
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of pyrrolo[1,2-a]quinolines and ullazines by visible light mediated one- and twofold annulation of N-arylpyrroles with arylalkynes†
Amrita
Das
,
Indrajit
Ghosh
* and
Burkhard
König
 *
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de
First published on 15th June 2016
Abstract
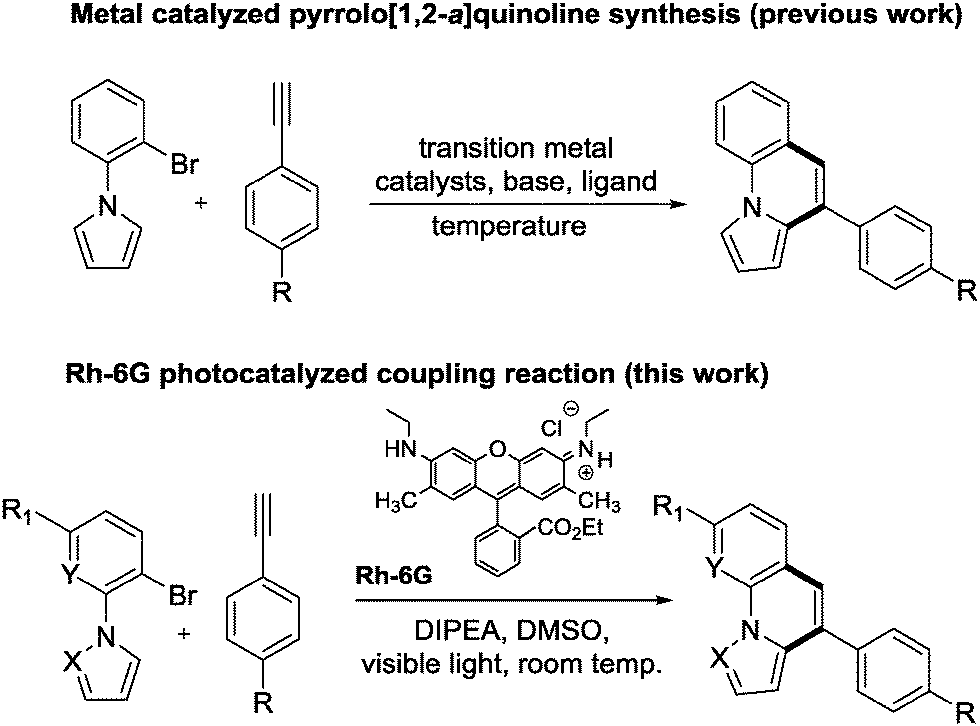
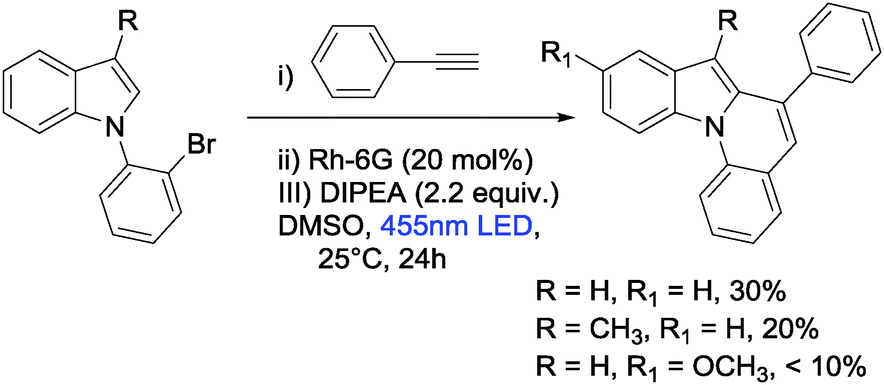
1-(2-Bromophenyl)-1H-pyrrole and 1-(2,6-dibromophenyl)-1H-pyrrole react in the presence of catalytic amounts of rhodamine 6G (Rh-6G) and N,N-diisopropylethylamine (DIPEA) under blue light irradiation with aromatic alkynes and subsequently cyclize intramolecularly to form pyrrolo[1,2-a]quinoline and ullazines. The reactions proceed at room temperature, avoid transition metal catalysts, and provide the target compounds in one pot in moderate to good yields. Mechanistic investigations suggest that the photo excited Rh-6G is reduced by DIPEA to form the corresponding radical anion Rh-6G˙−, which is again excited by 455 nm light. The excited radical anion of Rh-6G donates an electron to the aryl bromide giving an aryl radical that is trapped by aromatic alkynes. The intermediate vinyl radical cyclizes intramolecularly and yields the product after rearomatization.
Fused nitrogen containing heterocycles are structural elements of biologically active natural products1 or active pharmaceutical ingredients (APIs) and find applications in organic materials.2 Among them, pyrrolo[1,2-a]quinolines, pyrazolo[1,5-a]quinolines, and ullazines are of particular importance. Some of their derivatives show antitumor,3 antibacterial4 or antimicrobial5 activities; they activate caspases or induce apoptosis6 and are used as organic semiconductors,7 in host–guest chemistry8,9 or as liquid crystals.10,11 Also, the pyrazolo derivatives, e.g.; 4-phenylpyrazolo[1,5-a]quinoline show anti reproductive utility.12 Ullazines have found applications in optoelectronics, as organic sensitizer for solar-cell and show very good electron transport properties.13
Several methods for the synthesis of fused nitrogen-containing heterocycles have been reported; many apply direct arylation reactions. These include palladium-catalyzed reactions,14–17 1,2-alkyl migration,18 DDQ-mediated intramolecular cyclizations,19 flash vacuum pyrolysis,20 photosubstitution reactions,21 and alkyne-carbonyl metathesis.22 Lautens et al. reported a palladium catalyzed direct arylation of geminal dibromo-olefins with a boronic acid via tandem Suzuki–Miyura coupling reaction.23 Larock et al. reported a copper catalyzed tandem synthetic methodology for the synthesis of pyrrolo- and indolo[2,1-a]isoquinolines.24 Recently, Baxendale et al. developed a method for the synthesis of pyrrolo[1,2-a]-quinolines based on an allene cascade reaction in batch and flow mode.25 Miura et al. described the coupling of phenylazoles with internal alkynes in the presence of a rhodium catalyst and a copper oxidant.26 Also, the same group reported the formation of indolo[1,2-a][1,8]naphthyridines by rhodium catalyzed dehydrogenative coupling via rollover cyclometallation.27 Dumitrescu et al. developed the synthesis of pyrrolo[2,1-a]isoquinolines by multicomponent 1,3-dipolar cycloaddition.28 However, all the current synthetic methods require base, specific ligands, high temperature and transition metal catalysts, multi-step processes, and in some cases both cyclized and non-cyclized products are formed, which are difficult to separate. Transition metal free visible light photoredox catalysis is an attractive mild, selective and efficient alternative for the synthesis of pyrrolo[1,2-a]quinolines, pyrazolo[1,5-a]quinolines, and ullazines by one and two fold annulation of N-aryl pyrroles/pyrazoles with aryl alkynes. We report here a one-step visible light mediated metal free direct arylation of 1-(2-bromophenyl)-1H-pyrrole, 1-(2-bromophenyl)-1H-pyrazole, 3-bromo-2-(1H-pyrrol-1-yl)pyridine and 1-(2,6-dibromophenyl)-1H-pyrrole with simple aromatic alkynes providing pyrrolo[1,2-a]quinolines, pyrazolo[1,5-a]quinolines, pyrrolo[1,2-a][1,8]naphthyridine and ullazine compounds, respectively. This method utilizes blue light, the organic dye rhodamine 6G (Rh-6G) as photocatalyst and N,N-diisopropylethylamine (DIPEA) as electron donor (Scheme 1).
 | ||
| Scheme 1 Pyrrolo[1,2-a]quinoline syntheses. | ||
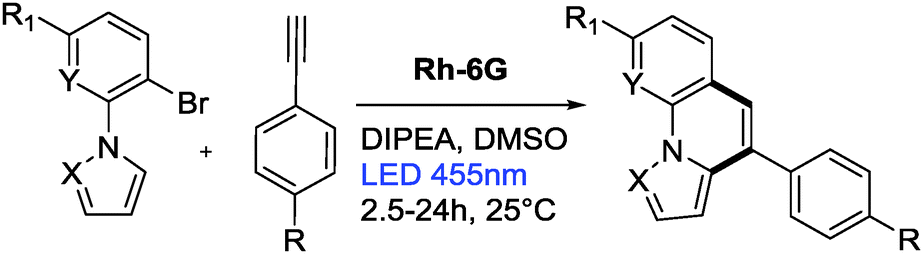
First, the annulation reaction of aryl halide 1a with aryl alkyne 2a (entry 1) in the presence of Rh-6G (20 mol%) as the photocatalyst in DMSO at 25 °C under visible light irradiation (blue LEDs: λmax = 455 ± 15 nm) was investigated. Under the reaction conditions Rh-6G photo-bleaches and 20 mol% catalyst loading was required. The annulated pyrrolo[1,2-a]quinoline 3a was obtained in 60% yield in 24 hours. This synthetic approach uses the high reduction power of the excited stable radical anion Rh-6G˙−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29,30 obtained upon photoirradiation of the xanthene dye under nitrogen with visible light in the presence of N,N-diisopropylethylamine (DIPEA). Our photocatalytic method allows the activation of electron rich heteroarenes with a redox potential up to ca. −2.4 V vs. SCE, which is not accessible using common photocatalysts,31,32 for C–C bond formation. The synthesis of fused heterocycles combines an intermolecular radical addition to alkynes with an intramolecular cyclization reaction. To improve the efficiency of this method, the reaction conditions were optimized trying various solvents, electron donors, varying the amount of catalyst and of the starting materials. DMSO was found to be the best solvent for the photoreaction using 0.07 mol L−1 substrate concentrations, 20 equiv. of the alkyne and 2.2 equiv. of DIPEA. Notably, excess amount of alkynes (unreacted alkynes were recovered during isolation) were used to avoid the dehalogenated byproduct30,33 formed upon hydrogen atom abstraction of the generated aryl radical from the radical cation of DIPEA or from the solvent.34,35 The cyclized product was obtained in good yield with 20 mol% of the photocatalyst. We used a thin layer (1.0 mm) glass reactor with 1.5 ml volume to have an optimal exposure of the reaction mixture for irradiation (for details see ESI†). Control reactions confirmed that light, the photocatalyst, and an electron donor are needed for the reaction to occur. Having identified the optimized reaction conditions, we examined the scope of the reaction with substituted aryl halides and alkynes. The products were obtained in moderate to good yields (Table 1; for the crystal structures of 3e and 3i, see Fig. 1). It is observed that the reaction is much faster with neutral and electron rich alkynes, and comparatively slower in the presence of electron withdrawing alkynes. The products (entries 5–13) in Table 1 were obtained in shorter time, but the reaction stops before full conversion. Adding one extra nitrogen to the system increases the redox potential of the aryl halide, hence, slows down the reaction. 1-(2-Bromophenyl)-1H-pyrazole (1d) has a redox potential of ∼−2.4 V vs. SCE (Table 1, entries 14 and 15) and 3-bromo-2-(1H-pyrrol-1-yl)pyridine (1e) has a redox potential of ∼−2.1 V vs. SCE (Table 1, entries 16 and 17). The observed low yields may be attributed to the slow reaction rates owing to the high reduction potentials of the substrates36 representing the limit of the photocatalyst scope, or due to the competing light absorption (i.e., the inner filter effect) of the colored products with the catalyst and/or its radical anion (see Fig. 2). In addition, trace amount of dehalogenated starting material was isolated as by-product.34,35
29,30 obtained upon photoirradiation of the xanthene dye under nitrogen with visible light in the presence of N,N-diisopropylethylamine (DIPEA). Our photocatalytic method allows the activation of electron rich heteroarenes with a redox potential up to ca. −2.4 V vs. SCE, which is not accessible using common photocatalysts,31,32 for C–C bond formation. The synthesis of fused heterocycles combines an intermolecular radical addition to alkynes with an intramolecular cyclization reaction. To improve the efficiency of this method, the reaction conditions were optimized trying various solvents, electron donors, varying the amount of catalyst and of the starting materials. DMSO was found to be the best solvent for the photoreaction using 0.07 mol L−1 substrate concentrations, 20 equiv. of the alkyne and 2.2 equiv. of DIPEA. Notably, excess amount of alkynes (unreacted alkynes were recovered during isolation) were used to avoid the dehalogenated byproduct30,33 formed upon hydrogen atom abstraction of the generated aryl radical from the radical cation of DIPEA or from the solvent.34,35 The cyclized product was obtained in good yield with 20 mol% of the photocatalyst. We used a thin layer (1.0 mm) glass reactor with 1.5 ml volume to have an optimal exposure of the reaction mixture for irradiation (for details see ESI†). Control reactions confirmed that light, the photocatalyst, and an electron donor are needed for the reaction to occur. Having identified the optimized reaction conditions, we examined the scope of the reaction with substituted aryl halides and alkynes. The products were obtained in moderate to good yields (Table 1; for the crystal structures of 3e and 3i, see Fig. 1). It is observed that the reaction is much faster with neutral and electron rich alkynes, and comparatively slower in the presence of electron withdrawing alkynes. The products (entries 5–13) in Table 1 were obtained in shorter time, but the reaction stops before full conversion. Adding one extra nitrogen to the system increases the redox potential of the aryl halide, hence, slows down the reaction. 1-(2-Bromophenyl)-1H-pyrazole (1d) has a redox potential of ∼−2.4 V vs. SCE (Table 1, entries 14 and 15) and 3-bromo-2-(1H-pyrrol-1-yl)pyridine (1e) has a redox potential of ∼−2.1 V vs. SCE (Table 1, entries 16 and 17). The observed low yields may be attributed to the slow reaction rates owing to the high reduction potentials of the substrates36 representing the limit of the photocatalyst scope, or due to the competing light absorption (i.e., the inner filter effect) of the colored products with the catalyst and/or its radical anion (see Fig. 2). In addition, trace amount of dehalogenated starting material was isolated as by-product.34,35
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aryl bromidea | R1 | Alkynea | R | Product | Yieldb,c |
| a The reaction was performed with 1(a–e) (0.1 mmol), 2(a–e) (20 equiv.), DIPEA (2.2 equiv.), and Rh-6G (20 mol%) in 1.5 mL of DMSO. b Isolated yields after purification by flash column chromatography using silica gel. c For the reaction times see ESI. d X, Y = C (entries 1–13). e X = N, Y = C (entries 14 and 15). f X = C, Y = N (entries 16 and 17). | ||||||
| 1 | 1a | H | 2a | H | 3a | 60 |
| 2 | 1a | H | 2b | CH3 | 3b | 51 |
| 3 | 1a | H | 2c | OCH3 | 3c | 50 |
| 4 | 1a | H | 2d | F | 3d | 48 |
| 5 | 1b | COCH3 | 2a | H | 3e | 56 |
| 6 | 1b | COCH3 | 2b | CH3 | 3f | 54 |
| 7 | 1b | COCH3 | 2c | OCH3 | 3g | 75 |
| 8 | 1b | COCH3 | 2d | F | 3h | 50 |
| 9 | 1b | COCH3 | 2e | Cl | 3i | 50 |
| 10 | 1c | CF3 | 2a | H | 3j | 52 |
| 11 | 1c | CF3 | 2b | CH3 | 3k | 56 |
| 12 | 1c | CF3 | 2c | OCH3 | 3l | 41 |
| 13 | 1c | CF3 | 2d | F | 3m | 41 |
| 14 | 1d | H | 2a | H | 3n | 39 |
| 15 | 1d | H | 2c | OCH3 | 3o | 35 |
| 16 | 1e | H | 2a | H | 3p | 49 |
| 17 | 1e | H | 2b | CH3 | 3q | 50 |
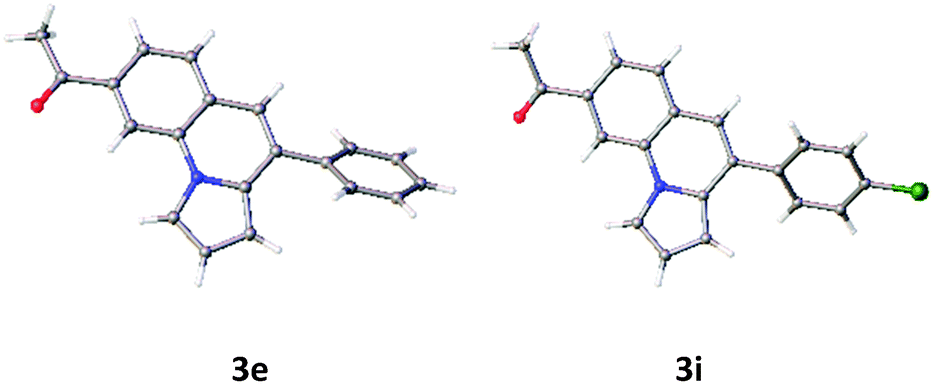 | ||
| Fig. 1 Crystal structures of compounds 3e and 3i. | ||
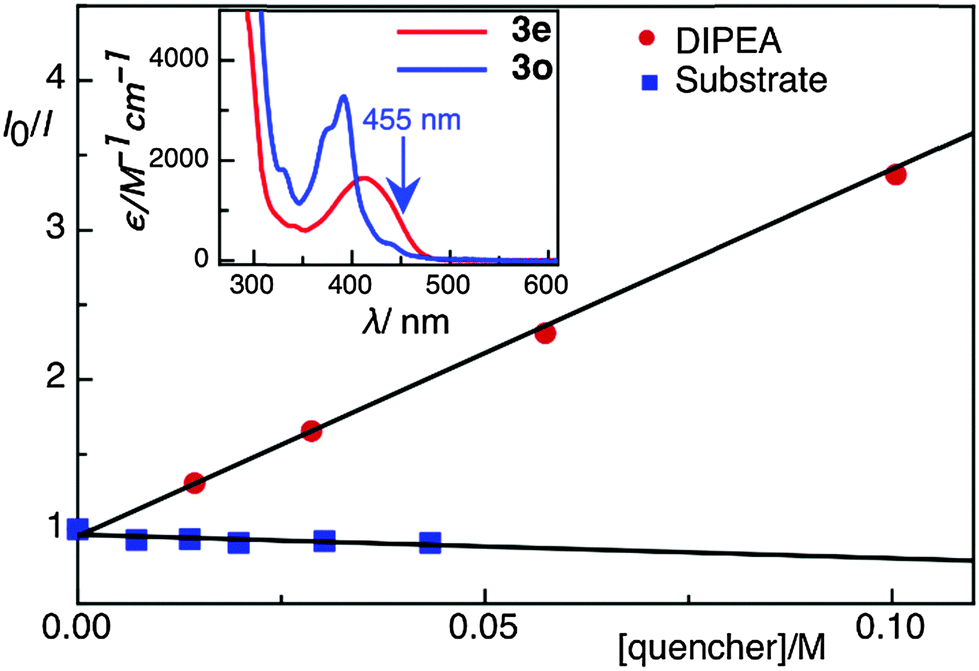 | ||
| Fig. 2 Stern–Volmer quenching plot of Rh-6G in the presence of DIPEA and 1b (test substrate). In the inset, absorption spectra of compounds 3e, and 3o are shown. | ||
Extending the scope of the reaction by replacing the pyrrole moiety by indole resulted in low yields for the transformation. A likely rational for the lower reactivity and yields of indoles in the cyclization reaction is the enforced unfavourable substitution at the 2 position (Scheme 2).37
 | ||
| Scheme 2 Indolo[1,2-a]quinoline syntheses. | ||
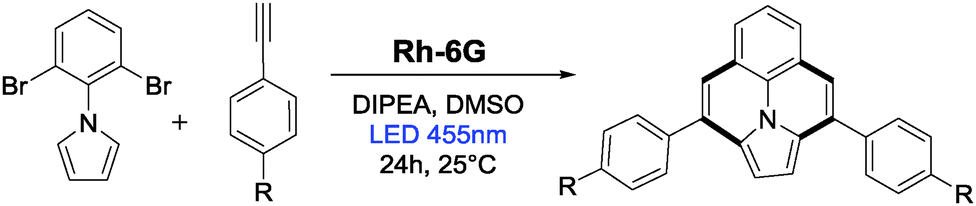
Next, we explored the photocatalytic synthesis of ullazines. First derivatives of these class of compounds were prepared in 1983 by Balli et al.38 in 9 steps with very poor yields. Takahashi route39 showed no regioselectivity and required harsh conditions. Grätzel's synthesis40 uses InCl3 and requires four steps. The photoredox reaction yields ullazines in one pot. Using the optimized reaction condition, the annulation reaction was carried out between the dibromo compound 1f and aryl alkynes. The results are summarized in Table 2. The observed low yields are due to the extremely high reduction potentials of quinoline intermediates and intensively colored products that inhibit the reaction by competing in absorption with the catalyst and/or it's radical anion (see Fig. 2). Although not quantified, most of the unreacted starting materials could be recovered during the purification process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R | Alkyne | Product | Yielda |
| The reaction was performed with 1f (0.1 mmol), 2(a–c) (20 equiv.), DIPEA (2.2 equiv.), and Rh-6G (0.2 equiv.) in 1.5 mL of DMSO.a Isolated yields after purification by flash column chromatography using silica gel. | |||||
| 1 | 1f | H | 2a | 3r | 45 |
| 2 | 1f | CH3 | 2b | 3s | 30 |
| 3 | 1f | OCH3 | 2c | 3t | 30 |
Based on previous results,30 spectroscopic investigations, and our experimental observations we propose the following mechanism (Scheme 3) for the cyclization reaction. Upon photoexcitation with blue light (λmax = 455 nm),41Rh-6G is photoreduced in the presence of DIPEA to the stable radical anion Rh-6G˙−, which is again excited by blue light and transfers an electron to the N-aryl bromide 1a forming Ar-Br˙− while regenerating the neutral Rh-6G. While the radical anion of rhodamine 6G (Rh-6G˙−) can also be generated with green light, the blue light excitation is required to photoexcite it again. Using blue light for both steps simplifies the experimental set up. The reduction potential of ground state Rh-6G˙− (ca. −1.0) is not sufficient to reduce the N-aryl bromide substrates investigated here. Fragmentation of the radical anion Ar-Br˙− generates the aryl radical, which reacts intermolecularly with the alkyne 2a to form the vinyl radical A. This vinyl radical cyclizes intramolecularly giving the annulated product 3a after oxidation and rearomatization. The formation of the dehalogenated product is due to hydrogen atom abstraction by the intermediate aryl radical either from the radical cation of DIPEA or from the solvent.
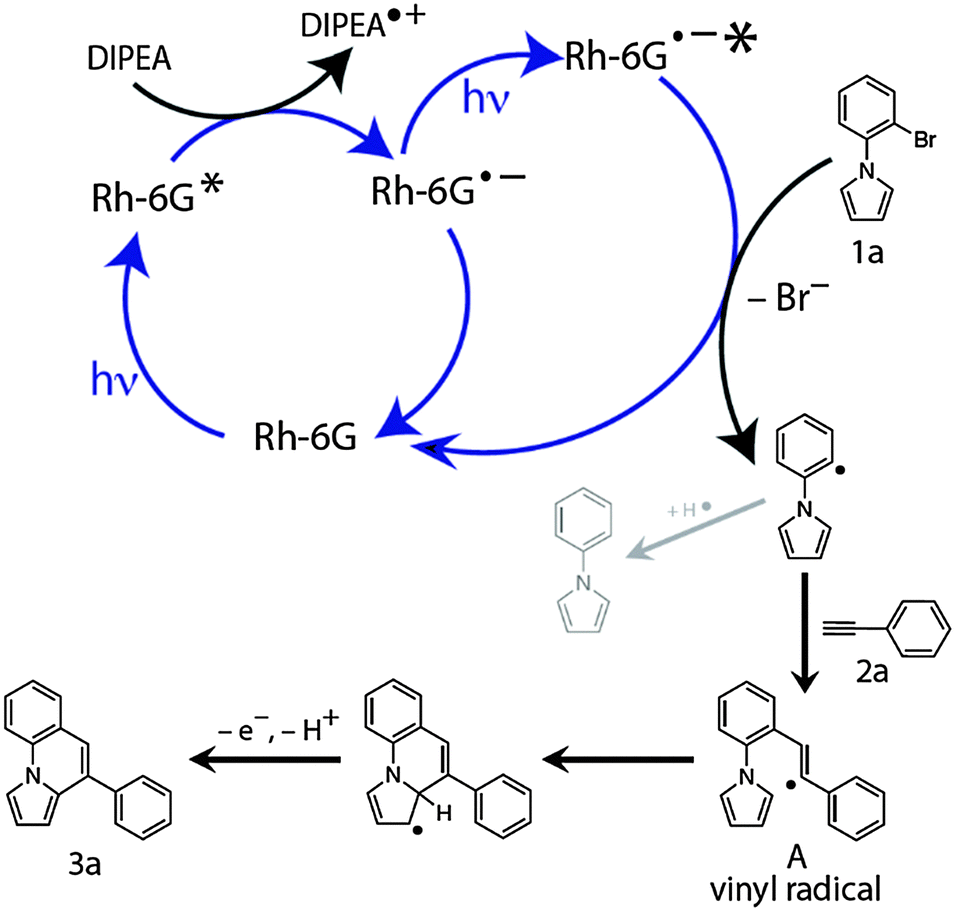 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In conclusion, the first photocatalytic synthesis of pyrrolo[1,2-a]quinolines and ullazines was accomplished starting from N-aryl halides and aryl alkynes. This method provides in a single step mild and efficient access to different types of substituted pyrrolo[1,2-a]quinolines, pyrazolo[1,5-a]quinolines, pyrrolo[1,2-a][1,8]naphthyridine and ullazines avoiding transition metal catalysts, strong bases, ligands, and high temperature.
We thank the Deutsche Forschungsgemeinschaft (GRK 1626) and Deutsche Forschungsgemeinschaft (DFG) for financial support, and Dr R. Vasold and Ms R. Hoheisel for GC–MS and CV measurements respectively.
Notes and references
- G. W. Gribble, Comprehensive Heterocyclic Chemistry II, Pergamon, 2nd edn, 1996, p. 207 Search PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
- A. Carbone, M. Pennati, B. Parrino, A. Lopergolo, P. Barraja, A. Montalbano, V. Spanò, S. Sbarra, V. Doldi, M. De Cesare, G. Cirrincione, P. Diana and N. Zaffaroni, J. Med. Chem., 2013, 56, 7060 CrossRef CAS PubMed.
- K. Tsuji, H. Tsubouchi and H. Ishikawa, Chem. Pharm. Bull., 1995, 43, 1678 CrossRef CAS PubMed.
- S. M. Gomha and K. M. Dawood, J. Chem. Res., 2014, 38, 515 CrossRef CAS.
- S. Cai, J. Drewe, S. Jiang, S. Kasibhatla, J. Kuemmerle, N. Sirisoma and H. Z. Zhang, Substituted 1-benzoyl-3-cyano-pyrrolo [1,2-a] quinolines and analogs as activators of caspases and inducers of apoptosis, US20050014759 A1, 2005 Search PubMed.
- L. Zhu, E.-G. Kim, Y. Yi, E. Ahmed, S. A. Jenekhe, V. Coropceanu and J.-L. Brédas, J. Phys. Chem. C, 2010, 114, 20401 CrossRef CAS.
- E. B. Schwartz, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1992, 114, 10775 CrossRef CAS.
- P. J. Dijkstra, M. Skowronska-Ptasinska, D. N. Reinhoudt, H. J. Den Hertog, J. Van Eerden, S. Harkema and D. De Zeeuw, J. Org. Chem., 1987, 52, 4913 CrossRef CAS.
- S. Chandrasekhar and G. S. Ranganath, Rep. Prog. Phys., 1990, 53, 57 CrossRef CAS.
- K. Praefcke, B. Kohne and D. Singer, Angew. Chem., Int. Ed., 1990, 29, 177 CrossRef.
- G. Winters, G. Odasso, G. Galliani and L. J. Lerner, 2-Phenyl-pyrazolo-[1,5-a]quinoline compounds, US4024149 A, 1977 Search PubMed.
- A. Dualeh, R. Humphry-Baker, J. H. Delcamp, M. K. Nazeeruddin and M. Grätzel, Adv. Energy Mater., 2013, 3, 496 CrossRef CAS.
- S. P. Shukla, R. K. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 10382 CrossRef CAS PubMed.
- D. G. Hulcoop and M. Lautens, Org. Lett., 2007, 9, 1761 CrossRef CAS PubMed.
- C. Baik, D. Kim, M.-S. Kang, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2009, 65, 5302 CrossRef CAS.
- D. M. Schultz and J. P. Wolfe, Org. Lett., 2010, 12, 1028 CrossRef CAS PubMed.
- J. Takaya, S. Udagawa, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 4906 CrossRef CAS PubMed.
- E. Ahmed, A. L. Briseno, Y. Xia and S. A. Jenekhe, J. Am. Chem. Soc., 2008, 130, 1118 CrossRef CAS PubMed.
- A. Ohsawa, T. Kawaguchi and H. Igeta, Synthesis, 1983, 1037 CrossRef CAS.
- K. M. Wald, A. A. Nada, G. Szilágyi and H. Wamhoff, Chem. Ber., 1980, 113, 2884 CrossRef CAS.
- M. Nayak and I. Kim, Org. Biomol. Chem., 2015, 13, 9697 Search PubMed.
- D. I. Chai and M. Lautens, J. Org. Chem., 2009, 74, 3054 CrossRef CAS PubMed.
- A. K. Verma, T. Kesharwani, J. Singh, V. Tandon and R. C. Larock, Angew. Chem., Int. Ed., 2009, 48, 1138 CrossRef PubMed.
- M. Baumann and I. R. Baxendale, J. Org. Chem., 2015, 80, 10806 CrossRef CAS PubMed.
- N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13 CrossRef CAS PubMed.
- R. Morioka, K. Nobushige, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2015, 17, 3130 CrossRef CAS PubMed.
- F. Dumitrascu, E. Georgescu, F. Georgescu, M. Popa and D. Dumitrescu, Molecules, 2013, 18, 2635 CrossRef CAS PubMed.
- S. van de Linde, A. Loschberger, T. Klein, M. Heidbreder, S. Wolter, M. Heilemann and M. Sauer, Nat. Protoc., 2011, 6, 991 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201602349.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. Konig, Science, 2014, 346, 725 CrossRef CAS PubMed.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051 CrossRef CAS PubMed.
- A. H. Jackson and P. Smith, Chem. Commun., 1967, 264, 10.1039/C19670000264.
- H. Balli and M. Zeller, Helv. Chim. Acta, 1983, 66, 2135 CrossRef CAS.
- K.-i. Kanno, Y. Liu, A. Iesato, K. Nakajima and T. Takahashi, Org. Lett., 2005, 7, 5453 CrossRef CAS PubMed.
- J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Graetzel, Angew. Chem., Int. Ed., 2013, 52, 376 CrossRef CAS PubMed.
- S. van de Linde, I. Krstic, T. Prisner, S. Doose, M. Heilemann and M. Sauer, Photochem. Photobiol. Sci., 2011, 10, 499 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, materials, methods and analytical data. CCDC 1481743 and 1481744. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04366f |
| This journal is © The Royal Society of Chemistry 2016 |