, other hydrogen transfer initiated reactions were scarcely disclosed in the literature. The first report on a metal catalyst variant of the above mentioned transformation only appeared in 2006 by a work from the group of Nishibayashi in Japan devoted to the enantioselective reductive alkylation of acetophenones with primary alcohols.35 They proposed a sequential one-pot operation with two complementary metal catalysts involved successively in each reductive step of the transformation (Scheme 18). The first achiral iridium complex in combination with KOH promoted the borrowing hydrogen ketone alkylation via a Claisen–Schmidt condensation, while the second chiral ferrocenyl ruthenium catalyst added subsequently induced the enantioselective full reduction of the transient unsaturated ketone by hydrogen transfer from the added isopropanol. By sequentially combining those two distinct catalysts, elongated chiral alcohols were obtained in 88 to 98% ee. The important aspect for the reaction to work with good levels of enantiocontrol is the ability of the ruthenium complex to act independently from the iridium complex in the enantioselective ketone transfer hydrogenation. It must be pointed out that the use of inorganic base (KOH or others) in this process as well as in the following ketone alkylation is crucial to promote the C–C bond-forming event by aldolization.
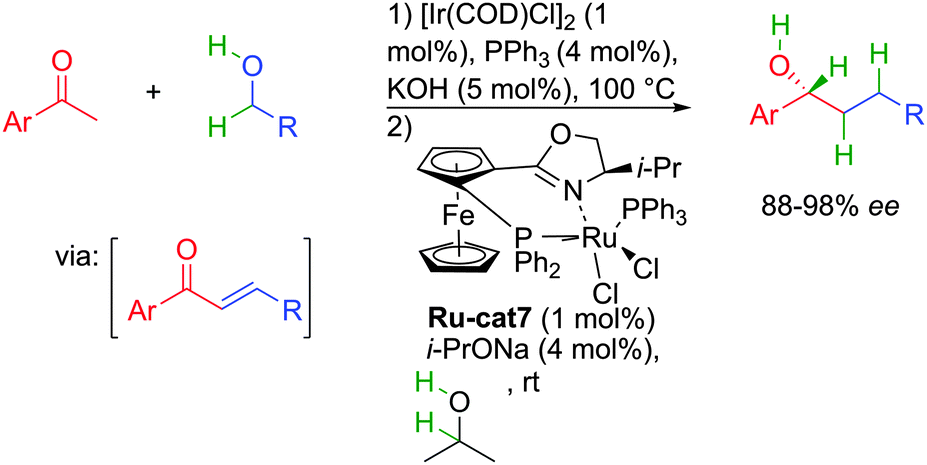 | ||
| Scheme 18 Sequential ketone alkylation reduction sequence. | ||
The challenge associated with the discovery of a single metal catalytic system for the direct use of alcohols in alkylation was tackled in 2007 by the group of Williams in the United Kingdom by combining a Wittig reagent with chiral BINAP-iridium complexes in a borrowing hydrogen alcohol alkylation (Scheme 19).36 In this process, the phosphorus ylide adds to the in situ generated reactive ON aldehyde providing an α-,β-unsaturated ester enantioselectively hydrogenated by the iridium–hydrogen complex. This represented the first success in this strategy even thought the final chiral product was obtained in a moderate 58% yield and 87% ee. It must be pointed out here that as in many cases of borrowing hydrogen, the reaction is performed at 110 °C, a temperature at which a lot of enantioselective processes provide lower enantiocontrol. This crucial point implies that the development of more efficient processes goes through the discovery of catalysts either being able to significantly decrease the reaction temperature or being highly enantioselective regardless of the conditions.
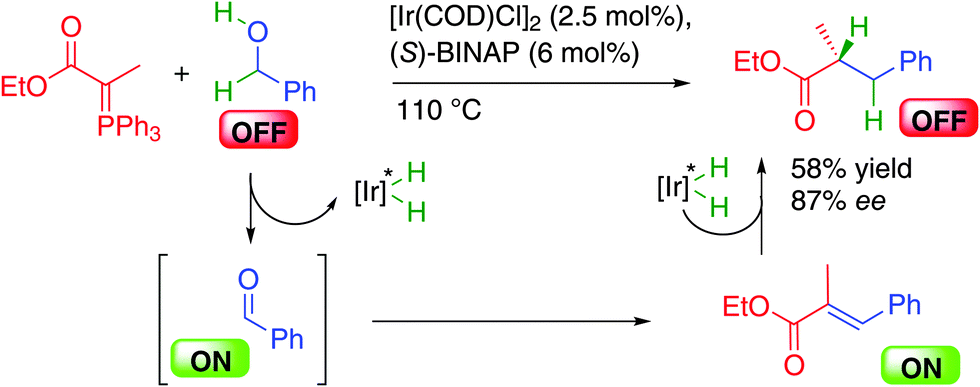 | ||
| Scheme 19 Hydrogen auto-transfer Wittig type reaction. | ||
In this context, the group of Adolfsson in Sweden proposed in 2014 a ruthenium based methodology performed between 40 and 65 °C for the reductive alkylation of acetophenones with primary alcohols (Scheme 20).37 Contrary to the work of Williams36 and more related to the Nishibayashi report,35 they did not attempt to perform final alkene enantioselective hydrogenation but focused more on ketone reduction as the stereoselective step. Using an excess of alcohol, in the presence of a chiral amino alcohol ligand, acetophenones are alkylated through the auto-transfer mechanism. Thanks to the chiral ruthenium complex, the excess starting alcohol subsequently promoted the enantioselective transfer hydrogenation of the ketone providing chiral alcohols in 57 to 89% ee. It is important to note that the same ruthenium complex is able to promote both the borrowing hydrogen as well as the enantioselective ketone transfer hydrogenation even though the yields remain moderate and the ee remains below 90%.
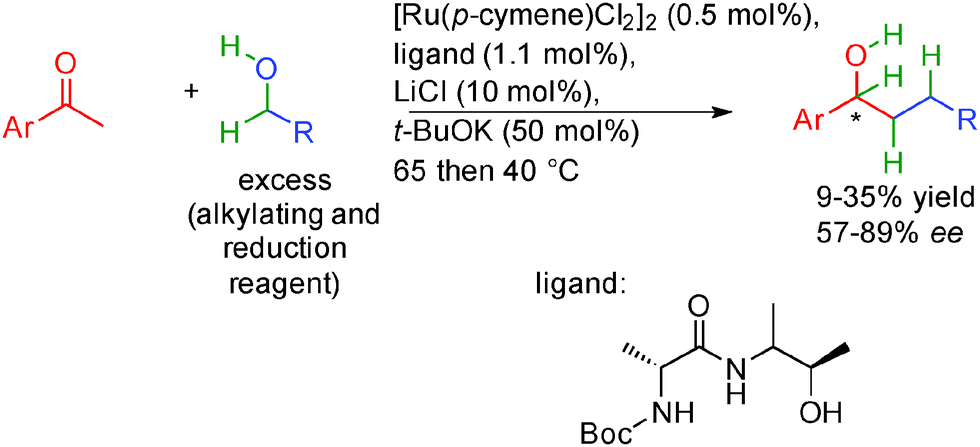 | ||
| Scheme 20 Hydrogen auto-transfer ketone alkylation and enantioselective transfer hydrogenation. | ||
From all these examples, it clearly appears that promoting direct enantioselective borrowing hydrogen C–C bond formation through hydrogenation remains a challenging task. Notably, finding a single catalytic system able to promote high reactivity in the reversible hydrogen transfer for efficient C–C bond formation and enantioselective hydrogenation remain difficult. Given the number of known hydrogenation catalysts, and given the amount of research on the racemic version of this highly eco-compatible reaction, rapid progresses can be expected in the near future.
(b) Dehydrogenation as the enantioselective step
The challenging direct enantioselective alkylation of ketones involving the final stereodetermining hydrogenation has been circumvented in 2013 by the group of Suzuki in Japan developing instead an enantioselective dehydrogenative desymmetrization of meso-1,3-diols in the presence of an excess of aldehyde.38a They first proposed an auto-transfer approach with a chiral diamino iridium complex in the presence of KOH for the generation of the optically active ON hydroxy ketone. Subsequent crotonization, followed by a final hydrogen transfer provided the chiral α-alkylated ketone in an excellent 92% ee and >99:1 dr albeit in moderate 33% yield (Scheme 21a). Indeed, some unsaturated ketone (14%) is also isolated as a side product probably due to the loss of hydrogen by competitive addition to the excess aldehyde. To solve this problem, a sequential alternative involving the addition of an extra hydrogen source (alcohols) after borrowing hydrogen allows to cleanly form the enantioenriched hydroxy ketones in good yields, perfect diastereocontrol and 88 to 94% ee (Scheme 21b).
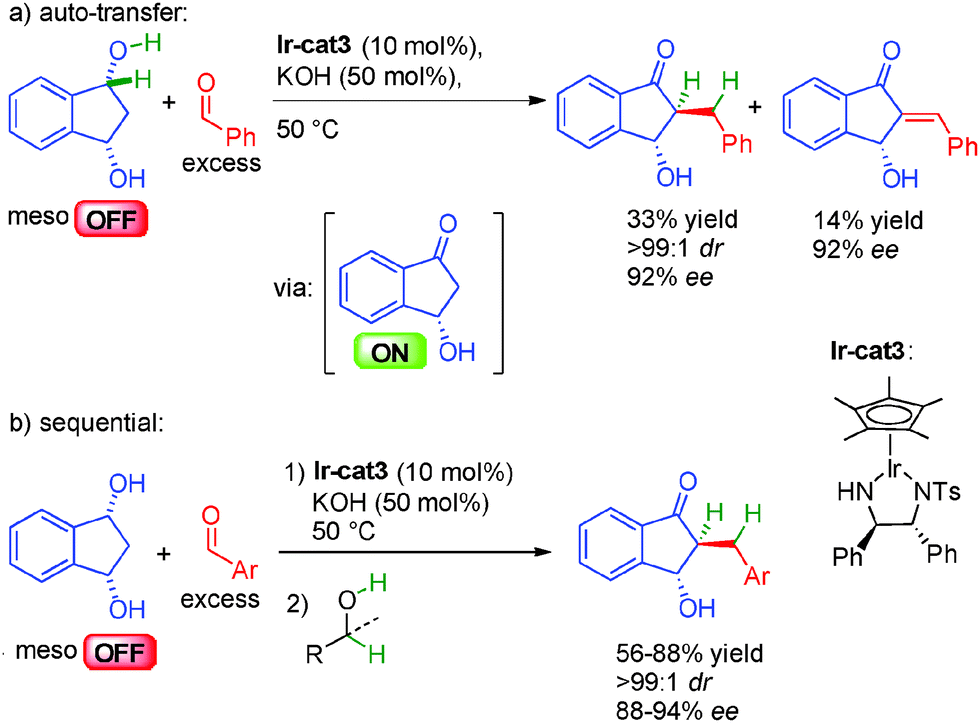 | ||
| Scheme 21 Ketone alkylation by enantioselective dehydrogenation. | ||
Building on these results, the same group reported in 2015 that this type of reactivity could be applied to meso-1,4-diols in a purely dehydrogenative process (Scheme 22).38b Hydrogen was transferred enantioselectively from the 1,4-diol to the excess aldehyde in-situ forming the key optically ON hydroxy ketone involved in the final aldolization/dehydration one-pot sequence promoted by CsOH·H2O delivering the enantioenriched hydroxy enones in 92 to 99% ee.
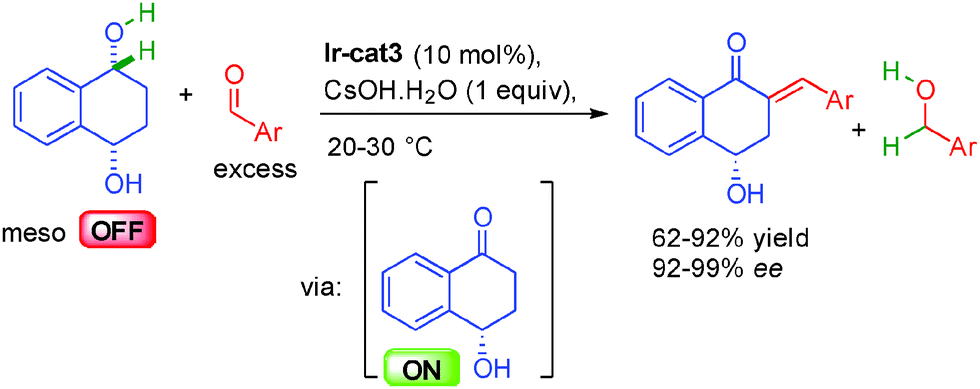 | ||
| Scheme 22 Dehydrogenative crotonisation. | ||
From these two examples, it appears that dehydrogenation is a valuable approach in terms of enantiocontrol for alkylation processes even though an excess of aldehyde as a hydrogen acceptor is required for better reactivity. Therefore future developments should be devoted to the design of more efficient acceptorless dehydrogenation systems to get rid of the required excess aldehyde.
4. Allylic alcohol functionalization by borrowing hydrogen
The precedent examples highlight the challenges facing the development of enantioselective borrowing hydrogen transformations notably in terms of efficient enantiocontrol. A breakthrough in the use of alcohols for an enantioselective borrowing hydrogen C–C bond-forming process came in 2013 from the group of Quintard and Rodriguez in France. To overcome the difficulty in performing both the reversible hydrogen transfer and the enantioselective hydrogenation with a chiral organometallic complex, they proposed a dual catalytic system involving a metal catalyzed borrowing hydrogen and an organocatalytic enantioselective reaction (Scheme 23). Starting from an OFF allylic alcohol, an easily available achiral iron complex [Fe]39 promoted dehydrogenation to the crucial ON α,β-unsaturated aldehyde. A chiral secondary amine organocatalyst subsequently promoted the iminium-type enantioselective Michael addition forming an enantioenriched aliphatic aldehyde able to be reduced by the iron hydride to form directly the enantioenriched alcohol. The interest in introducing an organocatalytic step in the borrowing hydrogen mechanism lies in the possible easy insertion of an enantiocontrol point as well as displacing the overall hydrogen transfer equilibrium towards product formation. As a result, the process using the iron complex occurs at relatively low temperatures (10–25 °C).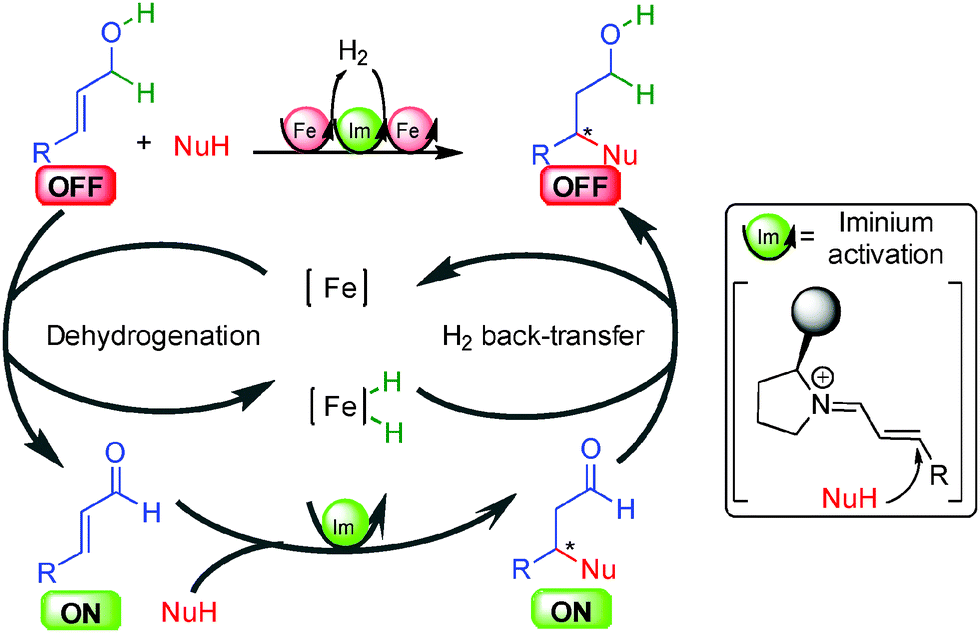 | ||
| Scheme 23 Concept of the dual allylic alcohols functionalization. | ||
In an initial study, the addition of variously substituted keto-esters to allylic alcohols was performed. The Knölker complex Fe-cat1 (activated by CO decoordination with Me3NO)40 in combination with diarylprolinol silyl ether O-cat1 was efficiently combined to give rise to the products with good enantiocontrol (79 to 90% ee) (Scheme 24).
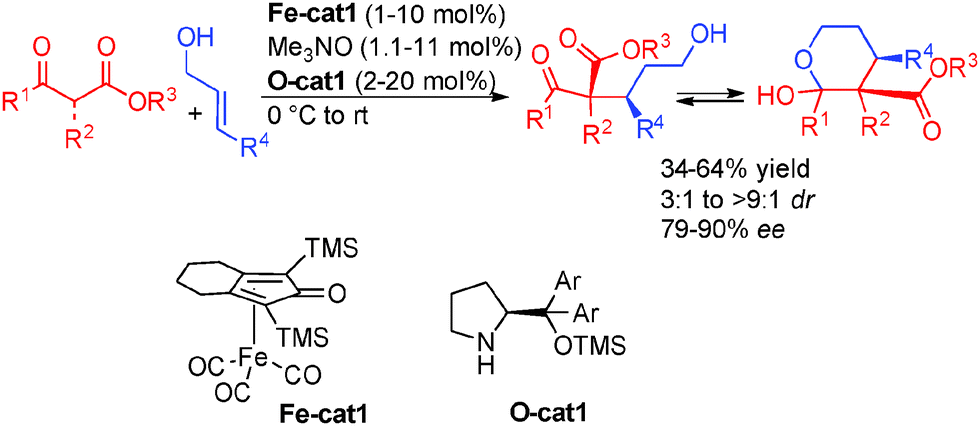 | ||
| Scheme 24 Enantioselective addition of keto-esters to allylic alcohols. | ||
It is interesting to note that the chiral linear alcohols obtained from this transformation are in equilibrium with the closed lactol form. This led to a following study from the same group to attempt a subsequent cascade fragmentation from the lactol to directly form linear protected 3-alkylpentanol scaffolds 2 (Scheme 25). These structures are present in numerous pheromones41 and used in various natural product synthesis. Unfortunately, their preparation highlights the challenge associated with the synthesis of products containing relatively inert functions (protected alcohols, unreactive ketones) from raw materials. As a result, the reported literature procedures are poorly eco-compatible and require multiple redox as well as protecting group manipulations.
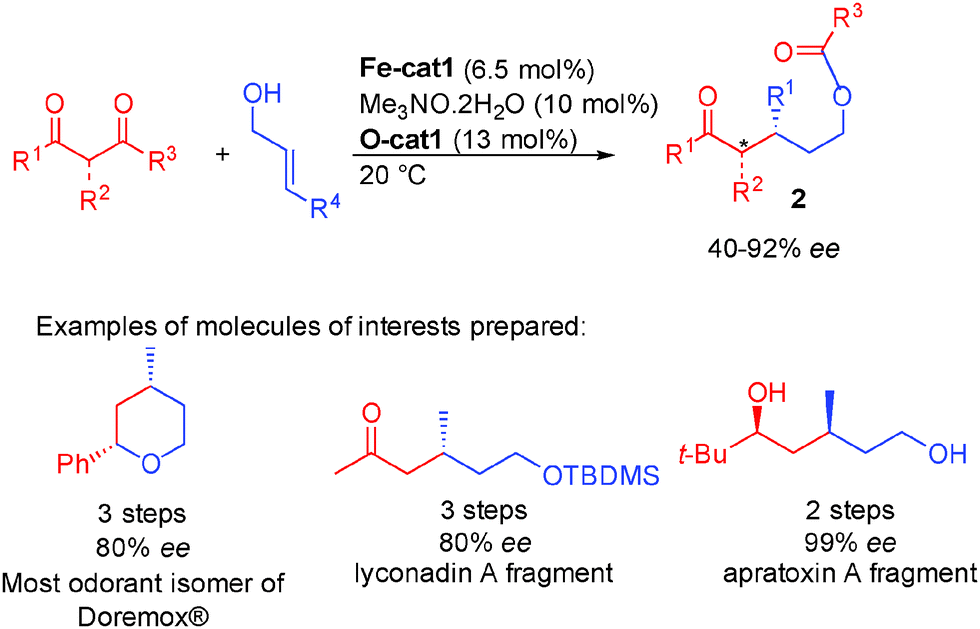 | ||
| Scheme 25 Borrowing hydrogen cascade fragmentation. | ||
With simple diketones instead of keto-esters, the designed cascade proceeds smoothly providing directly the optically active protected alcohols 2 from inexpensive starting materials. Using the combination between the iron complex and diarylprolinol silyl ethers, the expected protected 3-alkylpentanols were obtained in 40 to 92% ee depending on the substitution pattern (Scheme 25).42 This cascade strategy allows the rapid preparation of crucial synthetic intermediates that would otherwise require lengthy synthesis. For example, Doremox (an odorant rose scent) and key fragments of lyconadin A or apratoxin A were prepared in 2–3 steps vs. 5 to 11 steps by the known literature approaches.43
In a more recent study, the mechanism of the iron-organo-catalyzed transformation was studied in more detail leading to the development of an innovative triple catalyzed process involving iron-, copper- and organo-catalysis (Scheme 26).42b In this transformation, the additional copper catalyst [5 mol% Cu(acac)2] is used to activate the diketone, modulating the transition state of the Michael addition. The cooperative role of this added catalyst allows for a systematic improvement of the enantiomeric excess in the allylic alcohol functionalization.
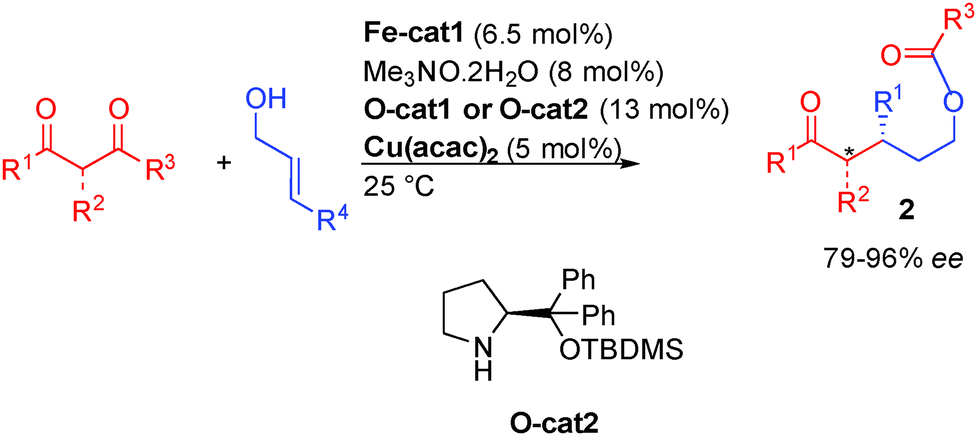 | ||
| Scheme 26 Triple catalysis for the enantioselective allylic alcohol functionalization. | ||
5. Amine alkylation by alcohols using borrowing hydrogen
The direct amine alkylation by alcohols is another key borrowing hydrogen reaction.44 A wide variety of metal complexes notably based on iridium or rhodium have been used for this attractive reaction avoiding stoichiometric leaving group utilization and forming only water as the side product. Even though the enantioselective preparation of chiral amines is a crucial industrial reaction, the first report on the enantioselective version of this eco-compatible transformation only appeared in 2013 by a work from the group of Oe in Japan (Scheme 27).45 In the presence of a secondary amine, racemic 1,2-diols are fully oxidized by abstraction of two hydrogen molecules with a ruthenium complex providing crucial reactive ON glyoxaldehyde imines 3. A final enantioselective back hydrogen transfer controlled by a chiral ferrocenyl diphosphine ligand forms the corresponding enantioenriched β-amino alcohols with moderate enantioselectivities (<78% ee). Even though acyclic amines showed low reactivity and no enantiocontrol, a broad range of chiral amino alcohols could be synthesized with five- and six-membered ring amines in 25 to 99% yield.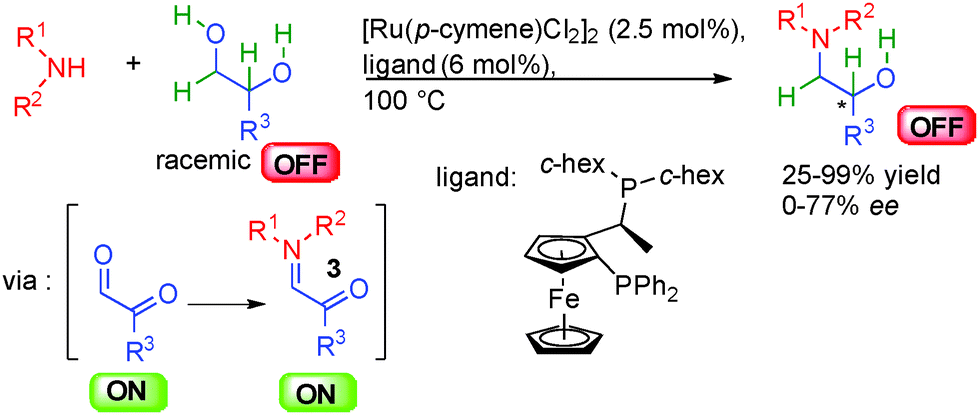 | ||
| Scheme 27 Amino alcohols synthesis by ruthenium catalysis. | ||
Soon after, the group of Zhao in Singapore discovered an innovative solution for the successful enantioselective amine alkylation (Scheme 28).46 Based on the recent discoveries on enantioselective counterion catalysis,47 they hypothesized that a combination between a chiral metal hydrogenation complex and a chiral phosphoric acid might provide a synergistic transition state for the key enantioselective reduction improving the stereocontrol of these transformations.
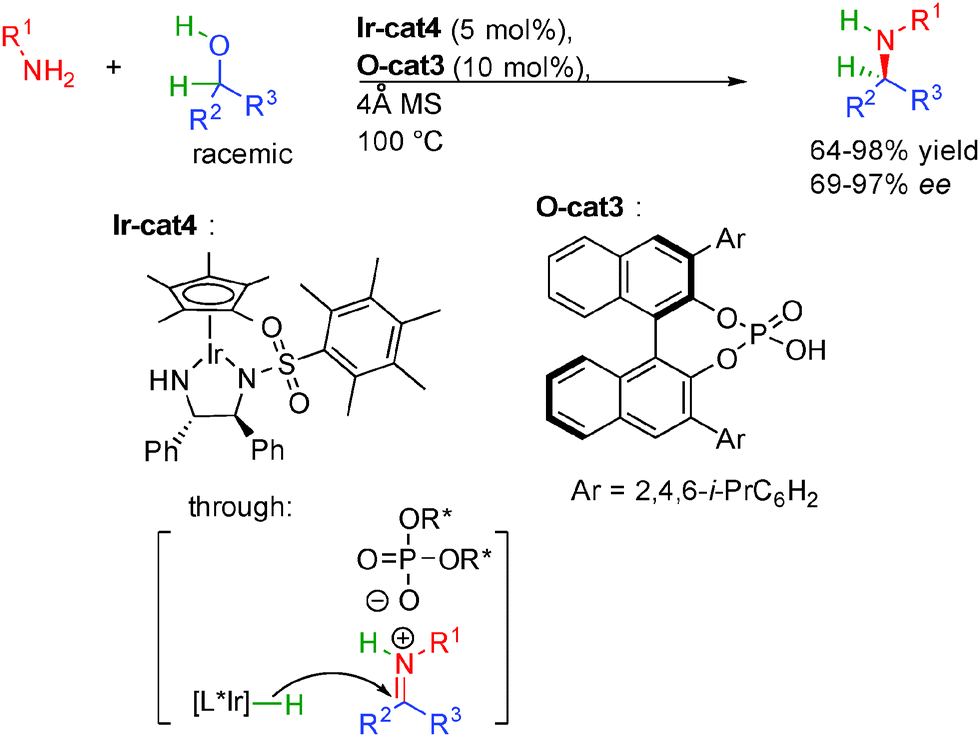 | ||
| Scheme 28 Dual iridium phosphoric acid catalysis in amine alkylation by alcohols. | ||
Extensive screening of metal-complex/phosphoric acid systems revealed that the best results were obtained by using a combination between Ir-cat4 and O-cat3. Under optimized conditions, the dual catalysis allows the formation of diversely functionalized amines in 64 to 98% yield and up to 97% ee. The use of molecular sieves is necessary to remove the formed water molecule from the reaction mixture. The remarkable enantioselectivity obtained by the synergistic catalysis at rather high temperature (100 °C) accounts for a well-defined transition state opening new opportunities for other borrowing hydrogen strategies.
Building on this initial report, the same team reported a dynamic kinetic resolution for the construction of α-branched amines possessing two contiguous stereocenters (Scheme 29).48 Using a combination of the same iridium complex with a spirocyclic chiral phosphoric acid, the final amines are constructed in excellent diastereo- and enantiocontrol (9:1 to >49:1 dr and 96 to 98% ee). Starting from a mixture of four stereoisomers of the alcohol, the efficient system converges to the formation of one single major diasteroisomer with high enantioselectivity. This enantioselective DKR is made possible by the fast equilibrium between the intermediate iminium ion through enamine formation prior to enantioselective hydrogenation.
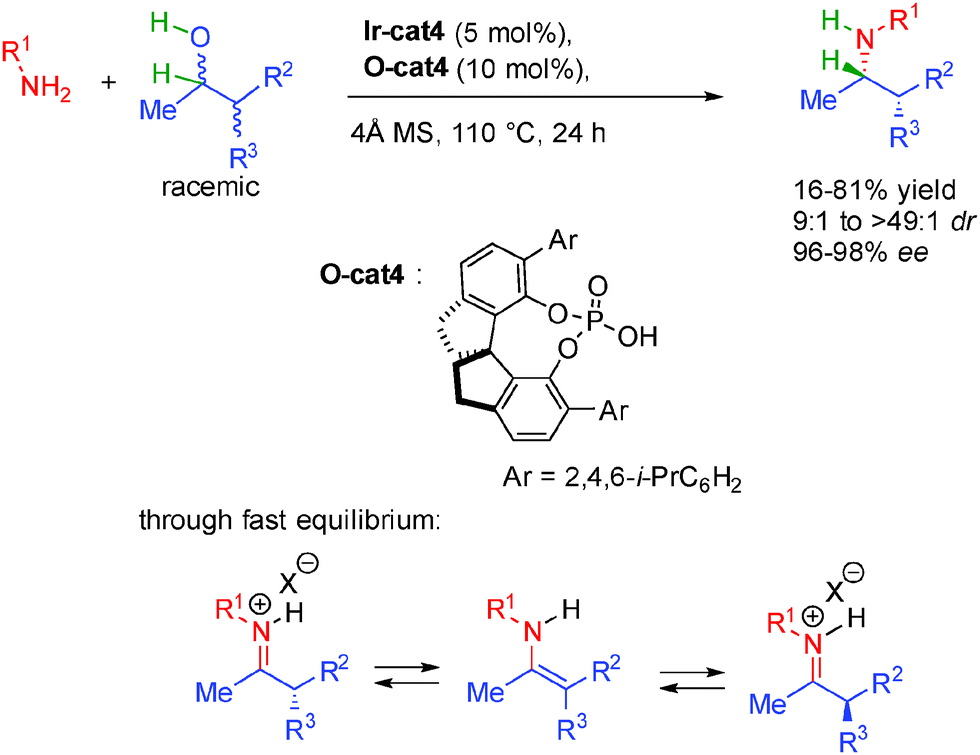 | ||
| Scheme 29 Amino-alcohols synthesis by DKR through borrowing hydrogen. | ||
Very recently, the group of Beller in Germany showed that racemic 1,2-diols could be condensed with urea to generate valuable oxazolidinones and only ammonia and water as by-products (Scheme 30).49 Interestingly, the use of a chiral biphosphine ligand (R)-(+)-MeOBIPHEP in combination with Ru3(CO)12 provided the final oxazolidones in good 82 to 92% ee. This is notable given the relatively high temperature required for the borrowing hydrogen (150 °C), highlighting the robustness of the catalytic system used.
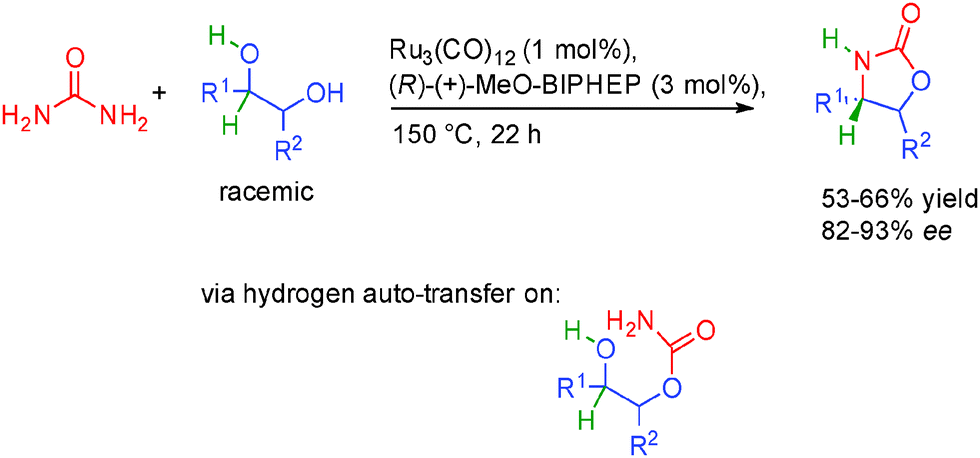 | ||
| Scheme 30 Synthesis of oxazolidinones from racemic diols through borrowing hydrogen. | ||
Finally, it must be added that the diastereoselective variants of amine alkylation by alcohols notably using Ellman's sulfinamide or -1-phenylethylamine as chiral auxiliaries were reported by several groups in the United States.50 The diastereoselective reactions using achiral iridium or ruthenium complexes provided chiral amines with 70:30 to >95:5 dr.
From this part, it is clear that enantioselective borrowing hydrogen transformations are still in their infancy but recent results notably based on multicatalysis clearly show that these highly eco-compatible reactions can be run with high stereocontrol, opening avenues for the development of other related processes.
6. Hydrogen-promoted catalytic metal hydride insertion forming reactive organometallics
Reactive organometallic intermediates such as Grignard, organozinc, or boron reagents are highly useful to introduce elongated carbon chains on various functional groups (aldehydes, imines, epoxides…). Unfortunately, their preparation requires the generation of a large amount of waste and these products are often associated with pyrophoric behavior. On the contrary, use of hydrogen, a simple non-heavy molecule, to promote a metal hydride insertion into an unsaturated substrate is an eco-compatible alternative to these classical organometallic reactions and recent powerful developments on this topic will be outlined in the following sections.51(a) Hydroformylation
Enantioselective metal catalyzed hydroformylation is a typical example of activation by metal hydrides formed upon hydrogen transfer. It received a full coverage in recent reviews and as a result is not presented in detail herein.52 The metal of choice for this transformation is rhodium in combination with chiral phosphorus donor ligands and the general concept relies on the insertion of a metal hydride into an alkene (Scheme 31).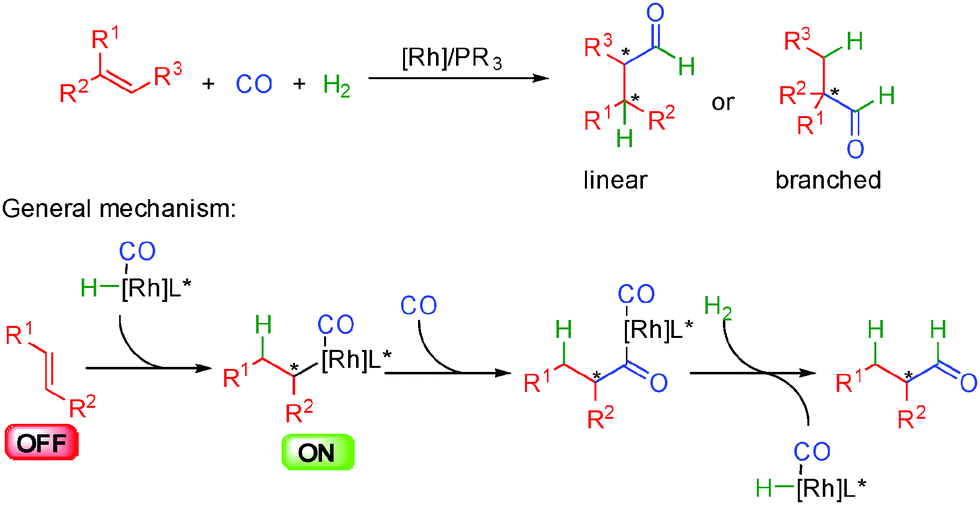 | ||
| Scheme 31 General pathway for enantioselective hydroformylation. | ||
Under a positive pressure of carbon-monoxide, the resulting reactive organometallic transient species evolves by CO insertion to form a crucial acyl-metal intermediate. Finally, under the hydrogen pressure, the chiral aldehyde is released after H2 oxidative insertion and reductive elimination giving back the catalytically active metal hydride complex.
One of the biggest challenges in hydroformylation reactions concerns the regioselectivity of metal insertion at a less or more hindered position delivering either linear or branched aldehydes. Huge efforts have been devoted in the past twenty years to solve this challenge and a wide array of chiral ligands, notably chiral phosphines, phosphites or phosphoramidites have been developed to promote selectively the formation of either products. In addition, thanks to the design of these ligands, excellent stereocontrol can be obtained in these enantioselective processes.
Besides this highly useful general method to access branched saturated aldehydes recent extensions to the insertion of other classes of functional groups will be presented in more detail in the following sections. Indeed, by distracting the process from the final CO insertion to the addition to other chemical functions, a variety of structurally diverse scaffolds can be created in an eco-compatible manner.
(b) Metal hydride insertion into unsaturated systems and subsequent functionalization
The group of Krische in the United States has considerably increased the scope of metal hydride insertions resulting in an incredible improvement of the potential of hydrogen mediated coupling reactions for the synthesis of complex molecules.53 This has led to the development of a broad range of original and eco-compatible reactions combining different donor–acceptor systems.The first enantioselective developments in these transformations were performed by inserting the metal hydride onto alkynes and trapping the resulting organometallic complex with unsaturated acceptors such as alkenes, aldehydes or imines (Scheme 32). Subtle differences exist between the catalytic systems used and the donor–acceptor combination but the general principles highlighted in this scheme prevail. Insertion of the metal-hydride into the alkyne forms a nucleophilic organometallic ON intermediate able to react with an acceptor (Scheme 32a). Under H2 pressure, oxidative addition occurs forming a metal hydrogen complex ready for a reductive elimination providing a functionalized alkene with concomitant release of the catalytically active initial metal-hydride. Alternatively, the reaction might in some cases occur through initial cycloaddition between the alkyne and the acceptor from a cationic rhodium complex (Scheme 32b). The resulting metallacycle evolves by hydrogen insertion and a reductive elimination closes the catalytic cycle.
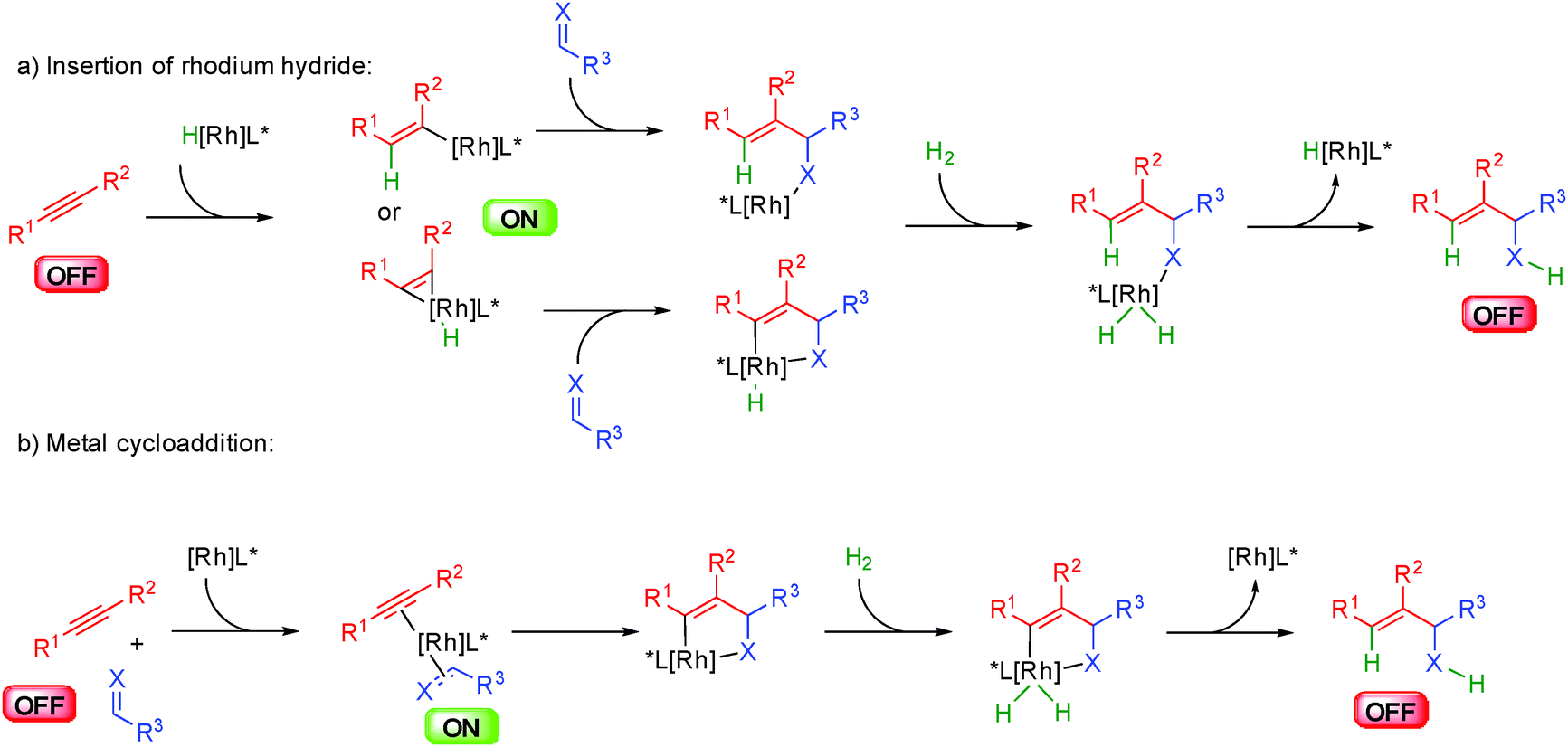 | ||
| Scheme 32 General mechanism for hydrometallation of alkynes. | ||
Following these lines, a wide range of alkynes can be activated simply by rhodium(I) complexes in combination with chiral bisphosphine ligands while an acid co-catalyst is sometimes required to modulate the counterion around the rhodium catalyst (Scheme 33). Only 1 atmosphere of hydrogen is sufficient to promote reductive coupling and the metal hydride formed can be added to aldehydes or imines in typically >90% enantiocontrol (products 4a–4d).54 The intramolecular version allows the use of aldehydes or even alkenes as potential acceptors in 98–99% ee (products 4e–4f).55 Moreover, highly reactive keto-esters are also good substrates for this transformation forming dienes such as 4g in 93% ee.56 Finally, if widely available acetylene is used as the starting alkyne, dimerization occurs before condensation to the acceptor (aldehydes or imines) forming valuable dienes such as 4h or 4i.57
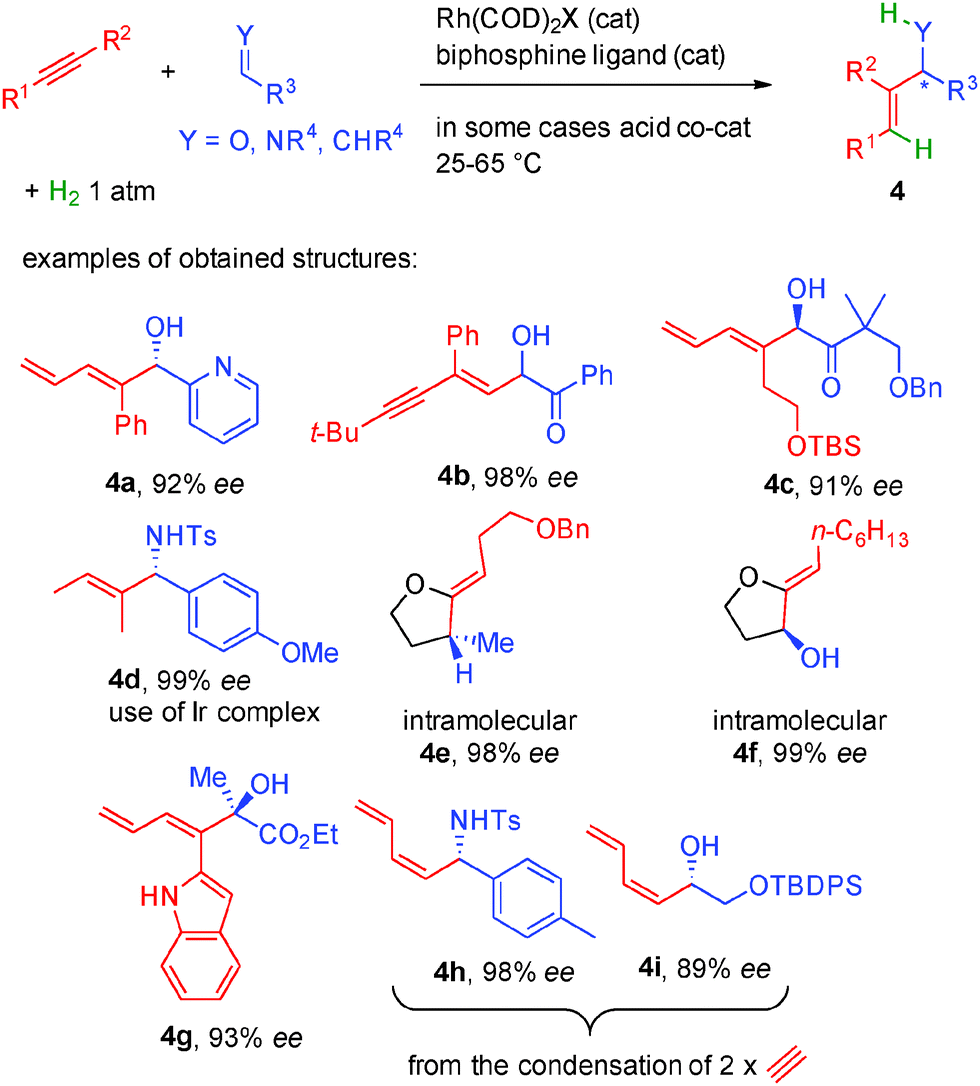 | ||
| Scheme 33 Alkyne activation by chiral rhodium complexes in the presence of 1 atmosphere of H2. | ||
Beside alkynes, enones also perform efficiently in these transformations (Scheme 34).58 In this case, hydrogenation of the double bond generates a chiral rhodium enolate involved in a stereoselective aldolization with the aldehyde leading to the expected syn aldol product. Compared to other aldolizations neither an excess of reagents nor the participation of stoichiometric additives is required except 1 atmosphere of hydrogen. In addition, a total regioselectivity for α-branched aldol products is observed.
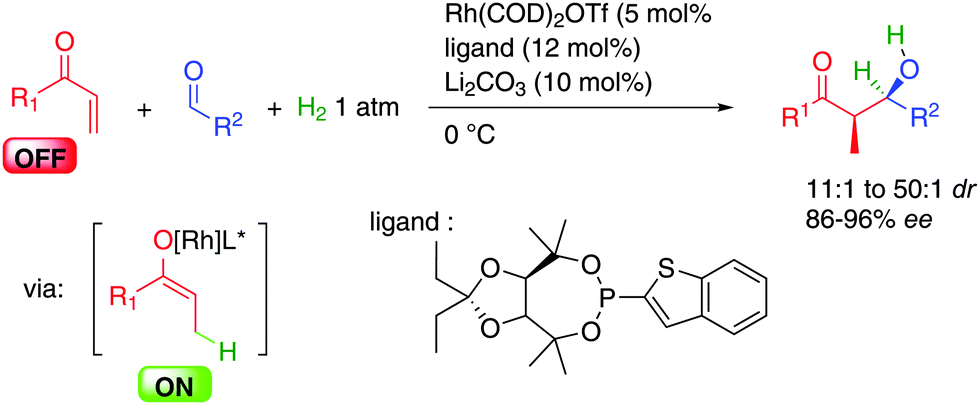 | ||
| Scheme 34 Enones in enantioselective aldolization. | ||
Another major breakthrough came with the discovery of the specific reactivity of functionalized allylic (propargylic) derivatives or simple allenes. With these OFF substrates, reactive allylic nucleophiles can be easily generated by oxidative addition to a metal opening new avenues for the synthesis of eco-compatible complex molecules. As compared to classical allylation methodologies employing allylic silane, stannane or borane reagents, this chemistry considerably decreases the waste generated to form metal allyl complexes. Most notably, the group of Krische has discovered that these metal hydride insertions could be promoted either as shown before using an aldehyde in combination with a H2 source (gaz or isopropanol) or directly in an eco-compatible auto-transfer from an alcohol (Scheme 35).59 In this case, the alcohol acts both as the hydrogen source for metal hydride insertion but also in situ generates a reactive aldehyde avoiding the use of positive hydrogen pressure.
 | ||
| Scheme 35 Scope of the hydrogen mediated allyaltion. | ||
Mechanistically, this allylation catalyzed by iridium(III) complexes in the presence of chiral phosphine ligands follows the general pathway shown in Scheme 36. An initial active iridacyclic π-allyl catalyst arising from insertion into an aromatic carboxylic acid co-catalyst, coordinates to the alcohol leading to a key metal alkoxide. Hydride insertion liberates the reactive ON aldehyde and the iridium(III) hydride in equilibrium with the anionic iridium(I). This Ir(I) complex adds to the allylic partner liberating the leaving group and forming the nucleophilic π-allyl metal. Nucleophilic addition to the electrophilic aldehyde enantioselectively creates a new carbon–carbon bond and final ligand exchange by another alcohol molecule closes the catalytic cycle.
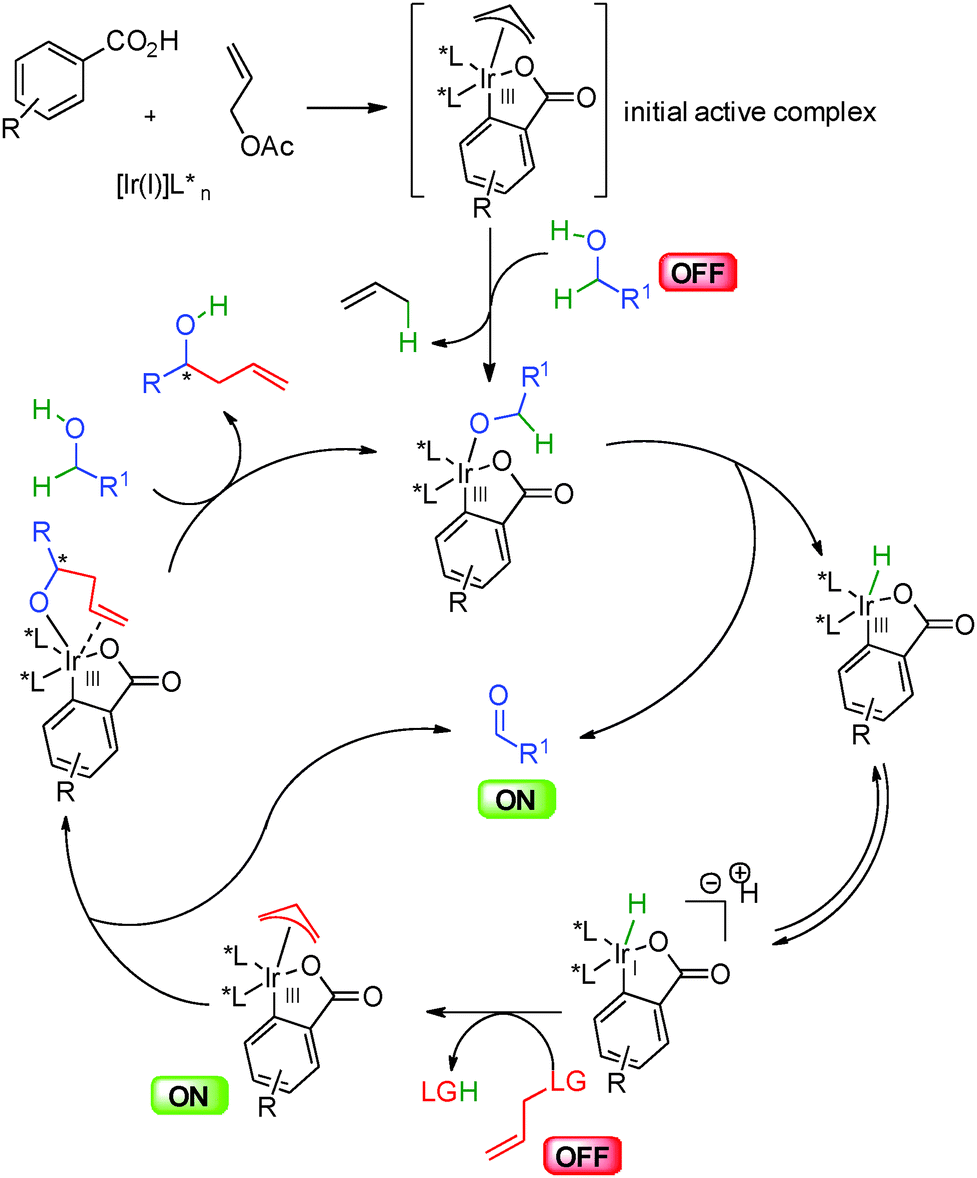 | ||
| Scheme 36 General mechanism of hydrogen mediated allylation. | ||
In most cases the iridium complexes derived from [Ir(COD)Cl]2, biphosphine ligand such as BIPHEP or SEGPHOS, and activated benzoic acids (e.g.: m-NO2-BzOH) are the most efficient in this type of chemistry (Scheme 37). In the presence of allyl acetate, the active iridium complex Ir-cat5 is formed. This bisphosphine iridium(III) complex features an iridacycle from CH insertion into the aromatic carboxylic acid and an allyl moiety coming from insertion in allyl acetate with concomitant acetic acid release. This stable complex can be isolated and is now commercially available.
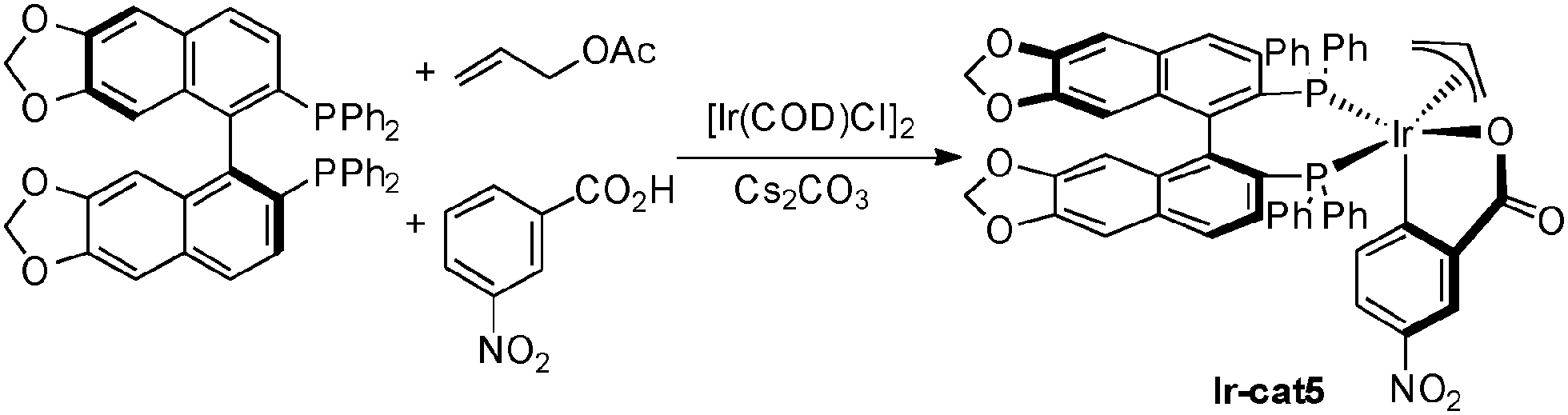 | ||
| Scheme 37 Example of active iridium complex formation. | ||
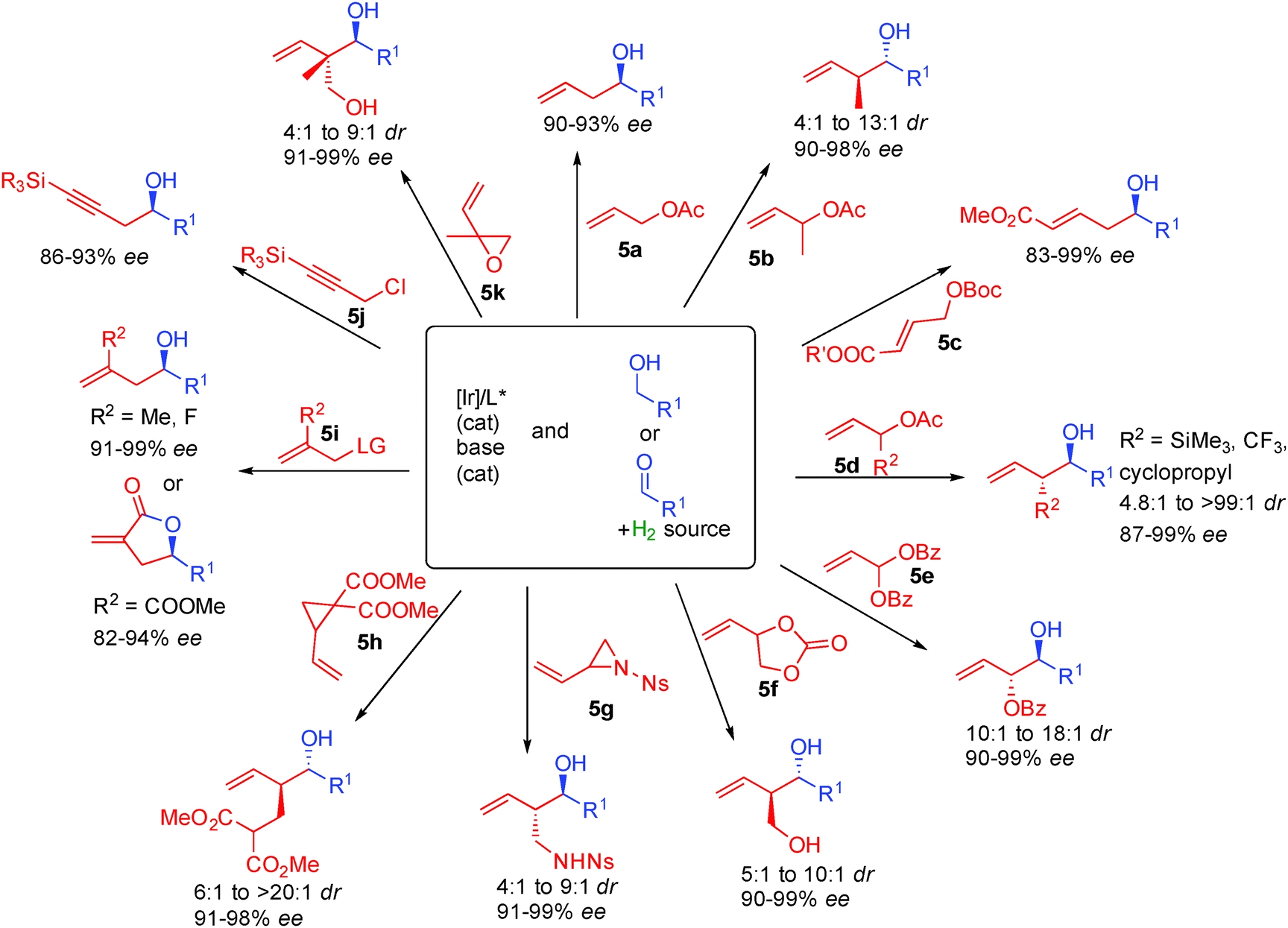
Following this general catalytic activation, a wide range of substrates are amenable to hydrogen initiated coupling (Scheme 35). Starting from either the aldehyde level in combination with a hydrogen source or starting directly from the alcohol, numerous functional groups can be incorporated increasing the toolbox available in such types of transformations. Different substituted allylic esters such as 5a–5f allow the creation of one or two contiguous stereocenters in 83 to 99% ee.59 The high level of enantiocontrol is another interesting aspect of this chemistry and a crucial point for its implementation in total synthesis.
Using allylic diester 5e, anti-1,2-diols are prepared in >10:1 dr and >90% ee.60 Starting from cyclic carbonate 5f, 1,3-diols are also prepared in typically excellent enantiocontrol.61 Allylic aziridine 5g is also amenable to the reductive coupling creating enantioenriched amino-alcohols.62 Cyclopropane 5h can also be opened by metal hydrides introducing an ester containing a carbon chain with excellent dia- and enantiocontrol (6:1 to >20:1 dr and >91% ee).63 Substitution on the internal position of the alkene of the allylic partner is also possible as in 5i providing useful entries to γ-butyrolactones for example.64 Alternatively, propargyl chlorides can also be used as in 5j, inserting a propargyl chain into chiral alcohols.65 Finally, vinyl epoxide 5k proved excellent to introduce 1,3-diols possessing a stereogenic quaternary center in >91% ee.66
Interestingly, ketones also performed well in these types of transformations67 and for example variously substituted isatins react with allylic acetates or allenes in the presence of isopropanol as the hydrogen source forming tertiary chiral allylic alcohols in 80 to 96% ee (Scheme 38). It must be pointed out that opposite absolute stereochemistry at the newly formed chiral carbon is observed starting from allyl acetate (R2 = H) and 1,1-dimethylallene (R2, R3 = Me) suggesting two different transition states for nucleophilic addition.
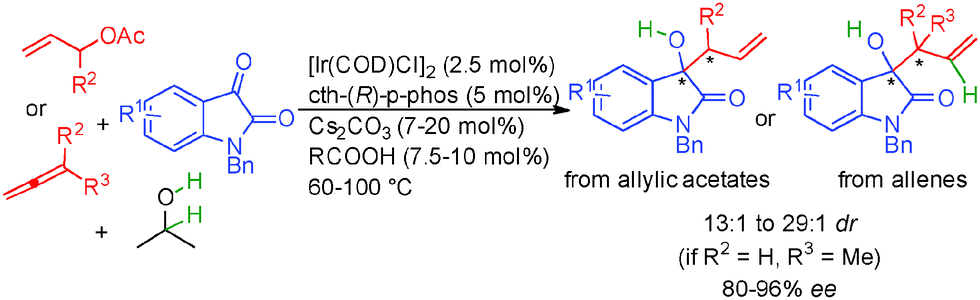 | ||
| Scheme 38 Use of isatins in enantioselective allylations. | ||
At this stage, we must highlight a particular example of this efficient hydrogen coupling chemistry: the use of 1,3-propane-diols as OFF substrates in bi-directional condensations. The corresponding oxidized 1,3-dialdehydes are poorly stable and cannot be applied in stereoselective synthesis. However, thanks to their in situ generation from 1,3-diols, a nucleophile-electrophile pair is efficiently combined allowing the direct access to chiral 1,3-diols (Scheme 39a).68
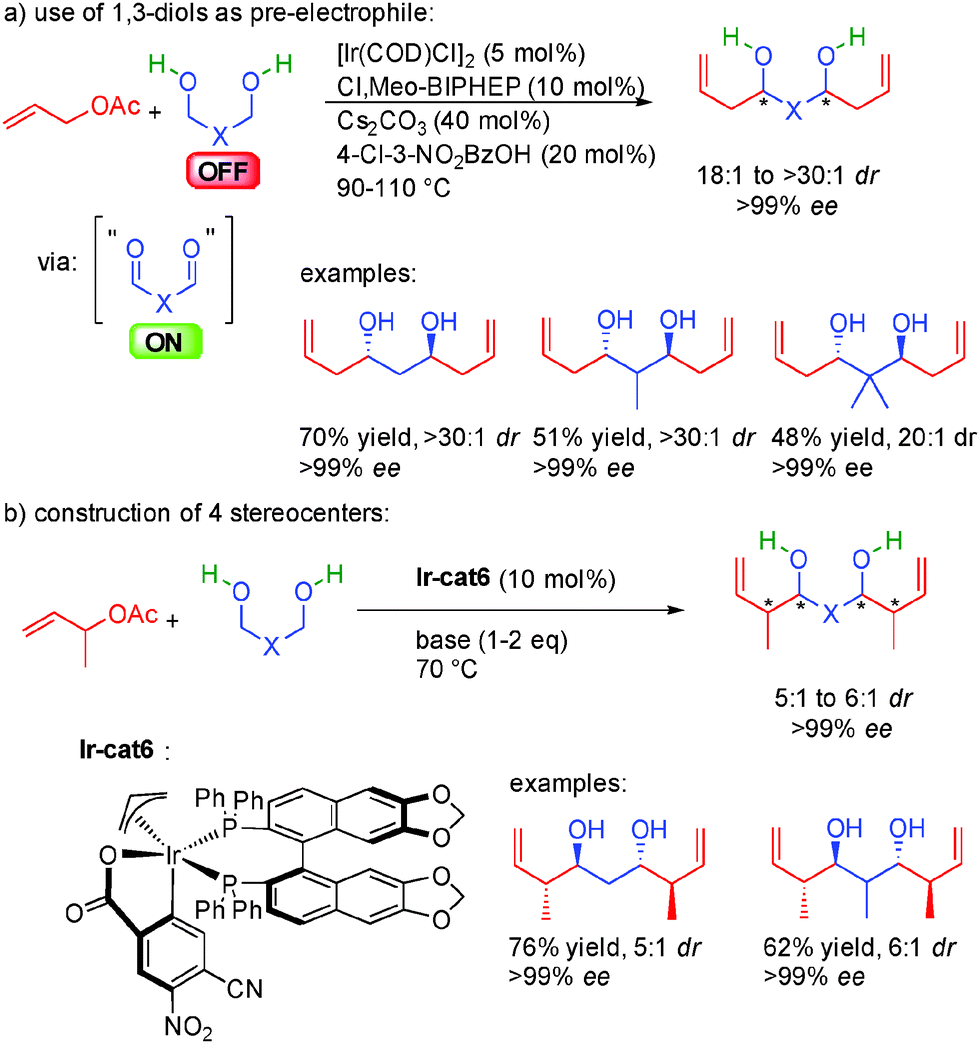 | ||
| Scheme 39 Direct enantioselective synthesis of 1,3-diols. | ||
More impressively, using substituted allylic acetates, the four acyclic stereogenic centers created in one single operation are obtained in 5:1 to 6:1 dr and perfect enantiocontrol (>99% ee) and ready for subsequent synthetic transformations (Scheme 39b). Chiral 1,3-diols (polyketides and polypropionates) are pivotal to an almost infinite number of complex natural molecules possessing a broad range of different bioactivities. Unfortunately, until then, their enantioselective preparation usually required iterative step-wise processes often requiring several redox group inter-conversion and protecting group manipulation.69 The discovery of the hydrogen initiated coupling of 1,3-diols constitutes a new scientific paradigm in the construction of these polyols and have been successfully applied in the synthesis of different complex natural products.70 For example, thanks to this approach, the synthesis of key fragments of Bryostatin 7, Roxaticin or Zincophorin methyl ester has been considerably shortened.71
Finally, the last breakthrough in this type of chemistry came from the discovery that reactive π-allyl complexes can also be generated from non-prefunctionalized conjugated alkenes or alkynes allowing access to an all new range of chiral alcohols by enantioselective addition to simple aldehydes. Interestingly, in this chemistry, depending on the substrate used, alternatives to iridium such as less expensive ruthenium complexes can also be applied successfully. Dienes 6a or 6c as well as enyne 6b are efficient starting materials providing chiral alcohols possessing an adjacent stereogenic center with >3.8:1 dr and 86 to 99% ee (Scheme 40).72 Interestingly, allenes can also be used in this chemistry either directly like in the case of 6e or generated from easily available alkynes 6d through hydrogen transfer.73 In the case of 1,1-disubstituted allene 6e the reactive π-allyl metal reacted regioselectively generating chiral alcohols with challenging contiguous quaternary stereocenters. Finally, propargylic alcohols 6f or 6g are also able to promote metal π-allyl formation through internal hydride transfer. This results in the formation of silyl enol ethers or 1,4 diols after subsequent reduction.74
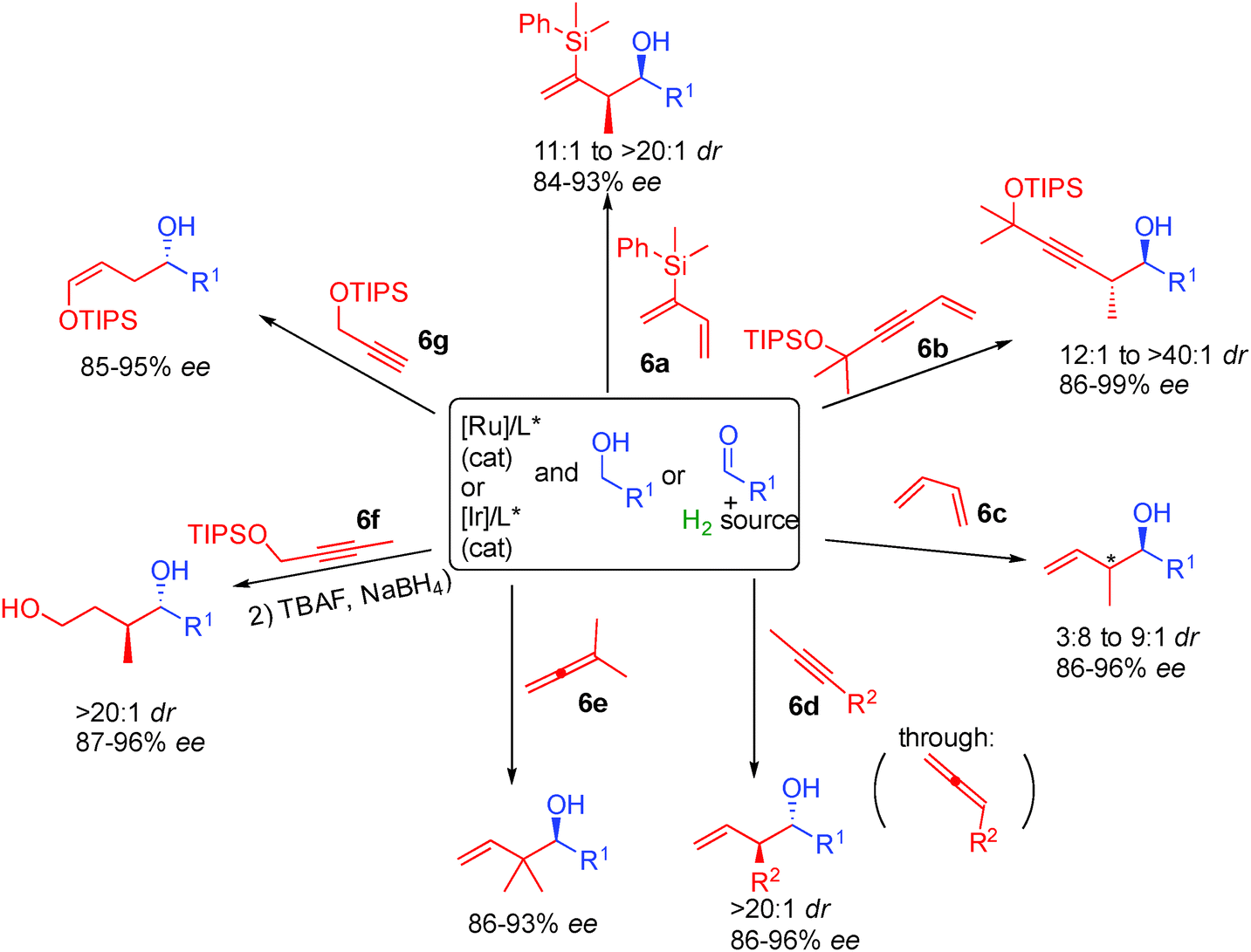 | ||
| Scheme 40 Scope of alkene and allene hydrogen mediated addition to aldehydes. | ||
From all these substrates, the use of butadiene, a feedstock reagent produced industrially is particularly interesting for the synthesis of complex molecules starting from raw materials and features a valuable diastereodivergent process depending on the nature of the acidic co-catalyst used. Indeed, the group of Krische showed that by using an achiral bisphosphine ligand (dppf) in combination with a chiral counterion (binol derived phosphoric acid), the anti-allylation product was formed predominantly (Scheme 41a).72c On the contrary, using a combination of chiral SEGPHOS and TADDOL derived phosphoric acid as a counterion, the opposite syn-allylation adduct was formed highlighting the potential of this chemistry in the synthesis of all possible steroisomers of a given molecule at hand (Scheme 41b).72d
 | ||
| Scheme 41 Diastereodivergent allylation using butadiene. | ||
In conclusion to this part, by applying the principle of hydrogen transfer metal hydride insertion to unsaturated carbon bonds and trapping the in situ generated organometallics with a wide variety of acceptors, an extraordinary rich chemistry has been developed allowing rapid and eco-compatible access to key enantioenriched building blocks. This strategy has already found applications in the efficient synthesis of numerous natural products considerably short-cutting existing routes.70 Given the generated economies and the excellent stereocontrol observed, a broad range of other transformations should be available based on this concept. Alternatively, because of the strong economic pressure related to the use of noble metals (Ir, Rh, Ru), future research studies should be devoted to the development of cheap metals as shown notably by recent progresses in hydrogen transfer reactions catalyzed by iron complexes.75 In addition, nickel76 or copper complexes also seem to be viable alternatives and through research studies innovative ways need to be found to avoid the use of stoichiometric reducing additives such as boranes or dialkylzinc.77
7. Conclusion
The current worldwide preoccupation on waste decrease and energy economy pushes the chemistry community at a point in its history requiring a dramatic change in the current approaches to construct complex chiral molecules by introducing greener processes in the synthetic routes. The initiation of eco-compatible transformations by hydrogen transfer appears to be an attractive method to this end. By applying simple hydrogen transfers, a wide range of molecules can be activated efficiently without the generation of any side products through a reversible OFF ↔ ON activation pathway providing genuine solutions for enantioselective catalysis.Long limited to alcohol or amine dynamic resolution as well as chiral lactone formation, recent developments in multi-catalysis and metal hydride insertion-addition have considerably increased the potential of this approach providing a broad range of eco-compatible tools for the synthesis of complex building blocks.
Considering the many advantages of these methods, the future should see an explosion in the development of related transformations notably through the discovery of innovative activation modes. In particular, the field should benefit from the huge amount of research done on the development of new complexes for transfer hydrogenations.78 In this field, an explosion in the alternative use of non-noble metals such as iron should additionally increase the interest in this approach in the context of eco-compatibility.
Acknowledgements
The Agence Nationale pour la Recherche (ANR-13-PDOC-0007-01), the Centre National de la Recherche Scientifique (CNRS) and the Aix-Marseille Université are gratefully acknowledged for financial support of our work on OFF ↔ ON systems.Notes and references
- B. M. Trost, Science, 1991, 254, 1471 CAS.
- P. A. Wender, F. C. Bi, G. G. Gamber, F. Gosselin, R. D. Hubbard, M. J. C. Scanio, R. Sun and T. J. Williams, Pure Appl. Chem., 2002, 74, 25 CrossRef CAS.
- (a) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed; (b) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC.
- (a) R. A. Sheldon, Chem. Ind., 1992, 903 CAS; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- (a) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Science, 2013, 341, 6143 CrossRef PubMed; (c) F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed.
- Reactions involving a single hydride transfer such as hydroacylation or using stoichiometric reductants such as Hantzch esters to initiate subsequent bond formation do not enter the field of the chemistry detailed herein. In order to facilitate the reading, solvents were omitted from all chemical equations.
- D. Hollmann, ChemSusChem, 2014, 7, 2411 CrossRef CAS PubMed.
- For selected reviews on multicatalysis: (a) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC; (b) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (c) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC.
- For the pioneering report on borrowing hydrogen: (a) M. C. R. Guerbet, Acad. Sci., 1909, 49, 129 Search PubMed . For selected recent reviews on borrowing hydrogen:; (b) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Willimas, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS; (c) T. D. Nixon, M. K. Wittlesey and J. M. J. Williams, Dalton Trans., 2009, 753 RSC.
- For the initial development of this chemistry requiring stoichiometric amount of ketone as additive: (a) P. M. Dinh, J. A. Howarth, A. R. Hudnott and J. M. Williams, Tetrahedron Lett., 1996, 37, 7623 CrossRef CAS; (b) A. L. E. Larsson, B. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 1997, 36, 1211 CrossRef CAS.
- For the first preparation of Shvo complex: (a) Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459 CrossRef CAS; (b) Y. Shvo, D. Czierkie, Y. Rahamin and D. F. Ghodosh, J. Am. Chem. Soc., 1986, 108, 7400 CrossRef CAS . For a review on Shvo catalyst; (c) B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294 CrossRef CAS PubMed.
- O. Pàmies and J. E. Bäckvall, Chem. Rev., 2003, 103, 3247 CrossRef PubMed.
- For the mechanism of racemization see: B. Martin-Matute, M. Edin, K. Bogar, F. B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817 CrossRef CAS PubMed.
- For the first report form the group of Bäckvall without added ketone as additive: B. A. Persson, A. L. E. Larsson, M. Le Ray and J.-E. Bäckvall, J. Am. Chem. Soc., 1999, 121, 1645 CrossRef CAS.
- (a) J. H. Choi, Y. H. Kim, S. H. Nam, S. T. Shin, M.-J. Kim and J. Park, Angew. Chem., Int. Ed., 2002, 41, 2373 CrossRef CAS; (b) J. H. Choi, Y. K. Choi, Y. H. Kim, E. S. Park, E. J. Kim, M.-J. Kim and J. Park, J. Org. Chem., 2004, 69, 1972 CrossRef CAS PubMed; (c) B. Martin-Matute, M. Edin, K. Bogar and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2004, 43, 6535 CrossRef CAS PubMed; (d) M. Päiviö, D. Mavrynsky, R. Leino and L. T. Kanerva, Eur. J. Org. Chem., 2011, 1452 CrossRef.
- For an alternative photo-activated ruthenium complex avoiding the use of base see: Y. Do, I.-C. Hwang, M.-J. Kim and J. Park, J. Org. Chem., 2010, 75, 5740 CrossRef CAS PubMed.
- (a) A. C. Marr, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 3283 CrossRef CAS; (b) Y. Sato, Y. Kayaki and T. Ikariya, Chem. Commun., 2012, 48, 3635 RSC.
- For reviews on this approach see: (a) B. Martin-Matute and J. E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226 CrossRef CAS PubMed; (b) P. Hoyos, V. Pace and A. R. Alcantara, Adv. Synth. Catal., 2012, 354, 2585 CrossRef CAS; (c) J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999 CrossRef CAS.
- (a) K. Leijondahl, L. Borén, R. Braun and J.-E. Bäckvall, J. Org. Chem., 1999, 64, 5237 CrossRef; (b) M. Edin, J. Steinreiber and J.-E. Bäckvall, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5761 CrossRef CAS PubMed; (c) B. Martin-Matute, M. Edin and J.-E. Bäckvall, Chem. – Eur. J., 2006, 12, 6053 CrossRef CAS PubMed; (d) M. Edin, B. Martin-Matute and J.-E. Bäckvall, Tetrahedron: Asymmetry, 2006, 17, 708 CrossRef CAS.
- (a) O. Pàmies and J.-E. Bäckvall, Adv. Synth. Catal., 2002, 344, 947 CrossRef; (b) O. Pàmies and J.-E. Bäckvall, J. Org. Chem., 2002, 67, 9006 CrossRef; (c) A.-B. L. Fransson, L. Borén, O. Pàmies and J.-E. Bäckvall, J. Org. Chem., 2005, 70, 2582 CrossRef CAS PubMed.
- (a) K. Bogar, P. H. Vidal, A. R. Alcantara Leon and J.-E. Bäckvall, Org. Lett., 2007, 9, 3401 CrossRef CAS PubMed; (b) M. C. Warner, A. Nagendiran, K. Bogar and J.-E. Bäckvall, Org. Lett., 2012, 14, 5094 CrossRef CAS PubMed; (c) R. Lhammar, R. Millet and J.-E. Bäckvall, J. Org. Chem., 2013, 78, 12114 CrossRef PubMed.
- D. Strübing, P. Krumlinde, J. Piera and J.-E. Bäckvall, Adv. Synth. Catal., 2007, 349, 1577 CrossRef.
- For examples of synthetic applications of this methodology: (a) K. Leijondahl, L. Borén, R. Braun and J. E. Bäckvall, J. Org. Chem., 2009, 74, 1988 CrossRef CAS PubMed; (b) P. Krumlinde, K. Bogar and J.-E. Bäckvall, J. Org. Chem., 2009, 74, 7407 CrossRef CAS PubMed; (c) E. V. Johnston, K. Bogar and J. E. Bäckvall, J. Org. Chem., 2010, 75, 4596 CrossRef CAS PubMed; (d) A. Träff, R. Lihammar and J.-E. Bäckvall, J. Org. Chem., 2011, 76, 3917 CrossRef PubMed; (e) M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen and C. Meier, Chem. – Eur. J., 2012, 18, 11046 CrossRef CAS PubMed; (f) N. Jana and S. Nanda, Tetrahedron: Asymmetry, 2012, 23, 802 CrossRef CAS.
- R. M. Haak, F. Berthiol, T. Jerphagnon, A. J. A. Gayet, C. Tarabiono, C. P. Postema, V. Ritleng, M. Pfeffer, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2008, 130, 13508 CrossRef CAS PubMed.
- S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149 CrossRef CAS PubMed.
- Y. K. Choi, M. J. Kim, Y. Ahn and M.-J. Kim, Org. Lett., 2001, 3, 4099 CrossRef CAS PubMed.
- T. Ohkuma, M. Kitamura and R. Noyori, Tetrahedron Lett., 1990, 31, 5509 CrossRef CAS.
- (a) K. Everaere, J.-L. Scheffler, A. Mortreux and J.-F. Carpentier, Tetrahedron Lett., 2001, 42, 1899 CrossRef CAS For other examples of lactone synthesis under hydrogenation or transfer hydrogenation:; (b) T. Benincori, S. Rizzo, T. Pilati, A. Ponti, M. Sada, E. Pagliarini, S. Ratti, C. Giuseppe, L. de Ferra and F. Sanicolo, Tetrahedron: Asymmetry, 2004, 15, 2289 CrossRef CAS; (c) E. V. Starodubtseva, O. V. Turova, M. G. Vinogradov, L. S. Gorshkova, V. A. Ferapontov and M. I. Struchkova, Tetrahedron, 2008, 64, 11713 CrossRef CAS; (d) B. Zhang, M.-H. Xu and G.-Q. Lin, Org. Lett., 2009, 11, 4712 CrossRef CAS PubMed; (e) Y. Su, Y.-Q. Tu and P. Gu, Org. Lett., 2014, 16, 4204 CrossRef CAS PubMed.
- T. Suzuki, K. Morita, Y. Matsuo and K. Hiroi, Tetrahedron Lett., 2003, 44, 2003 CrossRef CAS.
- K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329 CrossRef CAS PubMed.
- S. K. Murphy and V. M. Dong, J. Am. Chem. Soc., 2013, 135, 5553 CrossRef CAS PubMed.
- For reviews see: (a) G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358 CrossRef CAS PubMed; (b) Y. Obora, ACS Catal., 2014, 4, 3972 CrossRef CAS.
- (a) T. Itoh, Y. Takagi and T. Fujisawa, Tetrahedron Lett., 1989, 30, 3811 CrossRef CAS; (b) C. Fuganti, G. Pedrocchi-Fantoni and S. Servi, Tetrahedron Lett., 1990, 31, 4195 CrossRef CAS; (c) A. J. Smallridge, A. Ten and M. A. Trewhella, Tetrahedron Lett., 1998, 39, 5121 CrossRef CAS; (d) V. Gotor, J. R. Delhi and F. Rebolledo, J. Chem. Soc., Perkin Trans. 1, 2000, 307 RSC; (e) J. R. Dehli and V. Gotor, Tetrahedron: Asymmetry, 2001, 12, 1485 CrossRef CAS.
- G. Onodera, Y. Nishibayashi and S. Uemora, Angew. Chem., Int. Ed., 2006, 45, 3819 CrossRef CAS PubMed.
- D. J. Shermer, P. A. Slatford, D. D. Edney and J. M. J. Williams, Tetrahedron: Asymmetry, 2007, 18, 2845 CrossRef CAS.
- O. O. Kovalenko, H. Lundberg, D. Hübner and H. Adolfsson, Eur. J. Org. Chem., 2014, 6639 CrossRef CAS.
- (a) T. Suzuki, Y. Ishizaka, K. Ghozati, D.-Y. Zhou, K. Asano and H. Sasai, Synthesis, 2013, 2134 CrossRef CAS; (b) T. Suzuki, Ismiyarto, Y. Ishizaka, D.-Y. Zhou, K. Asano and H. Sasai, Org. Lett., 2015, 17, 5176 CrossRef CAS PubMed.
- For a review on this iron complex see: (a) A. Quintard and J. Rodriguez, Angew. Chem., Int. Ed., 2014, 53, 4044 CrossRef CAS PubMed; (b) H.-J. Knölker and J. Heber, Synlett, 1993, 924 CrossRef; (c) H.-J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064 CrossRef.
- (a) A. Quintard, T. Constantieux and J. Rodriguez, Angew. Chem., Int. Ed., 2013, 52, 12883 CrossRef CAS PubMed . For an account on this chemistry; (b) M. Roudier, T. Constantieux, J. Rodriguez and A. Quintard, Chimia, 2016, 70, 97 CrossRef PubMed.
- Y. Zou and J. G. Millar, Nat. Prod. Rep., 2015, 32, 1067 RSC.
- (a) M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, Org. Lett., 2014, 16, 2802 CrossRef CAS PubMed; (b) M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, ACS Catal., 2016 DOI:10.1021/acscatal.6b01102.
- (a) E. Brenna, C. Fuganti, S. Ronzani and S. Serra, Can. J. Chem., 2002, 80, 714 CrossRef CAS; (b) D. C. Beshore and A. B. Smith, III, J. Am. Chem. Soc., 2007, 129, 4148 CrossRef CAS PubMed; (c) J. Chen and C. J. Forsyth, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12067 CrossRef CAS PubMed; (d) D. Ma, B. Zou, G. Cai, X. Hu and J. O. Liu, Chem. – Eur. J., 2006, 12, 7615 CrossRef CAS PubMed.
- For reviews on the topic see: (a) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed; (b) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853 CrossRef; (c) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC . For an example of enantioselective reaction using enzyme see:; (d) F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525 CrossRef CAS PubMed.
- A. E. Putra, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6146 CrossRef.
- Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 1399 CrossRef CAS PubMed.
- For reviews see: (a) R. J. Phillips, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef PubMed; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- Z.-Q. Rong, Y. Zhang, R. H. B. Chua, H.-J. Pan and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4944 CrossRef CAS PubMed.
- M. Peña-Lopez, H. Neumann and M. Beller, Angew. Chem., Int. Ed. DOI:10.1002/anie.201600698.
- (a) R. Miao, S. C. Dimaggio, H. Shu and M. L. Trudell, Org. Lett., 2009, 11, 1579 CrossRef PubMed; (b) N. J. Oldenhuis, V. M. Dong and Z. Guan, J. Am. Chem. Soc., 2014, 136, 12548 CrossRef CAS PubMed . For an example using other chiral amine see; (c) K.-i. Fujita, T. Fujii and R. Yamagushi, Org. Lett., 2004, 6, 3525 CrossRef CAS PubMed.
- Hydroacylation is a reaction with a metal hydride not involving H2 transfer and as a result is not discussed in this review. Another approach to this enantioselective metal hydride insertion creating subsequent C–C bond is to combine allylic alcohol isomerization and C–C bond formation by enamine catalysis: A. Quintard, A. Alexakis and C. Mazet, Angew. Chem., Int. Ed., 2011, 50, 2354 CrossRef CAS PubMed.
- (a) M. Diéguez, O. Pàmies and C. Claver, Tetrahedron: Asymmetry, 2004, 15, 2113 CrossRef; (b) A. Gual, C. Godard, S. Castillon and C. Claver, Tetrahedron: Asymmetry, 2010, 21, 1135 CrossRef CAS; (c) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed; (d) Y. Deng, H. Wang, Y. Sun and X. Wang, ACS Catal., 2015, 5, 6828 CrossRef CAS.
- For reviews on this chemistry: (a) M.-Y. Ngai, J.-R. Kong and M. J. Krische, J. Org. Chem., 2007, 72, 1063 CrossRef CAS PubMed; (b) J. F. Bower, I. S. Kim, R. L. Patman and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 34 CrossRef CAS PubMed; (c) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142 CrossRef CAS PubMed.
- (a) V. Komanduri and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 16448 CrossRef CAS PubMed; (b) R. R. Huddleston, H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 11488 CrossRef CAS PubMed; C.-W. Cho and M. J. Krische, Org. Lett., 2006, 8, 3873 Search PubMed; (c) C.-W. Cho and M. J. Krische, Org. Lett., 2006, 8, 891 CrossRef CAS PubMed; (d) M.-Y. Ngai, A. Barchuk and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 12644 CrossRef CAS PubMed.
- (a) H.-Y. Jang, F. W. Hughes, H. Gong, J. Zhang, J. S. Brodbelt and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 6714 Search PubMed; (b) J. U. Rhee and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 10674 CrossRef CAS PubMed.
- J.-R. Kong, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 718 CrossRef CAS PubMed.
- (a) E. Skucas, J. R. Kong and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 7242 CrossRef CAS PubMed; (b) J. R. Kong and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 16040 Search PubMed.
- C. Bee, S. B. Han, A. Hassan, H. Lida and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 2746 CrossRef CAS PubMed.
- (a) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6240 Search PubMed; I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891 Search PubMed; (b) I. S. Kim, S. B. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2514 CrossRef CAS PubMed; (c) A. Hassan, J. R. Zbieg and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 3493 CrossRef CAS PubMed; (d) S. B. Han, X. Gao and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 9153 CrossRef CAS PubMed; (e) X. Gao, Y. J. Zhang and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 4173 CrossRef CAS PubMed; (f) R. Tsutsumi, S. Hong and M. J. Krische, Chem. – Eur. J., 2015, 21, 12903 CrossRef CAS PubMed.
- S. B. Han, H. Han and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 1760 CrossRef CAS PubMed.
- Y. J. Zhang, J. H. Yang, S. H. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 4562 CrossRef CAS PubMed.
- G. Wang, J. Franke, C. Q. Ngo and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 7915 CrossRef CAS PubMed.
- J. Moran, A. G. Smith, R. M. Carris, J. S. Johnson and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 18618 CrossRef CAS PubMed.
- (a) A. Hassan, I. A. Townsend and M. J. Krische, Chem. Commun., 2011, 47, 10028 RSC; (b) A. Hassan, T. P. Montgomery and M. J. Krische, Chem. Commun., 2012, 48, 4692 RSC; (c) T. P. Montgomery, A. Hassan, B. Y. Park and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 11100 CrossRef CAS PubMed.
- S. K. Woo, L. M. Geary and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 7830 CrossRef CAS PubMed.
- J. Feng, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2014, 136, 8911 CrossRef CAS PubMed.
- J. Itoh, S. B. han and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 6313 CrossRef CAS PubMed.
- (a) Y. Lu, I. S. Kim, A. Hassan, D. J. Del Valle and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 5018 CrossRef CAS PubMed; (b) A. Hassan, Y. Lu and M. J. Krische, Org. Lett., 2009, 11, 3112 CrossRef CAS PubMed; (c) X. Gao, H. Han and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 12795 CrossRef CAS PubMed.
- S. E. Bode, M. Wolberg and M. Muller, Synthesis, 2006, 557 CAS.
- For a review see: A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504 RSC.
- (a) S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559 CrossRef CAS PubMed; (b) Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876 CrossRef CAS PubMed; (c) Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900 CrossRef CAS PubMed.
- (a) J. R. Zbieg, J. Moran and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 10582 CrossRef CAS PubMed; (b) L. M. Geary, S. K. Woo, J. C. Leung and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 2972 CrossRef CAS PubMed; (c) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324 CrossRef CAS PubMed; (d) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628 CrossRef CAS PubMed.
- (a) T. Liang, K. D. Nguyen, W. Zhang and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 3161 CrossRef CAS PubMed; (b) S. B. Han, I. S. Kim, H. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 6916 CrossRef CAS PubMed.
- (a) T. Liang, W. Zhang, T.-Y. Chen, K. D. Nguyen and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 13066 CrossRef CAS PubMed; (b) T. Liang, W. Zhang and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 16024 CrossRef CAS PubMed.
- (a) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (b) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 12, 243 RSC; (c) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (d) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (e) A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28 CrossRef CAS PubMed . For an example see; (f) K. T. Sylvester and P. J. Chirik, J. Am. Chem. Soc., 2009, 131, 8772 CrossRef CAS PubMed.
- For a review on Nickel coupling: (a) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890 CrossRef CAS PubMed For examples of racemic coupling; (b) C. C. Baush, R. L. Patman, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 5687 CrossRef PubMed; (c) A. Köpfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1816 RSC , For racemic examples using stoichiometric reductants; (d) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065 CrossRef CAS; (e) G. M. Mahandru, G. Liu and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 3698 CrossRef CAS PubMed For nickel catalyzed enantioselective variants using stoichiometric reductant; (f) K. M. Miller, W.-S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442 CrossRef CAS PubMed; (g) K. M. Miller and T. F. Jamison, Org. Lett., 2005, 7, 3077 CrossRef CAS PubMed; (h) Y. Yang, S.-F. Zhu, C.-Y. Zhou and Q.-L. Zhou, J. Am. Chem. Soc., 2008, 130, 14052 CrossRef CAS PubMed; (i) C.-Y. Zhou, S.-F. Zhu, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 10955 CrossRef CAS PubMed; (j) Z. Tan, X. Wan, Z. Zhang, Q. Qian, W. Deng and H. Gong, Chem. Commun., 2014, 50, 3827 RSC Using cobalt; (k) C.-H. Wei, S. Mannathan and C.-H. Cheng, J. Am. Chem. Soc., 2011, 133, 6942 CrossRef CAS PubMed For racemic variant of this type of chemistry using alcohol as initial hydrogen transfer agent see for example; (l) K. Nakai, Y. Yoshida, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2014, 136, 7797 CrossRef CAS PubMed.
- For examples of copper catalyzed enantioselective variant using stoichiometric reductant: (a) J. Deschamp, O. Chuzel, J. Hannedouche and O. Riant, Angew. Chem., Int. Ed., 2006, 45, 1292 CrossRef CAS PubMed; (b) D. Zhao, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 14440 CrossRef CAS PubMed; (c) H. W. Lam and P. M. Joenssu, Org. Lett., 2005, 7, 4225 CrossRef CAS PubMed; (d) B. H. Lipshutz, W. Chrisman, K. Noson, P. Papa, J. A. Sclafani, R. W. Vivian and J. M. Keith, Tetrahedron, 2000, 56, 2779 CrossRef CAS; (e) J. Yun and S. L. Buchwald, Org. Lett., 2001, 3, 1129 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |