
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHost–guest chemistry that directly targets lysine methylation: synthetic host molecules as alternatives to bio-reagents
Fraser
Hof
Department of Chemistry, University of Victoria, Victoria, V8W3V6 Canada. E-mail: fhof@uvic.ca
First published on 29th June 2016
Abstract
Post-translational methylation is a chemically simple modification, but regulates the function of hundreds of proteins in profound ways. This Feature Article will report on the basic aspects of protein methylation, and will offer a personal perspective on our recent efforts at making supramolecular hosts that can bind and discriminate among post-translationally methylated partners. The article highlights several general lessons drawn from these efforts and related work by other groups. It also describes some ways in which supramolecular approaches are inherently well suited to provide tools that drive new research in the life sciences.
 Fraser Hof | Fraser Hof was born in 1976, raised in Medicine Hat, Alberta, Canada. He earned his BSc at the University of Alberta, carrying out research with David Bundle, Derrick Clive, and Neil Branda. In 1998 he moved to the Scripps Research Institute, La Jolla, to study supramolecular chemistry with Julius Rebek, Jr. After obtaining his PhD at Scripps, he was a Novartis Foundation (2003) and Human Frontier Science Program (2004) post-doctoral fellow studying medicinal chemistry with François Diederich at ETH. He took up his position at the University of Victoria in 2005, where he is currently Professor and Canada Research Chair in Supramolecular and Medicinal Chemistry. His research interests involve supramolecular chemistry, medicinal chemistry, assay development, and proteomics studies related to epigenetic methylation pathways. |
Introduction
Some of the very first synthetic host–guest molecular recognition systems—the crown ethers—were discovered accidentally by Charles Pedersen as minor synthetic byproducts. In addition to reporting their discovery, his initial communication and full paper thoroughly described the ion–dipole interactions, complementary sizes and shapes of host and guest, electronic effects, and solvation effects that drive selective complexation in dozens of crown ether hosts.1,2 The intervening decades gave rise to many more lessons learned from host–guest systems. Every fundamental kind of weak interaction that governs molecular recognition has been proven, probed, explored, and expanded using host–guest chemistry.Supramolecular chemistry was linked to biology shortly after its first invention, and the connection to the life sciences has remained strong. Both are sciences dominated by weak interactions and molecular recognition. All varieties of weak interactions, except those that involve unnatural functional groups, are encoded and used by natural molecules themselves. Biomolecules have always been common as ‘guests’ in host–guest chemistry. One could argue that even Na+ (one target of Pedersen's original host–guest chemistry) is a guest that is important to biology. But as host–guest chemistry has developed, we have increasingly thought of making hosts to target proteins, nucleic acids, carbohydrates, and complex metabolites.
The motivations for this work are most often fundamental—in spite of our encyclopedic knowledge of weak interactions, it is still often hard to answer the basic question, “can we make a host for such a complicated biological guest?” But there are also applied reasons for doing host–guest chemistry. The term technoscience—the pursuit of new, useful tools, while also pushing for new fundamental knowledge—is an apt description for much of the research that occurs in the area of host–guest chemistry. Ours is one of many research programs that operate under a common premise: that host–guest chemistry can solve critical problems in the applied biosciences. One such set of problems involves the recognition and binding of methylated amino acids, peptides, and proteins. This set of targets is important—their rich functional biology has been the subject of multiple volumes. Of particular note for this article, methylated peptide and protein targets are particularly hard to address with antibody-based affinity reagents.3–6
This Feature Article will review our work on creating supramolecular hosts for binding methylated targets, and will also offer a personal perspective on related science by other groups. The main biological motivations for our research program will be summarized. Both fundamental and applied lessons in molecular recognition will be discussed, along with the evolution of the host systems that we have created. We are not dogmatic about the use of calixarenes (as opposed to other macrocycles or other host families), but they have proven particularly useful for our targets and feature prominently in our research program. We will offer some examples that show how fundamental host–guest chemistry can lead to the creation of much-needed research reagents. We hope also to offer generally useful insight into the strategies and future prospects for host–guest chemistry on biological targets.
Post-translational methylation
“What's going on?” – Marvin GayeThe programmed addition of groups on to protein side chains is called post-translational modification. Such groups (collectively called PTMs) are diverse in chemical structure. Many interesting chemical questions arise when one considers what kinds of groups go on to proteins, what their roles are in the cell, and how they do their jobs at a molecular level.
There are >100 known PTMs,7 but from a chemical point of view PTMs can be put into three broad classes: those that involve changes of side-chain functional groups (e.g. phosphorylation, acetylation, and methylation), those that involve the grafting of large recognition modules onto side chains (e.g. ubiquitylation, SUMOylation, and many forms of glycosylation), and those modifications that involve conformational switching (e.g. proline cis–trans isomerization).
Many forms of biological control circuits have evolved (Fig. 1). The control of cellular processes occurs first at the level of the gene. Genetic sequences offer coded instructions: mutations can change the nature of the translated product, and multiple pathways that regulate the amount of mRNA present for a given gene control its protein expression levels. PTMs offer additional control over the functions of translated proteins, and are ubiquitous in all kingdoms of life. The manually annotated SwissProt database lists >28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 phosphorylation sites, >4900 acetylation sites, >2500 glycosylation sites, and >400 methylation sites in the human proteome.8 Other databases report much higher numbers.9 Regardless of the exact numbers, the numbers of PTM-labeled sites listed in various databases are greater than the number of discrete translated proteins that are thought to exist in any given human tissue. Evolution is complicated, but we can speculate that in many cases PTM pathways have evolved either because of a need for on/off functional switches that can respond more quickly than transcription–translation controls, or because a PTM offers functions that are impossible to achieve with the 20 canonical amino acids.
000 phosphorylation sites, >4900 acetylation sites, >2500 glycosylation sites, and >400 methylation sites in the human proteome.8 Other databases report much higher numbers.9 Regardless of the exact numbers, the numbers of PTM-labeled sites listed in various databases are greater than the number of discrete translated proteins that are thought to exist in any given human tissue. Evolution is complicated, but we can speculate that in many cases PTM pathways have evolved either because of a need for on/off functional switches that can respond more quickly than transcription–translation controls, or because a PTM offers functions that are impossible to achieve with the 20 canonical amino acids.
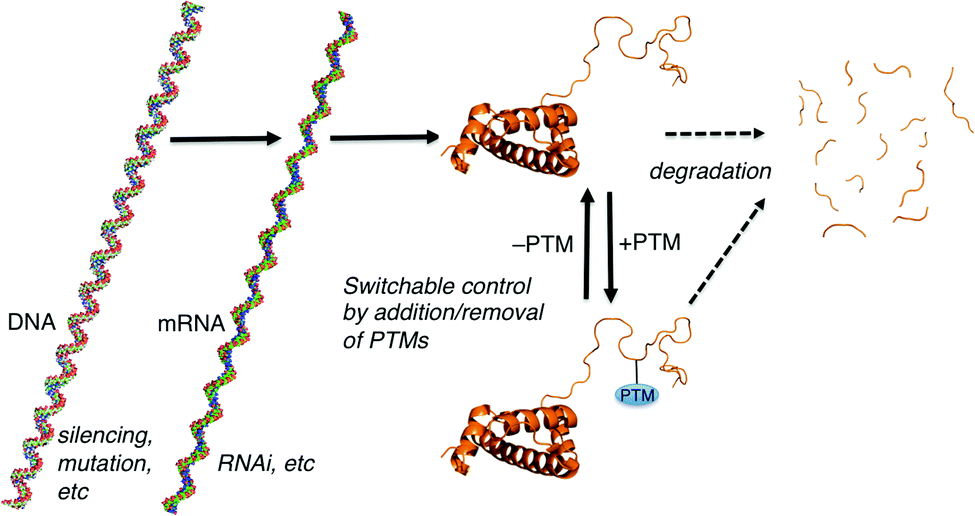 | ||
| Fig. 1 Paradigms for cellular control over protein-mediated processes. The DNA, mRNA, and protein form of histone 3 illustrate the general concepts. Structures generated using make-na (DNA, RNA) and pdb code 1KX5 (protein). | ||
Our work in the area focuses exclusively on PTMs that are relatively small functional groups (Fig. 2), and our initial curiosity was how relatively small chemical groups can cause dramatic changes in the cell. Protein phosphorylation is one such example, and it is often the first post-translational modification encountered by a chemistry or biochemistry student. The most common examples involve transfer of a phosphate group onto the alcohol side chains of serine, threonine, or tyrosine residues. Installation and removal is done by kinases and phosphatases with some degree of specificity for their targeted proteins and residues. Phosphorylation installs an anionic group onto a neutral side chain. It confers the ability to coordinate metal ions and to form salt bridges. Some proteins change their folded state upon phosphorylation, some become targets for binding and regulation by other phospho-recognizing proteins, and some have interactions with regulatory partners blocked upon phosphorylation. In all cases, the introduction of a formal charge is central to the functions of phosphorylation. Lysine acetylation and arginine citrullination are similar, in that in each case a formally cationic side chain is rendered neutral by the chemical modification.
 | ||
| Fig. 2 Common post-translational modifications. Kme1: monomethyllysine. Kme2: dimethyllysine. Kme3: trimethyllysine. MMA: monomethylarginine. sDMA: symmetric dimethylarginine. aDMA: asymmetric dimethylarginine. pS: phosphoserine. pT: phosphothreonine. pY: phosphotyrosine. | ||
Methylation is different than these other PTMs in some intriguing ways.
The term methylation in this context actually refers to multiple functionally different modifications. Lysine and arginine are the most commonly methylated residues, but even restricting the discussion only to these two side chains produces a long list of ‘methyl’ modifications. Lysine can be methylated 1–3 times. Arginine can be mono- or dimethylated, and two isomeric forms of dimethylarginine exist.
In biology, every methyl matters. Dimethyllysine can signal a different functional outcome than trimethyllysine. Asymmetric dimethylarginine (aDMA) and its isomeric PTM, symmetric dimethylarginine (sDMA), are installed by different enzymes, and one can produce a biological effect completely opposite to the effect of the other. The enzymes that install methylation marks are highly specific for a given target protein, a given residue number within that target protein, and the number of methyl groups that are installed. Examples exist in which an enzyme mutant that is poor at creating dimethyllysine, but is hyperactive for converting dimethyllysine to trimethyllysine, causes critical changes in tumor biology and patient survival. Demethylases have similarly high specificities for the sites and degrees of methylation that are their substrates and products.
Methylation is also unique among PTMs from a physicochemical point of view. It is the physically smallest group that can be added to a biomolecule. It also leaves cationic lysine or arginine residues in their native, positive states, with only small perturbations in pKa.10 This holds true for all degrees of methylation at any given residue.
How can we reconcile the subtlety of methylation events with the major biological changes ascribed to them? Because we don't expect large protein folding changes to result from, for example, the change from dimethyllysine to trimethyllysine, the differences in outcome must arise mainly from proteins that can recognize and distinguish between these PTMs. PTM-binding proteins are collectively called reader proteins, because of their ability to read out the code of different modifications.11 Methyl reader proteins are a subset of these that offer a master class in molecular recognition.
Methyl reader domains exist in at least seven distinct protein families.12 Some have shallower, surface-exposed binding sites, and others use deep pockets to engage the methylated side chain.13 All handle the recognition of neighbouring residues in different, mostly unremarkable ways… those that require high sequence and site specificity have more contacts with neighbouring side chains, and those that require lower sequence specificity have fewer peripheral contacts.14 But all methyllysine readers have evolved a common kind of binding pocket, the aromatic cage, that is the home to some particularly interesting molecular recognition.
Aromatic cages are small pockets that contain 2–4 aromatic residues, capable of forming cation–pi interactions with the methyllysine or methylarginine side chain (Fig. 3).13 Some methylation state selectivity comes from the size of the pocket. Mono- and dimethyllysine modifications are normally also engaged by a carboxylate, which forms a salt bridge to the side chain's remaining –NH atom(s). Trimethyllysines have no –NH hydrogen bond donors, and as such the aromatic cages of their reader proteins normally don't include carboxylates. In some examples, domains that have evolved to bind a certain methylation state have been converted to domains selective for another state simply by adding/removing H-bonding carboxylates from the aromatic cage (Fig. 3a).15–18
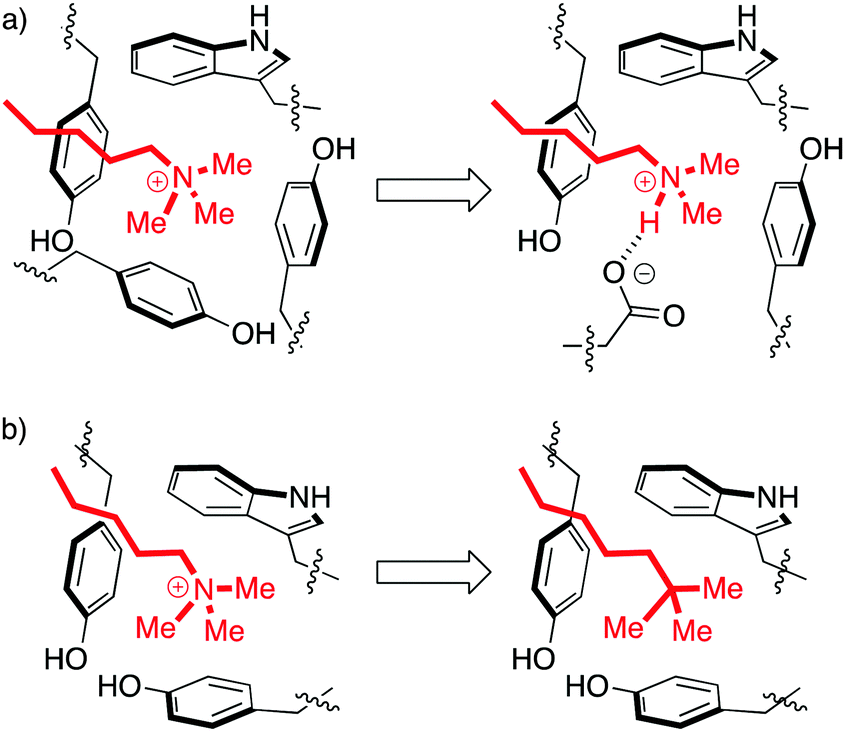 | ||
| Fig. 3 Perturbations of the aromatic cage motif provide insight into molecular recognition. (a) The aromatic cage of BPTF binds a Kme3 residue. Mutation of a tyrosine to a glutamate made the protein selective for Kme2.15 (b) The aromatic cage of HP1 binds a Kme3 residue. A ligand with a neutral t-Bu isostere of the Kme3 side chain has similar shape, surface area, and polarizability, but can't form cation–pi interactions and therefore binds 30-fold more weakly.19 | ||
Solvation is also important. The group of Waters used the chromodomain of HP1 to show that an isosteric neutral side chain bound >30-fold more weakly than the native peptide ligand, which contains a cationic Kme3 in the same position (Fig. 3b).19 This shows that hydrophobicity, dispersion forces, and shape complementarity are not sufficient for molecular recognition by readers, and shows the specific role of the cation–pi interaction in binding. But it is also true that the unmethylated lysine residues are capable of forming cation–pi interactions, yet are strongly solvated and are rejected by this pocket. In the same study of HP1, the binding of the analogous unmethylated peptide ligand was ≥100-fold weaker than that of the Kme3 ligand. This suggests an additional role for hydrophobicity in discriminating among more or less methylated ligands. Mecinovic recently confirmed the role of the cation–pi interactions in methyl reader protein binding pockets using a combination of experimental and theoretical approaches, while also proving that high-energy water molecules occupy the aromatic cage and are displaced upon Kme3 binding.20
Supramolecular hosts that target post-translational methylation
“H-2-O, No flow without the other. Oh but how, Do they make contact with one another?” – RushThe physical organic chemistry lessons on display in methyl reader proteins were the reason for our entry into this field. It started as a challenge… could supramolecular hosts be made that achieve protein-like affinity and selectivity for post-translational methylation sites? What could we learn from them? Could they serve as the basis for new methyl-targeting reagents useful for research in chemical biology?
Supramolecular hosts that would bind post-translationally methylated targets face some challenges:
– the ability to function in biologically relevant solvent (warm, salty water)
– the need to bind weakly to unmethylated parent amino acids in spite of their similar charge states
– the ability to bind strongly enough to out-compete native reader proteins
– the ability to function on amino acids, peptides, and/or whole proteins
– the need to discriminate among the different methylation states
Our first efforts to make synthetic hosts for post-translational methylation started with tryptophan—a building block that is both an electron-rich cation–pi donor, and a hydrophobic binding surface for alkyl ammonium ions. We proposed that the addition of an extended aromatic surface onto tryptophan itself, as in N-benzyl tryptophan, would provide a modular amino acid building block that could be incorporated into various host structures. Simple dipeptide hosts 1–3 were made, and tested for binding to guests with RNMe3+ groups similar to that of trimethyllysine (Fig. 4).21 Binding in buffered water was measurable, but very weak (Table 1). The increase of binding affinity in going from 1–3, along with NMR chemical shift perturbations, suggested that the binding was occurring through contacts between N-benzyl tryptophan and the cationic RNMe3+ element as predicted by molecular models (Fig. 4b), but we were unable to identify scaffolds within which this N-benzyl tryptophan binding element would offer useful guest binding.
 | ||
| Fig. 4 Two sets of hosts for quaternary ammonium ions based on the flexible N-benzyl indole moiety. (a) Simple dipeptides with zero, one, or two N-benzyl tryptophan residues bound weakly to methylammonium salts in water. (b) A molecular model of host 3 in complex with Me4N+. (c) Tris-indole hosts that bind to methylammonium salts in CDCl3 (4) and in water (5 and 6). Linking ethylene groups in 6 are highlighted in red due to their large influence on guest affinity. (d) Molecular model of host 4 in complex with acetylcholine. (e) Tetrazolate host 7 also binds Me4N+ weakly in water. | ||
| Host | Guest | Solvent | K assoc (M−1) |
|---|---|---|---|
| a Phosphate-buffered D2O (50 mM Na2HPO4/NaH2PO4 at pH 8.0). b Phosphate-buffered D2O (50 mM Na2HPO4/NaH2PO4 at pH 7.0). | |||
| 1 | ACh | D2Oa | <1 |
| 2 | ACh | D2Oa | 3 ± 1 |
| 3 | ACh | D2Oa | 14 ± 6 |
| 4 | ACh | CDCl3 | 5 ± 5 |
| Et4N+ | CDCl3 | 2 ± 1 | |
| Pr4N+ | CDCl3 | <1 | |
| Bu4N+ | CDCl3 | <1 | |
| 5 | ACh | D2Ob | 30 ± 6 |
| Kme3 | D2Ob | 55 ± 27 | |
| Et4N+ | D2Ob | 80 ± 85 | |
| Pr4N+ | D2Ob | 70 ± 30 | |
| Bu4N+ | D2Ob | 145 ± 55 | |
| 6 | ACh | D2Ob | 120 ± 8 |
| Kme3 | D2Ob | 250 ± 9 | |
| Et4N+ | D2Ob | 180 ± 10 | |
| Pr4N+ | D2Ob | 1100 ± 210 | |
| Bu4N+ | D2Ob | 7060 ± 2100 | |
This motif was modified and incorporated within a small set of threefold-symmetric host structures, including compounds 4–6 (Fig. 4).22,23 These hosts all use 1,3,5-trisubstituted benzene24 as a scaffold that presents three indoles around a central aromatic ring, presenting a larger and more organized binding surface for ammonium ion binding. Selected binding data are shown in Table 1. The role of the hydrophobic effect in these binding events is supported by multiple lines of evidence. Host 6 binds ACh in water, but organic-soluble host 4 shows >10-fold weaker binding to the same guest in CDCl3. In the series of increasingly hydrophobic ammonium ions Et4N+ to Bu4N+, host 6 shows dramatic increases in binding, up to >104 M−1. These increases are absent for host 4 in CDCl3. Surprisingly, they are also completely absent for host 5 in water, which differs from 6 only in the absence of linking CH2CH2 groups. This indicates that the behavior of host 6 is dominated by hydrophobic contacts to the alkyl linkers, rather than to the indoles that are the programmed binding surfaces for the alkylammonium guests.
These efforts, and related work using anionic tetrazoles as cation-binding elements (as in 7),25 were educational on the supramolecular aspects of this system, but ultimately failed to produce hosts for –NMe3+ groups with affinities strong enough to be useful in a biochemical setting.
p-Sulfonatocalix[4]arene binds post-translational methylations
“All I want to get is… a little bit closer” – Tegan and SaraA comparison to reader proteins was helpful in moving the program forward. NMR data for all of the above hosts showed major rearrangements of host conformations upon guest binding.21–23 In contrast, some reader proteins offer preorganized aromatic binding pockets that provide strongly oriented groups for molecular recognition.13 In some methyllysine reader proteins, a significant driving force for binding is the release of trapped water molecules.20 Increased host rigidity was needed, and so we turned to the use of macrocyclic host molecules.
An alkylated p-sulfonatocalix[6]arene was the first water-soluble calixarene host.26 Its more rigid counterpart, p-sulfonatocalix[4]arene (8, Fig. 5),27 has become one of the most widely employed hosts in aqueous solutions. A substructure search shows that there are >1100 SciFinder ‘substance’ records that indicate different salts, synthetic derivatives in which 8 is an explicit substructure, and/or host–guest complexes. Biochemical applications of 8 and related compounds have been reviewed.28,29
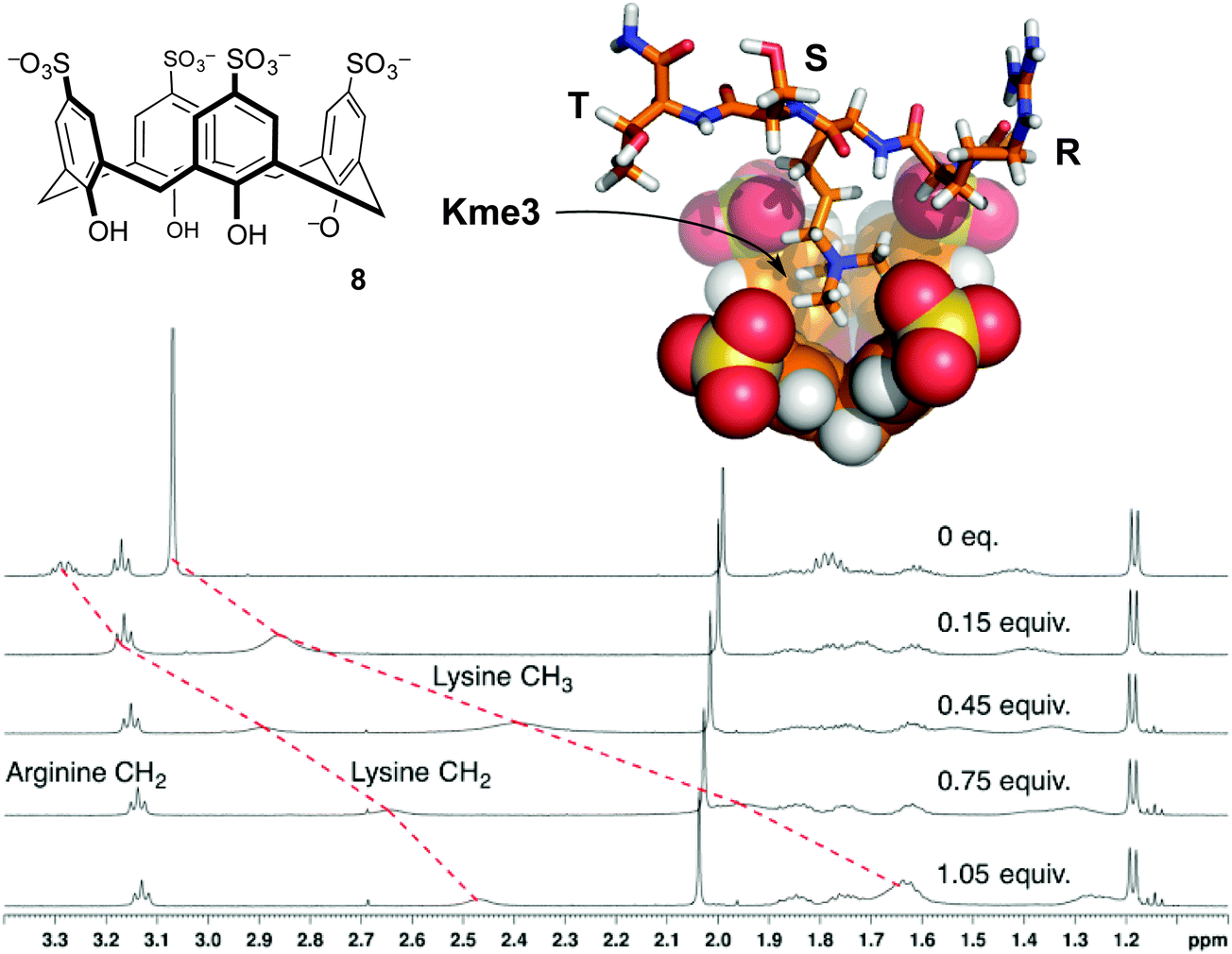 | ||
| Fig. 5 p-Sulfonatocalix[4]arene (8) binds the trimethyllysine side chain. NMR chemical shift data for peptide R-Kme3-S-T upon addition of 8 show upfield movement for the N-methyl group and the neighboring N-CH2 group on the trimethyllysine side chain, but minimal movement up to 1 equivalent of 8 for any other resonances.32 Data were collected in 40 mM NaH2PO4 buffer at pH 7.4. A molecular model of the complex shows the binding mode supported from NMR chemical shift data. | ||
The first characterized solution-phase host–guest complex using 8 was with trimethylanilinium, which showed a Kassoc of 5400 M−1 in phosphate-buffered water.30 Many reports of this host binding acetylcholine, various ammonium ions, and amino acids followed.31 Given the RNMe3+ groups present in the above-mentioned guests, the use of 8 as a starting point for binding trimethyllysine was an obvious thing to try. We found that it formed strong complexes with Kme3, and offered a >100-fold improvement on the affinity achieved by any other host for Kme3 up until that time.32
Compound 8 had already been studied as a host for almost all amino acids,28 so we had little expectation of it binding Kme3 with much selectivity. Our first set of studies on 8 showed that its affinity for the free amino acid trimethyllysine is higher than that of trimethylanilinium, and in fact >70-fold higher than its affinity for any other amino acid measured under identical conditions (Table 2).32 This host–guest complex also tolerated a variety of salt and buffer conditions,34 making it a promising starting point considering the simplicity of the host.
| Guest | Sequence | Solvent | K assoc (M−1) |
|---|---|---|---|
| a D2O containing 100 mM Na2HPO4/NaH2PO4 at pH 7.7. b D2O containing 40 mM Na2HPO4/NaH2PO4 at pH 7.4.32 c H2O containing 10 mM Na2HPO4/NaH2PO4 at pH 8.0.33 | |||
| Phenylalanine | F | D2Oa | 6.3 × 101 |
| Histidine | H | D2Oa | 2.0 × 101 |
| Arginine | R | D2Ob | 3.3 × 102 |
| Arginine | R | H2Oc | 1.5 × 103 |
| Lysine | K | D2Ob | 5.2 × 102 |
| Lysine | K | H2Oc | 7.4 × 102 |
| Trimethyllysine | Kme3 | D2Ob | 3.7 × 104 |
| H3K9me3 | R-Kme3-S-T | D2Ob | 9.7 × 105 |
Compound 8 also binds to methyllysines in the context of peptides. The short peptide R-Kme3-S-T, which represents a common lysine methylation site on histone 3, was used as a guest in NMR and ITC experiments.32 Addition of 1 equivalent of 8 caused upfield shifts mainly on the N-CH3 and ε-CH2 resonances. Similar shifts occurred for the free amino acids, proving that the structure of the complex in each case involves the quaternary RNMe3+ groups buried in the shielding environment of the host's binding pocket (Fig. 5). The affinity of the host for the peptide is even higher than its affinity for the free amino acid. It seems that the host's sulfonates can make additional favorable contacts with the neighboring polar side chains in the peptide. The backbone elements of the free amino acid Kme3 are strongly hydrated and zwitterionic, and therefore offer less in the way of electrostatic and/or hydrophobic driving force for host–guest binding. Later studies by Macartney showed that cucubit[7]uril was an incredibly strong binder of the free amino acid Kme3 (Kassoc = 1.9 × 106 M−1).35 Anecdotally, it seems that cucubit[7]uril does not strongly bind Kme3 in the context of peptides, suggesting that it relies on favorable contacts with the N-terminal –NH3+ group of the amino acid Kme3 in a way that 8 does not.
Could a simple host like 8 bind strongly enough to out-compete a native reader protein? One such target is the complex of reader protein Chromobox Homolog 7 (CBX7) with a trimethyllysine at site 27 on the tail of histone 3 (H3K27me3). The intrinsic affinity of this protein–protein interaction is relatively low, having been measured at a Kd of 10–100 μM in different biochemical assays.36,37 We were already pursuing inhibitors of this reader protein using a more traditional medicinal chemistry approach—targeting the binding pocket of CBX7 with peptidomimetic inhibitors.38 The idea of inhibiting this protein–protein interaction by targeting the key Kme3 side chain is relatively unusual, in that it involves targeting a protruding structure with a host, rather than occupying a concave binding pocket with a drug. We found that 8 was able to disrupt the CBX7–H3K27me3 interaction using a genetically encoded FRET biosensor, created by our collaborators in Robert Campbell's group.39 The related host, 9, was inactive in the same assay due to the conformational flexibility introduced by its glycol ether groups at the lower rim (Fig. 6). A rigidified analog 10, and a similar host bearing tetrazolate rings as recognition elements at the upper rim (11), were both able to disrupt the CBX7–H3K27me3 complex. Even though 8 and its analogs are known to bind promiscuously to cationic patches on protein surfaces,28,40–42 they are still able to target and obstruct the single Kme3 residue contained within the >50 kDa biosensor construct used in this study.39
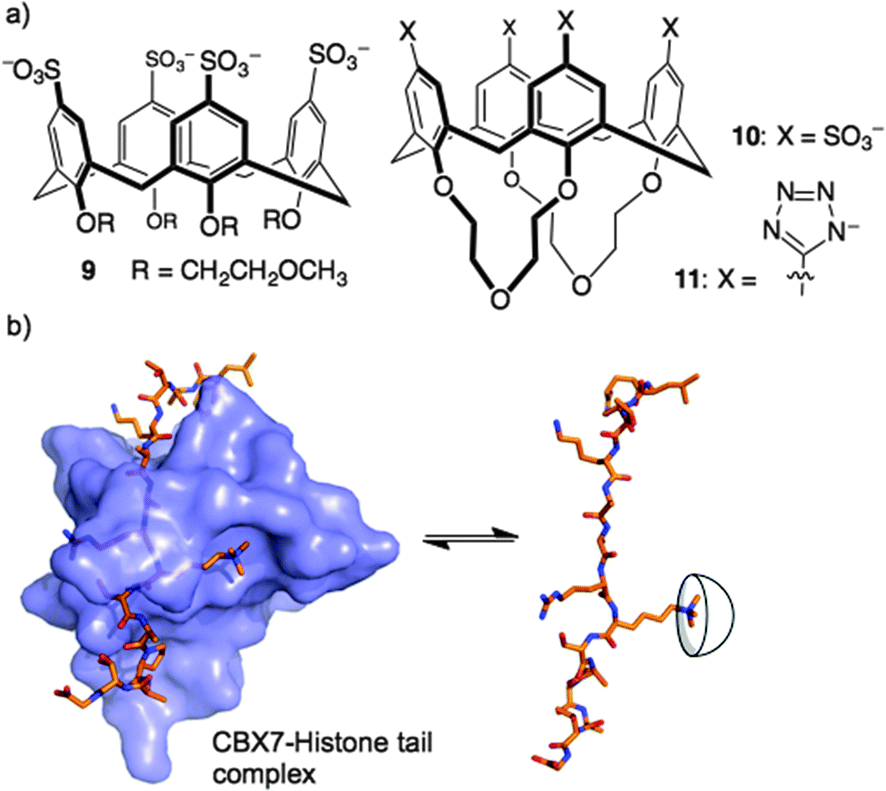 | ||
| Fig. 6 Trimethyllysine-targeting hosts used in experiments to show that a supramolecular host can out-compete a complex between a reader protein (CBX7) and its partner histone 3, lysine 27 trimethylated (H3K27me3). (Host 9 is inactive due to its conformational flexibility.) | ||
Histone tails are inherently unstructured. The peptides used for the above binding studies, as well as the trimethyllysine-containing tract of the FRET biosensor, are based on histone tail sequences and therefore all lack local structure around the methylated residue. We were interested in studying compound 8 in the context of methyllysine side chains presented on a well-folded protein. Our choice of model system was lysozyme, which we chemically dimethylated on all lysine side chains to present a set of Kme2 side chains projected from the folded protein in different local environments. (We had previously shown that Kme2, while weaker than Kme3, is also a good guest for 8.32) During a visit to Galway, we learned that the group of Peter Crowley was working on the same model construct at the same time—an unexpected meeting of the minds. Together with Irina Paci, we found through computation, NMR, and crystallography that 8 chooses a single Kme2 residue (K116me2) on the surface of the globally dimethylated lysozyme construct (Fig. 7).43 Selectivity in the context of a folded protein arises from varying degrees of side chain accessibility, as well as different peripheral contacts between sulfonates and neighboring residues. This is different than the relatively low level of selectivity displayed by 8 between Kme3 residues presented in different unstructured peptide sequences.39,44 The complexes in all cases are selective for methylated residues, and are mainly driven by the binding of methylammonium ions within the pocket of 8. But the strength and influence of secondary interactions that govern guest selectivity depends strongly on the degree of structure within the guest itself. These varying kinds of selectivity will return as a useful tool later in this article.
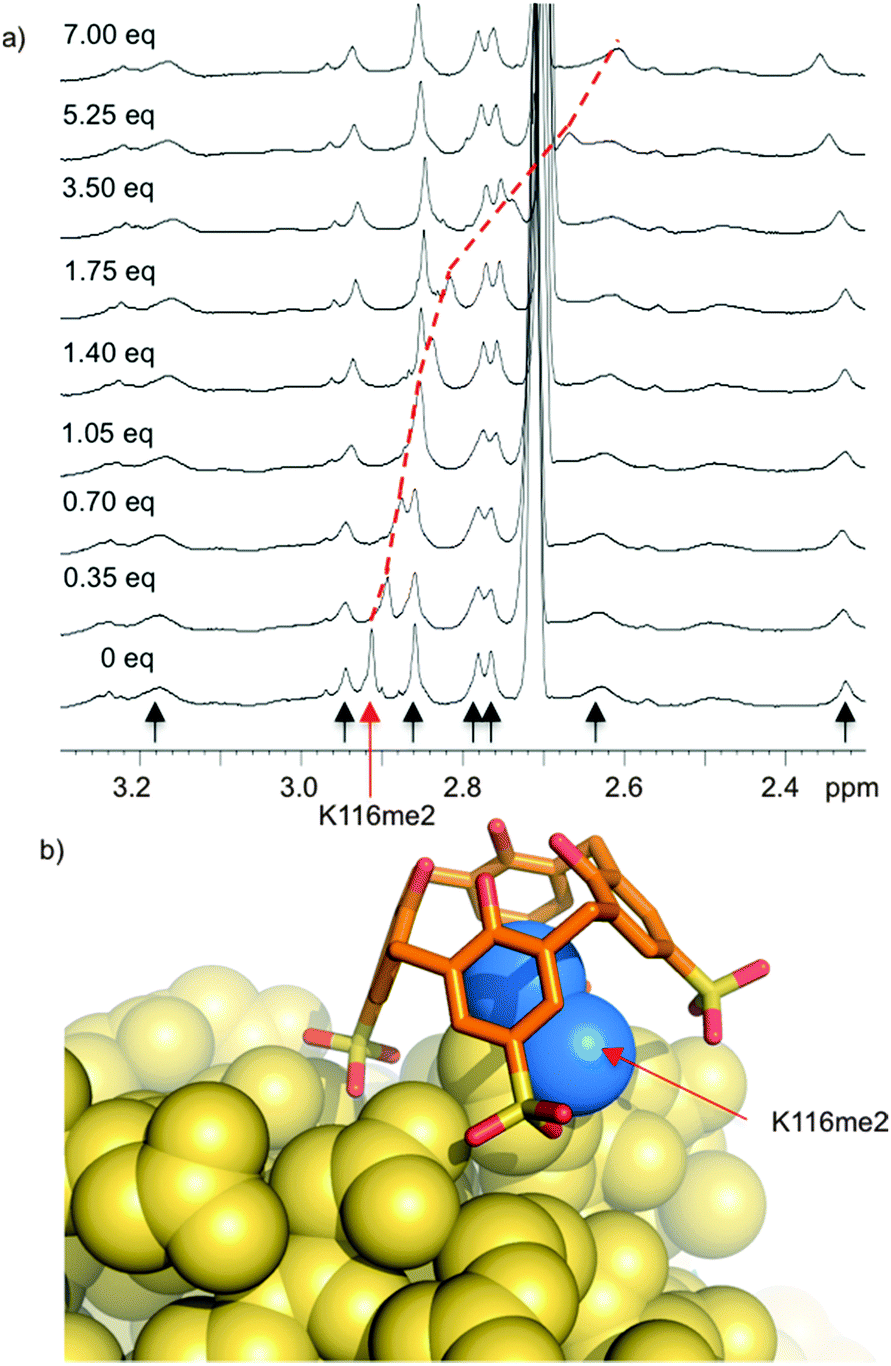 | ||
| Fig. 7 Compound 8 binds selectively to one Kme2 residue on the surface of a folded protein. (a) 1H NMR spectrum of Kme2-lysozyme. Each methyl resonance is indicated with an arrow, and position was confirmed by 1H–13C HSQC spectra on 13CH3-labeled protein. Only K116me2 (red arrow) is bound upon addition of 8. (b) X-ray structure (pdb 4PRU) shows 8 exclusively bound to K116me2. The methyl groups (blue) are bound within the pocket of 8. Additional contacts with neighboring residues define the specificity of the binding site. | ||
Expanding calixarene capabilities through synthesis of analogs
“I know the pieces fit” – ToolMany concave host systems have been developed to target biological binding partners: macrocyclic cucurbiturils, cyclodextrins, and calixarenes, as well as various C-shaped tweezers and clips, all have inherent advantages and disadvantages. While calixarenes are almost always lower-affinity reagents than, say, cucurbiturils, they are easier to modify than many other compound classes. And synthesis is one of the central powers that sets chemistry apart from purely observational sciences. Many students who choose chemistry as a career do so because they enjoy creation of new matter (tinkering) more than they enjoy the pure observation of nature (birdwatching).
Calixarenes are fully addressable by installation of different groups at lower- and upper-rim positions. In host–guest applications where the concave aromatic pocket is the primary binding surface, lower-rim modifications have strong effects through changing the conformation of the binding pocket. Upper-rim modifications directly change the shape and functionality of the binding surface—a feat that is not easily achieved for hosts like cucurbiturils, where all bond vectors are directed out and away from the guest. The chemistry used to modify calixarene upper-rim positions with high degrees of selectivity often uses elegant reactions that use the proximity of adjacent rings in 3-dimensional space to provide specificity that is impossible on a typical, flat organic molecule.45–48
Upper-rim modifications were key to tuning the affinity for trimethyllysine. Gutsche's method was used to differentiate a single upper-rim position, by high-yielding and selective installation of de-activating ester groups at three of the four lower-rim hydroxyls.49 Subsequent nitration50 or bromination51 by the methods of Nam and Harvey provides key, mono-functionalized intermediates 12 and 13 that we used to prepare a series of trisulfonate calix[4]arenes each presenting a different functional binding element on the fourth upper-rim position (Fig. 8).52
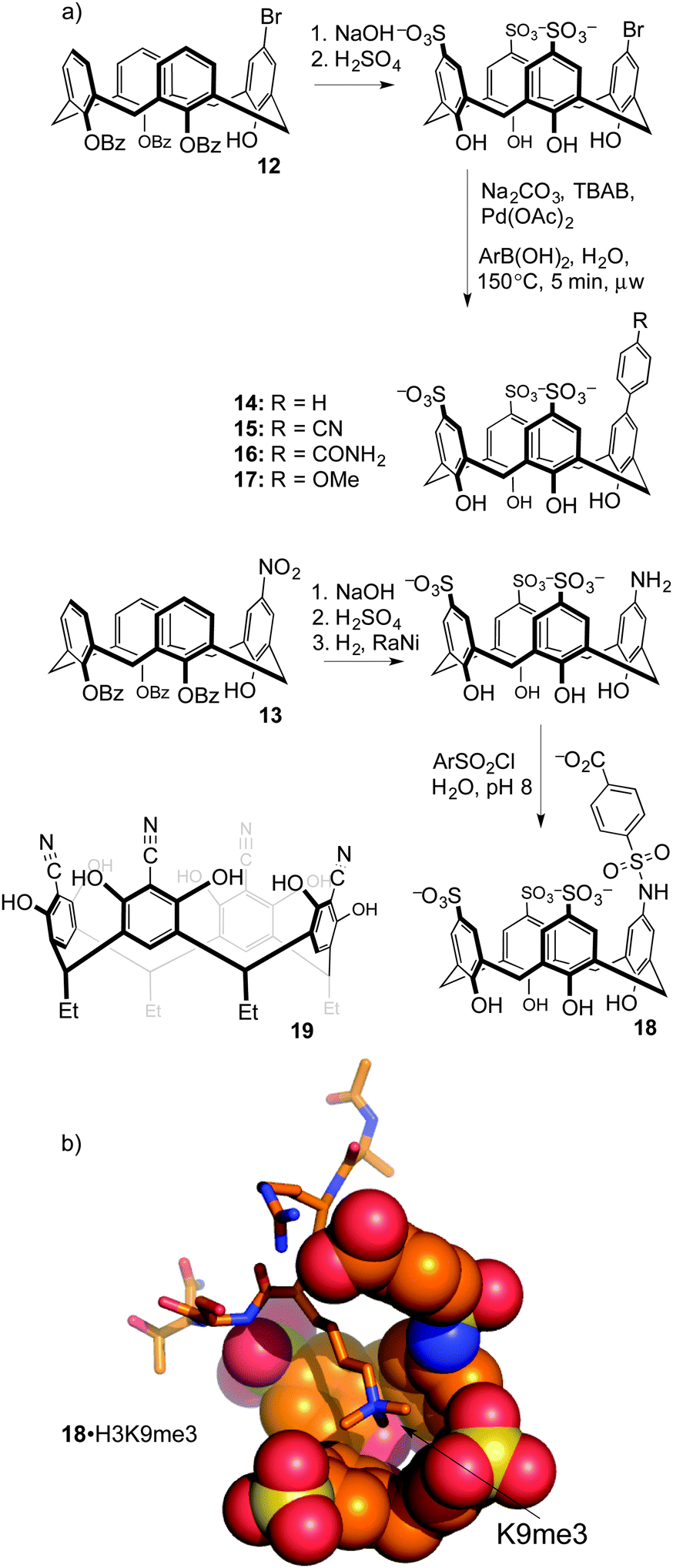 | ||
| Fig. 8 (a) Synthetic calixarene derivatives contain substitutions that tune their affinities for methylated targets. Functionalized resorcinarene derivative 19 is also shown. (b) A molecular model shows extended contacts with a trimethyllysine residue by host 18. | ||
This substitution pattern provided a small family of hosts with potencies for binding trimethyllysine that varied in unexpected ways (Table 3). The cation–pi interaction is important for contacts with N-CH3 and ε-CH2 groups in trimethyllysine,54,55 so we expected that hosts with electron-donating ring substituents would form stronger complexes with Kme3. This prediction was completely wrong. The addition of a phenyl ring (14) increased affinity for Kme3, as expected. The addition of polar, electron-donating groups (MeO, 17: tested on a Kme3 peptide), or polar, electron-withdrawing groups (e.g. CN, 15), each caused decreases in affinity, suggesting that hydrophobicity and not electrostatics dominates the influence of the appended binding element. This trend was confirmed by adding other polar groups that are strongly hydrated, but do not strongly influence ring electronics (e.g. CONH2, 16); they also decreased binding to Kme3 by >10-fold. In our experience, this sort of result is common when studying host–guest systems that operate in water. The influence of hydration, and especially how it plays with other classes of weak interaction, remains one of the hardest things to predict in the world of molecular recognition.
The list of mono-substituted hosts was further expanded, and they were next challenged with a variety of protein–peptide complexes. As with the parent compound, the higher affinity monosubstituted hosts are able to disrupt the CBX7–H3K27me3 interaction.44 Again, these agents operated by out-competing the methyl reader protein off of its methyllysine-containing binding partner.
When the agents were used to target the complexes of chromodomain helicase DNA-binding protein 4 (CHD4), a unique kind of selectivity was observed. The reader domain of CHD4 can form complexes with histone 3 in the region around lysine 9, whether the lysine is trimethylated or not (Fig. 9). Each complex has a distinct role in the cell. The complex of CHD4 with the trimethyllysine-containing partner, (H3K9me3, Kd = 0.9 μM) is stronger than that with the unmethylated partner (H3K9, Kd = 19 μM). A full protein NMR characterization, backed up with pulldown experiments, showed that monofunctionalized calixarene 18 was able to disrupt the (stronger) complex of CHD4–H3K9me3, while leaving the (weaker) complex of CHD4–H3K9 untouched.56 This is a different result than could be achieved with the traditional medicinal chemistry approach, which would target the concave binding pocket of CHD4 and would disrupt both complexes of CHD4 equally well. (In any case, small molecule inhibitors of CHD4 have never been reported.) This result highlights how supramolecular approaches can put unique tools in the hands of researchers.
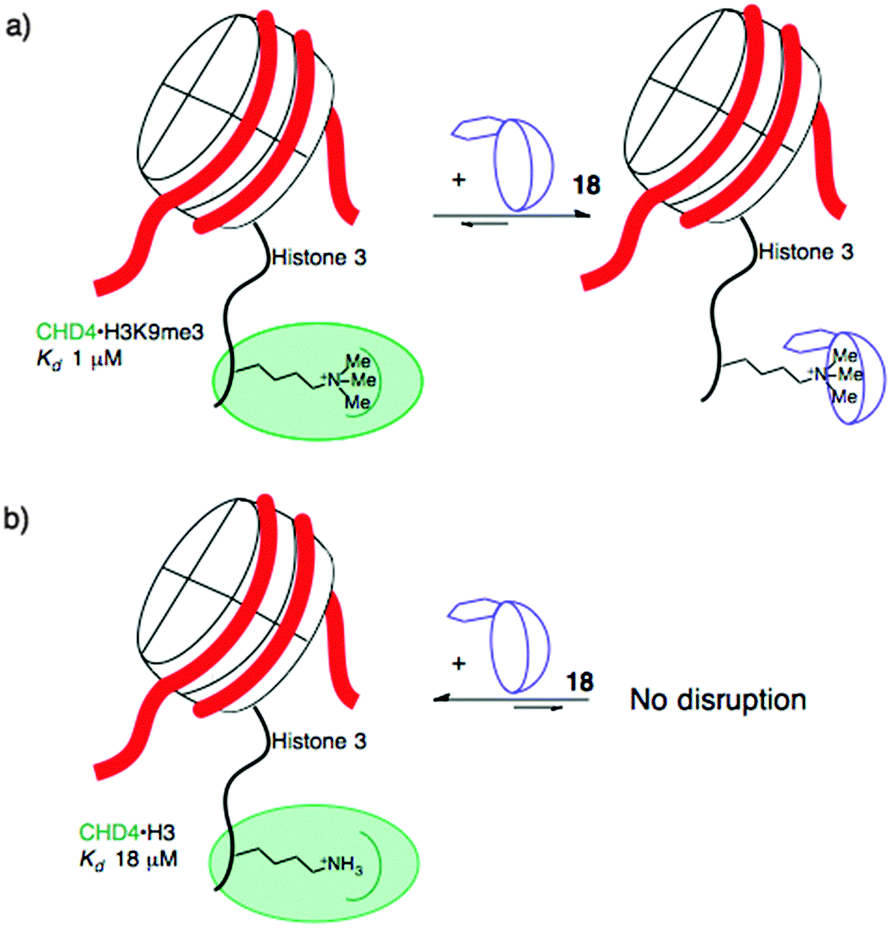 | ||
| Fig. 9 Selective disruption of the stronger of two related protein–protein complexes formed by CHD4 and histone tails. A supramolecular host targets the trimethyllysine residue in the stronger complex (a), and has no effect on the weaker complex (b). | ||
In more recent work, synthetic modification of resorcinarenes has also been used to target post-translational methylation. Hamilton's group prepared a substituted resorcinarene 19 (Fig. 8), in which the upper-rim cyano groups provide an extended pi surface for guest binding, while also inducing the deprotonation of multiple upper-rim resorcinol hydroxyl groups to create an anion binding pocket.53 Its association constants for binding to Kme3 (Kassoc = 4.7 × 104 M−1) and to the short peptide R-Kme3-S-T (Kassoc > 1.0 × 105 M−1) were stronger than those of 8. NMR studies show that it is also more site selective than 8, in that it does not produce chemical shifts in resonances arising from the cationic arginine side chain adjacent to the Kme3 site even after multiple equivalents are added. The authors also showed that the action of demethylase enzyme JMJD2A on its native substrate H3K9me3 was inhibited by the presence of 19 (IC50 = 64 μM) or 8 (IC50 = 10 μM). This amounts to enzyme inhibition through substrate binding—another relatively uncommon mode of action made possible by supramolecular approaches.
Discovery (not design) of desirable binding agents
“Reaching out to embrace the random, reaching out to embrace whatever may come” – ToolThe synthetic calixarene analogs discussed above were the products of rational design. It can be argued that it's a bad idea to target individual kinds of methylated protein residues using rational design. It is hard to create perfect designs that can deal with the subtle structural differences between peptides bearing zero, one, two, or three methyl groups. In addition, entropy remains a confounding factor in many host designs. Schmidtchen was discussing the complexities of solvent entropy in anion recognition when he asked provocatively “Is there a sign to resign from design?”57 His answer to his own rhetorical question was, “No”—but it remains true that the entropy of recognition processes is hard to predict.58 The entropy of binding for something as simple as 8 binding to the free amino acid Kme3 in a given concentration and pH of sodium phosphate buffer is beyond the predictive abilities of modern computational chemistry. These complexities provide strong motivation for approaches that at least partially give up the need to design and control each and every atom–atom contact in a host–guest complex.
The group of Waters used dynamic combinatorial chemistry to address the problem of methyllysine recognition. Their macrocyclic disulfide-containing hosts are built from dithiol reagents derived from those introduced by Sanders and Otto. The dynamic combinatorial approach allows each guest to select for its own host, by templating the formation of different reversibly formed hosts under equilibrium conditions (Fig. 10). Under the influence of a small dipeptide (Kme3-G), host 20 was amplified >10-fold from a mixture potentially containing dozens of different hosts. Host 20 binds to the peptide H3K9me3 with Kassoc = 4 × 104 M−1.59 The iterative redesign of this receptor library to include new building blocks subsequently led to host 21, also discovered by amplification during a dynamic combinatorial chemistry experiment. Host 21 has affinities for trimethyllysine peptides based on either the H3K9me3 sequence or on the H3K36me3 sequence of >3 × 106 M−1, and also has improved selectivity over unmethylated lysines relative to 20.60
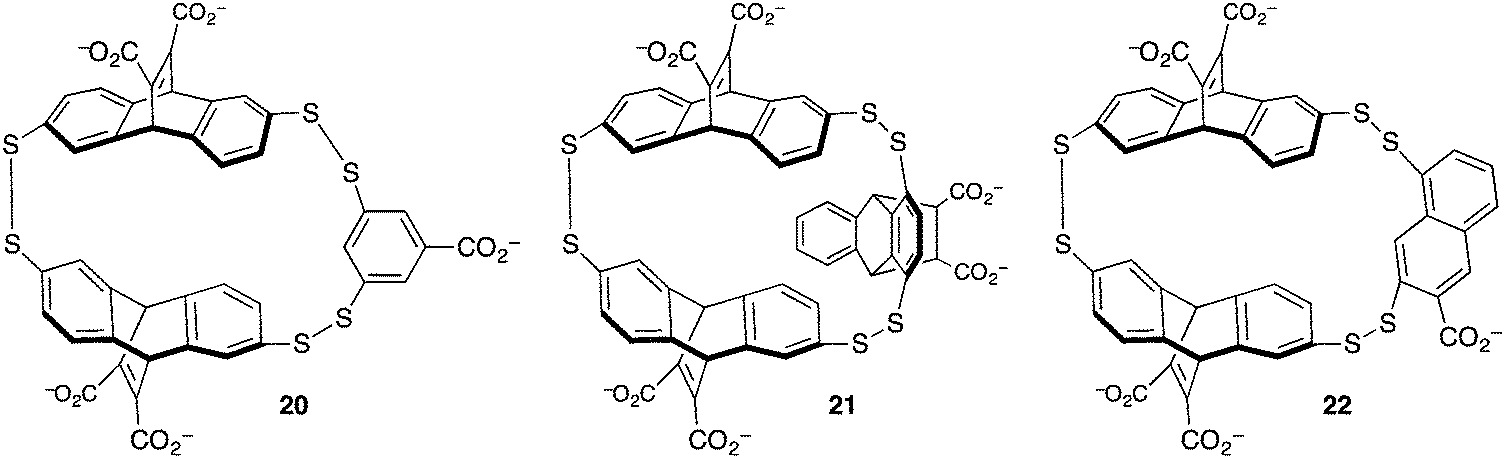 | ||
| Fig. 10 Hosts derived from dynamic combinatorial chemistry experiments to achieve trimethyllysine and dimethylarginine recognition. | ||
The challenge of methylarginine recognition was also addressed using this dynamic, responsive system. Host 22 was amplified 5-fold from a dynamic library upon addition of an asymmetric-dimethylarginine peptide.61 No amplification occurred in the presence of an equivalent symmetric-dimethylarginine peptide. A variety of aDMA containing peptides were bound by 22 with Kassoc values from 1.5 × 105 to 1.1 × 106 M−1, and selectivities over sDMA peptides of between 2.5- and 7.5-fold.
A comparison of the three similar-looking hosts in Fig. 10 shows highlights how their differing guest affinities and selectivities could not have been predicted by a priori design. These results also showcase one of the main powers of dynamic combinatorial chemistry—that a single equilibrating dynamic library can be used to identify more than one different useful species, when it changes differently under the templating influence of different guests. This work also shows how small changes in host structure can profoundly affect guest recognition in ways that are beyond the abilities of rigorous host designers to predict.62,63
To be fair, these examples from the Waters group are not really ‘random’ discovery… there is much thoughtful design behind each dynamic combinatorial chemistry experiment. One example of completely accidental piece of host–guest chemistry did arise from work in our lab. The aforementioned hosts 14–17 were prepared in order to tune selectivity for Kme3. One analog, host 14, showed unexpected upfield shifts in NMR spectra taken in water (but not in polar organic solvents), indicative of some sort of aggregation that depended on the phenyl group being bound as a guest within a calixarene cavity. A small series of para-alkyl analogs 23–27 (Fig. 11) were prepared, and each showed the same trend of concentration-dependent chemical shifts suggesting similarly ordered assemblies.64 The structure of the aggregate is a self-assembled homodimer (Fig. 11). This structure was indicated by X-ray crystallography of 26, and 1D-NMR data, NOESY data, and DOSY experiments confirmed the persistence of the same dimer in phosphate-buffered water for all of 14, 23–27.
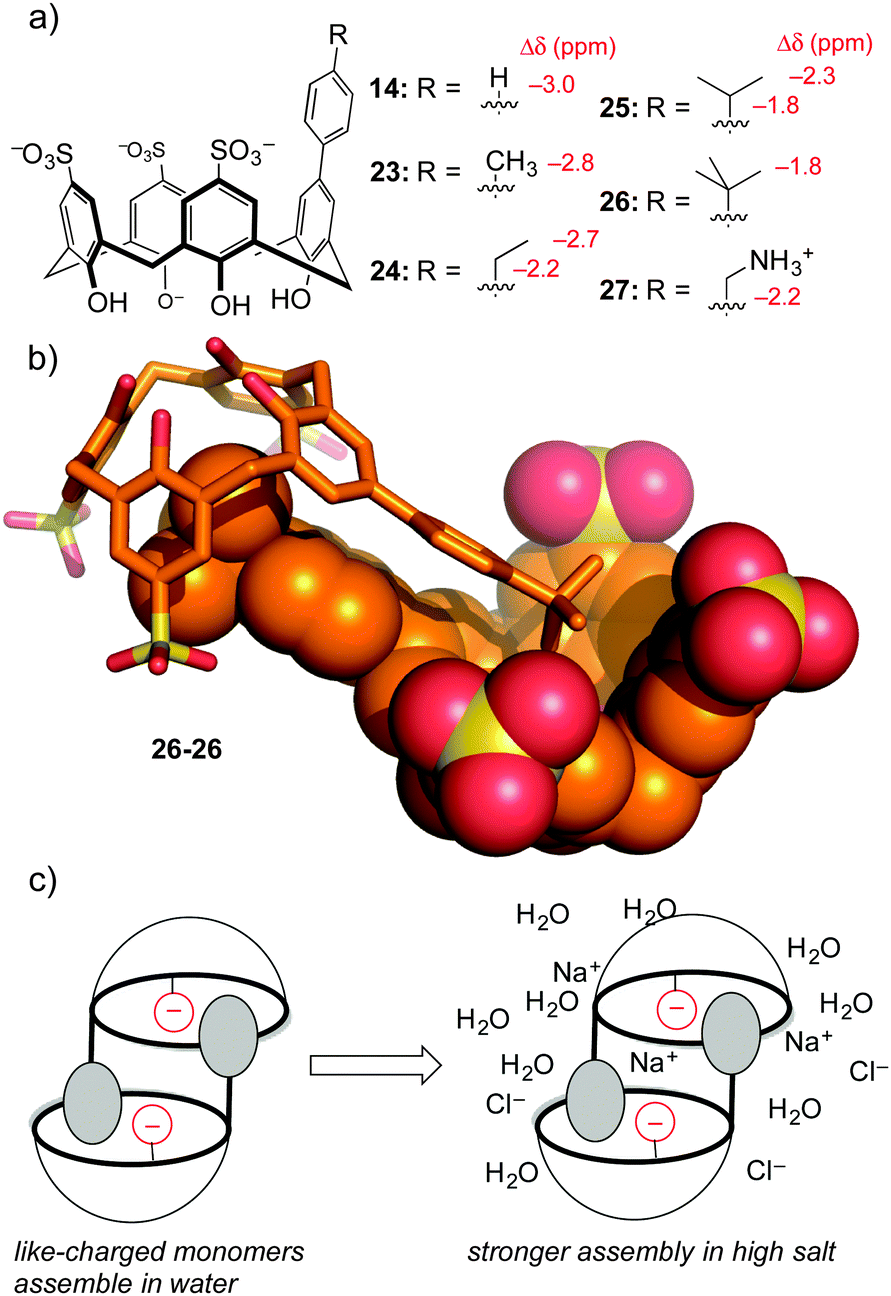 | ||
| Fig. 11 Self-assembling molecules developed after accidental discover of dimers formed by 14. (a) Chemical structures of the monomers, along with the characteristic upfield chemical shifts that arise upon dimerization in water. (b) X-ray structure of the homodimer of 26—this mode of assembly exists also in solution for all of 14, 23–27. (c) A model showing how monomers like 14, 23–26 that involve like-charged monomers get stronger under the influence of high salt. | ||
This lesson was at first only a curiosity in molecular recognition—we have little interest in self-assembling dimers, and we didn't set out to make them. But upon further study these compounds defied a trend with which many supramolecular chemists are familiar. Many biologically inspired recognition modules (including others that we've made) tend to operate in organic–aqueous solvent mixtures, before falling apart in pure water. Some that operate in pure water fall apart upon addition of biologically relevant levels of salt. Unlike all of our earlier efforts at programming recognition in polar media, we found by isothermal titration calorimetry that these dimers become stronger in high salt conditions. We could rationalize this result as being an outcome of using hydrophobic attraction tempered by mutual electrostatic repulsion—upon addition of salt, the reduced mutual repulsion between the highly charged monomers like tetra-anionic 26 leads to an overall stronger binding event.
The thermodynamics of these dimeric assemblies are intriguing (Table 4). The dimerization in 100 mM phosphate buffer is driven strongly by enthalpy, with unfavorable entropy (e.g. for 26 ΔH = −11.0 kcal mol−1, and TΔS = −6.9 kcal mol−1). Each dimerizing molecule responds to added salt by forming a stronger overall dimer, while doing something that looks like an unusual form of enthalpy–entropy compensation: the enthalpy gets less favourable, while the entropy gets more favorable by a greater extent (e.g. for 26 ΔH = −10.3 kcal mol−1, and TΔS = −5.9 kcal mol−1). Compound 27, which carries a cationic group and therefore suffers from less like-charge repulsion between monomers, behaves more like a normal system, and gets weaker upon addition of salt.
| K d (mM) | ΔG (kcal mol−1) | ΔH (kcal mol−1) | (−)TΔS (kcal mol−1) | |||||
|---|---|---|---|---|---|---|---|---|
| PHOS | PBS | PHOS | PBS | PHOS | PBS | PHOS | PBS | |
| a Values reported are the average of triplicate ITC dilution titrations. PHOS is 100 mM NaH2PO4 phosphate buffer at pH 7.4, and PBS is the same phosphate buffer containing NaCl, KCl, and MgCl2 at the levels found in human serum. All standard deviations for Kd and ΔH were ≤10% of the reported values. | ||||||||
| 14 | 8.1 | 4.2 | −2.9 | −3.3 | −14.4 | −14.4 | 11.5 | 11.1 |
| 23 | 7.0 | 4.5 | −3.0 | −3.3 | −10.8 | −10.6 | 7.8 | 7.4 |
| 24 | 4.7 | 3.3 | −3.2 | −3.5 | −12.7 | −10.3 | 9.5 | 6.9 |
| 25 | 1.5 | 1.1 | −3.6 | −4.1 | −12.8 | −12.0 | 6.2 | 7.9 |
| 26 | 1.0 | 0.73 | −4.2 | −4.4 | −11.0 | −10.3 | 6.9 | 5.9 |
| 27 | 1.1 | 1.4 | −4.1 | −4.0 | −8.4 | −8.9 | 4.3 | 4.9 |
The lessons learned with this accidental set of dimers seem to be general. A few other like-charged host–guest systems that operate in salty water were found in the literature, and some of them showed the ability to survive and even get stronger in high salt and/or physiological buffers.64–66 As we pushed the limits of our system, we found that the calixarene-based dimers were able to remain faithfully assembled even in real biological fluids like undiluted, untreated urine. This discovery is among our newest—we hope in the future to develop systems that use the key features of these accidental dimers in hosts and sensors for diverse biological applications.
Applied bioscience using calixarenes
“What have you done for me lately?” – Janet JacksonThere are also, increasingly, efforts to find applications for supramolecular hosts in the life sciences. These all require the ability to target complex biological molecules in physiologically relevant media—a feat that sulfonatocalixarenes achieve routinely. Applications and research in the areas of chemical biology and biochemistry each have different demands, and each can offer distinct new lessons.
Cell-based studies
“Got to swim in the sea of life… to be tested in the sea of life” – Bran Van 3000The discussion up to now have focused on agents selective for trimethyllysines over other post-translational methylation states… but what about targeting certain trimethyllysine sites over others? We were interested in identifying agents with selectivity for H3K4me3 over H3K9me3. These two methylation marks are located close to each other on the tail of histone 3, but produce opposite biological functions. H3K4me3 is associated with transcriptional activation, and recruits methyl reader proteins to chromatin in order to carry out this program. H3K9me3 drives transcriptional repression, and does so by recruiting a distinct set of silencing reader proteins.
We identified a small set of calixarenes that could bind an H3K4me3 peptide with small preference over an H3K9me3 peptide. We first discovered good candidates from a small screen using a fluorescence indicator displacement assay.67 Our collaborators in the group of Tatiana Kutateladze used fluorescence polarization, protein NMR, and pulldown experiments to show that these agents were able to disrupt the complex of H3K4me3 with multiple of its native reader proteins, including the PHD domain of the protein Mixed Lineage Leukemia-5 (MLL5).67
Sulfonated calixarenes look like terrible candidates for cell-based studies. They violate most of Lipinski's rules.68 They are highly charged at neutral pH. In fact, they fail on almost every structural predictor of drug-likeness except in having a low number of rotatable bonds.69 And yet some of these compounds defy low expectations—there are a handful of reports of calixarenes and resorcinarenes being able to enter and engage their targets inside of cells and/or liposomes.29,56,70–74
Our collaborators in the Strahl lab used a proximity ligation assay that probes the co-localization of H3K4me3 and MLL5 in C2C12 cells (Fig. 12).67 Surprisingly, the most effective compound was the unsubstituted parent—8. This compound is ∼8-fold selective for H3K4me3 over H3K9me3 in the peptide binding assay. The true specificity of 8 for H3K4me3 within cells can't be determined—there are hundreds of known trimethylation sites in the mouse proteome, and many of them must be bound by 8 within the living cell. The studies reported in this work prove that 8 can enter cells, gain access to the nucleus, and disrupt a protein recognition event that is specifically dependent on lysine trimethylation.
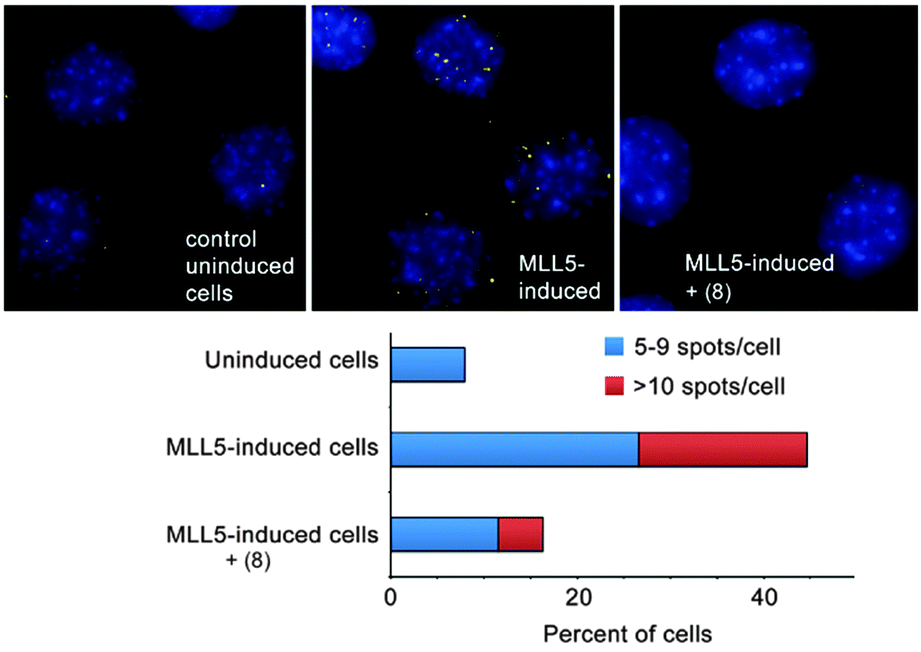 | ||
| Fig. 12 Compound 8 inhibits the interaction of H3K4me3 and MLL5, as measured by a DuoLink proximity ligation assay in C2C12 mouse myoblasts with inducible MLL5 expression. These images were originally published in J. Biol. Chem. M. Ali, K. D. Daze, D. E. Strongin, S. B. Rothbart, H. Rincon-Arano, H. F. Allen, J. Li, B. D. Strahl, F. Hof and T. G. Kutateladze Molecular insight into inhibition of the methylated histone-plant homeodomain complexes by calixarenes. J. Biol. Chem., 2015, 290, 22919–22930. © the American Society for Biochemistry and Molecular Biology. | ||
The list of examples of host–guest chemistry working inside of living cells grows longer each year, but a comprehensive review is beyond the scope of this Feature. There are, however, some examples from other labs involving 8 that provide useful context for our own work. Host 8 was reported to do indicator displacement assays inside of lipid membrane constructs,71 and also inside of living cells.70 This work involved initial treatment with host 8 (≥250 μM) and the fluorescent dye lucigenin (50 μM). (This reporter pair was first reported in other work by Nau in 2011.75) Subsequent treatment with different cell-penetrant molecules produced increases in intracellular fluorescence only when the added molecules were good guests for 8. Again, the key finding is not one of perfect specificity… but that the fidelity of the host–dye complex is sufficient for it to enter cells, persist within cells, and ultimately that host–guest chemistry can occur and be detected within the living cells. An additional finding of interest in this work is that the presence of 8 improves the uptake of lucigenin into cells in the first place.70 This result is reminiscent of the use of 8 to drive dye uptake and transport across a layer of epithelial cells,76 and also of the use of 8 to rescue experimental animals from poisoning with methyl viologen.77,78 The collective lesson of these and other studies with diverse supramolecular hosts is that low perceived drug-likeness is a poor reason not to try innovative host–guest chemistry in living systems.
Biochemical assays
“Send out the signals deep and loud” – Peter GabrielOne of the central analytical challenges facing those who work on post-translational modifications is simply telling one PTM state apart from another. These challenges can take many forms: discriminating different methylation states (e.g. mono-, di-, and trimethyllysine) in a common peptide sequence, discriminating similar methylation states at different sites (e.g. H3K4me3 from H3K9me3), and even discriminating among analytes bearing multiple nearby modifications. These challenges have almost exclusively been tackled using antibodies and related, engineered proteins, that have been selected for their high specificity for a given modification.
We took up this challenge using a chemical sensor array driven by host–guest chemistry. This chemical sensor array approach does not require specific lock-and-key molecular recognition. Instead, it uses a set of relatively promiscuous sensor elements that each must only provide a different pattern of responses to a given analyte. This makes the whole sensor array approach very well suited to being deployed in host–guest settings. Different aspects of this approach have been thoroughly reviewed.79,80
Based on the above results, sulfonato calixarenes were an obvious set of receptors to use, when trying to discriminate post-translationally methylated analytes. A set of promiscuous sensors is required for pattern-based sensing. We used indicator displacement assays based on Shinkai's work with 2881 and Nau's work with lucigenin75 (previously discussed). Each dye was partnered with a small set of hosts that were expected to be more-or-less promiscuous for this set of targets: 8, the desymmetrized synthetic analog 29, and p-sulfonatocalix[6]arene 30 (Fig. 13).82
 | ||
| Fig. 13 (a) Schematic representation of a chemical sensor array based on indicator displacement. (b) Sensor array components: calixarenes 8, 29, and 30 were combined with either lucigenin or dye 28 to create three-element sensor arrays for analysis of post-translationally modified amino acids and peptides. | ||
This simple three-element sensor array proved to be capable of multiple kinds of discrimination.82 In each case, the sensor array was trained on a set of known post-translationally modified peptide analytes. The intensity of fluorescence generated upon release of lucigenin from each host was quantified, and the three-value pattern was processed by known chemometric analyses in order to generate a reduced scatterplot of values from replicates and unknown samples. The sensor array was able to discriminate with high confidence between multiple different post-translational modifications (Fig. 14a). Asymmetric and symmetric isomers of dimethylarginine could be differentiated when present in the same peptide sequence without needing a highly specific host for either analyte in the sensor array (Fig. 14b). In separate experiments, the sensor array was able to discriminate between the same modification (trimethyllysine) on different peptide sequences—including closely related peptides like H3K9me3 and H3K27me3 that are sometimes confused by antibodies.
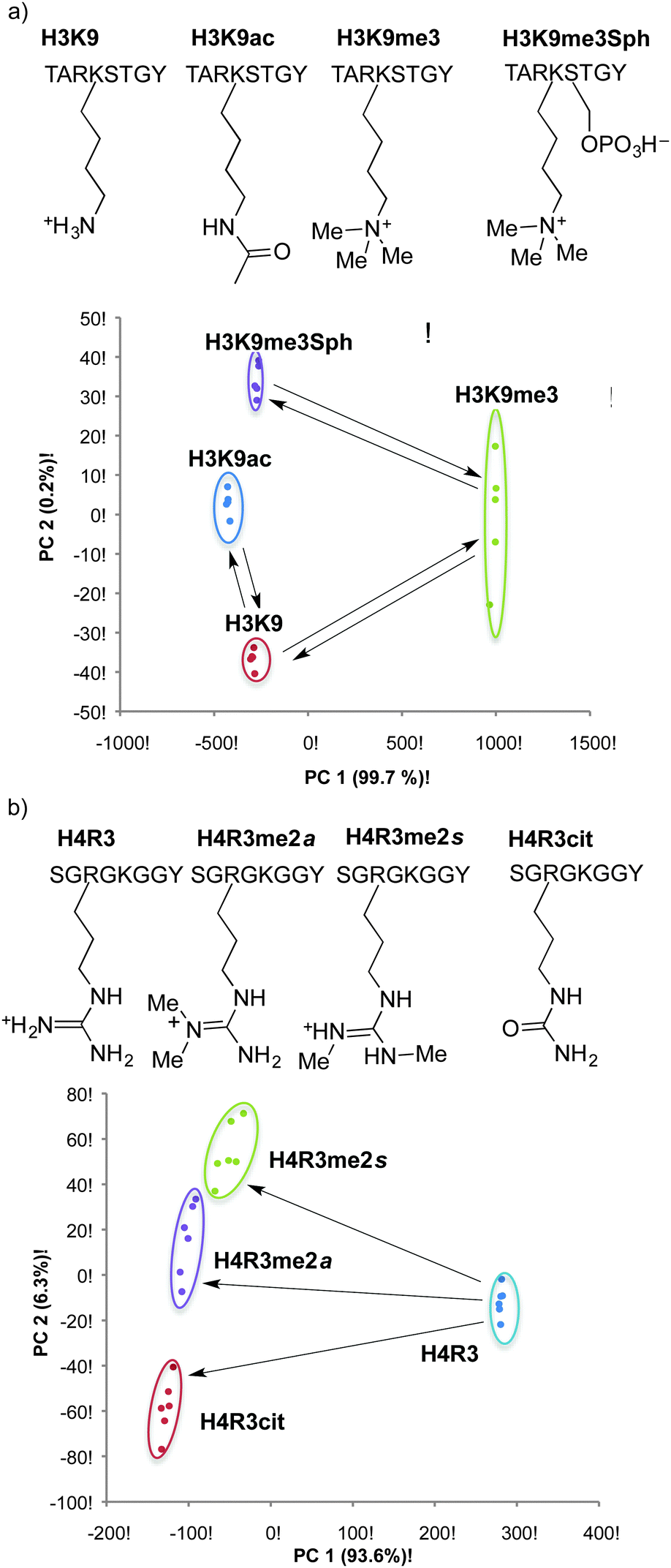 | ||
| Fig. 14 Examples in which post-translationally modified peptides are identified by a simple, three-calixarene sensor array after principle component analysis. Arrows indicate peptides that are related to each other as the starting materials and products of known enzymatic transformations. | ||
The real-time analysis of methyl transferase and demethylase enzyme kinetics presents a surprisingly hard problem in bioanalytical chemistry. Many assays for these enzymes exist. But the vast majority of them rely on developing signal using antibody- or radioisotope-driven detection schemes, and therefore can't report on reaction progress in real time.
We showed using our sensor array that a mocked up set of unmethylated and methylated analytes that represented the substrate and product of various enzymes. The idea and the power of this approach was to monitor either methyl transferase or demethylase reactions, while being able to discriminate in each case between multiple different products that could be formed by a given enzyme. Jeltsch and Nau showed that 8-lucigenin could report on methylation of H3K9 by the methyl transferase, Dim5, in real time.83 Our sensor array would potentially provide richer information, in that conversion to either H3K4me3 or H3K9me3 (or, for that matter, partial methylation to the mono- or dimethyllysine products) would generate distinct outputs. In proof-of-concept work, the array was able to analyze reproducibly the ‘reaction progress’ toward each possible product in mock samples.82 We anticipate that the combination of Nau's real-time analysis and our sensor array approach will create a continuous assay that is more information-rich than any currently available type of assay in the field of post-translational modification enzymes.
Selective, specific, pan-specific: a promising area of applications for host–guest chemistry
“Style's kinda different, but let's be specific” – Funkmaster FlexTrue specificity is hard to achieve using supramolecular host–guest systems. One of my professors said to me during a 4th year BSc oral exam that, “Supramolecular chemists always want to make something as specific as an enzyme… but by the time you've added all of the complex functionality to the host that is needed to get there… you might as well have made an enzyme instead.” This is one perspective on the fact that supramolecular hosts, by nature, have smaller interaction surface areas than do their naturally evolved protein counterparts. 20 years after this comment was made, we must admit that although we have in-depth knowledge of every kind of weak interaction, we remain unable to create by de novo design a supramolecular host that is perfectly selective for a single biological target.
While it is true that perfect specificity is hard, one can conceive of many forms of selectivity that are possible. Several examples are on display in the results discussed above. Other schemes like the chemical sensor arrays work by intentionally giving up on achieving perfect specificity, and instead taking advantage of selectivity patterns in order to do useful analysis.
The field of PTMs brings another kind of specificity to the discussion. “Pan-specific” reagents are those that can bind a given kind of modification (and not other modification types), regardless of its surrounding peptide sequence (Fig. 15a). Such reagents are critical for prospecting of post-translationally modified analytes, where all analytes marked with a given modification are the targets for binding and enrichment. Pan-specific reagents for phosphorylated peptides are the archetypal examples.81 In a typical protocol, biological samples are proteolyzed, and the resulting mix of thousands of peptides is run over a TiO2 or immobilized metal affinity chromatography (IMAC, Fig. 15b) column, which trap all phosphopeptides through strong phosphate–metal coordination. Subsequent analysis of the enriched samples allows observation of phosphopeptides that would not otherwise be detectable. Such pan-specific reagents are central to phosphoproteomics, and are used every day in hundreds of labs. Pan-specific reagents for other PTMs like citrullination have also been developed.20,21
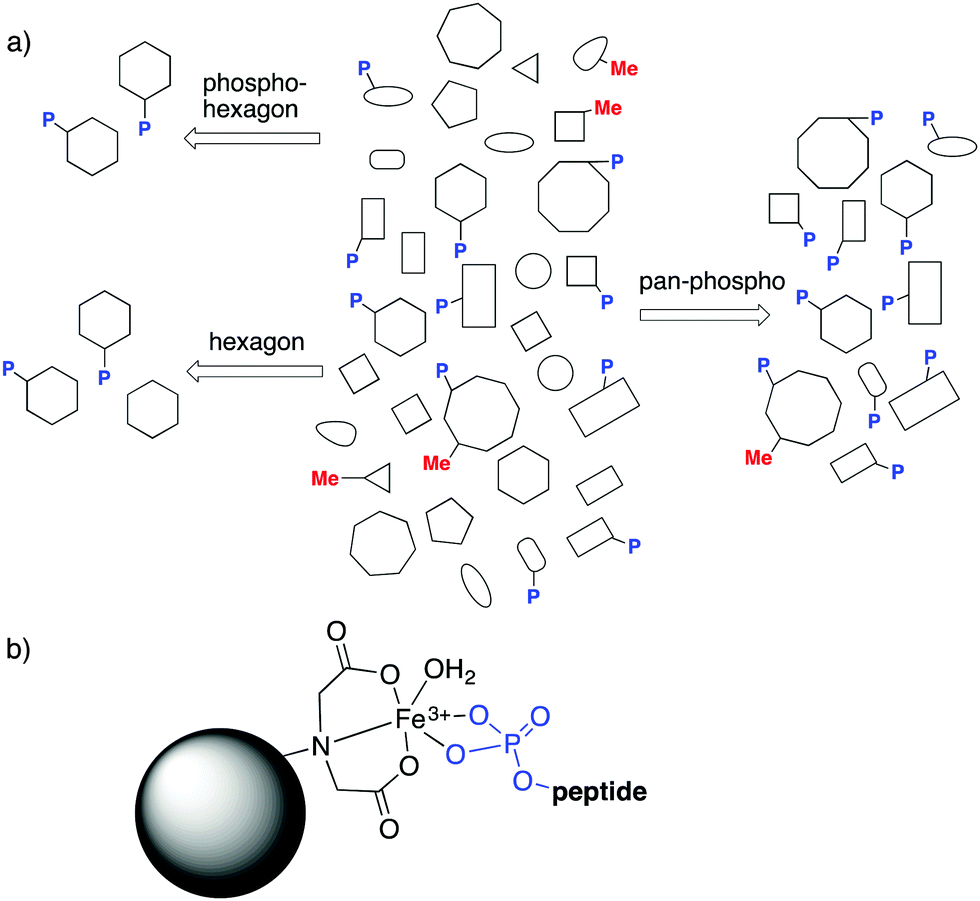 | ||
| Fig. 15 (a) The kinds of specificity, selectivity, and pan-specificity that might apply in different proteomics prospecting experiments. (b) Immobilized metal affinity chromatography (IMAC) is an example of a pan-specific method for enriching all phosphorylated peptides. | ||
The physicochemical properties of methyllysines, described earlier, make them especially challenging targets for selective enrichment. The pan-specific reagents that have been developed for methyllysines are exclusively antibody- or protein-based. Most are polyclonal antibodies that suffer from poor performance and significant batch variability bad enough that it has spawned a series of scholarly articles that examine just how bad the performance of PTM antibodies can be.4,6,84,85 Making and testing more antibodies will probably overcome these limitations, but it remains true that antibody- and protein-based binding interfaces are intrinsically not well suited to the problem of pan-specific PTM enrichment. They have large contact surface areas, and only a small portion of the binding surfaces can contact the key methyl groups that define the closely related analytes from each other.
When the specific application requires pan-specificity, we propose that supramolecular hosts have inherent advantages over biomolecular affinity agents. Their small binding surface are well suited to binding individual functional groups while ignoring surrounding structures on the targeted analyte. In addition, they can be synthesized and purified to homogeneity, avoiding the liabilities associated with batch reproducibility in bio-reagents.
We proved this concept using a calixarene-based affinity reagents for trimethyllysine.86 The PTM-binding abilities of a small set of hosts were determined in solution-based fluorescence indicator displacement experiments. An agent (18) with good affinity for multiple Kme3-containing peptides was chosen, and coupled to an agarose solid support to create 18-aga (Fig. 16a). This solid phase showed small selectivity for Kme3-containing peptides in pulldown experiments, which was amplified into excellent performance when the same solid phase was packed into a column and used for affinity chromatography.
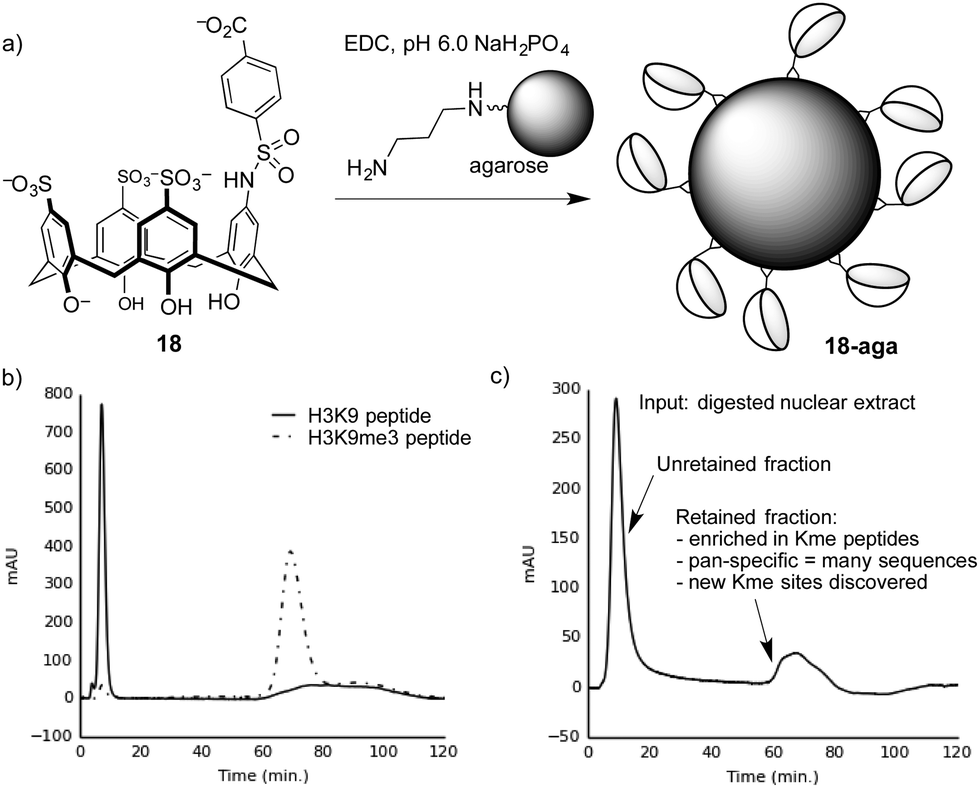 | ||
| Fig. 16 (a) Preparation of a trimethyllysine-binding stationary phase. (b) A chromatogram showing selective retention by stationary phase 18-aga of a trimethyllysine-containing peptide over an unmethylated analog. (c) Chromatogram resulting from separation of a proteolyzed extract containing dozens of nuclear proteins on 18-aga. The results from proteomics analysis of the retained material86 are summarized. | ||
Columns packed with 18-aga retain Kme3-containing peptides much more strongly than identical peptides with an unmodified lysine residue (Fig. 16b). It operates via a strong cation-exchange mechanism (thanks to its multiple sulfonates), and requires very strong salt conditions (gradients running up to >2 M NH4Cl) in order to achieve elution. We showed separately that a traditional sulfonate-based strong cation-exchange column can't achieve any separation of methylated and unmethylated peptides, because they each bear the same net charge. We conclude that the column 18-aga retains analytes by a mixture of ion exchange and host–guest mechanisms, which are broken up as salt concentrations increase.
The pan-specificity of 18-aga makes it useful for PTM enrichment and prospecting. A nuclear extract of calf thymus, being a heterogeneous mixture containing methylated proteins, was proteolyzed and the resulting digest was separated on a column of 18-aga (Fig. 16c).86 The retained material was collected and analyzed by a normal proteomics LC-MS/MS analysis. The enriched material had higher concentrations of methyllysine-containing peptides than untreated controls, and also than a set of samples that we enriched with a commercial methyllysine antibody. This enrichment made some known, low-abundance methylation marks detectable. It also revealed some trimethyllysine sites on nuclear proteins that had never been identified in any data set, whether collected with or without antibody enrichment. This first-of-a-kind demonstration of chemical enrichment for methyllysines bodes well for the use of diverse host–guest chemistry approaches in biological prospecting.
Conclusions and outlook
“The chemistry means everything to me, (the) long progression's why” – Red Hot Chili PeppersWe all first learn about noncovalent interactions one at a time, but in the long progression of experience we learn that they never operate in isolation. This has important effects on how we practice supramolecular chemistry. Reductionist labels for weak interactions are needed to discuss them in a simple way. In practice, the combination of those interactions can sometimes provide results that are programmable and understandable on an atom-by-atom basis. But properties often emerge that can't be accounted for by simple addition of non-covalent contacts. The success of reductionist thinking, and atom-by-atom designs, seems to be particularly poor for host–guest systems in salty aqueous solutions, where entropy effects become relatively large and unpredictable. Reductionist labels and atom-by-atom designs are unable to provide answers to our most interesting problems in biomolecular recognition.
The supramolecular hosts described in this article are inspired by nature, and aimed at natural targets. But a relatively small, rigid, and simple macrocyclic host will never encode the subtle combinations of structure, dynamics, and solvation that proteins have evolved naturally. Because of this, the pursuit of antibody-like specificity using a synthetic host molecule is an uphill battle. Better, then, to take aim at targets and applications that don't require ultimate specificity, and that are better suited to the inherent properties of supramolecular hosts. One such example is found in the creation of pan-specific reagents, but other applications also exist that rely on selectivity, and not specificity, in a way that plays to the natural strengths of supramolecular hosts.
It is not necessary to give up on fundamental discoveries while doing applied science. Biological targets set up many difficult challenges. Some of those challenges arise from their complex structures. Others arise simply from the wide array of salts and co-solutes that exist in biological solutions. All of those challenges make this a very rewarding area for basic discovery.
It is important to consider the field of bio-supramolecular chemistry within the broader context of emerging automation, increasing computational power, big data, artificial intelligence, and the promise of all of these things to solve intractable problems with little human assistance. Computational approaches have revolutionized supramolecular chemistry, have greatly helped us to understand host–guest binding, and will continue to do even more amazing things in the coming years. But, for now, we remain unable to predict the influence of complex solute structures, solvent, salt, and co-solutes on host–guest chemistry. We can't yet design the perfect host for a task in molecular recognition without running through multiple iterations of design, synthesis, and testing. There are still countless stable organic molecular structures that have never been synthesized. And, like some other ‘wet sciences’, the practice of organic synthesis and purification has so far resisted efforts at end-to-end automation except in very limited circumstances. It is encouraging to me as a scientist that I work in a field that still relies heavily on human ingenuity and skill to make important advances, and whose most difficult problems can't yet be solved by simply applying more computing power.
Acknowledgements
I am deeply grateful to all co-workers, colleagues, and collaborators, past and present.References
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- C. Conner, I. Cheung, A. Simon, M. Jakovcevski, Z. Weng and S. Akbarian, Epigenetics, 2010, 5, 1–4 CrossRef.
- A. Jeltsch, I. Bock, A. Dhayalan, S. Kudithipudi, O. Brandt and P. Rathert, Epigenetics, 2011, 6, 256–263 CrossRef.
- B. D. Strahl and S. M. Fuchs, Epigenomics, 2011, 3, 247–249 CrossRef PubMed.
- S. B. Rothbart, B. M. Dickson, J. R. Raab, A. T. Grzybowski, K. Krajewski, A. H. Guo, E. K. Shanle, S. Z. Josefowicz, S. M. Fuchs, C. D. Allis, T. R. Magnuson, A. J. Ruthenburg and B. D. Strahl, Mol. Cell, 2015, 59, 502–511 CrossRef CAS PubMed.
- A. Bateman, M. J. Martin, C. O'Donovan, M. Magrane, R. Apweiler, E. Alpi, R. Antunes, J. Ar-Ganiska, B. Bely, M. Bingley, C. Bonilla, R. Britto, B. Bursteinas, G. Chavali and E. Cibrian-Uhalte, et al. , Nucleic Acids Res., 2015, 43, D204–D212 CrossRef PubMed.
- E. Boutet, D. Lieberherr, M. Tognolli, M. Schneider, P. Bansal, A. J. Bridge, S. Poux, L. Bougueleret and I. Xenarios, Methods Mol. Biol., 2016, 1374, 23–54 Search PubMed.
- P. V. Hornbeck, B. Zhang, B. Murray, J. M. Kornhauser, V. Latham and E. Skrzypek, Nucleic Acids Res., 2015, 43, D512–D520 CrossRef PubMed.
- D. L. Sparks, M. C. Phillips and S. Lundkatz, J. Biol. Chem., 1992, 267, 25830–25838 CAS.
- T. Jenuwein and C. D. Allis, Science, 2001, 293, 1074–1080 CrossRef CAS PubMed.
- L. Liu, X. T. Zhen, E. Denton, B. D. Marsden and M. Schapira, Bioinformatics, 2012, 28, 2205–2206 CrossRef CAS PubMed.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CAS.
- S. M. Carlson, K. E. Moore, E. M. Green, G. M. Martin and O. Gozani, Nat. Protoc., 2014, 9, 37–50 CrossRef CAS PubMed.
- H. Li, W. Fischle, W. Wang, E. M. Duncan, L. Liang, S. Murakami-Ishibe, C. D. Allis and D. J. Patel, Mol. Cell, 2007, 28, 677–691 CrossRef CAS PubMed.
- Y. Guo, N. Nady, C. Qi, A. Allali-Hassani, H. Zhu, P. Pan, M. A. Adams-Cioaba, M. F. Amaya, A. Dong, M. Vedadi, M. Schapira, R. J. Read, C. H. Arrowsmith and J. Min, Nucleic Acids Res., 2009, 37, 2204–2210 CrossRef CAS PubMed.
- R. J. Eisert and M. L. Waters, ChemBioChem, 2011, 12, 2786–2790 CrossRef CAS PubMed.
- B. J. Pieters, E. Meulenbroeks, R. Belle and J. Mecinovic, PLoS One, 2015, 10, e0139205 Search PubMed.
- R. M. Hughes, K. R. Wiggins, S. Khorasanizadeh and M. L. Waters, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11184–11188 CrossRef CAS PubMed.
- J. J. Kamps, J. Huang, J. Poater, C. Xu, B. J. Pieters, A. Dong, J. Min, W. Sherman, T. Beuming, F. Matthias Bickelhaupt, H. Li and J. Mecinovic, Nat. Commun., 2015, 6, 8911 CrossRef CAS PubMed.
- C. S. Beshara and F. Hof, Can. J. Chem., 2010, 88, 1009–1016 CrossRef CAS.
- A. L. Whiting and F. Hof, Org. Biomol. Chem., 2012, 10, 6885–6892 CAS.
- A. L. Whiting, N. M. Neufeld and F. Hof, Tetrahedron Lett., 2009, 50, 7035–7037 CrossRef CAS.
- X. Wang and F. Hof, Beilstein J. Org. Chem., 2012, 8, 1–10 CrossRef CAS PubMed.
- D. J. Mahnke, R. McDonald and F. Hof, Chem. Commun., 2007, 3738–3740 RSC.
- S. Shinkai, S. Mori, T. Tsubaki, T. Sone and O. Manabe, Tetrahedron Lett., 1984, 25, 5315–5318 CrossRef CAS.
- S. G. Bott, A. W. Coleman and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 610–611 CrossRef CAS.
- F. Perret, A. N. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425–2438 RSC.
- D. S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef CAS PubMed.
- S. Shinkai, K. Araki and O. Manabe, J. Am. Chem. Soc., 1988, 110, 7214–7215 CrossRef CAS.
- G. Arena, A. Contino, F. G. Gulino, A. Magri, F. Sansone, D. Sciotto and R. Ungaro, Tetrahedron Lett., 1999, 40, 1597–1600 CrossRef CAS.
- C. S. Beshara, C. E. Jones, K. D. Daze, B. J. Lilgert and F. Hof, ChemBioChem, 2010, 11, 63–66 CrossRef CAS PubMed.
- N. Douteau-Guével, A. W. Coleman, J.-P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 1999, 629–633 RSC.
- K. D. Daze, C. E. Jones, B. J. Lilgert, C. S. Beshara and F. Hof, Can. J. Chem., 2013, 91, 1072–1076 CrossRef CAS.
- M. A. Gamal-Eldin and D. H. Macartney, Org. Biomol. Chem., 2013, 11, 488–495 CAS.
- E. Bernstein, E. M. Duncan, O. Masui, J. Gil, E. Heard and C. D. Allis, Mol. Cell. Biol., 2006, 26, 2560–2569 CrossRef CAS PubMed.
- L. Kaustov, H. Ouyang, M. Amaya, A. Lemak, N. Nady, S. Duan, G. A. Wasney, Z. Li, M. Vedadi, M. Schapira, J. Min and C. H. Arrowsmith, J. Biol. Chem., 2011, 286, 521–529 CrossRef CAS PubMed.
- C. Simhadri, K. D. Daze, S. F. Douglas, T. T. H. Quon, A. Dev, M. C. Gignac, F. N. Peng, M. Heller, M. J. Boulanger, J. E. Wulff and F. Hof, J. Med. Chem., 2014, 57, 2874–2883 CrossRef CAS PubMed.
- K. D. Daze, T. Pinter, C. S. Beshara, A. Ibraheem, S. A. Minaker, M. C. F. Ma, R. J. M. Courtemanche, R. E. Campbell and F. Hof, Chem. Sci., 2012, 3, 2695–2699 RSC.
- F. Perret and A. W. Coleman, Chem. Commun., 2011, 47, 7303–7319 RSC.
- L. Memmi, A. N. Lazar, A. Brioude, V. Ball and A. W. Coleman, Chem. Commun., 2001, 2474–2475 RSC.
- R. E. McGovern, A. A. McCarthy and P. B. Crowley, Chem. Commun., 2014, 50, 10412–10415 RSC.
- R. E. McGovern, B. D. Snarr, J. A. Lyons, J. McFarlane, A. L. Whiting, I. Paci, F. Hof and P. B. Crowley, Chem. Sci., 2015, 6, 442–449 RSC.
- S. Tabet, S. F. Douglas, K. D. Daze, G. A. E. Garnett, K. J. H. Allen, E. M. M. Abrioux, T. T. H. Quon, J. E. Wulff and F. Hof, Bioorg. Med. Chem., 2013, 21, 7004–7010 CrossRef CAS PubMed.
- M. Ciaccia, I. Tosi, R. Cacciapaglia, A. Casnati, L. Baldini and S. Di Stefano, Org. Biomol. Chem., 2013, 11, 3642–3648 CAS.
- K. Flidrova, S. Bohm, H. Dvorakova, V. Eigner and P. Lhotak, Org. Lett., 2014, 16, 138–141 CrossRef CAS PubMed.
- P. Slavik, K. Flidrova, H. Dvorakova, V. Eigner and P. Lhotak, Org. Biomol. Chem., 2013, 11, 5528–5534 CAS.
- K. Flidrova, P. Slavik, V. Eigner, H. Dvorakova and P. Lhotak, Chem. Commun., 2013, 49, 6749–6751 RSC.
- C. D. Gutsche and L. G. Lin, Tetrahedron, 1986, 42, 1633–1640 CrossRef CAS.
- K. C. Nam and D. S. Kim, Bull. Korean Chem. Soc., 1994, 15, 284–286 CAS.
- M. Vezina, J. Gagnon, K. Villeneuve, M. Drouin and P. D. Harvey, Organometallics, 2001, 20, 273–281 CrossRef CAS.
- K. D. Daze, M. C. F. Ma, F. Pineux and F. Hof, Org. Lett., 2012, 14, 1512–1515 CrossRef CAS PubMed.
- H. Peacock, C. C. Thinnes, A. Kawamura and A. D. Hamilton, Supramol. Chem., 2016, 28, 575–581 CrossRef CAS.
- R. M. Hughes and M. L. Waters, J. Am. Chem. Soc., 2005, 127, 6518–6519 CrossRef CAS PubMed.
- R. M. Hughes, M. L. Benshoff and M. L. Waters, Chem. – Eur. J., 2007, 13, 5753–5764 CrossRef CAS PubMed.
- H. F. Allen, K. D. Daze, T. Shimbo, A. Lai, C. A. Musselman, J. K. Sims, P. A. Wade, F. Hof and T. G. Kutateladze, Biochem. J., 2014, 459, 505–512 CrossRef CAS PubMed.
- F. P. Schmidtchen, Coord. Chem. Rev., 2006, 250, 2918–2928 CrossRef CAS.
- “Ah, entropy… Just when I think I understand it, it slips through our fingers”, Ken Shimizu, University of South Carolina.
- L. A. Ingerman, M. E. Cuellar and M. L. Waters, Chem. Commun., 2010, 46, 1839–1841 RSC.
- N. K. Pinkin and M. L. Waters, Org. Biomol. Chem., 2014, 12, 7059–7067 CAS.
- L. I. James, J. E. Beaver, N. W. Rice and M. L. Waters, J. Am. Chem. Soc., 2013, 135, 6450–6455 CrossRef CAS PubMed.
- N. K. Pinkin, A. N. Power and M. L. Waters, Org. Biomol. Chem., 2015, 13, 10939–10945 CAS.
- N. K. Pinkin, I. Liu, J. D. Abron and M. L. Waters, Chem. – Eur. J., 2015, 21, 17981–17986 CrossRef CAS PubMed.
- G. A. E. Garnett, K. D. Daze, J. A. P. Diaz, N. Fagen, A. Shaurya, M. C. F. Ma, M. S. Collins, D. W. Johnson, L. N. Zakharov and F. Hof, Chem. Commun., 2016, 52, 2768–2771 RSC.
- R. S. Carnegie, C. L. D. Gibb and B. C. Gibb, Angew. Chem., Int. Ed., 2014, 53, 11498–11500 CrossRef CAS PubMed.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276–279 CrossRef CAS PubMed.
- M. Ali, K. D. Daze, D. E. Strongin, S. B. Rothbart, H. Rincon-Arano, H. F. Allen, J. Li, B. D. Strahl, F. Hof and T. G. Kutateladze, J. Biol. Chem., 2015, 290, 22919–22930 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- A. Norouzy, Z. Azizi and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 792–795 CrossRef CAS PubMed.
- G. Ghale, A. G. Lanctot, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762–2765 CrossRef CAS PubMed.
- Y. Ghang, M. Schramm, F. Zhang, R. Acey, C. David, E. Wilson, Y. Wang, Q. Cheng and R. Hooley, J. Am. Chem. Soc., 2013, 135, 7090–7093 CrossRef CAS PubMed.
- A. Mueller, R. Lalor, C. M. Cardaba and S. E. Matthews, Cytometry, Part A, 2011, 79, 126–136 CrossRef PubMed.
- R. Lalor, H. Baillie-Johnson, C. Redshaw, S. E. Matthews and A. Mueller, J. Am. Chem. Soc., 2008, 130, 2892–2893 CrossRef CAS PubMed.
- D. S. Guo, V. D. Uzunova, X. Su, Y. Liu and W. M. Nau, Chem. Sci., 2011, 2, 1722–1734 RSC.
- E. Roka, M. Vecsernyes, I. Bacskay, C. Felix, M. Rhimi, A. W. Coleman and F. Perret, Chem. Commun., 2015, 51, 9374–9376 RSC.
- K. Wang, D. S. Guo, H. Q. Zhang, D. Li, X. L. Zheng and Y. Liu, J. Med. Chem., 2009, 52, 6402–6412 CrossRef CAS PubMed.
- G. F. Wang, X. L. Ren, M. Zhao, X. L. Qiu and A. D. Qi, J. Agric. Food Chem., 2011, 59, 4294–4299 CrossRef CAS PubMed.
- L. You, D. J. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. P. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595–2626 CrossRef CAS PubMed.
- K. N. Koh, K. Araki, A. Ikeda, H. Otsuka and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 755–758 CrossRef CAS.
- S. A. Minaker, K. D. Daze, M. C. F. Ma and F. Hof, J. Am. Chem. Soc., 2012, 134, 11674–11680 CrossRef CAS PubMed.
- M. Florea, S. Kudithipudi, A. Rei, M. J. Gonzalez-Alvarez, A. Jeltsch and W. M. Nau, Chem. – Eur. J., 2012, 18, 3521–3528 CrossRef CAS PubMed.
- B. D. Strahl, S. M. Fuchs, K. Krajewski, R. W. Baker and V. L. Miller, Curr. Biol., 2011, 21, 53–58 CrossRef PubMed.
- T. A. Egelhofer, A. Minoda, S. Klugman, K. Lee, P. Kolasinska-Zwierz, A. A. Alekseyenko, M. S. Cheung, D. S. Day, S. Gadel, A. A. Gorchakov, T. Gu, P. V. Kharchenko, S. Kuan, I. Latorre and D. Linder-Basso, et al. , Nat. Struct. Mol. Biol., 2011, 18, 91–93 CAS.
- G. A. E. Garnett, M. J. Starke, A. Shaurya, J. Li and F. Hof, Anal. Chem., 2016, 88, 3697–3703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |