
Dynamic, conjugated microporous polymers: visible light harvesting via guest-responsive reversible swelling†
K.
Venkata Rao
a,
Ritesh
Haldar
a,
Tapas Kumar
Maji
*ab and
Subi J.
George
*a
aSupramolecular Chemistry Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur P.O., Bangalore 560064, India. E-mail: george@jncasr.ac.in; tmaji@jncasr.ac.in; Fax: +91 80 22082760; Tel: +91 80 2208296
bChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur P.O., Bangalore 560064, India
First published on 12th November 2015
Abstract
The light-harvesting properties of two fluorescent dynamic conjugated microporous organic polymers (Py-PP and Py-BPP) rendered with pyrene chromophores are described. The hydrophobic and dynamic nature of these porous frameworks allows the selective capture of various organic solvents by instantaneous swelling at room temperature. Moreover, the dynamic nature of these frameworks indicates the swelling process with visible volume expansion and enhanced fluorescence. This was further explored for the rapid encapsulation of various fluorescent chromophoric guests at room temperature and investigated for photoinduced energy transfer process. The resultant host–guest antenna materials showed efficient light-harvesting and funnelling of excitation energy of host framework towards the entrapped guest molecule. This process further yielded solid-state luminescent materials with tunable emission. This work holds a great promise on the design of smart porous organic solids from π-conjugated small molecules for optoelectronics, sensing and separation.
Introduction
Much attention has been devoted in recent years to the design of synthetic host materials that can capture and entrap guest molecules with specific host–guest interactions.1–3 It has been shown that the host materials can alter the physical and chemical properties of the guest molecules there by creating conditions for new phenomena within the confines of their cavities.4,5 These strategies are technologically important for sensing, catalysis, separation and purification of specific targets. In this context, discrete molecular hosts such as calixarenes,6 molecular capsules7 and metal–organic cages8 were exploited for various applications. Interestingly, the discovery of extended porous materials such as crystalline molecular flasks,9 metal–organic frameworks (MOFs)10,11 and periodic mesoporous organosilica (PMO)12–14 with extended 3D cavities has brought many advances in this field compared to discrete molecular cages. However, these porous framework materials are not ideal candidates for rapid and reversible capture of organic solvents and guest molecules due to their rigid structure and poor chemical and hydrothermal stability.15,16 Moreover, encapsulation of appropriate guest molecules in these rigid host frameworks often requires harsh and lengthy procedures.17 In this context, porous organic polymers (POPs)18–24 synthesized from pure organic linkers would be promising owing to their high thermal and chemical stability and tunable surface areas.25–27 However, most of the POPs reported so far contain non-dynamic backbones and display very few guest encapsulation properties. We envisage that imparting dynamic functionality into these robust materials would enable their guest encapsulation properties and hence could be exploited in wide range of applications. In this regard, the dynamic soft porous MOF developed by Kitagawa et al. is an impressive example.28,29 These dynamic MOFs serve as excellent gas sensors even under low pressures of analyte molecules.30 We recently observed that POPs derived from pyrene chromophores (Py-PP) comprise a dynamic framework.31 Owing to the porous dynamic backbone, Py-PP showed multifunctional behaviour such as guest-responsive reversible swelling, enhanced fluorescence and super-absorbent nature in organic solvents that were previously unknown for POPs. Recently similar design principles have been applied to various POPs and exploited for sensing32,33 and photocatalysis.34 Moreover, strong visible light absorption, luminescence and good wettability with various organic solvents of Py-PP make it a potential material for the production of H2 gas from water under visible light.34Design of artificial light-harvesting materials35–43 continues to be an important area of research as they are key ingredients in advanced photovoltaic and optoelectronic devices. Light-harvesting and energy-transfer processes were extensively investigated in porous materials such as MOFs44–50 and PMOs38,51 in the context of tunable luminescence and photocatalysis. However, encapsulation of an acceptor guest (luminescent trap) in these porous materials is challenging. It has been shown that conjugated luminescent POPs can serve as versatile donor scaffolds for excitation energy-transfer due to efficient exciton migration along conjugated backbones.52–54 To date very few conjugated POPs have been exploited for energy-transfer processes mainly due to their non-dynamic and poor guest-encapsulation properties. It is highly reasonable to use luminescent, dynamic and superabsorbent POPs as a novel class of donor scaffolds for excitation energy-transfer because they facilitate easy and quick encapsulation of acceptor dye molecules under ambient conditions. Moreover, apart from gas storage and catalysis,55 functional properties that are characteristics of POPs made from π-conjugated backbones such as luminescence56,57 and energy transfer are seldom reported.
Here we describe the light-harvesting properties of two dynamic microporous polymer networks (Py-PP and Py-BPP) rendered with green fluorescent pyrene scaffold (Scheme 1). The unique swelling and dynamic nature of these polymers in various organic solvents enabled a facile, room-temperature encapsulation of red-emitting 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran (DMDP) and Nile red (NR) dyes (Scheme 1b). The resulting host–guest antenna materials showed efficient light harvesting and tunable solid-state emission (Scheme 1c).58 The ability of these polymers to absorb a wide range of guest molecules ranging from small solvent molecules to big organic dyes reiterates the importance of dynamic porous materials as they can undergo structural reorganization in response to incoming guest molecules.
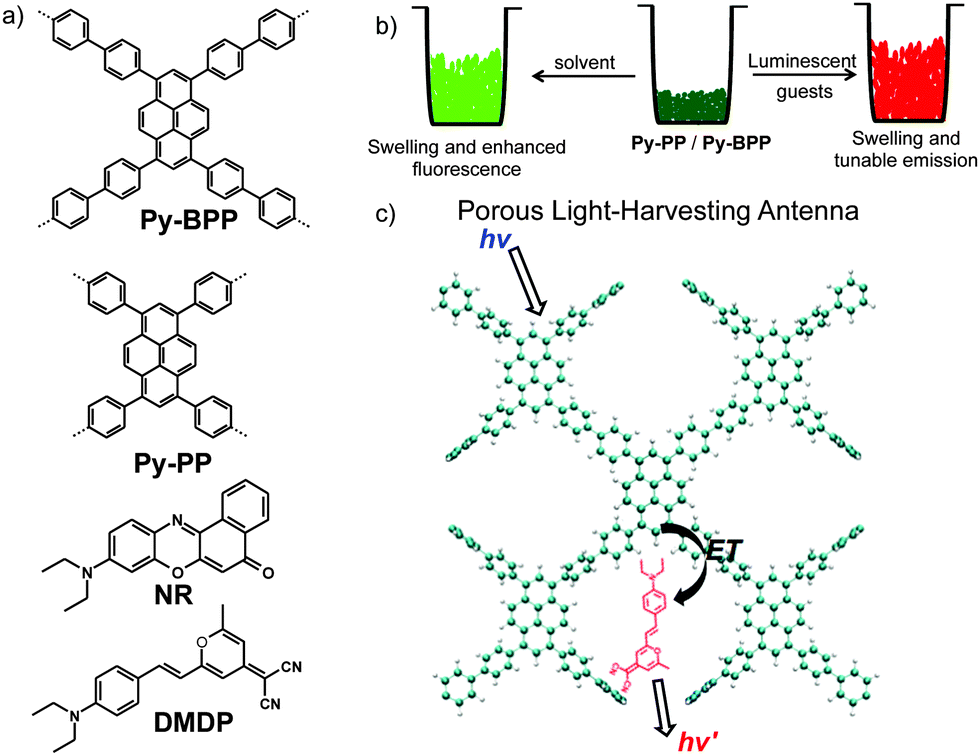 | ||
| Scheme 1 (a) Molecular structures of microporous polymers (Py-PP and Py-BPP) and guest molecules (DMDP and NR). (b) Schematic of solvent-induced swelling and facile guest encapsulation into dynamic porous polymer frameworks. (c) Schematic representation of light-harvesting process from porous polymer framework to entrapped chromophores. | ||
Experimental section
General methods
Electronic emission spectra were recorded on a PerkinElmer Ls55 Luminescence Spectrometer. Fluorescence spectra of solid powders were recorded in front-face geometry with 380 nm excitation wavelength. Fluorescence lifetime decay was recorded in a time-correlated single-photon-counting spectrometer by Horiba-Jobin Yvon. The fluorescent quantum yields were measured using an absolute photoluminescence quantum yield measurement system. Solid-state 13C NMR CPTOSS measurements were performed on a Bruker Avance 400 (400 MHz) spectrometer with an MAS rate of 5 kHz. Infrared (IR) spectra were recorded on a small quantity of the samples embedded in KBr pellets using a Bruker FT-IR spectrometer. Thermogravimetric analysis (TGA) was carried out (Mettler Toledo) in nitrogen atmosphere (flow rate 50 mL min−1) in the range of 30–600 °C (heating rate 5 °C min−1). CHNS analyses were carried out using the Thermo Scientific Flash 2000 Elemental Analyzer. Powder XRD patterns of the compounds were recorded by a Bruker D8 Discover (40 kV, 30 Ma) instrument using Cu Kα radiation (2θ = 0.8–60°).Computational details
The tetrabiphenyl pyrene monomer was optimized using a Gaussian-09 suite of programs.59–61 Optimization was implemented based on density functional theory (DFT) using a B3LYP hybrid exchange–correlation functional and the 6-31G basis set. The optimized geometries were visualised using visual molecular dynamics (VMD).62Adsorption measurements
N2 gas adsorption study of the degassed samples (degassed at 210 °C for 18 h under vacuum) was carried out using a Quantachrome Quadrasorb-SI analyzer at 77 K. The adsorbates were charged in the sample tube, and then the pressure change was monitored and the degree of adsorption determined by the reduction in pressure at the equilibrium state. All operations were computer controlled and automatic.Adsorption of toluene vapour at 293 K was measured for the desolvated PY-BPP using a BELSORP-aqua-3 analyzer. A sample of about ∼100–150 mg was prepared by heating at 210 °C for about 18 h under vacuum (1 × 10−1 Pa) prior to measurement of the isotherms. The solvent used to generate the vapour was degassed fully by repeated evacuation. Dead volume was measured with helium gas. The adsorbate was placed into the sample tube, and then the change of the pressure was monitored and the degree of adsorption was determined by pressure reduction at the equilibrium state. All operations were computer controlled and automatic.
Synthesis
Synthesis of Py-BPP. A mixture of TBP (0.19 mmol) and 4,4′-biphenyldiboronic acid bis(pinacol) ester (BDPE, 0.19 mmol) in DMF (20 mL) was degassed by four freeze–pump–thaw cycles. To this mixture 2 M K2CO3 in water (2 mL) and tetrakis(triphenylphosphine)-palladium(0) (45 mg, 38.9 μmol) were added followed by degassing by four freeze–pump–thaw cycles. Next the resultant mixture was purged with Ar 3 times and stirred at 130 °C in a Schlenk flask for 36 h. After cooling to room temperature, the mixture was poured into water and filtered. The precipitate was washed with methanol:dichloromethane and dried in vacuum. The precipitate was further purified by Soxhlet extractions with methanol, dichloromethane, toluene and tetrahydrofuran for 12 h each to give the product as a green solid (140 mg). Py-BPP polymer is practically insoluble in all common organic solvents and hence cannot be characterized using conventional methods for soluble polymers. Solid-state 13C NMR (100 Mz, δ; ppm): 126.5, 132.7, 138.4; FT-IR (ν; cm−1): 3025, 2913, 1602, 1485, 1359, 1145, 1095, 1004, 823, 728; elemental analysis (%) calculated for C40H22: C 95.617, H 4.383; found: C 89.47; H 4.42.
Results and discussion
We previously reported the synthesis, swelling and gas storage properties of Py-PP.31Py-BPP is synthesized via a Suzuki cross-coupling reaction between 1,3,6,8-tetrabromopyrene (TBP) and 4,4′-biphenyldiboronic acid bis(pinacol) ester (BDPE).63 The topologies of these two polymers were first prepared by Jiang et al. using Yamamoto coupling that resulted in statistical copolymers.64 However, in the present case, Suzuki heterocoupling reactions ensure perfectly alternating monomer sequence in resulting polymer structures. As a result, the photophysical properties of these polymers significantly vary depending on synthesis conditions. The structure of Py-BPP is characterized by solid-state 13C-CP TOSS NMR, FT-IR and thermogravimetric analysis (TGA) (Scheme 2).63 The appearance of two sets of peaks at 138–133 ppm and 126.5 ppm in 13C-CP TOSS NMR corresponds to two types of unsubstituted and substituted phenyl carbon atoms, respectively, and hence consistent with the proposed alternating structure of Py-BPP.63 Pyrene-phenylene linkages in the polymer backbone are also evident in the FT-IR measurements that showed signals corresponding to aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch (1595 cm−1) and CC vibrational modes (1390 and 1480 cm−1) of the substituted phenyl rings and aromatic C–H stretch (3030–3010 cm−1).63Py-BPP failed to show diffraction peaks due to its amorphous nature similar to several other organic microporous polymers.63 The high thermal stability of Py-BPP was confirmed using thermogravimetric analysis (TGA).63 The TGA profile showed that Py-BPP is thermally stable up to 350 °C and the observed weight loss at the initial stages of heating (∼200 °C) could be attributed to trapped solvent molecules. Surface area and porosity of Py-BPP were evaluated using N2 gas adsorption experiments at 77 K (Fig. 1a).63 The activated polymer at 210 °C showed a typical type-I profile isotherm with steep uptake at low-pressure regions, symptomatic of the permanent microporous nature of the networks; the evaluated BET (Brunauer–Emmett–Teller) surface area was 350 m2 g−1. The sudden increase in N2 uptake at P/P0 > 0.8 by the polymer is attributed to the interparticulate porosity associated with the meso- and macro-structures of the sample in the bulk.63 Using the nonlocal DFT (NLDFT) method, we also calculated the differential pore volume distributions for Py-BPP as a function of pore width. The Py-BPP has a pore size of less than 25 Å, indicating its microporous nature.63 BET surface area of Py-PP (1070 m2 g−1) is much higher than Py-BPP. This clearly indicates that, with judicious choice of the linker, it is possible to tune the surface area, porosity and photophysical properties of resultant polymers.65,66 Interestingly, BET surface areas of the perfectly alternating polymers (Py-PP and Py-BPP) reported here closely match with statistical copolymers reported by Jiang et al.64
C stretch (1595 cm−1) and CC vibrational modes (1390 and 1480 cm−1) of the substituted phenyl rings and aromatic C–H stretch (3030–3010 cm−1).63Py-BPP failed to show diffraction peaks due to its amorphous nature similar to several other organic microporous polymers.63 The high thermal stability of Py-BPP was confirmed using thermogravimetric analysis (TGA).63 The TGA profile showed that Py-BPP is thermally stable up to 350 °C and the observed weight loss at the initial stages of heating (∼200 °C) could be attributed to trapped solvent molecules. Surface area and porosity of Py-BPP were evaluated using N2 gas adsorption experiments at 77 K (Fig. 1a).63 The activated polymer at 210 °C showed a typical type-I profile isotherm with steep uptake at low-pressure regions, symptomatic of the permanent microporous nature of the networks; the evaluated BET (Brunauer–Emmett–Teller) surface area was 350 m2 g−1. The sudden increase in N2 uptake at P/P0 > 0.8 by the polymer is attributed to the interparticulate porosity associated with the meso- and macro-structures of the sample in the bulk.63 Using the nonlocal DFT (NLDFT) method, we also calculated the differential pore volume distributions for Py-BPP as a function of pore width. The Py-BPP has a pore size of less than 25 Å, indicating its microporous nature.63 BET surface area of Py-PP (1070 m2 g−1) is much higher than Py-BPP. This clearly indicates that, with judicious choice of the linker, it is possible to tune the surface area, porosity and photophysical properties of resultant polymers.65,66 Interestingly, BET surface areas of the perfectly alternating polymers (Py-PP and Py-BPP) reported here closely match with statistical copolymers reported by Jiang et al.64
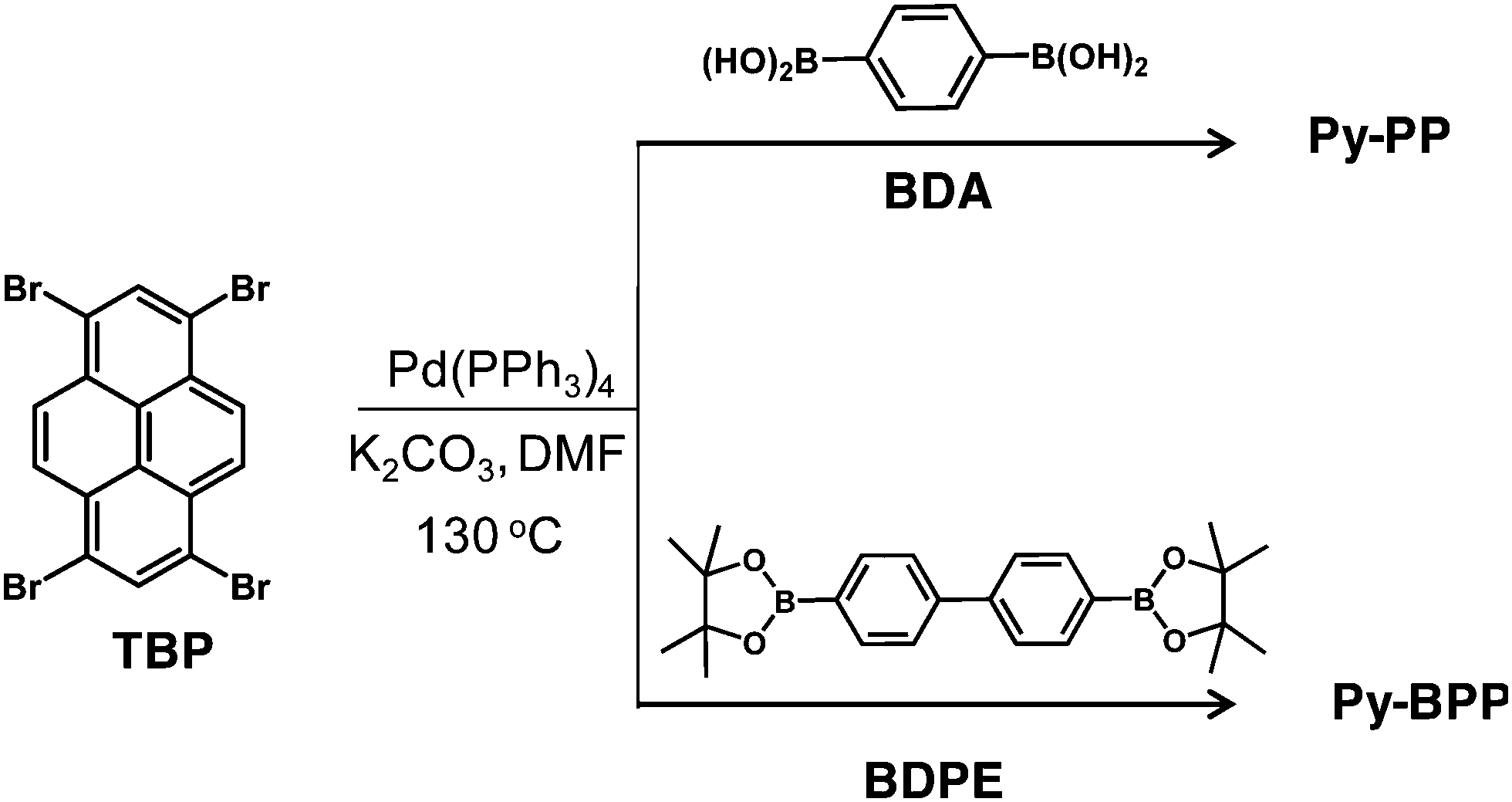 | ||
| Scheme 2 Synthetic schemes for Py-PP and Py-BPP. | ||
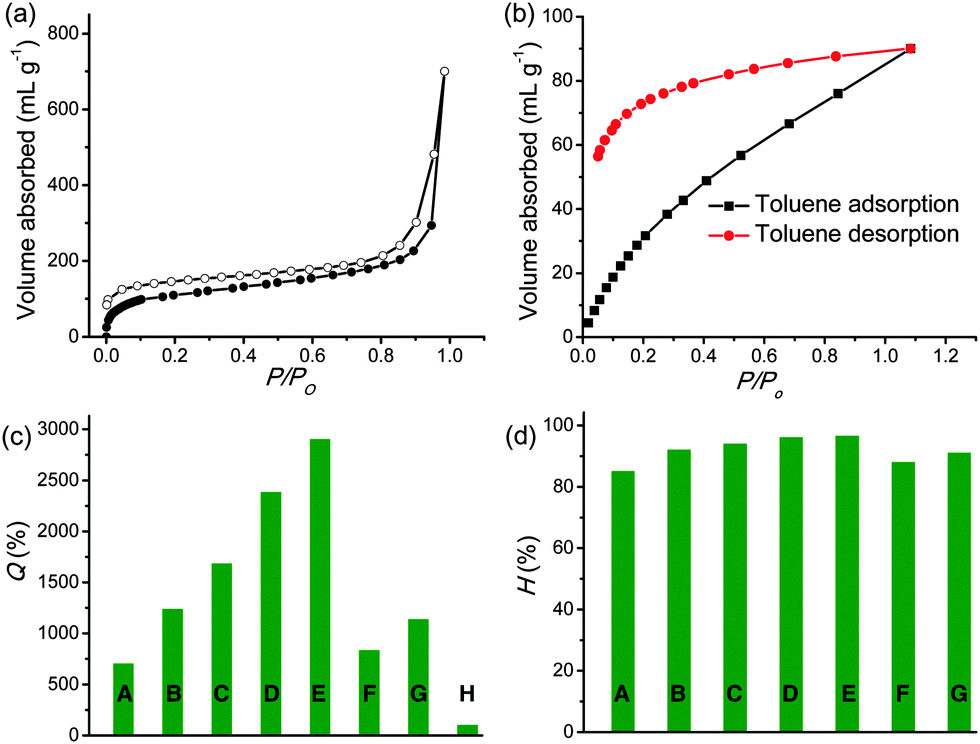 | ||
| Fig. 1 (a) N2 gas sorption isotherms (77 K) of the desolvated Py-BPP (closed symbols show the adsorption and open symbols show the desorption). (b) Toluene vapour adsorption isotherms of Py-BPP at 293 K; the sample was degassed at 483 K before measurement and the equilibrium time was 500 s. Swelling behaviour of Py-BPP expressed in terms of (c) Q (%) and (d) H (%), in various organic solvents with different polarities (A: hexane, B: diesel, C: toluene, D: ODCB, E: chloroform, F: ethanol, G: DMF, H: water). | ||
Similar to previously reported Py-PP, the biphenyl bridged Py-BPP framework also exhibits dynamic behaviour as evidenced in the toluene vapour adsorption measurements (Fig. 1b). Rapid uptake of solvent by Py-BPP at low relative pressures (P/P0 ∼ 0.2) followed by a monotonic pressure increase and ending without saturation at P/P0 ∼ 1 indicates the dynamic nature of the Py-BPP framework. Moreover, the incomplete desorption along with large hysteresis indicate strong interaction of the toluene molecules with the Py-BPP pore surface. We further quantified the solvent uptake by Py-BPP in terms of the equilibrium state of swelling parameter (Q%) and equilibrium solvent content (H%), calculated from the weight of dried and swollen polymers (Fig. 1c and d).63 These parameters reflect the ability of Py-BPP in absorbing various organic liquids under ambient conditions, which is of practical relevance. For various organic solvents, the Q% and H% vary from 900 to 3000 and 90 to 99, respectively, suggesting the superabsorbent nature of Py-BPP. Similar to Py-PP, Py-BPP displayed higher uptake for medium polar solvents like chloroform and ortho-dichlorobenzene (ODCB) than nonpolar (hexane) and polar (ethanol) solvents and failed to swell in water due to its hydrophobic nature. Differences in the amount of solvent uptake might also be dependent on the size and shape of guest molecules and specific π–π interactions between host–guest components. This is a quite remarkable property for Py-PP and Py-BPP polymers, compared to MOFs, which cannot show the superabsorbent nature for a wide range of organic solvents.
Py-BPP exhibits green emission (470–650 nm) with a maximum at 517 nm (λexc = 380 nm). Similar to Py-PP, Py-BPP also displayed red-shifted absorption and emission compared to the tetraphenyl pyrene chromophoric unit (λabs = 320 nm and λem = 450 nm),67 due to the presence of extended conjugation in the polymer. The absorption spectra of both Py-PP and Py-BPP are approximately blue shifted by 30 nm compared to their corresponding statistical copolymers reported by Jiang et al.31,64 However, in terms of fluorescence properties, Py-PP emission closely matches with its statistical copolymer, whereas Py-BPP shows a nearly 70 nm blue shift. These observations once again confirm that the photophysical properties of these polymers are highly dependent on synthesis conditions. The swelling of Py-BPP in various organic solvents is accompanied by fluorescence enhancement indicating the guest induced structural changes in the conjugated backbones of the polymer (Fig. 2a). The soaked polymers in various solvents showed visible colour changes and bright green emissions with enhanced fluorescence intensity at varying orders of magnitude depending on solvent nature and polarity. Interestingly, self-standing fluorescent gels were formed when the polymer was soaked in toluene indicating its soft and superabsorbent nature (Fig. 2b). Additionally, the emission spectra of the solvent-treated polymers were blue shifted by 10–25 nm with respect to the desolvated samples likely due to the unfolding of polymer backbones and reduced aggregation due to swelling. In the present case, fluorescence response of POPs towards organic solvents reiterates the dynamic nature of polymer framework. Maximum fluorescence enhancement (4–5 times) was observed for aromatic non-polar solvents like toluene, whereas the polar solvents like ethanol showed only 2 times enhancement in emissions and did not show any fluorescence response with water, further confirming the hydrophobic interior of the polymer. Hence emission changes with solvent molecules can be used as a probe to understand the swelling-induced structural changes of Py-BPP framework.
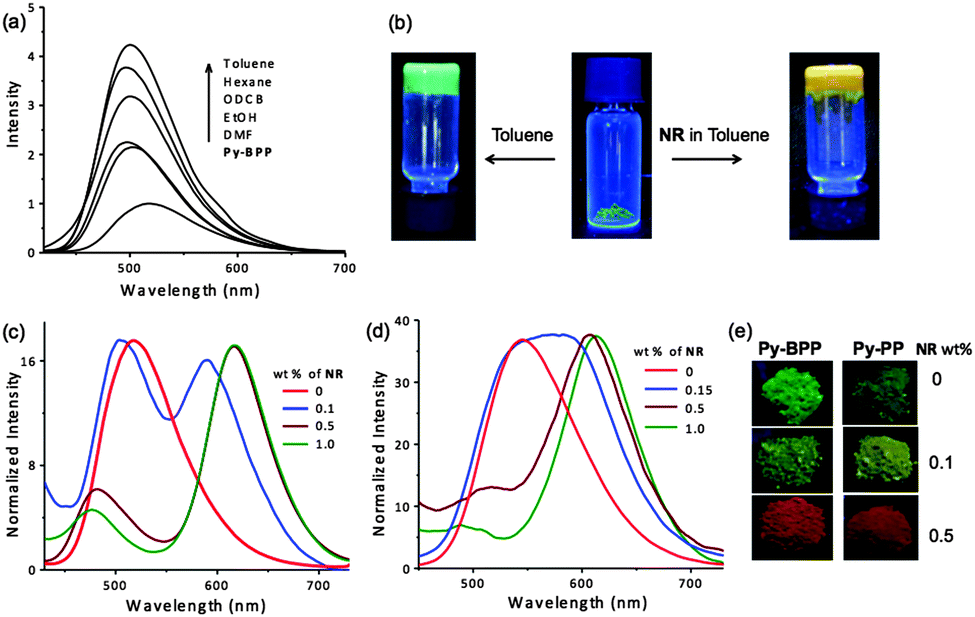 | ||
| Fig. 2 (a) Fluorescence enhancement of Py-BPP after treatment with different organic solvents. (b) Photographs showing swelling and gel formation by Py-BPP powder in toluene and toluene containing NR under 365 nm UV light. Normalized emission spectra of (c) Py-BPP and (d) Py-PP powders with different wt% of NR loading and their (e) corresponding photographs under 365 nm UV light. | ||
We envisaged that the instantaneous swelling of both Py-PP and Py-BPP in various organic solvents and their hydrophobic interior would enable the simple and rapid encapsulation of luminescent hydrophobic dye molecules inside their pores. For this purpose, both polymers were swollen in toluene with varying amounts of a well-known hydrophobic dye such as NR and dried under vacuum (Fig. 2b). Toluene was used as a mediating solvent for guest encapsulation due to good solubility of NR and excellent swelling behaviour of the polymers. A similar solution-state encapsulation was previously reported by Jiang et al. for encapsulating pyrene in the polypyrene porous network, and quenching the fluorescence of the host network was observed upon increasing the amount of guest pyrene.64 In the present study we took advantage of the swelling behaviour of our polymers for encapsulating photofunctional hydrophobic dyes for creating efficient light-harvesting antenna. Since our polymers exhibited a blue shift in their emissions upon solvent guest encapsulation (see above), we expected similar fluorescence changes from the host frameworks as the NR was becoming encapsulated. In contrast to organic solvents, the loading of NR led to quenching in host framework emissions with enhanced NR emission that could be due to energy transfer (see below) from host frameworks to the entrapped guest molecules (Fig. 2c and d). This further led to the tunable solid-state emission of both Py-BPP and Py-PP (Fig. 2e). However, residual emission of both polymer networks after loading with NR was used as a probe for guest encapsulation. Remarkably, the residual emission of both polymers was gradually blue shifted with increasing wt% of NR in toluene suggesting its encapsulation inside the porous cavities of Py-BPP and Py-PP (see above, Fig. 2c and d). Initially a 13 nm blue shift in the emission of Py-BPP was observed with 0.1 wt% of NR loading, which was further blue shifted to 36 nm with 0.5 wt% of NR, finally reaching 40 nm with 1 wt% loading of NR (517 nm to 477 nm). Similarly, blue shifts up to 45 nm were observed in Py-PP emission with 1 wt% of loading NR. When Py-BPP was treated with various solvents, a maximum 15 nm blue shift was observed due to swelling. The observed large blue shifts in NR-loaded polymers compared to the organic solvents was attributed to the large size of NR, which could minimize aggregation of the backbone to a greater extent.
The ability of these polymers to absorb a wide variety of guest molecules irrespective of their size highlights the role of the dynamic porous framework as it can reorganize according to the incoming guest molecules. Moreover, the encapsulation of NR and tunable solid-state emission further encouraged us to swell these polymers with appropriate luminescent acceptor molecules to facilitate efficient Förster-type energy transfer (FRET) from the pyrene frameworks to the guest molecules. Although the energy transfer was shown in porous organic polymers,52–54 encapsulation of guest molecules requires overnight stirring at high temperatures that can be made easier with dynamic porous polymers. Since visible light harvesting is preferred for many practical applications, we used red-emitting DMDP as the acceptor dye to harvest visible excitation energy from Py-PP and Py-BPP. The dynamic and swelling nature of these polymers allowed quick, controlled and room-temperature encapsulation of DMDP dye. Encapsulation of DMDP was performed in a similar way to NR, using toluene as a mediating solvent. In a typical experimental procedure, different amounts of dye were dissolved in 0.5 mL toluene and added to 30 mg of the polymer. The resultant swelled polymer-dye composites were kept at room temperature for 30 min and dried at room temperature as well as in vacuum to completely remove the toluene. The resultant powders loaded with different wt% of DMDP were used for optical measurements. The gradual ground-state colour and excited-state luminescence changes in polymer powders with increasing guest loading further confirm their encapsulation within the porous cavities of the polymers.
The good spectral overlap between emission of Py-BPP and Py-PP polymer powders with the absorption spectrum of DMDP dye indicates that it would be possible to make efficient light-harvesting antennae from these host–guest molecules (Fig. 3). Energy transfer experiments were performed with varying amounts of DMDP loading (0–10 wt%) inside the porous network of the polymers. As the amount of encapsulated DMDP increases, gradual quenching in the emission of both the donor frameworks with concomitant increase in DMDP emission (600–700 nm) intensity was observed (Fig. 4a and b).63 This gives an indication of excitation energy transfer from host frameworks (Py-BPP and Py-PP) to the entrapped dye molecules. To show the encapsulation of dye molecules, we measured the surface area before and after encapsulation of 10 wt% DMDP dye.63 However, in order to evaluate the surface area, we had to degas the dye-containing polymers at high temperature (210 °C) and high vacuum (10−1 Pa). Under such harsh conditions, the incorporated dye molecules can decompose or come out of the pores. This could be the reason why we did not observe a significant decrease in the micropore step.63 Alternatively, we showed that fluorescence could be used to prove the dye encapsulation. When solvent molecules are incorporated we see the blue shift in the fluorescence maximum of the polymer (see above). A similar blue shift in the polymer's fluorescence (up to ∼45 nm) maximum was observed after dye incorporation (Fig. 4a). This convincingly supports the encapsulation of dye molecules in the micropores of the polymer. Moreover, the absence of vibronic features in the DMDP emission when it was loaded into the pores of Py-BPP and Py-PP confirms the existence of orientational dipolar interactions between DMDP and aromatic shells of host frameworks.68 These kinds of dipolar interactions are indeed important to achieve efficient Förster-type energy transfer between donor and acceptor chromophores. This inherent property of DMDP to show orientational dipolar interactions68 with aromatic and polar solvents is an added advantage in addition to the dynamic behaviour of Py-BPP and Py-PP in the construction of present visible light-harvesting antenna. In these host–guest systems, 2.0–2.5 wt% of the guest was able to completely harvest the excitation energy from host frameworks indicating an efficient energy-transfer process. The observed gradual red shift in the emission maxima of encapsulated DMDP molecules (up to 30 nm) by increasing its concentration is attributed to the formation of DMDP aggregates inside the pores at higher amounts of loading (∼10 wt%).58 Emission spectra, collected for different wt% of DMDP-loaded polymers, indicates that at below 2 wt% of DMDP, energy-transfer is mainly to its isolated species (DMDP, λmax = 570 nm), where all DMDP molecules are spatially distributed inside the pore channels. On the other hand, above 2 wt% of DMDP loading, energy transfer happens mostly to its aggregated species (λmax = 604 nm). This was further rationalized by the observed amplified fluorescence of DMDP by energy-transfer in this host–guest system (Fig. 4c and d). In the case of Py-BPP loaded with 10 wt% of the DMDP, excitation at 380 nm (indirect excitation) resulted in a nearly 2 times higher DMDP emission due to energy transfer, compared to its direct excitation at 550 nm. Similarly, a 2.5 times greater amplification in the DMDP emission was observed in the case of Py-PP loaded with 2.5 wt% of DMDP. This clearly indicates that the enhancement in the emission of both isolated and aggregated DMDP molecules inside the pores is indeed due to efficient resonance-energy transfer from the host framework to the entrapped guest molecules. Furthermore, the controlled loading of DMDP molecules inside the polymers leads to the tunable emission from green to yellow to red. This is visually shown in the photographs taken under 365 nm UV irradiation of Py-BPP and Py-PP powders loaded with different wt% of DMDP (insets, Fig. 4a and b). As shown in the insets of Fig. 4a, Py-BPP alone is green emissive, with 1 wt% of DMDP loading leads to greenish yellow, at 2 wt% it is yellow, and finally at 10 wt% the polymer is orange-red emissive. Similar colour trends were seen in for Py-PP loaded with different wt% of DMDP (Fig. 4b). Energy-transfer in these host–guest antenna materials was confirmed by excitation spectra collected at both polymers (host) and DMDP (guest) emission wavelengths (Fig. 5). The appearance of absorption features of both polymer and DMDP in the excitation spectra collected for both Py-BPP–DMDP and Py-BPP–DMDP host–guest systems at DMDP emission (650 nm) wavelength, where polymers have negligible emission, unambiguously supports the population of DMDP-excited states via energy-transfer from host polymers.
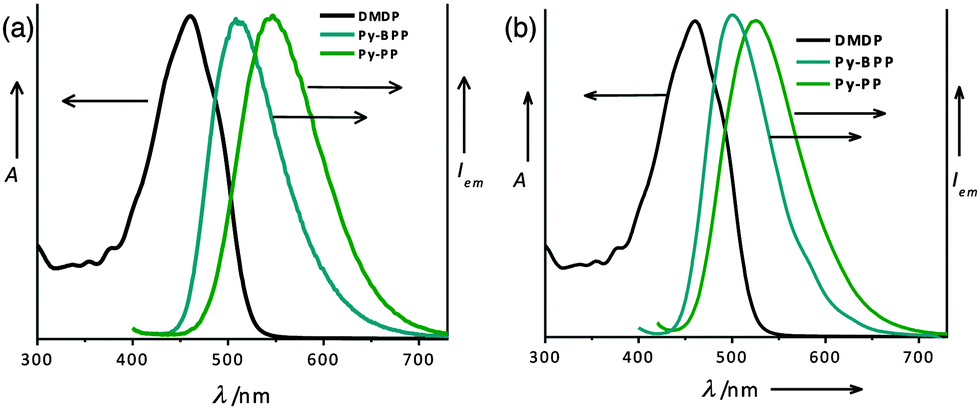 | ||
| Fig. 3 Normalized absorption spectra of DMDP (in toluene) with the emission spectra of Py-BPP and Py-PP (a) before and (b) after swelling in toluene. | ||
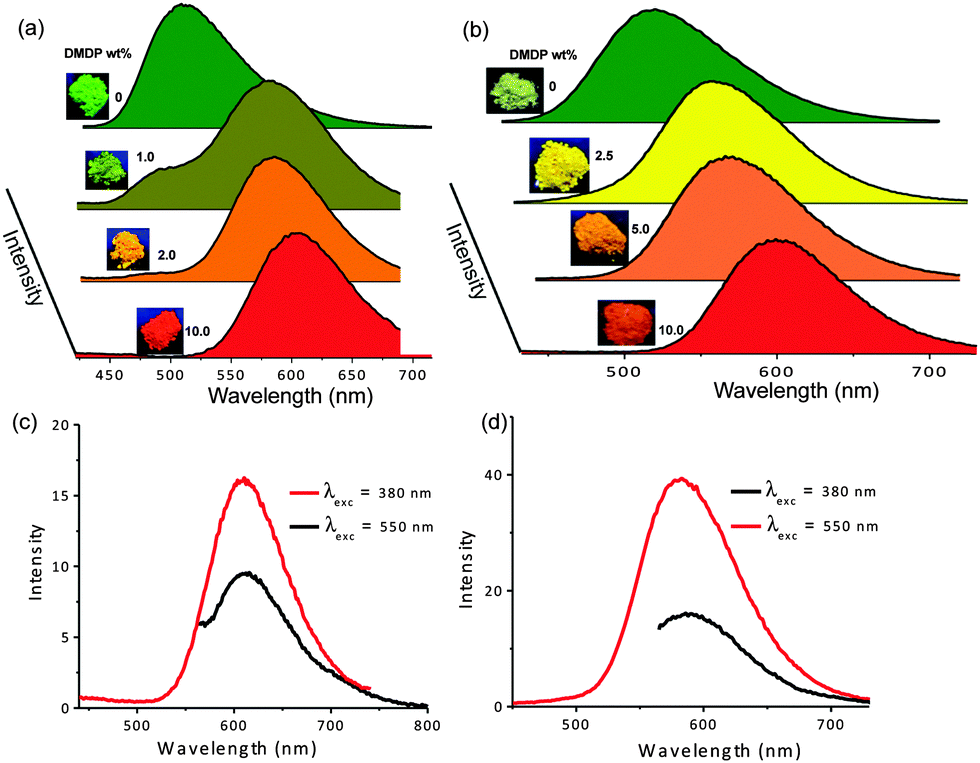 | ||
| Fig. 4 Normalized emission spectra of (a) Py-BPP and (b) Py-PP loaded with different wt% of DMDP. Insets show the photographs of Py-BPP (inset of a) and Py-PP (inset of b) powders under 365 nm UV light loaded with different wt% of DMDP (fluorescence measurements were done with a front-face geometry, λexc = 380 nm). Emission spectra of directly (λexc = 380 nm) and indirectly (λexc = 500 nm) excited (c) Py-BPP loaded with 10.0 wt%, and (d) Py-PP loaded with 2.5 wt% of DMDP dye. | ||
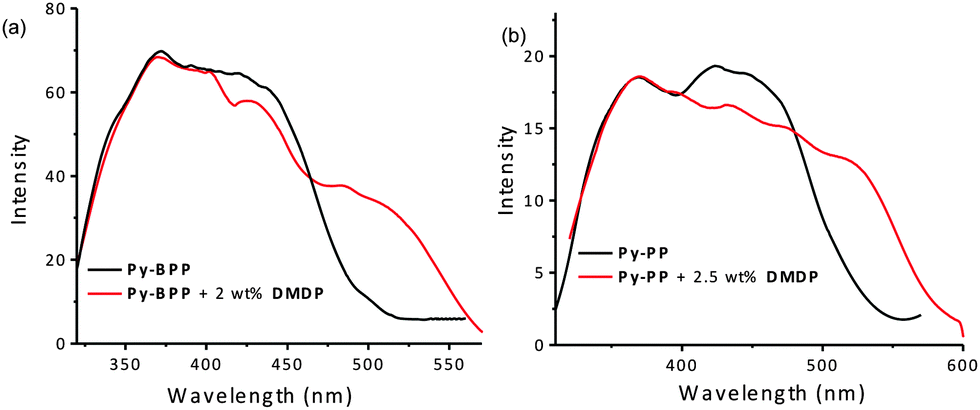 | ||
| Fig. 5 Excitation spectra of (a) Py-BPP powder with DMDP (monitored at 650) and without DMDP (monitored at 540). Excitation spectra of (b) Py-PP powder with DMDP (monitored at 650) and without DMDP (monitored at 560). | ||
Further investigations into fluorescence lifetime and absolute quantum yields of these host–guest antenna materials provided decisive proof for resonance energy transfer from host frameworks to the guest molecules without any radiation reabsorption process (Fig. 6). The lifetime decay profiles were fitted with triexponentials and average lifetime was considered for the analyses. The Py-BPP framework with an average lifetime of 2.49 ns in its solid state decreased to 2.1 ns (monitored at 500 nm) after loading with 2 wt% of DMDP. Similarly, the average lifetime of Py-PP was also shortened from 0.72 ns to 0.58 ns (monitored at 520 nm) with 2.5 wt% of DMDP loading. This decrease in the excited-state lifetimes of host frameworks upon encapsulation of DMDP is a clear indication of the excited-state energy-transfer mechanism for fluorescence quenching of host frameworks. Absolute quantum yield measurements provide further quantitative data for the efficiency of the energy transfer process. The high absolute quantum yield (ϕ = 0.56%) of DMDP (2 wt%) loaded Py-BPP compared to the individual Py-BPP (ϕ = 0.31%) and DMDP (ϕ = 0.24%) indicates efficient resonance-energy transfer without a radiation reabsorption process.
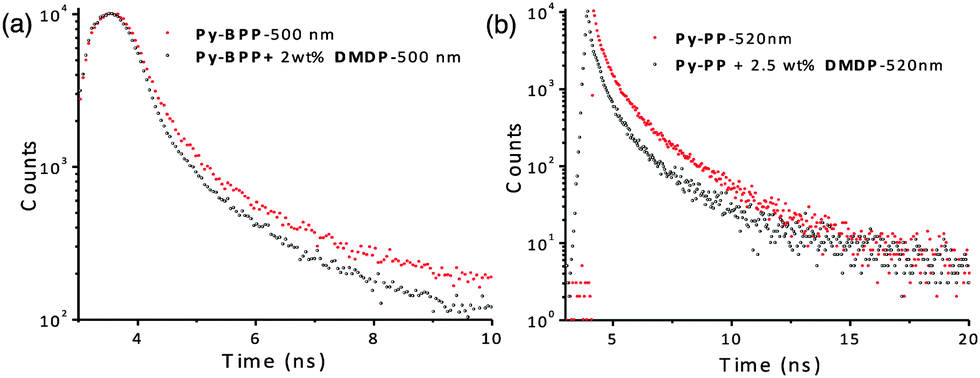 | ||
| Fig. 6 Lifetime decay profiles of (a) Py-BPP and (b) Py-PP alone and with DMDP (lifetime measurements were done with front-face geometry, λexc = 380 nm). Wavelengths in graphs indicate where decay profiles were monitored. | ||
Conclusions
In conclusion, this work introduces a novel class of multi-functional, conjugated organic microporous polymers, in which the porous fluorescent frameworks can undergo structural reorganization in the presence of hydrophobic and aromatic guest molecules. This further imparted unprecedented properties, such as super-absorbency and swelling in these polymers. The dynamic nature of these porous polymeric frameworks enabled facile encapsulation of guest molecules. Furthermore, the fluorescent pyrene scaffold combined with dynamic porosity facilitated guest-induced emission changes and encapsulation of luminescent hydrophobic guests led to tunable solid-state emission via light harvesting. The unique ability of these polymers to encapsulate various guest molecules with fluorescence response behaviour holds great promise for designing smart materials from simple building blocks for sensing, separation and photocatalysis.Acknowledgements
We thank Prof. C. N. R. Rao for his support and encouragement; the Jawaharlal Nehru Centre for Advanced Research (JNCASR) and Department of Science and Technology for financial support; and Profs. S. Balasubramanian and C. Kulkarni for the computational experiments. K.V.R. and R.H. thank Council of Scientific and Industrial Research for research fellowships.Notes and references
- P. D. Frischmann and M. J. MacLachlan, Chem. Soc. Rev., 2013, 42, 871–890 RSC.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed.
- K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC.
- Z. Laughreya and B. C. Gibb, Chem. Soc. Rev., 2011, 40, 363–386 RSC.
- R. Joseph and C. P. Rao, Chem. Rev., 2011, 111, 4658–4702 CrossRef CAS PubMed.
- J. Rebek Jr., Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349–358 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- N. Mizoshita, T. Taniab and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- B. V. V. S. P. Kumar, K. V. Rao, T. Soumya, S. J. George and M. Eswaramoorty, J. Am. Chem. Soc., 2013, 135, 10902–10905 CrossRef CAS PubMed.
- B. V. V. S. P. Kumar, K. V. Rao, S. Sampath, S. J. George and M. Eswaramoorty, Angew. Chem., Int. Ed., 2014, 53, 13073–13077 CrossRef CAS PubMed.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975–13983 CrossRef CAS PubMed.
- Y. Du, K. Mao, P. Kamakoti, P. Ravikovitch, C. Paur, S. Cundy, Q. Li and D. Calabro, Chem. Commun., 2012, 48, 4606–4608 RSC.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS; Z. Chang, D.-S. Zhang, Q. Chen and X.-H. Bu, Phys. Chem. Chem. Phys., 2013, 15, 5430–5442 RSC; Y. Li, S. Roy, T. Ben, S. Xu and S. Qiu, Phys. Chem. Chem. Phys., 2014, 16, 12909–12917 RSC.
- J.-X. Jiang, F. Su, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak, A. I. Cooper and A. Trewin, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- L. Chen, Y. Yang, Z. Guo and D. Jiang, Adv. Mater., 2011, 23, 3149–3154 CrossRef CAS PubMed.
- K. V. Rao, S. Mohapatra, C. Kulkarni, T. K. Maji and S. J. George, J. Mater. Chem., 2011, 21, 12958–12963 RSC.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818–1824 CrossRef CAS.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Chem. Mater., 2012, 24, 969–971 CrossRef CAS.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Polymer, 2014, 55, 1452–1458 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- T. K. Maji, K. Uemura, H.-C. Chang, R. Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 3269–3272 CrossRef CAS PubMed.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed.
- K. V. Rao, S. Mohapatra, C. Kulkarni, T. K. Maji and S. J. George, Chem. – Eur. J., 2012, 18, 4505–4509 CrossRef CAS PubMed.
- X. Liu, Y. Xu, Z. Guo, A. Nagai and D. Jiang, Chem. Commun., 2013, 49, 3233–3235 RSC.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- M. D. Ward, Chem. Soc. Rev., 1997, 26, 365–375 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC; A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332–335 CrossRef CAS PubMed; C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef; C. Vijayakumar, V. K. Praveen, K. K. Kartha and A. Ajayaghosh, Phys. Chem. Chem. Phys., 2011, 13, 4942–4949 RSC.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- S. Inagaki, O. Ohtani, Y. Goto, K. Okamoto, M. Ikai, K. Yamanaka, T. Tani and T. Okada, Angew. Chem., Int. Ed., 2009, 48, 4042–4046 CrossRef CAS PubMed.
- K. V. Rao, K. K. R. Datta, M. Eswaramoorty and S. J. George, Angew. Chem., Int. Ed., 2011, 50, 1179–1184 CrossRef CAS PubMed.
- S. S. Babu, K. K. Kartha and A. Ajayaghosh, J. Phys. Chem. Lett., 2010, 1, 3413–3424 CrossRef CAS.
- K. V. Rao, K. K. R. Datta, M. Eswaramoorthy and S. J. George, Adv. Mater., 2013, 25, 1713–1718 CrossRef CAS PubMed.
- K. V. Rao, K. K. R. Datta, M. Eswaramoorthy and S. J. George, Chem. – Eur. J., 2012, 18, 2184–2194 CrossRef CAS PubMed.
- K. V. Rao, A. Jain and S. J. George, J. Mater. Chem. C, 2014, 2, 3055–3064 RSC.
- X. Zhang, M. A. Ballem, Z.-J. Hu, P. Bergman and K. Uvdal, Angew. Chem., Int. Ed., 2011, 50, 5729–5733 CrossRef CAS PubMed.
- X. Zhang, M. A. Ballem, M. Ahrén, A. Suska, P. Bergman and K. Uvdal, J. Am. Chem. Soc., 2010, 132, 10391–10397 CrossRef CAS PubMed.
- X. Zhang, Z.-K. Chen and K. P. Loh, J. Am. Chem. Soc., 2009, 131, 7210–7211 CrossRef CAS PubMed.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed.
- R. Haldar, K. V. Rao, S. J. George and T. K. Maji, Chem. – Eur. J., 2012, 19, 5848–5852 CrossRef PubMed.
- R. Haldar, R. Matsuda, S. Kitagawa, S. J. George and T. K. Maji, Angew. Chem., Int. Ed., 2014, 53, 11772–11777 CrossRef CAS PubMed.
- M. V. Suresh, S. J. George and T. K. Maji, Adv. Funct. Mater., 2013, 23, 5585–5590 CrossRef.
- N. Mizoshita, Y. Goto, Y. Maegawa, T. Tani and S. Inagaki, Chem. Mater., 2010, 22, 2548–2554 CrossRef CAS.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- A. Patra and U. Scherf, Chem. – Eur. J., 2012, 18, 10074–10080 CrossRef CAS PubMed.
- V. M. Suresh, S. Bonakala, S. Roy, S. Balasubramanian and T. K. Maji, J. Phys. Chem. C, 2014, 118, 24369–24376 CAS.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- Y. Xu, A. Nagai and D. Jiang, Chem. Commun., 2013, 49, 1591–1593 RSC.
- A. Ajayaghosh, C. Vijayakumar, V. K. Praveen, S. S. Babu and R. Varghese, J. Am. Chem. Soc., 2006, 128, 7174–7175 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- See ESI†.
- J.-X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J.-X. Jiang, A. Trewin, F. B. Su, C. D. Wood, H. J. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
- T. Oyamada, S. Akiyama, M. Yahiro, M. Saigou, M. Shiro, H. Sasabe and C. Adachi, Chem. Phys. Lett., 2006, 421, 295–299 CrossRef CAS.
- S. L. Bondarev, V. N. Knyukshto, V. I. Stepuro, A. P. Stupak and A. A. Turban, J. Appl. Spectrosc., 2004, 71, 194–201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures. See DOI: 10.1039/c5cp05052a |
| This journal is © the Owner Societies 2016 |