
Rotational spectroscopy of the atmospheric photo-oxidation product o-toluic acid and its monohydrate†
Elijah G.
Schnitzler
,
Brandi L. M.
Zenchyzen
and
Wolfgang
Jäger
*
Department of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta T6G 2G2, Canada. E-mail: wolfgang.jaeger@ualberta.ca; Tel: +1-780-492-5020
First published on 23rd November 2015
Abstract
o-Toluic acid, a photo-oxidation product in the atmosphere, and its monohydrate were characterized in the gas phase by pure rotational spectroscopy. High-resolution spectra were measured in the range of 5–14 Hz using a cavity-based molecular beam Fourier-transform microwave spectrometer. Possible conformers were identified computationally, at the MP2/6-311++G(2df,2pd) level of theory. For both species, one conformer was identified experimentally, and no methyl internal rotation splittings were observed, indicative of relatively high barriers to rotation. In the monomer, rocking of the carboxylic acid group is a large amplitude motion, characterized by a symmetrical double-well potential. This and other low-lying out-of-plane vibrations contribute to a significant (methyl top-corrected) inertial defect (−1.09 amu Å2). In the monohydrate, wagging of the free hydrogen atom of water is a second large amplitude motion, so the average structure is planar. As a result, no c-type transitions were observed. Water tunneling splittings were not observed, because the water rotation coordinate is characterized by an asymmetrical double-well potential. Since the minima are not degenerate, tunneling is precluded. Furthermore, a concerted tunneling path involving simultaneous rotation of the water moiety and rocking of the carboxylic acid group is precluded, because the hilltop along this coordinate is a virtual, rather than a real, saddle-point. Inter- and intramolecular non-covalent bonding is discussed in terms of the quantum theory of atoms in molecules. The percentage of o-toluic acid hydrated in the atmosphere is estimated to be about 0.1% using statistical thermodynamics.
Introduction
In the atmosphere, photo-oxidation of volatile organic compounds (VOCs) is a significant source of carboxylic acids.1 These and other low-volatility oxygenated products form secondary organic aerosol (SOA),2 by condensing onto pre-existing particles3 or first participating in nucleation.4 For example, the addition of aromatic carboxylic acids to gas-phase mixtures of sulphuric acid and water increases the rate of nucleation;5 at the molecular scale, a ternary critical nucleus (from which further particle growth is spontaneous)6 may result from the inclusion of a single carboxylic acid molecule in an otherwise binary cluster.7 One step towards characterizing critical nuclei is the characterization of carboxylic acid–water clusters.Aromatic VOCs are the most important anthropogenic SOA precursors, accounting for up to 12% of global SOA.8 The structural isomers of xylene are emitted by transportation and industry, such as oil refining.9 For instance, the concentration of o-xylene measured over the Athabasca oil sands in Alberta is 85 times greater than the background concentration.10 SOA mass yields of about 10% to 25% have been measured for the oxidation of o-xylene by hydroxyl radical,11 and the reaction mechanism has been investigated thoroughly, both experimentally (in smog chambers)12,13 and theoretically (using ab initio calculations).14 Hydrogen abstraction from o-xylene by hydroxyl radical leads to the formation of o-tolualdehyde;15 further oxidation leads to the formation of o-toluic acid (OTA).16 OTA and its structural isomers, m- and p-toluic acid, have been detected in atmospheric aerosols sampled in the North China Plain, and the diurnal trend in their concentrations is similar to that of benzoic acid (BA),17 possibly indicating a mutual source in addition to photo-oxidation, such as primary engine exhaust emissions.18
OTA has been investigated in earlier spectroscopic studies. Ito et al. used ultraviolet absorption spectroscopy to demonstrate increasing formation of the hydrogen-bonded OTA–OTA complex in solution with decreasing temperature.19 More recently, Babu et al. investigated OTA in the solid phase using Fourier-transform infrared and Raman spectroscopies.20 Using X-ray diffraction, Polito et al. determined that the crystal structure of OTA is composed of hydrogen-bonded ribbons; in each monomer unit, the carbonyl oxygen atom, rather than that bonded to the acidic hydrogen atom, is neighbouring the methyl group.21 In the gas phase, the ionization energy of OTA has been measured by photo-ionization mass spectrometry;22 however, the structure of OTA in the gas phase has not been investigated. Furthermore, the OTA–H2O complex has not been investigated previously, either experimentally or theoretically.
Using high-resolution microwave spectroscopy, the structures (and, in some cases, dynamics) of hydrates of several atmospherically-relevant carboxylic acids have been characterized in the past, including BA,23 formic acid,24 and perfluorobutyric acid,25 a persistent organic pollutant. In the minimum energy conformers of these complexes, water is bound to the acid by two hydrogen bonds, participating in the formation of a six-membered intermolecular ring by acting as both proton donor and acceptor.23
Here, we investigate the structure and dynamics of OTA and OTA–H2O in the gas phase using high resolution microwave spectroscopy and ab initio calculations. Transitions of one conformer of each species were observed, and the spectra were fitted using a conventional semi-rigid rotor Hamiltonian. Internal dynamics, including large amplitude motions of the methyl group and the water moiety, are discussed in terms of calculated barrier heights. Hydrogen bond energies and dissociation energies are discussed in the context of the quantum theory of atoms in molecules (QTAIM) and symmetry adapted perturbation theory (SAPT). Finally, the percentage of OTA hydrated in the atmosphere is estimated on the basis of dissociation energies and partition functions, using statistical thermodynamics.
Methods
Experimental
A cavity-based molecular beam Fourier-transform spectrometer was used to measure the rotational spectra of OTA and OTA–H2O in the range of 5–14 GHz. The spectrometer has been described in detail previously.26,27 Briefly, the sample is injected through the nozzle, which is set near the centre of one of two aluminium mirrors that compose the microwave resonator. The difference in pressure across the nozzle results in a supersonic expansion, in the earlier stages of which the sample molecules are cooled to a (rotational) temperature of about 1 K by collisions with backing gas molecules. A near-resonant π/2 microwave excitation pulse causes polarization of the sample, after which molecules spontaneously emit radiation at the transition frequency. The emission signal is digitized in the time domain, averaged, and Fourier-transformed into the frequency spectrum. Because the resonator axis and supersonic expansion are oriented coaxially, lines are split into Doppler pairs. The average is used as the rest frequency. Automated scans for targeted transitions were collected in 0.2 MHz increments.Reagent grade OTA (Aldrich, 99%) was used without further purification. OTA was placed in a stainless steel container heated to about 110 °C, downstream of a heated coil of copper tubing. Backing gas, Ne (3–5 atm), was passed over the melted sample before reaching a pulsed solenoid nozzle, which was heated to about 250 °C in order to prevent deposition in the nozzle. To prepare OTA–H2O, we passed a mixture of about 0.1% water in Ne over the OTA sample.
Computational
Optimized structures of OTA and OTA–H2O were used to predict the rotational spectra of these species and to define frequency ranges to search for targeted transitions. Both density functional theory (DFT) and wavefunction-based electronic structure calculations were used in Gaussian09.28 For DFT calculations, the B3LYP functional was used.29 For wavefunction-based calculations, second order Møller–Plesset (MP2) perturbation theory was applied.30 Both methods were implemented with the Pople basis set 6-311++G(2df,2pd).31 Minimum energy structures were found using geometry optimizations, followed by frequency analyses to verify that no imaginary frequencies occurred. Zero-point energy (ZPE) corrections were also determined from frequency analyses. Transition states along coordinates associated with large amplitude motions were initially located using relaxed scans of the potential energy surface. Transition states were subsequently verified by geometry optimizations (“opt = ts”) and frequency analyses, which gave one imaginary frequency. For the complex, the Boys and Bernardi counterpoise correction was used to correct for basis set superposition error (BSSE).32 QTAIM,33 as implemented in Multiwfn,34 was used to analyse the calculated electron density distributions of OTA and OTA–H2O and to characterize inter- and intramolecular non-covalent bonding. In order to export the MP2-calculated wavefunctions to input files for Multiwfn, the keywords “density = current” and “output = wfx” were used. Atomic energies were corrected for errors resulting from numerical integration.35 SAPT,36 as implemented in PSI4,37 was used as an alternate method to characterize the non-covalent bonding. As a compromise between accuracy and computing time, we used zeroth-order SAPT (SAPT0) with the jun-cc-pVDZ basis set.38Rotational spectra were predicted from the calculated rotational constants using the PGOPHER program.39 In turn, the measured transition frequencies were input in PGOPHER to fit rotational and centrifugal distortion constants, using Watson's A-reduction Hamiltonian.40
Results and discussion
Spectrum, structure, and internal dynamics of the OTA monomer
Four conformers of the OTA monomer were located at the MP2/6-311++G(2df,2pd) level of theory, as illustrated in Fig. 1. In the two most stable conformers, OTA-I and OTA-II, the carboxylic acid group is rotated slightly (about 7°) out of the plane of the aromatic ring, and the acidic hydrogen atom is oriented in a syn-conformation with respect to the carbonyl oxygen atom. In OTA-I, the carbonyl oxygen atom, rather than that of the hydroxyl moiety, is adjacent to the methyl group. Rotating about the bond between the aromatic ring and the carboxylic acid group by roughly 180° results in OTA-II. In the two least stable conformers, OTA-III and OTA-IV, the acidic hydrogen atom is oriented in an anti-conformation. In OTA-III, the carbonyl group is rotated approximately 35° out of the plane of the aromatic ring, and in OTA-IV, it is rotated about 180° from that of OTA-III. Relative energies (including ZPE corrections), relative free energies, abundances, rotational constants, and dipole moments for the optimized structures are shown in Table 1.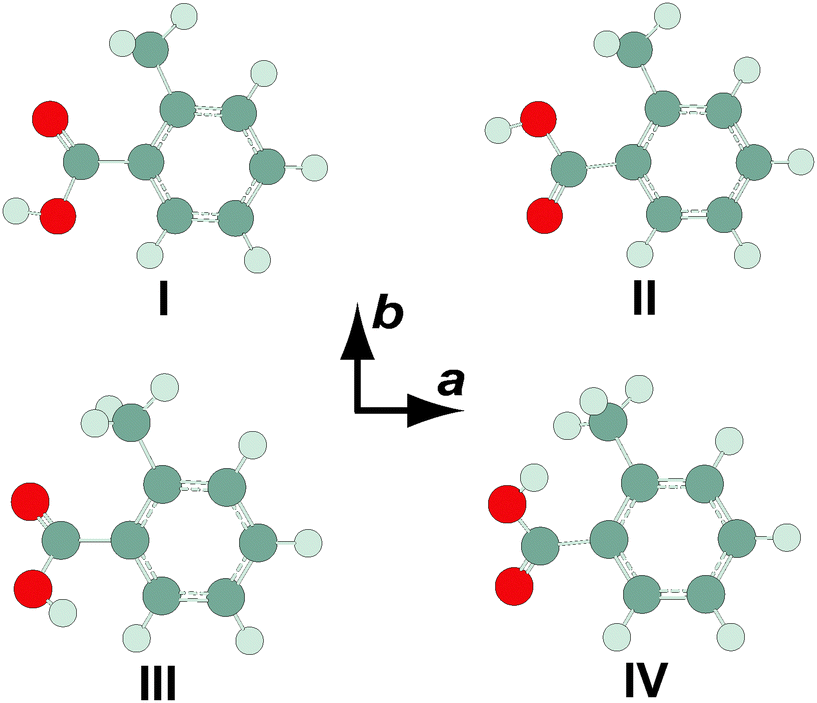 | ||
| Fig. 1 Conformers of o-toluic acid monomer, optimized at the MP2/6-311++G(2df,2pd) level of theory. | ||
| Constant | OTA-I | OTA-II | OTA-III | OTA-IV |
|---|---|---|---|---|
| a Total energy (sum of electronic and zero-point energies) relative to the minimum energy conformer. b Relative free energy. c Percent abundance (based on relative free energy) at 523 K, the nozzle temperature. d Asymmetry parameter, κ = (2B − A − C)/(A − C). | ||||
| ΔEa/kJ mol−1 | 0 | 5.0 | 20.2 | 25.1 |
| ΔGb/kJ mol−1 | 0 | 5.5 | 22.0 | 26.1 |
| P /% | 77.6 | 21.8 | 0.5 | 0.2 |
| A/MHz | 2230.28 | 2241.51 | 2236.61 | 2244.39 |
| B/MHz | 1218.58 | 1208.33 | 1188.66 | 1175.23 |
| C/MHz | 793.10 | 794.79 | 815.97 | 822.29 |
| κ | −0.41 | −0.43 | −0.48 | −0.50 |
| |μa|/D | 1.62 | 1.35 | 4.44 | 4.18 |
| |μb|/D | 1.03 | 1.99 | 1.31 | 2.85 |
| |μc|/D | 0.13 | 0.33 | 1.52 | 1.53 |
We began our spectral search by scanning for transitions of OTA-I and OTA-II, because OTA-III and OTA-IV are expected to be present at abundances of less than 0.5%, even at the elevated nozzle temperature. The predicted rotational constants of the two most stable conformers are very similar, so targeted transitions were predicted to vary in frequency only slightly. For example, we first targeted the JKa,Kc = 51,5–41,4 transition using an automated scan in the range of 8500–8750 MHz; for OTA-I and OTA-II, the predicted frequencies of this transition are about 8666 and 8670 MHz, respectively. In the experimental scan, however, only one candidate for this particular transition was observed. Among others, the 41,3–31,2 and 50,5–40,4 transitions were also in this range, and together these three lines were used in a preliminary spectroscopic fitting procedure. In all, 27 a-type transitions and 13 b-type transitions were observed (see Table S1, ESI†), whose frequencies were used to determine the rotational and centrifugal distortion constants shown in Table 2. A representative transition is shown in Fig. 2. The percent mean absolute discrepancies between predicted and experimental rotational constants are only 0.6 and 0.7% for OTA-I and OTA-II, respectively, so the rotational constants alone do not strictly allow assignment of the spectrum to either conformer. However, achieving optimal intensity for b-type transitions required a longer microwave pulse width than that required for a-type transitions, indicating that the b-dipole moment component is smaller than the a-dipole moment component. This observation is consistent with OTA-I, which has a predicted abundance of 77.6%, but not OTA-II. Conformer OTA-I has also been observed in the crystal structure of the acid.21 OTA-II has a predicted abundance of 21.8% at the elevated nozzle temperature, and it is separated from OTA-I by a significant barrier, 17.2 kJ mol−1 at the B3LYP/6-311++G(d,p) level of theory, which likely prevents conformational cooling in the molecular expansion. However, we failed to achieve adequate sensitivity to detect OTA-II, because (i) the gas-phase sample was difficult to generate, and (ii) the OTA spectrum is very dense – even at a rotational temperature of 1 K, many energy levels are populated.
| Constant | OTA | OTA–H2O |
|---|---|---|
| a One standard deviation in the last decimal place before the parentheses. b Asymmetry parameter, κ = (2B − A − C)/(A − C). c Number of transitions included in fit. d Root-mean-square deviation of fit. | ||
| A/MHz | 2216.3345(4)a | 2190.358(1) |
| B/MHz | 1210.6114(1) | 641.7891(1) |
| C/MHz | 787.99923(5) | 498.88178(8) |
| ΔJ/kHz | 0.0320(7) | 0.0208(4) |
| ΔJK/kHz | 0.013(4) | 0.077(5) |
| ΔK/kHz | 0.12(1) | 0.2(2) |
| δ J /kHz | 0.0111(4) | 0.0046(2) |
| δ K /kHz | 0.054(5) | 0.11(2) |
| κ | −0.41 | −0.83 |
| N | 40 | 36 |
| σ /kHz | 0.8 | 0.7 |
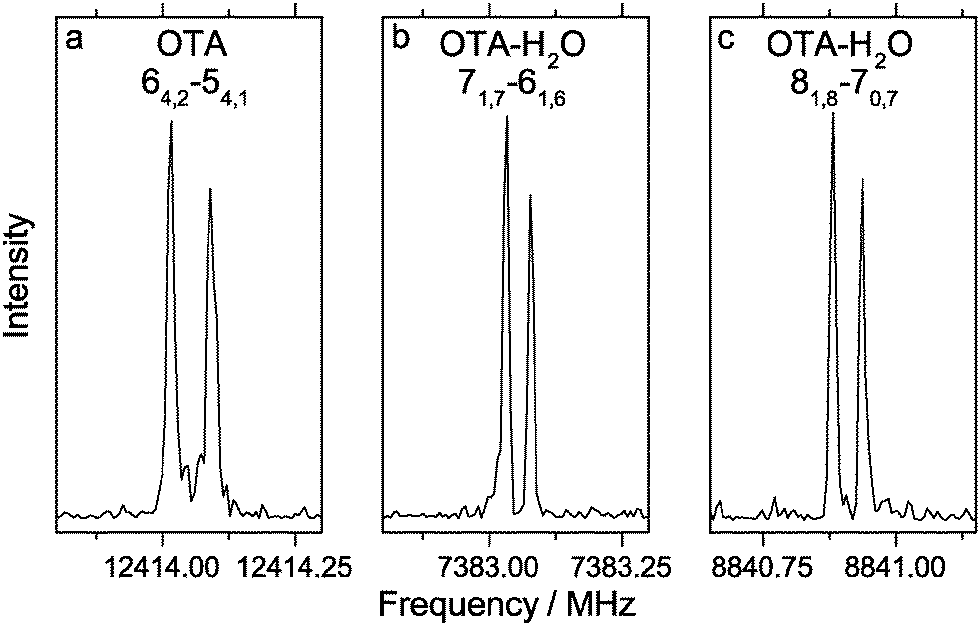 | ||
| Fig. 2 Representative transitions of the o-toluic acid monomer and its monohydrate, measured using 0.5 mW excitation pulses. Panel (a) illustrates an average of 100 cycles, measured with an excitation pulse length of 0.6 μs; panel (b) illustrates an average of 500 cycles, measured with an excitation pulse length of 0.8 μs; panel (c) illustrates an average of 3000 cycles, measured with an excitation pulse length of 1.5 μs. | ||
No methyl internal rotation splittings were observed. At the MP2/6-311++G(2df,2pd) level of theory, the height of the three-fold barrier to methyl rotation, V3, is 7.87 kJ mol−1. This barrier is relatively high; for example, the experimentally-determined barriers of o-chlorotoluene41 and o-fluorotoluene42 are about 5.58 kJ mol−1 and 2.72 kJ mol−1, respectively. In general, the more unsymmetrical the ring geometry on either side of the methyl substituent, the higher the barrier to methyl rotation, because of anisotropic steric repulsion.43 Thus, as the size of the substituent at the ortho position of toluene increases, the barrier height increases, as well. Based on the experimental rotational and centrifugal distortion constants and the calculated structural parameters and V3, the expected splittings can be predicted using the program XIAM.44 The values of the structural properties δ (the angle between methyl top axis and the a principal inertial axis), ε (the angle between the projection of the methyl top axis onto the bc plane and b principal inertial axis), and F0 (the methyl top rotation constant) are about 2.018 rad, 0.019 rad, and 161.8 GHz, respectively. The resulting splitting predicted for the 64,2–54,1 transition is 28 kHz. Since no splitting is observed (see Fig. 2a), the actual V3 is likely higher than calculated. If we increase V3 by 10%, to give 8.65 kJ mol−1, the predicted splitting (12 kHz) is roughly twice the experimental resolution, so we would be able to measure one point between the two peaks. Consequently, 8.65 kJ mol−1 is likely a lower limit of the barrier height.
In the equilibrium structure of OTA-I calculated at the MP2/6-311++G(2df,2pd) level of theory, the carboxylic acid group is not in the plane of the aromatic ring, so the c-dipole moment component is small but non-zero. However, despite many averages, no c-type transitions were observed. The lowest energy normal mode vibration (22 cm−1) is the large amplitude “rocking” motion of the carboxylic acid group. A transition state (whose one imaginary frequency normal mode corresponds to rocking of the carboxylic acid group) was found to connect the mirror-image minima. The symmetrical double-well potential along this coordinate is shown in Fig. 3. The small barrier lies below the ZPE of the minima, so the average geometry is that of the transition state. The transition state has Cs symmetry, resulting in a c-dipole moment component of zero, which is consistent with the absence of c-type transitions. We note that the B3LYP/6-311++G(2df,2pd) level of theory fails to predict a double-well potential (see Fig. 3). A similar observation has been made for isomers of dimethylbenzaldehydes.45
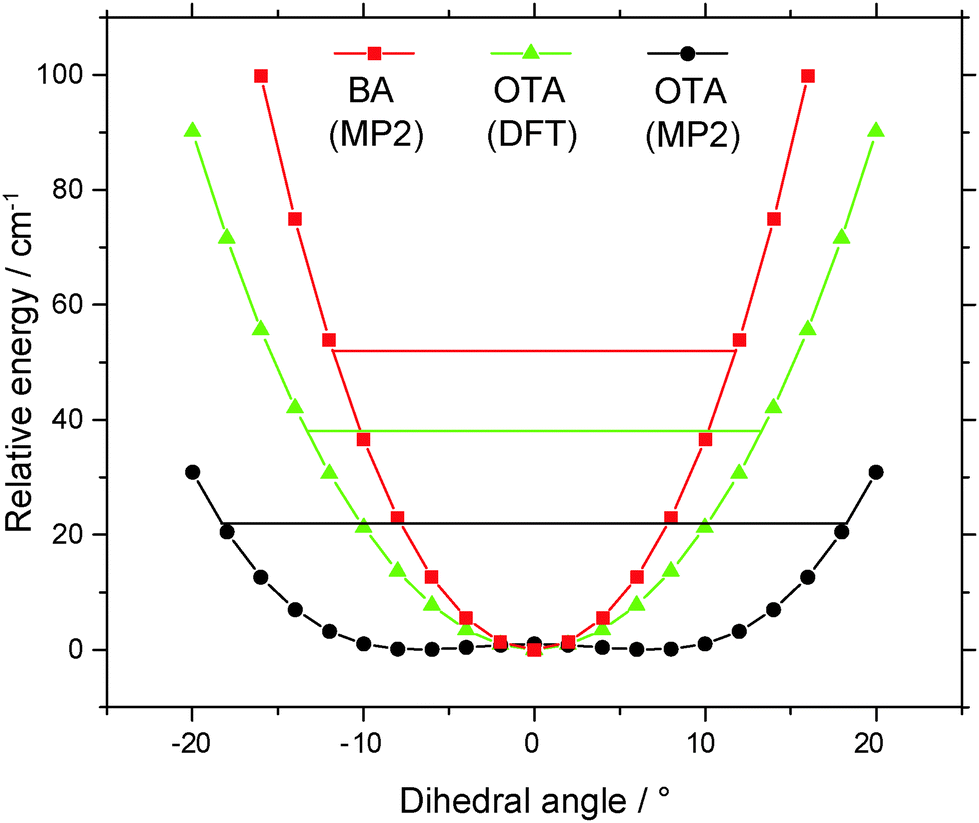 | ||
| Fig. 3 Potential energy curves of benzoic acid and o-toluic acid monomers along the “rocking” coordinate of the carboxylic acid group. | ||
In the average structure of OTA-I, all the atoms lie in the plane of symmetry, except the two staggered hydrogen atoms of the methyl group. Even molecules that have planar equilibrium geometries may still have non-zero mass distributions outside of the plane, because of low-lying zero-point vibrations. The inertial defect, Δ = Ic − Ib − Ia, is a measure of this mass distribution outside of the plane.46 For example, BA is calculated to have a planar equilibrium geometry at both the DFT and MP2 levels of theory, but it has a small negative inertial defect (−0.37 amu Å2).23,47 For non-planar molecules with a plane of symmetry, the inertial defect can be corrected for the masses of the out-of-plane atoms, so that it still reflects low-lying out-of-plane motions. For example, in molecules with methyl groups, the inertia of the out-of-plane hydrogen atoms must be added to the inertial defect to give a methyl-top-corrected inertial defect, Δcorr = Δ + 2∑mHcH2, where mH and cH are the mass and c-coordinate, respectively, of a given out-of-plane hydrogen atom.48 For the present purposes, we take the c-coordinates from the MP2-calculated geometry of the transition state along the rocking coordinate of the carboxylic acid group. The resulting value of Δcorr is −1.09 amu Å2, significantly greater in magnitude than the inertial defect of BA. This difference is consistent with the greater number of low-lying out-of-plane vibrational modes (<400 cm−1) predicted for OTA than BA, as shown in Table 3. Additionally, for the lowest-lying mode, rocking of the carboxylic acid group, the ZPE level intersects the potential energy curve at about ±18° in OTA and at about ±12° in BA, so this large amplitude motion involves greater out-of-plane displacement in OTA than in BA (see Fig. 3).
Spectrum, structure, and internal dynamics of OTA monohydrate
Four minimum energy conformers were located at the MP2/6-311++G(2df,2pd) level of theory for the OTA monohydrate (see Fig. 4). The two most stable conformers are complexes of OTA-I and II. In OTA–H2O-I, the acid moiety is in the geometry of OTA-I, and it is bound to the water moiety by two hydrogen bonds, forming a six-membered intermolecular ring. The oxygen atom and the unbound hydrogen atom of the water moiety are out of the plane of the carboxylic acid group, which is itself rotated slightly out of the plane of the aromatic ring, resulting in a predicted c-dipole moment component of 1.24 D. The two higher energy conformers of OTA–H2O are complexes of OTA-III and IV. These conformers are predicted to have abundances of about 0.1% even at the elevated nozzle temperature, so they are not our focus here. Relative energies (including ZPE corrections), relative free energies, abundances, rotational constants, and dipole moments for the optimized structures are shown in Table 4.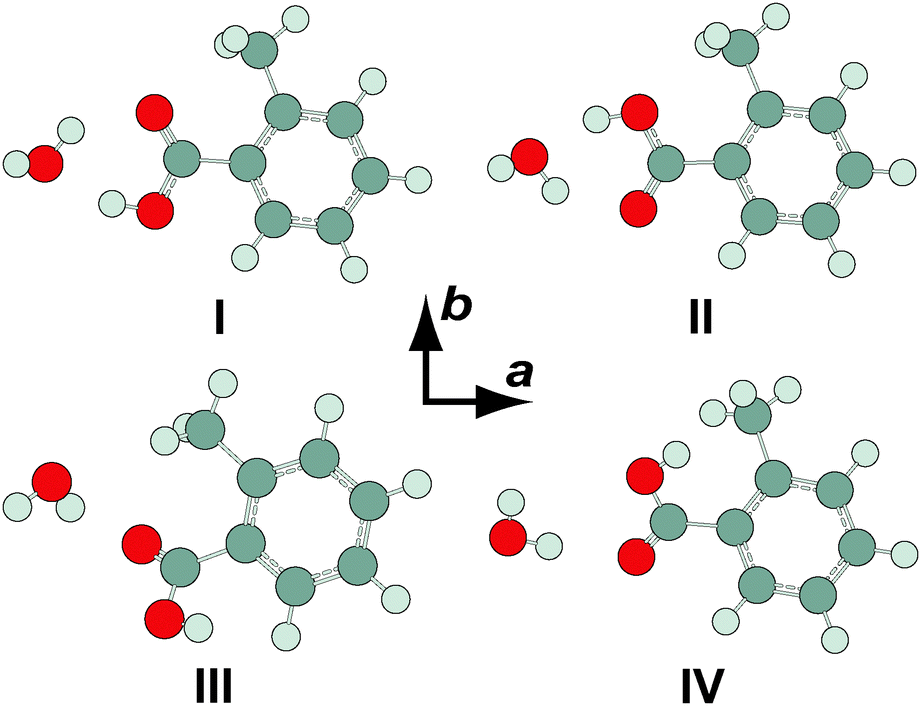 | ||
| Fig. 4 Conformers of o-toluic acid monohydrate, optimized at the MP2/6-311++G(2df,2pd) level of theory. | ||
| Constant | OTA–H2O-I | OTA–H2O-II | OTA–H2O-III | OTA–H2O-IV |
|---|---|---|---|---|
| a Total energy (sum of electronic energy, ZPE, and BSSE) relative to the minimum energy conformer. b Relative free energy. c Percent abundance (based on relative free energy) at 523 K, the nozzle temperature. d Asymmetry parameter, κ = (2B − A − C)/(A − C). | ||||
| ΔEa/kJ mol−1 | 0.0 | 3.6 | 33.3 | 39.4 |
| ΔGb/kJ mol−1 | 0.0 | 3.7 | 28.1 | 30.0 |
| P /% | 69.9 | 29.9 | 0.1 | 0.1 |
| A/MHz | 2203.87 | 2214.81 | 1814.48 | 2146.43 |
| B/MHz | 652.02 | 650.89 | 728.76 | 588.75 |
| C/MHz | 506.52 | 507.95 | 559.30 | 477.95 |
| κ | −0.83 | −0.83 | −0.73 | −0.87 |
| |μa|/D | 0.66 | 0.56 | 3.68 | 6.78 |
| |μb|/D | <0.01 | 0.18 | 1.46 | 2.86 |
| |μc|/D | 1.24 | 1.23 | 1.44 | 1.69 |
We first targeted the 71,6–61,5, 72,5–62,4, and 81,8–71,7 transitions of OTA–H2O-I and OTA–H2O-II in an automated scan in the range of 8300–8500 MHz. Using 125 cycles per step, we observed three very weak candidate lines, which were used in our preliminary fit. In all, 32 a-type transitions and 4 b-type transitions were observed (see Table S2, ESI†). Two representative transitions are shown in Fig. 2. On average, the rotational constants (see Table 2) are most consistent with the OTA–H2O-I conformer. At first glance, the presence of b-type transitions would not be expected for the minimum energy structure, which has only a very small ab initio b-dipole moment component (<0.01 D). However, as was the case for the monomer, rocking of the carboxylic acid group (see Fig. 5a) is barrier-less in the monohydrate. The average structure is more similar to the geometry of the transition state along this coordinate, in which the acid group is in the plane of the aromatic ring, than the minimum. This transition state has a larger b-dipole moment component (0.20 D) than the minimum energy structure. Based on the intensity of lines from this conformer under optimal conditions, we expect that observing the second conformer would require several hundred cycles per step, so we did not continue our conformational search.
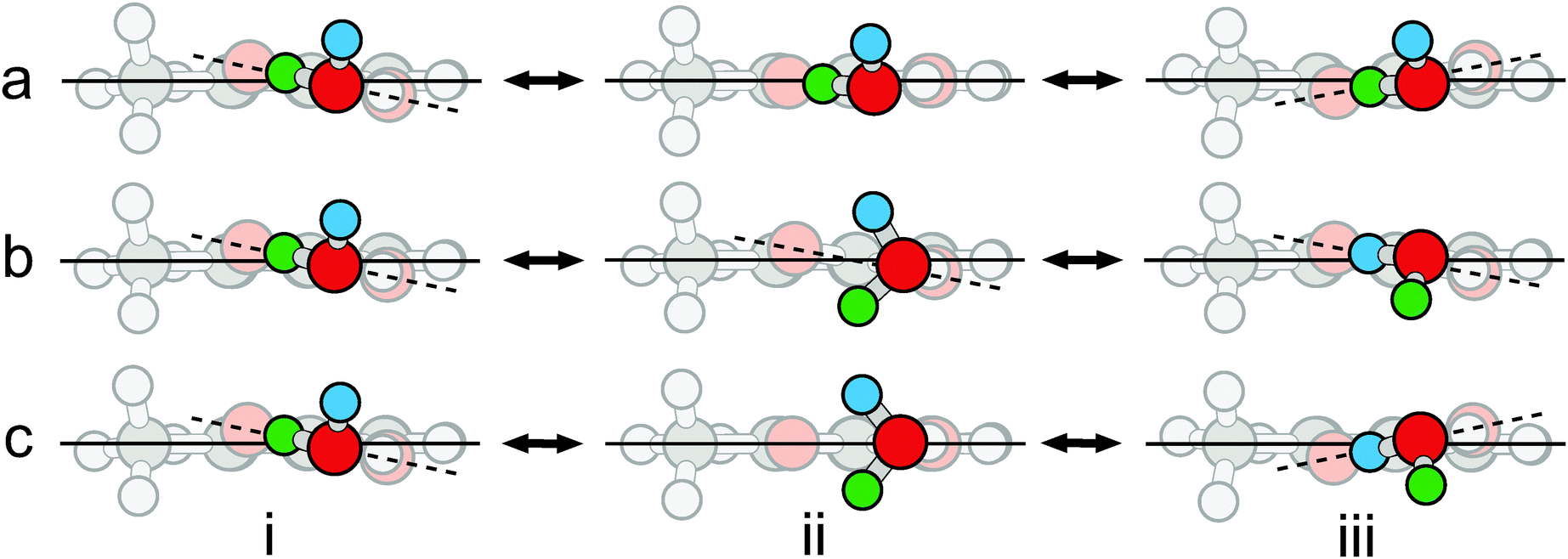 | ||
| Fig. 5 Three internal motions of o-toluic acid monohydrate: (a) rocking of the carboxylic acid group; (b) rotation of the water moiety; and (c) concerted rocking and rotation. The o-toluic acid moiety is faded to distinguish it from the water moiety. The point of view is in the plane of the aromatic ring, represented by the solid line. The plane defined by the four atoms of the carboxylic acid group is represented by the dashed line. The two inequivalent minima, as well as the transition states to the motions shown in (a) and (b), were optimized at the MP2/6-311++G(2df,2pd) level of theory. The structure bridging the equivalent minima illustrated in (c) was generated based on the above transition states, not optimized, and characterized using a single point calculation; it has two imaginary frequencies. | ||
We searched ±10 MHz about the frequencies at which c-type transitions were predicted to occur by the final fit, but no c-type transitions were observed. Their absence is inconsistent with the large calculated c-dipole moment component (1.24 D). We located a transition state in which the unbound hydrogen atom of water is in the plane of the carboxylic acid group. Though the electronic energy of the transition state is higher than that of the minimum, the ZPE of the minimum lies 10 cm−1 above the barrier. In the minimum energy structure, this large amplitude motion corresponds to a normal mode involving “wagging” of the unbound hydrogen atom of the water moiety (282 cm−1). Since both rocking of the acid group and wagging of the free hydrogen atom are barrier-less, the average structure has a plane of symmetry, and a c-dipole moment component of zero, explaining the absence of c-type transitions. Similarly, c-type transitions were not observed for propanoic acid–H2O49 and BA–H2O.23
The absence of tunneling splittings is striking, in particular in light of the, sometimes complex, observed splittings in other carboxylic acid hydrates,23,52 and warrants further consideration. Regarding only methyl internal rotation, their absence can be easily rationalized. Based on the calculated V3 of 7.59 kJ mol−1, as well as the values of δ, ε, and F0 derived from the calculated structure (1.939 rad, 0.027 rad, and 161.7 GHz, respectively), none of the measured transitions are predicted to have splittings greater than 10 kHz. The value of V3 is lower than the V3 lower limit proposed above for OTA monomer (8.65 kJ mol−1). For comparison, the experimentally-determined barrier of the acetic acid monohydrate is also lower than that of the acetic acid monomer.50 Considering the water moiety,51 however, the absence of tunneling splittings is more difficult to rationalize. For example, water tunneling splittings were observed for trifluoroacetic acid–H2O52 and BA–H2O.23 Since the current system is similar to these precedents, we discuss tunneling in more detail below, which may also shed light, for example, on the nature of the, as of yet unexplained, complex splittings observed in BA monohydrate.
For OTA–H2O, there are nominally two potential water motions, in addition to the wagging of the free hydrogen atom, which is barrier-less, as discussed above. The first motion is exchange of the hydrogen atoms involved in the intermolecular six-membered ring. However, this motion actually converts OTA–H2O-I into II. The second motion is rotation of water about the axis defined by the oxygen atom and its lone pair of electrons hydrogen-bonded to the acidic hydrogen atom. Along this coordinate (see Fig. 5b), the dihedral angle between the carboxylic acid and aromatic groups is fixed at about 11° – even though the average angle is zero. Since the angle does not change with water rotation, the minima are not degenerate. In the global minimum (see Fig. 5b, structure i), the free hydrogen atom is slightly further from the methyl group than in the local minimum (see Fig. 5b, structure iii). Since the ZPE levels of the two minima differ by 5 cm−1, localized eigenstates exist for each well,53 and no motion between the two will result in tunneling splittings. Hence, the water rotation coordinate does not lead to tunneling, though the barrier (6.16 kJ mol−1) is similar to that of BA–H2O (7.0 kJ mol−1 at the B3LYP/6-311++G(d,p) level of theory).
Despite the energy mismatch between the minima, rotation of water simultaneously accompanied by rocking of the carboxylic acid group could facilitate tunneling (see Fig. 5c), because it converts the global minimum to a structural degeneracy. A similar pathway has been proposed to explain splittings observed in the microwave spectrum of the glycolaldehyde–H2O complex.54 However, for the present complex, the structure connecting the degeneracies is not a true transition state, because it has two imaginary frequencies, each corresponding to one of the uncoupled motions; the hilltop is a virtual, rather than proper, saddle point of index = 2 – simply a geometrical superposition of two transition states.55 We conclude that the motions are not concerted.
Since we rationalize the absence of water tunneling splittings in terms of the asymmetric double-well potentials along the carboxylic acid rocking and water rotation coordinates of OTA–H2O-I, a rotational spectroscopic study of the m-toluic acid–H2O complex, which we have calculated to have single-well potentials along the same coordinates, like BA–H2O, would provide further insight in this regard.
Inter- and intramolecular non-covalent bonding
QTAIM33 is often used to investigate bonding.56,57 In QTAIM, molecules are partitioned into atomic basins whose boundaries are defined by interatomic surfaces of zero flux of the electron density gradient, ∇ρ. A given property of an individual atom can be determined by integrating over the volume of the atomic basin. Furthermore, the properties of critical points in the electron density distribution, where ∇ρ = 0, provide insight into the interactions in a molecule or complex. For instance, the electron density distribution at bond critical points (BCPs), which are maxima in two dimensions (the third is tangential to the bond path), corresponds directly to the strength of the bond.As shown in Table 5, the electron density distribution and its Laplacian (curvature) at the (intermolecular) bond critical points of OTA–H2O-I and BA–H2O (see Fig. S1, ESI†) are similar. The values for both properties fall within the typical ranges for hydrogen bonds: 0.002–0.034 a.u. for electron density, and 0.024–0.139 a.u. for the Laplacian.58 The OH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C BCP of OTA–H2O-I has more electron density that that of BA–H2O, and the r(H⋯O) distance is slightly shorter; in contrast, the OH⋯OH BCP of OTA–H2O-I has less electron density than that of BA–H2O, and the r(H⋯O) distance is longer. Integrated atomic properties of the involved hydrogen atoms (see Table 5) are also consistent with the criteria for hydrogen bonding; for example, the hydrogen atoms experience a decrease in electron population and atomic volume upon complexation.
C BCP of OTA–H2O-I has more electron density that that of BA–H2O, and the r(H⋯O) distance is slightly shorter; in contrast, the OH⋯OH BCP of OTA–H2O-I has less electron density than that of BA–H2O, and the r(H⋯O) distance is longer. Integrated atomic properties of the involved hydrogen atoms (see Table 5) are also consistent with the criteria for hydrogen bonding; for example, the hydrogen atoms experience a decrease in electron population and atomic volume upon complexation.
| Constant | OTA–H2O-I | BA–H2O | ||
|---|---|---|---|---|
| a Properties at BCPs: electron density, ρ; Laplacian of electron density, ∇2ρ; and electronic potential energy density, V(r). b Changes in integrated atomic properties of hydrogen atom that occur upon formation of hydrogen bonds: electronic population, N(H); atomic volume, v(H); and atomic energy, E(H). c Hydrogen bond energy, based on electronic potential energy density, V(r), at the BCP (calculated according to ref. 60). d Hydrogen bond energy, based on electron density, ρ, at the BCP (calculated according to ref. 61). e Interaction energy. f Counterpoise-corrected interaction energy. g Interaction energy based on SAPT0. h Dissociation energy, which is corrected for basis set superposition error and zero-point energy. | ||||
| Bonding | OH⋯OC |
OH⋯OH | OH⋯OC |
OH⋯OH |
| r(H⋯O)/Å | 1.930 | 1.772 | 1.933 | 1.765 |
| ρ /au | 0.0262 | 0.0355 | 0.0260 | 0.0362 |
| ∇2ρ/au | 0.0976 | 0.1072 | 0.0970 | 0.1082 |
| V(r)/au | −0.0228 | −0.0347 | −0.0226 | −0.0356 |
| ΔN(H)b/au | −0.0722 | −0.0434 | −0.0641 | −0.0535 |
| Δv(H)/au | −6.69 | −7.06 | −6.44 | −7.62 |
| ΔE(H)/au | 0.0532 | 0.0308 | 0.0463 | 0.0395 |
| E HB[V(r)]c/kJ mol−1 | 30.0 | 45.6 | 29.7 | 46.7 |
| E HB(ρBCP)d/kJ mol−1 | 23.1 | 32.4 | 22.9 | 33.2 |
| ΔEe/kJ mol−1 | −47.8 | −49.2 | ||
| ΔEBSSEf/kJ mol−1 | −40.6 | −41.9 | ||
| ΔESAPT0g/kJ mol−1 | −47.8 | −48.0 | ||
| D 0 /kJ mol−1 | 30.3 | 31.7 | ||
The hydrogen bond energy, EHB, can be estimated from QTAIM properties.59–61 We will consider two methods: estimating EHB based on (i) the electronic potential energy density at the BCP, V(r), according to the equation EHB = −0.5a03V(r), where a03 is the Bohr radius;60 and (ii) the electron density at the BCP, ρBCP, according to the equation, EHB = −3.09 + 239ρBCP.61 As shown in Table 5, EHB[V(r)] is greater for the OH⋯OH bond (45.6 kJ mol−1) than the OH⋯OC bond (30.0 kJ mol−1). The same ranking occurs for EHB(ρBCP), though the absolute energies are much smaller. These energies, as well as the r(H⋯O) distances (see Table 5), are characteristic of moderately strong hydrogen bonds.62 The sums of hydrogen bond energies based on V(r) and ρBCP are 75.6 and 55.5 kJ mol−1, respectively. For OTA–H2O-I, the raw interaction energy is −47.8 kJ mol−1. Though some discrepancy between the sum of hydrogen bond energies and interaction energy is expected, since atoms not directly involved in hydrogen bonding may still be stabilized or destabilized by complexation, the relatively good agreement between ΔE and the sum of the hydrogen bond energies based on ρBCP suggests that this method is more accurate than the method based on V(r). The ZPE- and BSSE-corrected dissociation energy of OTA–H2O-I (30.3 kJ mol−1) is very similar to that of BA–H2O (31.7 kJ mol−1),23 consistent with the similar hydrogen bond energies. For each acid, ΔESAPT0 (which is BSSE-free63) is in better agreement with ΔE than ΔEBSSE, so the BSSE may be over-correcting at this level of theory. Still, both QTAIM and SAPT predict slightly stronger intermolecular interactions for BA–H2O than OTA–H2O-I.
In passing, we note that BCPs are also present between the sp3 carbon atom of the methyl group and the carbonyl oxygen atom of both the OTA monomer and monohydrate (see Fig. S1, ESI†). Mani and Arunan coined the term “carbon bonding” to describe interactions between highly electronegative atoms and carbon atoms covalently bonded to electron withdrawing groups;64 the resulting non-covalent bonding is analogous to hydrogen bonding, and it meets the conventional criteria developed by Koch and Popelier.58 As shown in Table 6, the local BCP properties are typical of hydrogen bonds; for example, though the electron density of the C(sp3)⋯OC BCP of the OTA-I monomer (0.014 a.u.) is lower than that of the OH⋯OC BCPs discussed above (for example, 0.026 a.u. for OTA–H2O-I), it is still well within the typical range (0.002–0.034 a.u.).58 To estimate the effect of this bonding on integrated atomic properties, we take the methyl carbon atom of OTA-I to represent the final (bonding) scenario and that of p-toluic acid to represent the initial (non-bonding) scenario, so the presence of the aromatic ring and its carboxylic acid substituent is taken into account. The resulting differences in electron population, atomic volume, and atomic energy (see Table 6) are further evidence of carbon bonding, since they are consistent with the Koch and Popelier criteria.58 For example, the methyl carbon atom of OTA is slightly higher in energy than that of p-toluic acid; it is slightly destabilized by bonding with the nearby carbonyl oxygen atom in OTA. Consequently, we tentatively attribute the BCP to carbon bonding. Previously, intramolecular carbon bonding has also been identified in conformers of D-ribose.65
| Constant | OTA |
|---|---|
| a Properties at BCPs: electron density, ρ; Laplacian of electron density, ∇2ρ; and electronic potential energy density, V(r). b Changes in integrated atomic properties of hydrogen atom that occur upon formation of hydrogen bonds: electronic population, N(H); atomic volume, v(H); and atomic energy, E(H). c Hydrogen bond energy, based on electronic potential energy density, V(r), at the BCP (calculated according to ref. 60). | |
| Bonding | C(sp3)⋯OC |
| r(H⋯O)/Å | 2.773 |
| ρ /au | 0.0136 |
| ∇2ρ/au | 0.0652 |
| V(r)/au | −0.0113 |
| ΔN(H)b/au | −0.0136 |
| Δv(H)/au | −2.5 |
| ΔE(H)/au | 0.0010 |
| E HB[V(r)]c/kJ mol−1 | 14.8 |
However, this potential instance of carbon bonding has several distinctive characteristics. Previously studied species contain many electron withdrawing groups covalently bonded to the sp3 carbon (hydroxyl, amino, and cyano groups among them),64,66 but not the phenyl group. In organic synthesis, the methyl group is a common electron-donating group;67 its loss of electron density to the phenyl group in OTA makes it slightly more electropositive, facilitating carbon bonding with the electronegative carbonyl oxygen atom. Furthermore, previously studied C(sp3)⋯O bonds have been linear, but the C–C(sp3)⋯O angle in OTA-I is about 80°. Finally, the C(sp3)⋯OC BCP is present in the minimum energy structure along the coordinate defined by rotation of the methyl group, but not in the transition state – which contains a more conventional CH⋯CO BCP, instead.
Atmospheric abundance of OTA monohydrate
The equilibrium constant of complexation, Kp, can be calculated directly from the partition functions that underlie the free energy change, according to the following equation,where q⊖ is the total partition function of the species in parentheses, k is the Boltzmann constant, T is temperature in Kelvin, and NA is the Avogadro constant.68 The coefficient of two is included, because (at the average position of the carboxylic acid group) the wagging motion of the free hydrogen atom of the water moiety results in mirror images.49,69 Since the mole fractions of conformers III and IV are less than 1% at 298 K, we consider only conformers I and II.
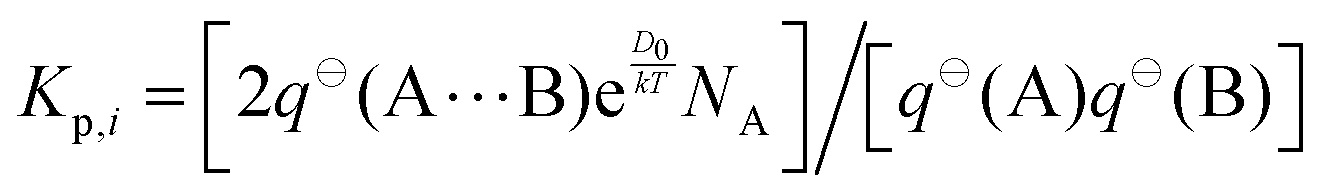
In addition to the dissociation energies, the partition functions must be calculated. The total partition function of each species is the product of translational, rotational, vibrational, and electronic partition functions.70 The translational partition function of a species can be calculated simply from its mass and the temperature. The rotational partition functions depend on temperature, rotational constants, and symmetry number. The rotational constants of water are known.71 The rotational constants of OTA-I and OTA–H2O-I are those reported above, and the rotational constants of OTA-II and OTA–H2O-II are those of the calculated minimum energy structures. The symmetry numbers for H2O, OTA, and OTA–H2O are 2, 1, and 1.69 The vibrational partition function, which is also dependent on temperature, is the product of the individual partition functions of each normal mode, which are most significant for modes below 400 cm−1. The normal mode frequencies for all three species were taken from the MP2-calculated frequency analyses. Since no excited electronic states are accessible at ambient temperature, the electronic partition function is one.
The percentage of conformer i hydrated in the atmosphere, χi, depends on the concentration of water vapour, and it can be calculated according to the following equation,
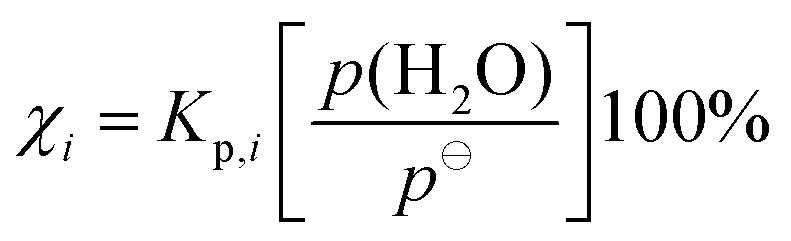
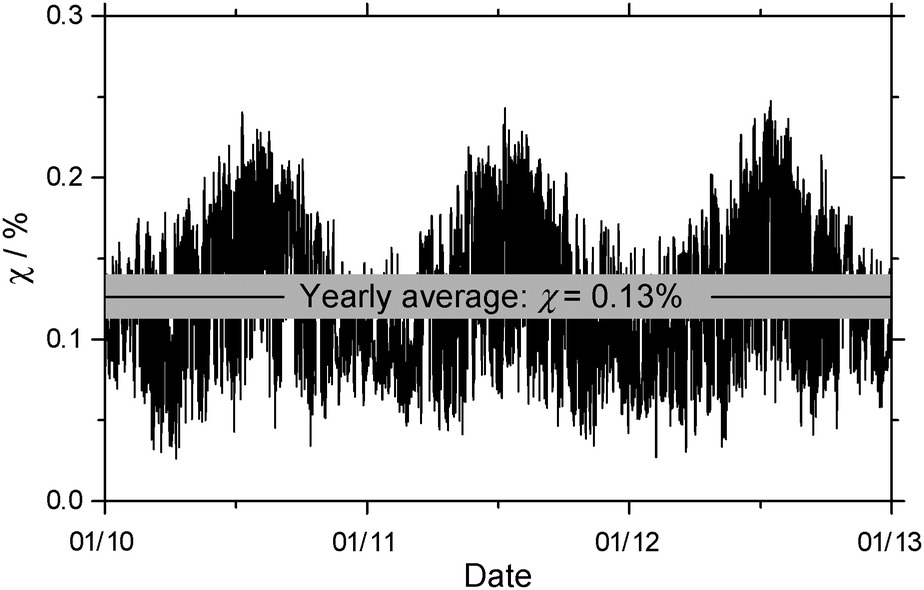 | ||
| Fig. 6 Seasonal fluctuation of the percentage of o-toluic acid hydrated in the atmosphere, under conditions reported in Calgary, Alberta. | ||
At the same level of theory, the yearly average of percentage of BA hydrated is about 1%.23 The decrease is due more to differences in the partition functions than the small difference between dissociation energies. Because the methyl group adds mass to OTA and OTA–H2O, they have slightly greater translational partition functions than BA and its monohydrate. The rotational partition functions also increase, because the rotational constants increase and – in the case of the monomer – the symmetry number decreases from 2 for BA to 1 for OTA. In all, the rotational partition function increases by about a factor of 3 for OTA. The increase in the number of atoms in OTA and OTA–H2O, compared to BA and BA–H2O, results in a greater number of vibrational modes, and greater vibrational partition functions. In each of the cases above, the relative increase is greater for the OTA monomer than the monohydrate, so Kp,i decreases. Though these calculations provide only a preliminary estimate, it is likely that less OTA is hydrated in the atmosphere than BA. Nonetheless, the interactions between OTA and water characterized by the above structural and energetic results further our understanding of gas-phase clusters containing organic acids and water in the atmosphere.
Conclusions
We have measured the rotational spectra of the monomer and monohydrate of OTA, an important product of photo-oxidation in the atmosphere. The lowest-energy conformers of both species in the gas phase have been determined for the first time; the lowest-energy conformer of the monomer is the same as that previously observed in the crystal structure.21 No methyl internal rotation splittings were observed in the spectra of either species, because the barriers are too high. The displacement of the carboxylic acid group during its rocking motion is greater in OTA than in BA, causing the inertial defect of OTA to be greater in magnitude than that of BA. For the monohydrate, minima on either side of the barrier to the same rocking motion are not degenerate, precluding water tunneling splittings. The hydrogen bond energies (based on a QTAIM analysis) and, consequently, dissociation energies of OTA and BA monohydrates are similar, but less OTA is predicted to be hydrated in the atmosphere, because the presence of the methyl group increases the partition functions of the monomer to a greater extent than those of the monohydrate.Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC). Computing resources were provided by Westgrid and Compute/Calcul Canada. We thank Nathan A. Seifert for discussions about the SAPT methodology and the anonymous referees for valuable comments regarding the methyl internal rotation. EGS thanks NSERC for a Postgraduate Scholarship, the University of Alberta for a President's Doctoral Prize of Distinction, and Environment Canada for an Atmospheric and Meteorological Graduate Supplement. WJ holds a Tier I Canada Research Chair in Cluster Science.References
- S. Liu, D. A. Day, J. E. Shields and L. M. Russell, Atmos. Chem. Phys., 2011, 11, 8321 CrossRef CAS.
- M. Ehn, et al., Nature, 2014, 506, 476 CrossRef CAS PubMed.
- E. G. Schnitzler, A. Dutt, A. M. Charbonneau, J. S. Olfert and W. Jäger, Environ. Sci. Technol., 2014, 48, 14309 CrossRef CAS PubMed.
- F. Riccobono, et al., Science, 2014, 344, 717 CrossRef CAS PubMed.
- R. Zhang, I. Suh, J. Zhao, D. Zhang, E. C. Fortner, X. Tie, L. T. Molina and M. J. Molina, Science, 2004, 304, 1487 CrossRef CAS PubMed.
- R. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Chem. Rev., 2012, 112, 1957 CrossRef CAS PubMed.
- R. McGraw and R. Zhang, J. Chem. Phys., 2008, 128, 064508 CrossRef PubMed.
- D. K. Henze, J. H. Seinfeld, N. L. Ng, T.-M. Fu, D. J. Jacob and C. L. Heald, Atmos. Chem. Phys., 2008, 8, 2405 CrossRef CAS.
- R. J. Bell, N. G. Davey, M. Martinsen, C. Collin-Hansen, E. T. Krogh and C. G. Gill, J. Am. Soc. Mass Spectrom., 2015, 26, 212 CrossRef CAS PubMed.
- I. J. Simpson, N. J. Blake, B. Barletta, G. S. Diskin, H. E. Fuelberg, K. Gorham, L. G. Huey, S. Meinardi, F. S. Rowland, S. A. Vay, A. J. Weinheimer, M. Yang and D. R. Blake, Atmos. Chem. Phys., 2010, 10, 11931 CrossRef CAS.
- C. Song, K. Na, B. Warren, Q. Malloy and D. R. Cocker, III, Environ. Sci. Technol., 2007, 41, 7403 CrossRef CAS PubMed.
- Y. Zhou, H. Zhang, H. M. Parikh, E. H. Chen, W. Rattanavaraha, E. P. Rosen, W. Wang and R. M. Kamens, Atmos. Environ., 2011, 45, 3882 CrossRef CAS.
- M. G. Vivanco, M. Santiago, A. Martínez-Tarifa, E. Borrás, M. Ródenas, C. Garciá-Diego and M. Sánchez, Atmos. Environ., 2011, 45, 708 CrossRef CAS.
- M. Huang, W. Zhang, Z. Wang, L. Hao, W. Zhao, X. Liu, B. Long and L. Fang, Int. J. Quantum Chem., 2008, 108, 954 CrossRef CAS.
- J. Yu, H. E. Jeffries and K. G. Sexton, Atmos. Environ., 1997, 31, 2261 CrossRef CAS.
- G. M. Clifford, A. Hadj-Aïssa, R. M. Healy, A. Mellouki, A. Muñoz, K. Wirtz, M. Martín-Reviejo, E. Borrás and J. C. Wenger, Environ. Sci. Technol., 2011, 45, 9649 CrossRef CAS PubMed.
- P. Q. Fu, K. Kawamura, J. Chen, J. Li, Y. L. Sun, Y. Liu, E. Tachibana, S. G. Aggarwal, K. Okuzawa, H. Tanimoto, Y. Kanaya and Z. F. Wang, Atmos. Chem. Phys., 2012, 12, 8359 CAS.
- K. Kawamura, L.-L. Ng and I. R. Kaplan, Environ. Sci. Technol., 1985, 19, 1082 CrossRef CAS PubMed.
- M. Ito, H. Tsukioka and S. Imanishi, J. Am. Chem. Soc., 1960, 82, 1559 CrossRef CAS.
- P. D. S. Babu, S. Periandy and S. Ramalingam, Spectrochim. Acta, Part A, 2011, 78, 1321 CrossRef PubMed.
- M. Polito, E. D'Oria, L. Maina, P. G. Karamertzanis, F. Grepioni, D. Braga and S. L. Price, CrystEngComm, 2008, 10, 1848 RSC.
- F. Gaie-Levrel, C. Gutlé, H.-W. Jochims, E. Rühl and M. Schwell, J. Phys. Chem. A, 2008, 112, 5138 CrossRef CAS PubMed.
- E. G. Schnitzler and W. Jäger, Phys. Chem. Chem. Phys., 2014, 16, 2305 RSC.
- D. Priem, T.-K. Ha and A. Bauder, J. Chem. Phys., 2000, 113, 169 CrossRef CAS.
- J. Thomas, A. Serrato, III, W. Lin, W. Jäger and Y. Xu, Chem. – Eur. J., 2014, 20, 6148 CrossRef CAS PubMed.
- Y. Xu, J. Van Wijngaarden and W. Jäger, Int. Rev. Phys. Chem., 2005, 24, 301 CrossRef CAS.
- Y. Xu and W. Jäger, J. Chem. Phys., 1997, 106, 7968 CrossRef CAS.
- M. J. Frisch, et al., Gaussian09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- R. Ditchfield, W. J. Hehrf and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- C. F. Matta and R. J. Boyd, in The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. F. Matta and R. J. Boyd, Wiley, New York, 2007, pp. 29–30 Search PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887 CrossRef CAS.
- J. M. Turney, et al. , WIREs Comput. Mol. Sci., 2012, 2, 556 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherill, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed.
- C. M. Western, PGOPHER, A Program for Simulating Rotational Structure, University of Bristol, http://pgopher.chm.bris.ac.uk.
- J. K. G. Watson, J. Chem. Phys., 1967, 46, 1935 CrossRef CAS.
- D. Gerhard, A. Hellweg, I. Merke, W. Stahl, M. Baudelet, D. Petitprez and G. Wlodarczak, J. Mol. Spectrosc., 2003, 220, 234 CrossRef CAS.
- S. Jacobsen, U. Anderson and H. Mäder, Struct. Chem., 2003, 14, 217 CrossRef CAS.
- K.-T. Lu, F. Weinhold and J. C. Weisshaar, J. Chem. Phys., 1995, 102, 6787 CrossRef CAS.
- H. Hartwig and H. Dreizler, Z. Naturforsch., 1996, 51A, 923 Search PubMed.
- M. Tudorie, I. Kleiner, M. Jahn, J.-U. Grabow, M. Goubet and O. Pirali, J. Phys. Chem. A, 2013, 117, 13636 CrossRef CAS PubMed.
- T. Oka, J. Mol. Struct., 1995, 352/353, 225 CrossRef CAS.
- P. D. Godfrey and D. McNaughton, J. Chem. Phys., 2013, 138, 024303 CrossRef PubMed.
- E. G. Schnitzler, B. L. M. Zenchyzen and W. Jäger, Astrophys. J., 2015, 805, 141 CrossRef.
- B. Ouyang and B. J. Howard, J. Phys. Chem. A, 2008, 112, 8208 CrossRef CAS PubMed.
- B. Ouyang and B. J. Howard, Phys. Chem. Chem. Phys., 2009, 11, 366 RSC.
- L. Evangelisti and W. Caminati, Phys. Chem. Chem. Phys., 2010, 12, 14433 RSC.
- B. Ouyang, T. G. Starkey and B. J. Howard, J. Phys. Chem. A, 2007, 111, 6165 CrossRef CAS PubMed.
- S. Albert, P. Lerch, R. Prentner and M. Quack, Angew. Chem., 2013, 125, 364 CrossRef.
- J.-R. Aviles-Moreno, J. Demaison and T. R. Huet, J. Am. Chem. Soc., 2006, 128, 10467 CrossRef CAS PubMed.
- D. Heidrich and W. Quapp, Theor. Chim. Acta, 1986, 70, 89 CrossRef CAS.
- M. Petković and M. Etinski, RSC Adv., 2014, 4, 38517 RSC.
- O. O. Brovarets, R. O. Zhurakivsky and D. M. Hovorun, J. Comput. Chem., 2014, 35, 451 CrossRef CAS PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747 CrossRef CAS.
- E. G. Schnitzler, M. R. Poopari, Y. Xu and W. Jäger, Phys. Chem. Chem. Phys., 2015, 17, 21942 RSC.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS.
- T. Yu. Nikolaienko, L. A. Bulavin and D. M. Hovorun, Phys. Chem. Chem. Phys., 2012, 14, 7441 RSC.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford, New York, 1997, pp. 12–16 Search PubMed.
- F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873 CrossRef CAS.
- D. Mani and E. Arunan, Phys. Chem. Chem. Phys., 2013, 15, 14377 RSC.
- L. M. Azofra, M. M. Quesada-Moreno, I. Alkorta, J. R. Avilés-Moreno, J. J. López-González and J. Elguero, New J. Chem., 2014, 38, 529 RSC.
- P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2014, 16, 17238 RSC.
- C. W. Anson and D. M. Thamattoor, J. Org. Chem., 2012, 77, 1693 CrossRef CAS PubMed.
- B. J. McClelland, Statistical Thermodynamics, Studies in Chemical Physics Series, Chapman and Hall, London, 1973, pp. 173–175 Search PubMed.
- A. Fernández-Ramos, B. A. Ellingson, R. Meana-Pañeda, J. M. C. Marques and D. G. Truhlar, Theor. Chem. Acc., 2007, 118, 813 CrossRef.
- P. Atkins, Physical Chemistry, Freeman, New York, 1998, ch. 20 Search PubMed.
- J. K. Messer, F. C. De Lucia and P. Helminger, Int. J. Infrared Millimeter Waves, 1983, 4, 505 CrossRef CAS.
- Clean Air Strategic Alliance Data Warehouse, http://www.casadata.org, accessed April 2013.
- R. W. Hyland and A. Wexler, ASHRAE Trans., 1983, 89, 500 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of observed transitions of OTA and OTA–H2O and molecular graphs of atoms-in-molecules analyses. See DOI: 10.1039/c5cp06073g |
| This journal is © the Owner Societies 2016 |