
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoswitchable metal organic frameworks: turn on the lights and close the windows
S.
Castellanos
,
F.
Kapteijn
and
J.
Gascon
*
Catalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: j.gascon@tudelft.nl
First published on 1st February 2016
Abstract
The ability of modulating the properties of metal–organic frameworks (MOF) on demand by external light-stimuli is a most appealing pathway to enhance their performance in storage and separation and to render novel advanced applications. Photoswitchable linkers of different nature have been inserted in several MOF structures either as integral parts of their scaffolds or as guests in the pores. In this highlight we analyse the different strategies and expose some aspects that should be considered in the design of new generations of photoswitchable MOFs.
 Sonia Castellanos | Sonia Castellanos (1983) received her PhD in Chemistry from the University of Barcelona in 2010 as a result of her research in small organic molecules for electronic applications, awarded with an Extraordinary Prize. During her postdoc in HU Berlin she became an Alexander von Humboldt fellow to investigate molecular photoswitches for efficient light-to-electron conversion. Since 2014 her research as a postdoc in TU Delft focused on the preparation of novel photoactive metal–organic frameworks. Her research work has been published in high-impact peer-reviewed journals. She is currently (February 2016) starting a tenure-track in the Amsterdam Research Center for Nanolithography (ARCNL). |
 Freek Kapteijn | Freek Kapteijn (1952), MSc. in Chemistry and Mathematics, received his PhD on ‘Metathesis of alkenes’ in 1980 at the University of Amsterdam. After post-doc positions (Coal Science) in Amsterdam and Nancy (ENSIC), he became Associate professor in Amsterdam. Moved to Delft University of Technology in 1992, became ‘Anthonie van Leeuwenhoek professor’ in 1999, and is since 2008 chair of Catalysis Engineering, with visiting professorships at ETH Zürich, Tianjin and Zhejiang Normal University. Research interest focuses on the interplay of catalysis and engineering, comprising structured and multifunctional catalysts, adsorption, separation and (catalytic) membranes. Co-authored over 500 publications in peer-reviewed journals and as book chapters. |
 Jorge Gascon | Jorge Gascon (1977) received his MSc. in Chemistry in 2002 and his PhD in Chemical Engineering in 2006, both at the University of Zaragoza (Spain). Starting as post-doc at TUDelft he is since 2014 ‘Anthonie van Leeuwenhoek Professor’ of Catalysis Engineering. Research interests include fundamental aspects and applications of new nano-structured materials and composites. He has co-authored over 120 publications, several patents and has edited the book: “Metal Organic Frameworks as Heterogeneous Catalysts”. He has been the recipient of the prestigious VENI (2010), VIDI (2013) and ERC Starting (2013) personal grants. Recently he received the 2013 ExxonMobil Chemical European Science and Engineering Award. |
Introduction
Metal organic frameworks (MOFs) are a fascinating class of materials. Their hybrid nature together with their highly open structures place them among the most studied structured solids nowadays.1 An emerging approach to optimize the performance of MOFs in applications like selective adsorption of guest molecules is the use of light to change their properties on demand.2–5 Indeed, light is a very accessible and easy to tune stimulus and providing MOFs with photoinduced response can render advanced materials for many applications.For instance, if the sorption capacity of the MOF can be increased and decreased at will through reversible changes in its structure by simple illumination (Fig. 1 top), the storage and release cycle would require less expensive procedures than traditional high temperature and low pressure conditions. Having two interconvertible pore configurations is also appealing for catalysis,6 for example, by gating the selective entrance of one or another reagent with light stimuli, block-copolymerization could be fully controlled (Fig. 1 center). Moreover, by promoting the desorption of the formed products due to changes inside the pore (polarity, geometry, etc.), one could mimic the natural activity of enzymes.
 | ||
| Fig. 1 Potential applications of MOFs provided with photoswitchable behavior. In this highlight, examples of MOFs are described in which sorbate uptake and release, as well as optical properties are modulated by means of light stimuli. | ||
In addition to host/guest interactions, there are other features arising from MOFs periodicity7 whose modulation by light can find applications in different technological fields. The capability of switching a material from an ON state to an OFF state, e.g. from a fluorescent to a not fluorescent state or from a conductive to a not conductive state, can be exploited for binary coding.8–10 It is known that photochromic small molecules do not perform efficiently in the solid state due to their motion restriction in the dense crystalline packing stabilized by intramolecular interactions.11,12 If photochromic units are embodied in the frameworks, their separated arrangement or their confinement in the pores can enhance their light-triggered response.
This highlight describes the strategies that have successfully provided MOFs with photoswitchable properties, that is, with the possibility of modifying in a reversible way the framework's features by means of external light stimuli. Despite the topic has been briefly addressed as part of previous reviews,2–5 here we make a global evaluation of each design considering the nature of the photoactive component and how it is integrated in the MOF. Further, some considerations about key issues for the optimization of this kind of photoresponsive materials are exposed, aiming at their future implementation in practical uses.
Strategies to endow MOFs with photoswitchable behaviour
Despite the simplicity and beauty of the concept of endowing MOFs with photoswitchable properties, its realization needs a careful design. A straightforward strategy consists of inserting entities that undergo reversible structural changes upon light irradiation. This kind of compounds are also known as molecular photoswitches, since they can switch from one isomeric form to the other by means of light exposure (Scheme 1). This is the case of azobenzenes (ABs),11 which interconvert from trans (E) to cis (Z) configuration via rotation of the aromatic rings around the central –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– double bond. Other photoswitchable compounds accompany geometrical changes with deep alterations in their electronic system, thus yielding drastic changes in their optical properties. Diarylethenes (DAEs)9 – especially dithienylethenes (DTEs), which can exist in open and closed forms, and spiropyrans (SPs),12 which convert to the zwitterionic merocyanine (MC) counterpart, are remarkable examples of this last group.
N– double bond. Other photoswitchable compounds accompany geometrical changes with deep alterations in their electronic system, thus yielding drastic changes in their optical properties. Diarylethenes (DAEs)9 – especially dithienylethenes (DTEs), which can exist in open and closed forms, and spiropyrans (SPs),12 which convert to the zwitterionic merocyanine (MC) counterpart, are remarkable examples of this last group.
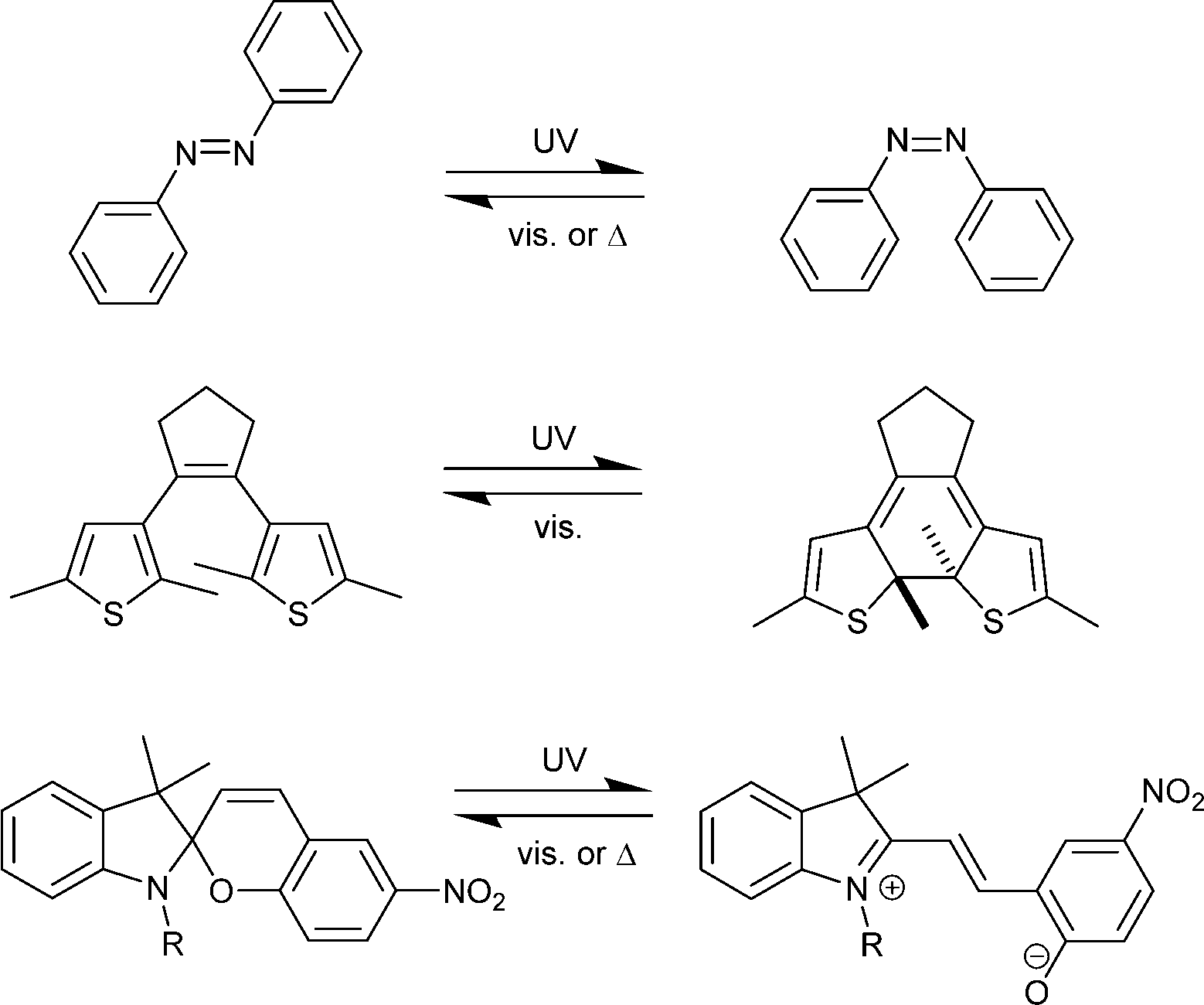 | ||
| Scheme 1 Prototypical structures of photoswitchable molecular compounds: top, azobenzene (AB); center, dithienylethene (DTE); bottom, spiropyrane/merocyanine (SP/MC). | ||
Coming back to the porous hybrid composition of MOFs mentioned above, one can think of incorporating photoswitchable units into MOFs as guests in the voids (extrinsically) or as linkers integrated as building blocks in the framework (intrinsically). Within this last approach, linkers that undergo changes along their backbone and thus alter the connection between the metal-based secondary building units (SBUs) will work fundamentally different from linkers that are provided with a photoresponsive side-group oriented towards the pore. The strategies that have proven successful in the literature are summarized in Table 1 and discussed in the following sections.
| Intrinsic photoresponse approach | ||||
|---|---|---|---|---|
| Design | MOF | Unit | Modulated properties | Ref. |
| Back-bone | Zn2(R-TC)(L-N,N′) | DTE | Fluorescence | 13 |
| Zn2(R-DC)2(L-N,N′) | DTE | CO2 sorption | 14 | |
| Zn2(R1-DC)2(R2-N,N′) | AB | CO2 sorption | 15 | |
| Isoret. UiO-66 (Zr) | AB | Inactive | 16 | |
| Side group | Isoret. ZIF-8 | AB | Light absorption | 17 |
| MOF-74 (Mg) | AB | Cargo release | 18 | |
| MIL-53 (Al), UiO-66 (Zr) | AB | CO2 sorption vis. absorption | 19 | |
| Cu2(L-DC)2(R-N,N′) | AB | Butandiol sorption | 20 | |
| Isoret. MIL-101 (Cr) | AB | Inactive | 21 | |
| Isoret. MIL-101 (Cr) | AB | Vis. absorption CH4 sorption | 22 | |
| Zn2(R-DC)2(L-N,N′) | AB | Vis. absorption | 23 | |
| M2(R-DC)2(L-N,N′) (M = Co, Cu, Zn) | AB | Vis. absorption | 24 | |
| Isoret. MOF-5 | AB | CO2 sorption | 25 | |
| UBMOF-1 (Zn) | DTE | Vis. absorption | 26 | |
| UBMOF-2 (Zn) | DTE | Vis. absorption | 27 | |
| Cu2(R-DC)2(L-N,N′) and Cu2(L-DC)2(R-N,N′) | AB | Vis. absorption, butandiol sorption | 28 | |
| Cu2(R-DC)2(L-N,N′) | AB | IR spectrum | 29 | |
A. Intrinsic photoswitching units
One might expect that greater structural changes will be attained via intrinsic backbone photoresponse. Notice, though, that not all MOF architectures might allow for a synergistic response of all photoactive linkers,16,23i.e. the geometrical changes in the linker might be such that they cannot occur simultaneously over the whole periodical network and thus might not afford a MOF capable of switching between two configurations. Indeed, very few examples of MOFs featuring photoswitchable backbone units have exhibited photoresponse, and the ones that did are composed of Zn-based frameworks built with two different kind of linkers,13–15 bipyridyl type and dicarboxylic type, where only one of them bears the photoisomerzable fragment, except for the MOF reported by Lyndon et al.,15 which contains both azobenzene and stilbene moieties (Table 1). It must be also remarked that for DTE photochromes the cyclization mechanisms implies less marked geometrical changes (planarization, rigidification and shrinking of the length along the thiophene units in around 0.1 Å)9,34 than for AB based ones (shrinking of the backbone length from 9 Å to 6 Å and change of dihedral angle in the order of from 0° to 100° thus implying important alterations in the –NN–C and –C–NN–C– angles during the transition states).35
Shustova et al. used DTEs featuring two terminal pyridines to pillar 2D frameworks based on Zn-porphyrin linkers and dimeric Zn-SBUs. While DTEs linkers are in the open form, the Zn-porphyrin linkers display fluorescence. In contrast, when the ring closure of the DTEs is induced by UV irradiation, non-radiative energy transfer to the closed DTEs occurs and fluorescent emission is quenched (Fig. 2).13
 | ||
| Fig. 2 Energy transfer to the DTE-based linker in it closed form quenches fluorescence of the porphirine-based linker. In this way, the MOF can be switched from an fluorescent to a non-fluorescent version by means of light. Reprinted with permission from ref. 13. Copyright 2014 American Chemical Society. | ||
The same kind of pyridine terminated DTEs were employed by Guo and coworkers.14 In this case the interpenetrated Zn-MOF exhibited reversible photochromism and could be switched between yellow to dark blue by UV exposure, and back to yellow through visible light irradiation. Importantly, the structural changes promoted by the light-induced ring-closure of DTE linkers resulted in a 4-fold enhancement of CO2 sorption capacity. In addition, CO2 uptake experiments showed that optimal gas uptake and release could be attained by adsorbing under UV irradiation and desorbing under 600 nm light. Another dual linker Zn-MOF prepared from dicarboxylic azobenzenes was also reported by Lyndon et al. to modulate CO2 sorption under dynamic conditions, but in this case CO2 desorption was more efficient under UV light exposure, instead.15
Judging from the number of publications, functionalizing the linkers with the photoswitchable functionality as a side-group, seems a more reliable approach. Furthermore, such design is accessible also via post-functionalization of the MOFs.21,22 This synthetic pathway already provides the desired framework and, at the same time, guarantees that the network will take only as many side groups as it can accommodate in its voids. Regardless of the synthesis method, light-induced configurational changes by azobenzene side-groups is so far the most efficient strategy to modulate the storage properties of MOFs, from small gas molecules like CO2 or CH4,19,23,25 to bigger ones, like butandiol20,28 or propidium iodide fluorescent dye.18 The reason is that not only the geometrical alterations upon trans/cis isomerization are fairly large but also the alterations in polarity can have a strong influence in the sorption capacities. As a matter of fact, Heinke et al. observed that trans to cis isomerization decreased butandiol permeability in AB-based SURMOFs.20 Intriguingly, the trend in the sorption modulation is not constant, i.e. the cis configuration in some cases increases the sorbant uptake capacity20,22,28 and in some others produces the opposite effect.25 In addition, isomerization of the AB side-groups can also be used to generate a dynamic situation to expel the guest in the pore under continuous irradiation of the MOF.18
The combination of photoactive side-group and crystalline lattice is vital to attain light-induced response. This involves not only assuring that the most thermodynamically stable form of the photoactive unit that protrudes from the linker fits in the pores but also that its light-triggered isomerization mechanism can occur and that the resulting isomeric form can be accommodated within the resulting lattice. Such questions have been addressed by Wang et al., who observed that small differences in the framework dimensions have shown to suppress the photoresponse of Cu-containing surface-attached metal–organic frameworks (SURMOF) due to the lack of a free trajectory for the photoisomerization mechanism of the azobenzene pending groups (Fig. 3).28 Further, depending on the length of these linkers, interpenetration may occur during synthesis, which normally results in lower accessible volume and smaller pore sizes than expected for the primary network.23,24
 | ||
| Fig. 3 Changes in the pore dimensions of SURMOFs arising from the different linkers lengths enable or disable the photoisomerization of azobenzene side-groups. Ref. 28 – Published by the PCCP Owner Societies. | ||
Steric hindrance is thus usually undesired but can though be used as a way to provide MOFs with gas uptake modulation by means of light. Castellanos and coworkers reported a MIL-53-like structure synthesized with terephthalic acids provided with fluorinated azobenzene side-group (Fig. 4).19 In this structure, the crowded packing of the side-groups in the pores was claimed to prevent isomerization. As a consequence, upon light absorption the only relaxation pathway for the linkers was via non-radiative vibrational processes, which increased the surface energy of the crystalline lattice, thus resulting in the decrease of CO2 sorption capacity.
 | ||
| Fig. 4 Terephthalic linkers featuring fluorinated azobenzene side-groups yielded a photochromic MOF isoreticular to UiO-66 (Zr) that switches E to Z upon green light irradiation and Z to E upon blue light exposure (a, b) and a MOF with similar to MIL-53 (Al) with sterically crowded channels (c, d). The impossibility to isomerize in the last case gave place to a photothermal effect that decreased CO2 sorption under visible light. Reprinted from ref. 19 with permission from John Wiley and Sons. | ||
Alternative to ABs, DTEs as side groups of the linkers have also been implemented in MOFs to yield photochromic properties, that is, the framework rendered a drastic change of color upon UV irradiation. However, the system could not revert to its original configuration by visible light exposure, contrarily to the typical DTE behavior, thus indicating that the closed form of the DTE was too stabilized in the crystalline lattice.26 Such lack of reversibility was overcome replacing the terminal methyl groups in the thiophene heterocycles with phenyl groups. This “minor” modification in the linker structure enabled the isoreticular MOF with back thermal ring-opening, yet with low fatigue resistance.27
B. Extrinsic photoswitching units
A less synthetically demanding approach is to confine photoswitchable molecules in MOFs' pores. However, this method implies a reduction in the pore volumes of the host MOF, which can be a drawback for some practical purposes. An outstanding example that compensates this blocking effect was reported by Yanai et al.32 In this work, the geometry changes triggered by UV irradiation on ABs in the pores of a flexible Zn-containing MOF induced large alterations of the hosting framework's crystalline cell, as evidenced by PXRD monitoring. Importantly, N2 sorption capacity was increased in 8.3-fold after the system was exposed to UV light. The authors claimed that thermal treatment (120 °C) of the material reproduced the N2 isotherms before illumination and that the behavior persisted after repetitive cycles.When the same hosting MOF built from terephthalate and triethyldiamine bifunctional linkers was used to embed DTEs, the guests aligned in the [001] direction. Upon UV irradiation and thus promotion of ring-closure of the DTEs, this directional preference led to a linear dichroism in the visible light absorption of the MOF, that is, absorption of polarized visible light was stronger in certain directions of the crystal.31
Hermann and coworkers studied the influence of the size and shape of the pore on the photoresponse degree by means of IR spectroscopy. Again, this study pointed out how the spatial restrictions of the MOF cavities are vital for the isomerization efficiency. Whereas AB hosted in MIL-69 (In or Ga) can isomerize in the pores, in MIL-53 (Al) the dense packing of ABs in the voids hindered its light-driven configurational changes.30
To the best of our knowledge, the only photoswitchable MOF system involving SP/MC couple in the literature have been prepared by encapsulating the SP molecule during the crystallization of an In-based MOF with cubic zeolitic MTN topology, named JUC-120 and analogous to MIL-100 (Al, Fe or Cr).33 Noteworthy, inside the pores the MC form is better stabilized and it only transforms into its SP counterpart upon UV irradiation. This behavior is opposite to the one usually observed for this kind of photochromic molecules and is known as reverse photochromism.
C. Alternative light-induced reversible processes
A particular and interesting photoswitching mechanism that yielded modulation of MOF's properties but did not involve structural modifications was reported by Sun et al.36 In this work, UV-triggered inter-linker electron transfer was favored by the pseudo-rotaxane assembly of a Eu-containing MOF. Viologen radicals resulted from this process, which conferred a blue color to the material and, consequently, quenched the fluorescence of the Eu(III) cations. Thus, whereas the non-illuminated MOFs showed a yellow appearance and displayed the characteristic fluorescent bands of Eu(III), upon UV irradiation the material turned blue and did not exhibited light-emission. Such alteration of the optical changes proved reversible via exposure to O2 in the dark and exhibited high fatigue resistance. The authors claimed that reoxidation of the radical anion was the origin of the bleaching.To be addressed in the future
Once the concept of modulation of optical or storage/release properties has been proven, optimization of light utilization is needed for a real implementation of photoswitchable MOFs. Like in any other light-induced process, a vital parameter governing the efficiency is the quantum yield, or the number of events triggered by an absorbed photon. Such parameter is, though, not easy to evaluate, since quantitative determination of the absorbed photons in the solid phase is not as straightforward as in solution.37,38 In addition, taking the quantum yield values of the isolated photoswitchable components as an estimation is not an option, since it can be very different once incorporated in the MOF, given that the coordination to the metal cations as well as the surroundings in the network can interfere in many ways.To begin with, other chromophores will take their share of light, which will decrease the photon flux arriving to the photoresponsive unit. Most importantly, the covalent attachment of the photoswitching molecule to the metals can give place to new excited states that may not relax toward the targeted configurational change. For instance, ligand-to-metal charge transfer can be promoted instead of a more linker-centered excited state. Even in the case of reaching the desired excited state, other relaxation pathways might compete with the isomerization mechanism, such as energy transfer or electron transfer. Computational studies can be a great tool to inspect the topology and energy of the crystalline molecular orbitals in order to comprehend the origin of their properties,39–41 as well as mechanistic aspects concerning the photoisomerization the building units28,42,43 and thus assist in the design of photoswitchable MOFs.
The competing light-triggered processes can be overcome by selectively irradiating the photoresponsive units, which requires that the light absorption spectrum of the photoresponsive building blocks does not overlap the bands associated with other optical transitions. As a matter of fact, photoswitchable MOFs that require UV light to be active usually exhibit low fatigue resistance.17,22,27 That is what makes MOFs displaying forth and back photoisomerization in the visible light regime remarkable.19 Of particular interest are materials that could undergo structural changes upon very low energetic light irradiation, such as near infrared, since this property would be most relevant for drug release biomedical applications.44
In addition to molecular design, crystal engineering can be an essential tool to improve the efficiency in the incident photons' usage. Light penetration could be in principle enhanced by increasing the surface to volume ratio of the MOF's particles, for instance by growing the crystalline frameworks as thin films, such us in SURMOFs. The epitaxial growth of the later further allows to implement the photoisomerizable struts only at the surface of the film. Such approach was nicely employed by Heinke and coworkers,20 who prepared SURMOFs with a basement layer of biphenyl dicarboxylic linkers and a roof layer of analogous AB-containing linkers. Indeed, this work proved that is not necessary to provide the whole bulk of the MOF with photoswitchable units to attain light-mediated sorption modulation. This result could thus be extrapolated to core–shell structures45 in the future.
The integration of SP in MOFs has also proven challenging. As reported for fully organic nanoporous materials,46 the switching between the SP neutral form and the charged MC form can allow for the modulation of several tasks. However, precisely this zwitterionic isomer might interact strongly with the metallic ions, and together with the relatively large molecular size of both SP and MC the number of suitable frameworks that could accept them is quite reduced. As a consequence, up to date only a zeolitic-like MOF hosting MC and exhibiting reverse photochromism has been reported,33 and SP/MC-based MOFs still remain unexplored.
Conclusions
Azobenzene and dithienylethene derivatives have proven suitable photoisomerizable compounds to impart photoswitching behavior to MOFs. Their incorporation in the backbone of the framework is limited due to constrains arising from the geometrical changes upon isomerization, especially for azobenzene-based linkers. Hence, the introduction of the functionality undergoing light-induced motion as a side group oriented toward the pores appears as the most efficient design so far. Light-driven uptake and release modulation of gas or cargo molecules has been repeatedly achieved, mainly with azobenzene containing MOFs. Dithienylethene-based linkers, on the other hand, emerge as prominent units to endow the MOFs with switchable optical properties. The ability to use light input to control on the ON and OFF readout in the solid state might have great potential in data storage applications.Despite some impressive modulation, the efficiency of light utilization could be improved by inducing the response with selective irradiation of the photoswitchable units. The use of visible light or even less energetic radiation is a most appealing target. Furthermore, the effects of other photochromic compounds involving greater polarity changes, such as spiropyrans, must still be pursued, since it is predicted that many properties in the MOF could be drastically modified when going from the neutral species to the charge merocyanine species. Last but not least, MOFs' crystalline nature and synthetic versatility should be exploited via smart crystal engineering47 that can lead to an optimized utilization of light.
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 335746, CrystEng-MOF-MMM.References
- J.-C. Tan and B. Civalleri, CrystEngComm, 2015, 17, 197–198 RSC.
- R. D. Mukhopadhyay, V. K. Praveen and A. Ajayaghosh, Mater. Horiz., 2014, 1, 572–576 RSC.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- M. Zhang, M. Bosch, T. Gentle Iii and H.-C. Zhou, CrystEngComm, 2014, 16, 4069–4083 RSC.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- R. Gostl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed.
- E. Orgiu, N. Crivillers, M. Herder, L. Grubert, M. Pätzel, J. Frisch, E. Pavlica, D. T. Duong, G. Bratina, A. Salleo, N. Koch, S. Hecht and P. Samorì, Nat. Chem., 2012, 4, 675–679 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- L. A. Frolova, P. A. Troshin, D. K. Susarova, A. V. Kulikov, N. A. Sanina and S. M. Aldoshin, Chem. Commun., 2015, 51, 6130–6132 RSC.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- D. E. Williams, J. A. Rietman, J. M. Maier, R. Tan, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2014, 136, 11886–11889 CrossRef CAS PubMed.
- F. Luo, C. B. Fan, M. B. Luo, X. L. Wu, Y. Zhu, S. Z. Pu, W.-Y. Xu and G.-C. Guo, Angew. Chem., Int. Ed., 2014, 53, 9298–9301 CrossRef CAS PubMed.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS PubMed.
- A. Schaate, S. Dühnen, G. Platz, S. Lilienthal, A. M. Schneider and P. Behrens, Eur. J. Inorg. Chem., 2012, 2012, 790–796 CrossRef CAS.
- S. Bernt, M. Feyand, A. Modrow, J. Wack, J. Senker and N. Stock, Eur. J. Inorg. Chem., 2011, 2011, 5378–5383 CrossRef CAS.
- J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart and O. M. Yaghi, Chem. Sci., 2013, 4, 2858–2864 RSC.
- S. Castellanos, A. Goulet-Hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn and D. Bléger, Chem. – Eur. J., 2016, 22, 746–752 CrossRef CAS PubMed.
- L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges, S. Bräse and C. Wöll, ACS Nano, 2014, 8, 1463–1467 CrossRef CAS PubMed.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053–12055 RSC.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2012, 41, 8690–8696 RSC.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2011, 40, 4217–4222 RSC.
- A. Modrow, M. Feyand, D. Zargarani, R. Herges and N. Stock, Z. Anorg. Allg. Chem., 2012, 638, 2138–2143 CrossRef CAS.
- J. Park, D. Yuan, K. T. Pham, J.-R. Li, A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 99–102 CrossRef CAS PubMed.
- D. G. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Chem. Commun., 2014, 50, 2653–2656 RSC.
- I. M. Walton, J. M. Cox, C. A. Benson, D. G. Patel, Y.-S. Chen and J. B. Benedict, New J. Chem., 2016, 40, 101–106 RSC.
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges, S. Brase, K. Reuter and C. Woll, Phys. Chem. Chem. Phys., 2015, 17, 14582–14587 RSC.
- X. Yu, Z. Wang, M. Buchholz, N. Fullgrabe, S. Grosjean, F. Bebensee, S. Brase, C. Woll and L. Heinke, Phys. Chem. Chem. Phys., 2015, 17, 22721–22725 RSC.
- D. Hermann, H. Emerich, R. Lepski, D. Schaniel and U. Ruschewitz, Inorg. Chem., 2013, 52, 2744–2749 CrossRef CAS PubMed.
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC.
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501–4504 CrossRef CAS PubMed.
- F. Zhang, X. Zou, W. Feng, X. Zhao, X. Jing, F. Sun, H. Ren and G. Zhu, J. Mater. Chem., 2012, 22, 25019–25026 RSC.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef PubMed.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS PubMed.
- J.-K. Sun, L.-X. Cai, Y.-J. Chen, Z.-H. Li and J. Zhang, Chem. Commun., 2011, 47, 6870–6872 RSC.
- B. C. Daglen, J. D. Harris, C. D. Dax and D. R. Tyler, Rev. Sci. Instrum., 2007, 78, 074104 CrossRef PubMed.
- G. Pariani, A. Bianco, R. Castagna and C. Bertarelli, J. Phys. Chem. A, 2011, 115, 12184–12193 CrossRef CAS PubMed.
- M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, J. Mater. Chem. B, 2014, 2, 766–774 RSC.
- K. T. Butler, C. H. Hendon and A. Walsh, ACS Appl. Mater. Interfaces, 2014, 6, 22044–22050 CAS.
- K. T. Butler, C. H. Hendon and A. Walsh, J. Am. Chem. Soc., 2014, 136, 2703–2706 CrossRef CAS PubMed.
- J. N. Amanda, P. Jinhee, Z. Vladmir, W. Hong, J. Pavel, V. P. Oleg, Z. Hong-Cai and P. L. James, J. Phys.: Condens. Matter, 2015, 27, 134208 CrossRef PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., 2013, 125, 4471–4475 CrossRef.
- P. A. Szilagyi, M. Lutz, J. Gascon, J. Juan-Alcaniz, J. van Esch, F. Kapteijn, H. Geerlings, B. Dam and R. van de Krol, CrystEngComm, 2013, 15, 6003–6008 RSC.
- P. K. Kundu, G. L. Olsen, V. Kiss and R. Klajn, Nat. Commun., 2014, 5, 3588 CrossRef PubMed.
- B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Coord. Chem. Rev., 2016, 307, 147–187 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |