
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The adsorbed state of a thiol on palladium nanoparticles
Scott M.
Rogers
ab,
Nikolaos
Dimitratos
ac,
Wilm
Jones
bc,
Michael
Bowker
bc,
Antonios G.
Kanaras
d,
Peter P.
Wells
ab,
C. Richard A.
Catlow
abc and
Stewart F.
Parker
*be
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
bUK Catalysis Hub, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0FA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
dSchool of Physics and Astronomy, Faculty of Physical Sciences and Engineering, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
eISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0QX, UK. E-mail: stewart.parker@stfc.ac.uk
First published on 5th April 2016
Abstract
In the present work, a combination of imaging, spectroscopic and computational methods shows that 1-dodecanethiol undergoes S-deprotonation to form 1-dodecanethiolate on the surface of palladium nanoparticles, which then self-assembles into a structure that shows a high degree of order. The alkyl chain is largely in the all-trans conformation, which occurs despite the small size of the nanoparticle, (mean diameter = 3.9 nm). Inelastic neutron scattering spectroscopy is readily able to characterise organic surface layers on nanoparticles; the nature of the material is irrelevant: whether the nanoparticle core is an oxide, a metal or a semiconductor makes no difference. Comparison to DFT calculations allows insights into the nature and conformation of the adsorbed layer.
Introduction
Nanoparticle size materials have attracted remarkable academic and industrial interest due to the high surface area to volume ratio.1,2 The main challenges of the academic and industrial researchers in the area of nanoparticles are the controllable syntheses of metal nanoparticles with a desired shape, size and structure.3 Solving these challenges will facilitate the commercial utilisation of nanoparticles in many areas ranging across chemical sensing, biolabeling, catalysis, photonics, and semiconductors. In the area of catalysis, the utilisation of supported metal nanoparticles (Au, Pd, Pt) has been shown to be particularly effective for a broad range of catalytic reactions such as oxidation, hydrogenation and combustion of volatile organic compounds (VOCs).4–9 These include the oxidation of CO,10,11 the selective oxidation of alcohols and polyols,12–14 the epoxidation of olefins,15,16 the hydrochlorination of ethyne,17,18 the selective hydrogenation of unsaturated carbonyl and nitro groups,19 and the direct synthesis of hydrogen peroxide from molecular hydrogen and oxygen.20,21 Typical methodologies for the synthesis of supported metal nanoparticles are based primarily on colloidal methods, and to a lesser extent impregnation methods. The main advantages of colloidal methods are (i) control of the particle size, shape and dispersity by tuning reaction conditions such as the metal salt-to-ligand ratio, concentration and nature of the reducing agent, solvent and temperature, (ii) the large variety of functionalised ligands that can be introduced and (iii) isolation, cleaning and redispersion of the particles in different solvents. However, some disadvantages and challenges remain such as; (i) the effective removal of ligands after the synthesis of metal nanoparticles, (ii) the excess use of solvents and (iii) the effective scale up production of nanoparticles using chemical liquid phase methods in industry.Nano-sized metal colloids are usually synthesised by an in situ reduction method from a suitable metal precursor, by chemical reduction (with NaBH4 or alcohol), photoreduction, electrochemical reduction or thermal decomposition.2 Small molecular ligands, surfactants and polymers have been used to stabilise the so-formed metallic colloids. In some cases, metal colloids are stable over long periods even without coagulation in a solvent. However, for prolonged stability in a sol state, and in order to obtain a uniform, controlled particle size, use of a stabilising ligand is essential. Typically, polymers such as polyvinyl alcohol,22,23 polyvinylpyrrolidone24–26 and ligands such as alkanethiols27–30 and alkylamines31 are employed. Among these systems, alkanethiolate-capped nanoparticles are considered as a well-defined system and many examples in the area of catalysis have shown the potential applications of Pd alkanethiolate-capped nanoparticles. However, because of their high ligand-surface coverage, they are not yet the most efficient catalytic materials for organic reactions, although a few examples have shown promising catalytic results such as C–C-coupling reactions,32 hydrogenation reactions,33 oxidation reactions34–36 and the isomerisation of allyl alcohols.37,38
To understand and control the effect of alkanethiolate ligands on the catalytic properties of nanoparticles, it is essential, therefore to know the structure and conformation of the alkanethiol ligand. Surface ligands play a critical role in catalysis because they determine the accessibility of the reactant to the nanoparticle surface. In this paper we present our results using a combination of techniques including inelastic neutron scattering (INS) spectroscopy. INS spectroscopy39–41 is an ideal technique for studying the interaction of a metal with ligands because the metal nanoparticle is essentially invisible to neutrons and therefore only the organic layer is visible. This approach builds on that used to characterise successfully the hydroxyl and water layers on PdO.42
Experimental
Synthesis
The synthesis employed in this study is based on the two-phase Brust–Schiffrin method27 that has been popularly used for the synthesis of thiol-protected gold nanoparticles. The Brust–Schiffrin method has been extensively documented and studied due to its ease of use and is still popular more than two decades after the first publication.43Potassium tetrachloropalladate (K2PdCl4; 0.0188 mol) was dissolved in 0.627 L of Milli-Q water. Tetraoctylammonium bromide (0.08356 mol) was dissolved in 1.67 L of toluene. Both solutions were mixed and continuously stirred until the organic layer turned dark orange and the aqueous layer cleared, indicating the completion of the phase transfer of [PdCl4]2− to the organic layer, 1-dodecanethiol (3.55 g) was then added to the organic phase. Afterward, a freshly prepared aqueous solution of sodium borohydride (0.4 M, 0.517 L) was slowly added to the vigorously stirred reaction mixture. A rapid color change to black was observed, indicating the formation of nanoparticles. Upon the completion of 8 h of continuous stirring, the aqueous layer was removed by using a separatory funnel and toluene was removed by vacuum. The resulting crude nanoparticles were washed by using 2 L of ethanol to remove excess thiol. The resulting nanoparticles were dried at room temperature. The preparation was repeated four times to produce 8 g of Pd nanoparticles.
TEM-EDX
Samples for examination by TEM were prepared by first dispersing 5 mg of the Pd nanoparticles in high purity ethanol using ultra-sonication. 40 μL of the black suspension was dropped on to a holey carbon film supported by a 300 mesh copper TEM grid before the solvent was evaporated. The sample was then examined using a JEOL JEM 2100 TEM model operating at 200 kV. Elemental analysis was performed using the mapping mode. Particle size analysis was carried out with ImageJ.44Inelastic neutron scattering (INS) spectroscopy
INS spectroscopy was carried out on the high resolution broadband spectrometer TOSCA45 and the direct geometry spectrometer MAPS46 at the ISIS Pulsed Neutron and Muon Facility47 (Chilton, UK). The sample, ∼5 g, was loaded into a thin-walled aluminium cell, which was inserted into the closed cycle refrigerator of the spectrometer and cooled to <20 K. The two spectrometers are complementary; for the present work the key features are that TOSCA provides good resolution spectra over 0–2000 cm−1 while MAPS enables the observation of the S–H and C–H stretch region. Raman spectra (785 nm excitation) were recorded simultaneously with the TOSCA INS spectra using a previously described system.48Computational studies
A five layer palladium slab with (111) termination was generated from the bulk structure of palladium using the Materials Studio49 package. S-Deprotonated 1-dodecanethiol was then added to one surface. A periodic system was created with a 30 Å vacuum gap between layers. The large gap was to ensure that there was at least 10 Å between the nearest atoms in different slabs. Two models of the thiol were investigated: one with the carbon backbone in the lowest energy all-trans configuration. For the second, 5000 molecular structures were generated via a molecular mechanics conformational search. This was done using the mixed Monte Carlo multiple minimization and Large Scale low mode method, as implemented in the MacroModel software (MacroModel v.10.3, Schrödinger).50 A high energy conformer with a significant number of gauche defects was then selected at random and used for the second configuration. Periodic density functional theory (periodic-DFT) calculations were carried out using the plane wave pseudopotential method as implemented in the CASTEP code.51,52 Exchange and correlation were approximated using the PBE functional. The plane-wave cut-off energy was 940 eV. Brillouin zone sampling of electronic states was performed on a 8 × 8 × 1 Monkhorst–Pack grid. The equilibrium structure, an essential prerequisite for lattice dynamics calculations was obtained by BFGS geometry optimization after which the residual forces were converged to zero within ±0.008 eV Å−1. Phonon frequencies were obtained by diagonalisation of dynamical matrices computed using density-functional perturbation theory53 and also to compute the dielectric response and the Born effective charges. The structure with the all-trans dodecanethiol had Cm (no. 8) symmetry, while the gauche structure had P1 (no. 1) symmetry. To make the phonon calculations more tractable for the latter, the constrained lattice dynamics method54 was used. In this case, all the Pd atoms except those in the top layer were “frozen”, in essence, the atoms are assigned a mass of infinity and the corresponding entries of the dynamical matrix are set to zero. It is not necessary to perform any computations for perturbation of these atoms considerably reducing the computational resources needed. The atomic displacements in each mode that are part of the CASTEP output enable the visualization of the modes to aid assignments and are also all that is required to generate the INS spectrum using the program ACLIMAX.55 We emphasise that for all the calculated spectra shown the transition energies have not been scaled.Results and discussion
Fig. 1 shows a TEM image of the Pd nanoparticles and a histogram of the resulting particle size distribution. It can be seen that the synthesis has resulted in small particles with a uniform distribution. The EDX data, Fig. 2, confirm that sulfur is uniformly spread over the particles.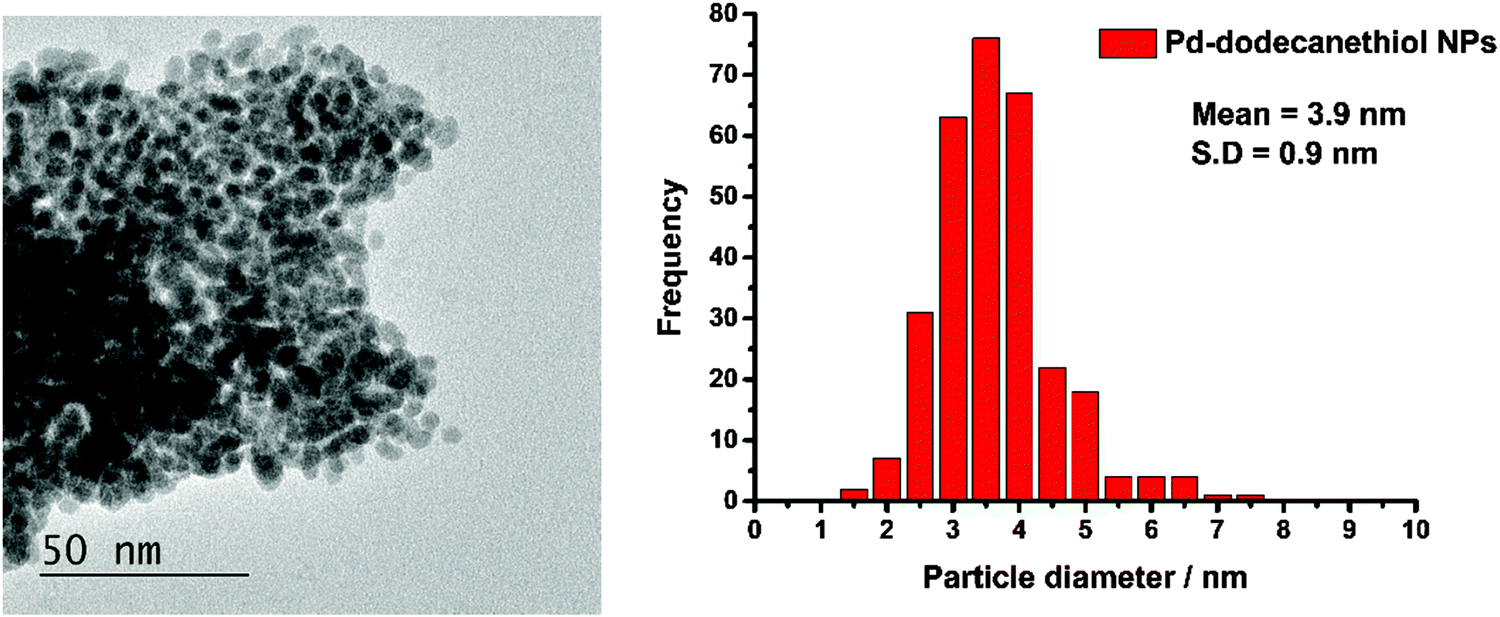 | ||
| Fig. 1 Left: The TEM image of the 1-dodecanethiol coated Pd nanoparticles and right: the particle size distribution derived from the image. | ||
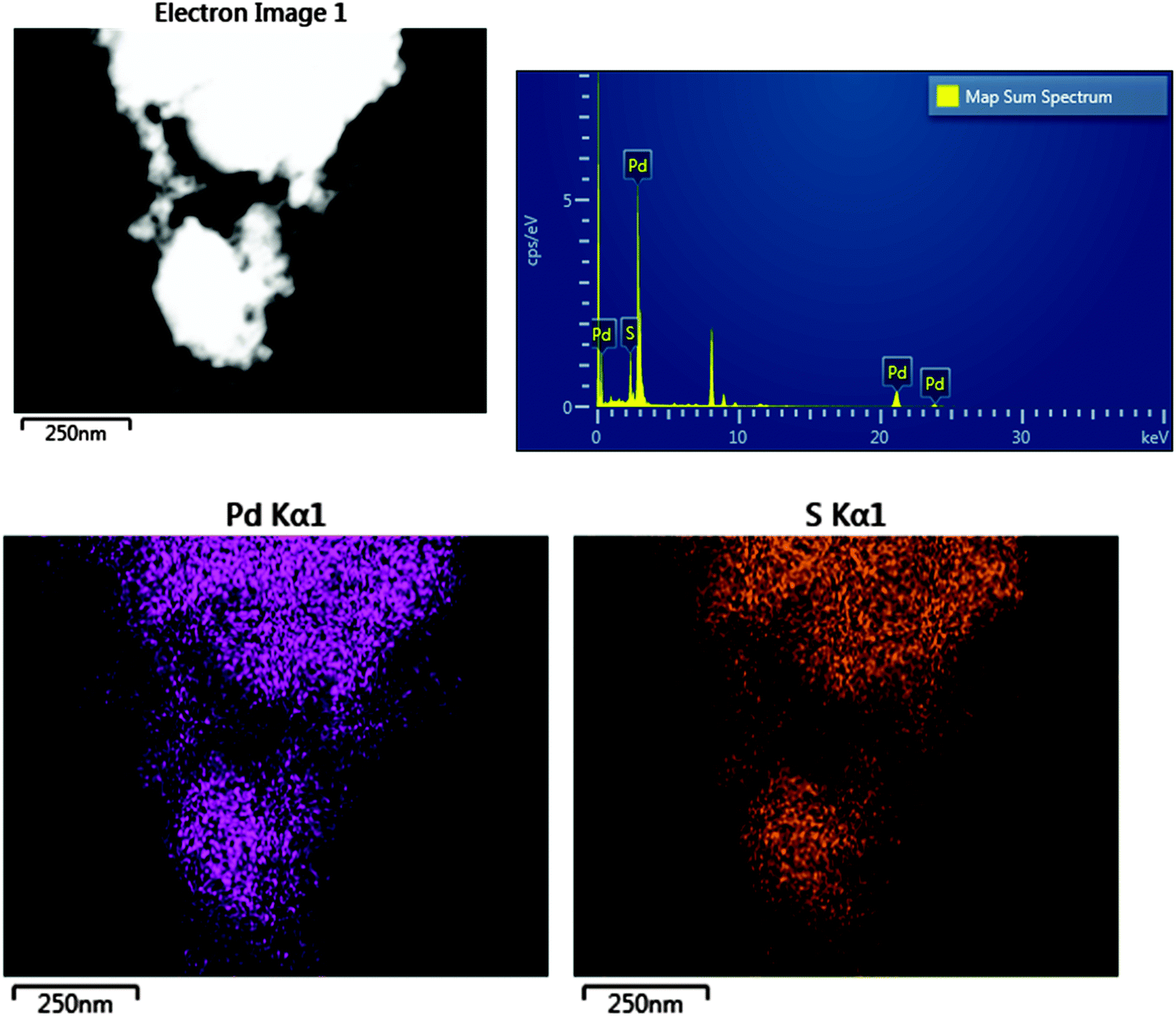 | ||
| Fig. 2 Top left; The dark field STEM image, top right; the EDX spectrum, bottom; Pd and S Kα1 elemental intensity maps. | ||
Fig. 3 shows the INS and Raman spectra in the S–H and C–H stretch region. Adsorption of the 1-dodecanethiol, Fig. 3a, on the Pd nanoparticles, Fig. 3b, results in a 20 cm−1 blueshift, which is more clearly seen in the derivative-like difference spectrum, Fig. 3c. This also shows a negative-going peak at 2545 cm−1 (arrowed) that, by comparison to the Raman spectrum of solid 1-dodecanethiol, Fig. 3d, is assigned to the S–H stretch. The peak is negative indicating loss of the thiol functionality, while the EDX shows the presence of sulfur, in combination the data show that 1-dodecanethiol, C12H25SH, is present as a thiolate, C12H25S.
 | ||
| Fig. 3 INS spectra recorded on MAPS at 20 K of: (a) solid 1-dodecanethiol, (b) 1-dodecanethiol coated Pd nanoparticles and (c) the scaled difference spectrum (b–a), (c) is ×2 ordinate expanded relative to (a). (d) The Raman spectrum of solid 1-dodecanethiol. | ||
Fig. 4 and 5 show the Raman and INS spectra of solid 1-dodecanethiol and the coated Pd nanoparticles respectively. The Raman spectrum of the nanoparticles, Fig. 4b, only shows one band attributable to the thiolate at 1075 cm−1. In contrast the INS spectrum, Fig. 5b, shows many more features. Comparison with the INS spectrum of 1-dodecanethiol, Fig. 5a, shows both similarities and differences.
 | ||
| Fig. 4 Raman spectra at 20 K of: (a) solid 1-dodecanethiol and (b) 1-dodecanethiol coated Pd nanoparticles and (c) the sapphire window of the sample cup. | ||
 | ||
| Fig. 5 INS spectra recorded on TOSCA at 20 K of: (a) solid 1-dodecanethiol and (b) 1-dodecanethiol coated Pd nanoparticles. | ||
A useful similarity is that both INS spectra exhibit the CH2 rocking mode near 720 cm−1. This band occurs when there are sequences of methylene groups in the all-trans conformation and is diagnostic of them.56 For an infinite polymethylene (CH2)n chain the band occurs at 720 cm−1 and shifts to higher energy as n decreases. For solid 1-dodecanethiol, the band occurs at 726 cm−1 and is at 728 cm−1 for the thiolate, suggesting a shorter sequence of all-trans methylene groups in the adsorbed material, indicating some disorder in the chemisorbed layer.
In order to analyse Fig. 5 it is necessary to understand the spectra of the thiol. To our knowledge, the structure of 1-dodecanethiol has not been reported in any phase. However, the crystal structure of 1,12-dodecanedithiol is known.57 In the crystal there is no evidence for hydrogen bonding, thus the molecule can reasonably be treated as an isolated system. Accordingly, one of the thiol groups was replaced by a hydrogen atom to generate 1-dodecanethiol from 1,12-dodecanedithiol. Using the computational techniques, the structure was then geometry optimised and the vibrational spectrum calculated. A comparison of observed and calculated INS spectra is a stringent test of the model and as Fig. 6 shows the agreement is excellent. By setting the cross section of all the atoms to zero, except for the hydrogen of the thiol group, it is possible to isolate the modes that involve the motion of this group. In particular, it can be seen that there are prominent modes at 217, 246 and 682 cm−1. Inspection of the mode visualisations shows that these are due to the out-of-plane C–S–H bend, the bend coupled to the methyl torsion and the in-plane C–S–H bend. The first and last of these are diagnostic of the thiol functionality and are observed in the experimental spectrum at 232 and 708 cm−1. Inspection of Fig. 5b shows no trace of these modes, consistent with Fig. 3 and adsorption as thiolate. None of the spectra of the adsorbed species show any evidence for unreacted thiol, our experience is that this means that, at most, ∼10% of the adsorbate could be unreacted thiol.
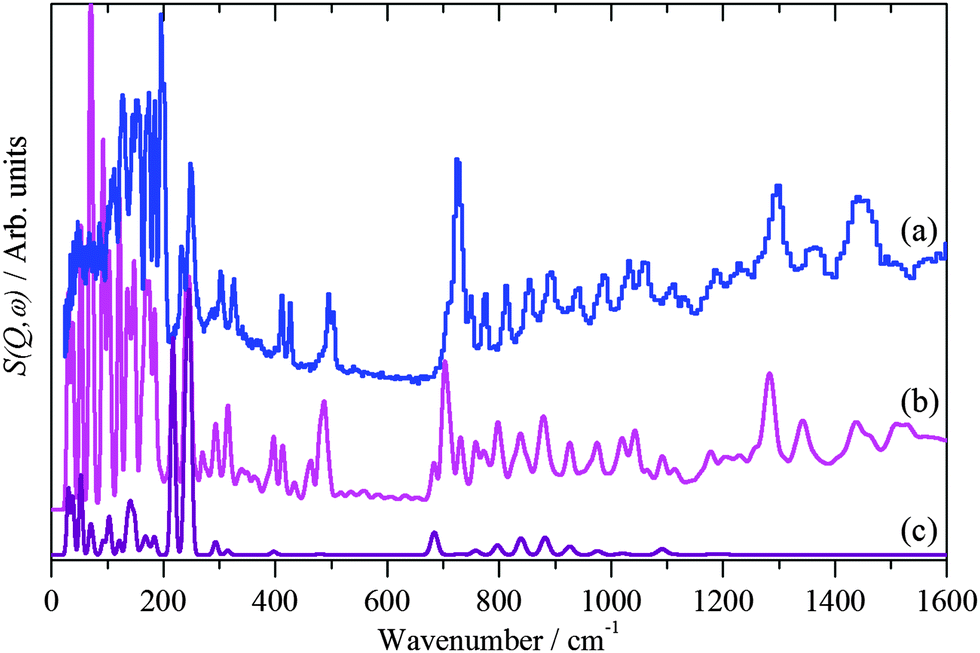 | ||
| Fig. 6 INS spectra recorded of solid 1-dodecanethiol: (a) experimental, (b) calculated by CASTEP and (c) as (b) but only including the modes that involve motion of the thiol proton. | ||
To model the adsorbed thiolate, the anion was placed with the sulfur atom above the threefold site and the C–S bond almost perpendicular to the surface, which resulted in a tilted structure with the carbon backbone at ∼30° to the surface normal. The geometry optimised structure is shown in the left part of Fig. 7. It can be seen that the thiolate occupies a twofold bridge site (C–S = 2.291 Å) with the carbon backbone almost perpendicular to the surface. To model a disordered system, a thiolate with a large number of gauche defects was used. To prevent intermolecular contacts, it was necessary to use a doubled unit cell. This structure optimised to that shown on the right of Fig. 7.
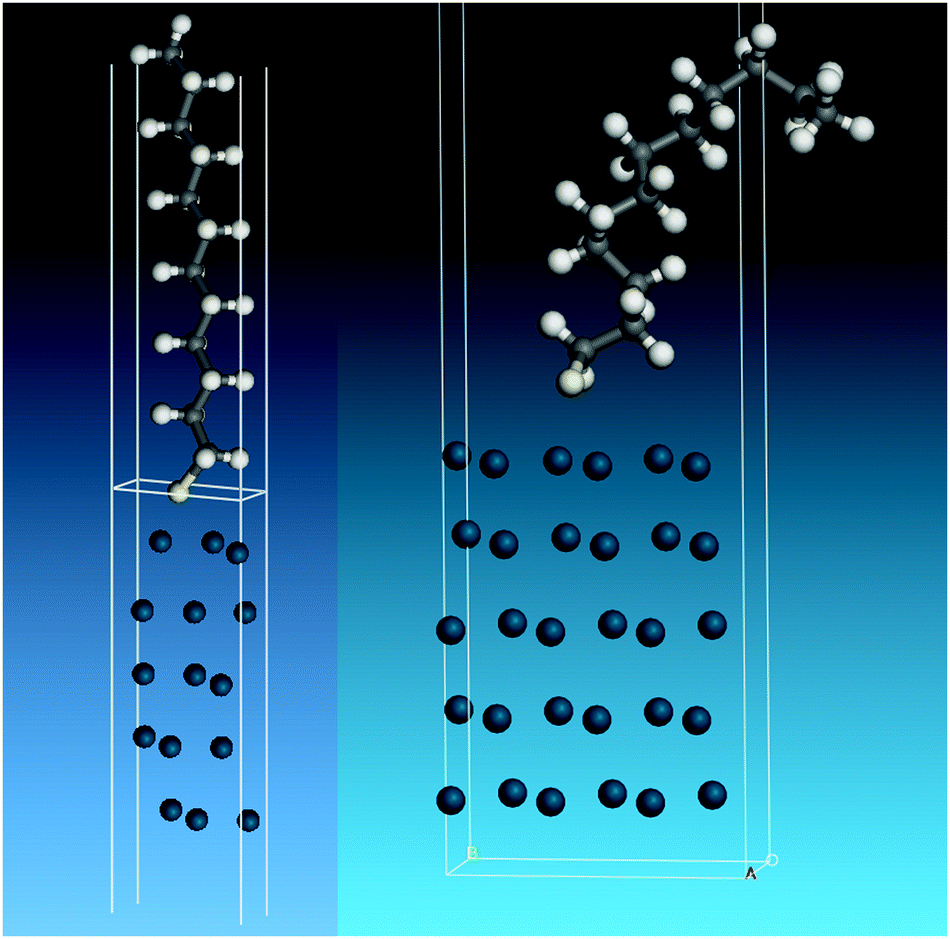 | ||
| Fig. 7 Structures used to model the adsorbed thiolate on Pd(111). Left: All-trans thiolate. Right: Thiolate with gauche defects. | ||
The calculated INS spectra of the two structures are compared to the experimental data in Fig. 8. It can be seen that neither result exactly matches the data, although the all-trans conformation, Fig. 8b, is the closer match. In particular, in the 0–600 cm−1 region the in-plane C–C–C bending modes (longitudinal acoustic modes, LAMs)58 occur and overlap with the lower energy, 0–200 cm−1, out-of-plane C–C–C bending modes (transverse acoustic modes, TAMs).59 As can be seen from Fig. 8b and c, these modes are conformationally sensitive. The disordered chain predicts a sequence of almost constant intensity modes up to the bandhead at 510 cm−1, whereas the envelope of the ordered chain more closely matches the experimental profile. We also note that the disordered model does not reproduce the shape of the CH2 rocking mode at 728 cm−1. If the adsorbed thiolate was completely in the all-trans conformation, then individual LAM modes in the 200–500 cm−1 region would be visible, as seen in Fig. 5a for the parent thiol. The disordered example used has only four gauche defects, yet this is sufficient to drastically alter the spectrum as compared to the all-trans arrangement, this indicates that there can only be one or two defects per chain. On balance, the modelling and the INS spectroscopy strongly suggest that the thiolate is largely, but not completely, ordered on the nanoparticle surface.
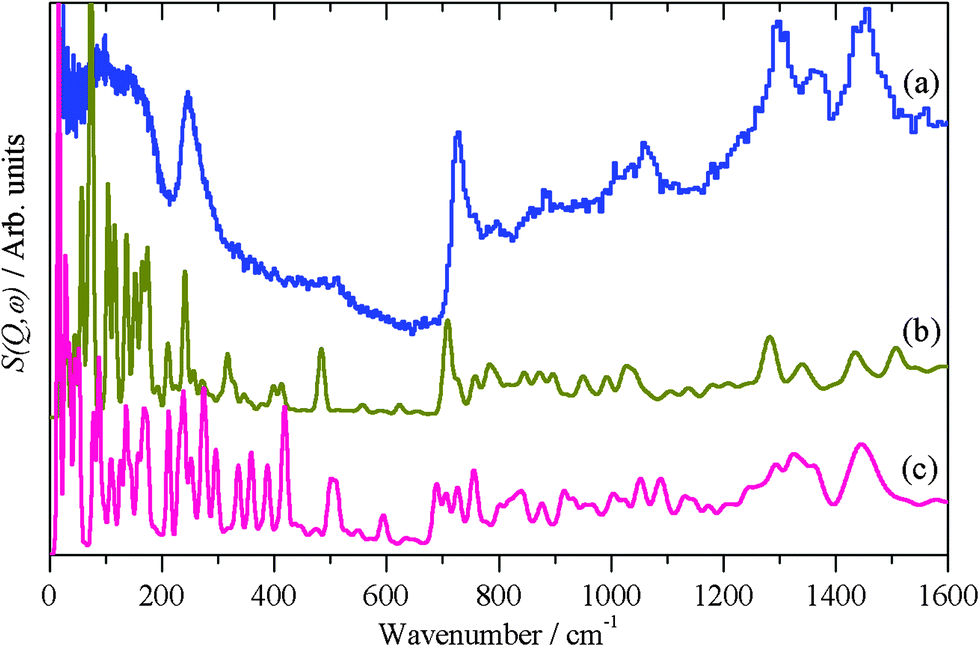 | ||
| Fig. 8 (a) Experimental INS spectrum of thiolate on palladium nanoparticles and as calculated by CASTEP for thiolate on Pd(111) in the (b) all-trans conformation and (c) gauche conformation. | ||
Conclusions
In the present work, a combination of imaging, spectroscopic and computational methods shows that 1-dodecanethiol undergoes S-deprotonation to form 1-dodecanethiolate on the surface of the nanoparticle, that then self-assembles into a structure that shows a high degree of order. The alkyl chain is largely in the all-trans conformation. This occurs despite the small size of the nanoparticle, (mean diameter = 3.9 nm), even for a perfect nanocube there would only be ∼100 molecules per face, the real material undoubtedly has fewer than this. The Hirshfeld analysis from the CASTEP results shows that there is a small positive charge (+0.03 electron) on the hydrogens and a small negative charge (−0.07 electron) on the carbons, providing a very weak electrostatic attraction. The ordered structure maximises this attraction which provides a driving force for the order.In this paper, we have shown that INS spectroscopy is readily able to characterise organic surface layers on nanoparticles; the nature of the material is irrelevant: whether the nanoparticle core is an oxide, a metal or a semiconductor makes no difference. This is because the scattered intensity depends on the inelastic cross section, which is independent of the electronic state of the atom. Since the scattering is dominated by modes involving hydrogen motion, provided that the nanoparticle core is actually (or almost) non-hydrogenous, the core will be invisible to INS spectroscopy. The capability to view just the adlayer is a major advantage of INS spectroscopy that is under-exploited. Comparison to DFT calculations allows insights into the nature and conformation of the adsorbed layer.
Acknowledgements
The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/I038748/1, EP/I019693/1, EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). This research has been performed with the use of facilities at the Research Complex at Harwell. The authors would like to thank the Research Complex for access and support to these facilities and equipment. The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities. Computing resources (time on the SCARF compute cluster for the CASTEP calculations) were provided by STFC's e-Science facility. We would like to thank Dr Svemir Rudić (ISIS Facility, UK) for help with the MacroModel software.References
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- G. C. Bond and D. T. Thompson, Catal. Rev.: Sci. Eng., 1999, 41, 319–388 CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- T. Akita, M. Kohyama and M. Haruta, Acc. Chem. Res., 2013, 46, 1773–1782 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855–863 CrossRef CAS PubMed.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546–1548 CrossRef CAS PubMed.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- B. Nkosi, M. D. Adams, N. J. Coville and G. J. Hutchings, J. Catal., 1991, 128, 378–386 CrossRef CAS.
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137, 14548–14557 CrossRef CAS PubMed.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- J. K. Edwards, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512–8515 CrossRef CAS PubMed.
- A. Villa, D. Wang, N. Dimitratos, D. S. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15 CrossRef CAS.
- G. L. Brett, P. J. Miedziak, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, R. Tiruvalam, C. J. Kiely, D. W. Knight, S. H. Taylor, D. J. Morgan, A. F. Carleya and G. J. Hutchings, Catal.: Sci. Technol., 2012, 2, 97–104 CAS.
- S. Albonetti, T. Pasini, A. Lolli, M. Blosi, M. Piccinini, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, G. J. Hutchings and F. Cavani, Catal. Today, 2012, 195, 120–126 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- I. Hussain, Z. X. Wang, A. I. Cooper and M. Brust, Langmuir, 2006, 22, 2938–2941 CrossRef CAS PubMed.
- C. K. Yee, R. Jordan, A. Ulman, H. White, A. King, M. Rafailovich and J. Sokolov, Langmuir, 1999, 15, 3486–3491 CrossRef CAS.
- M. Schulz-Dobrick, K. V. Sarathy and M. Jansen, J. Am. Chem. Soc., 2005, 127, 12816–12817 CrossRef CAS PubMed.
- F. Lu, J. Ruiz and D. Astruc, Tetrahedron Lett., 2004, 45, 9443–9445 CrossRef CAS.
- M. Biswas, E. Dinda, M. H. Rashid and T. K. Mandal, J. Colloid Interface Sci., 2012, 368, 77–85 CrossRef CAS PubMed.
- J. Llorca, M. Dominguez, C. Ledesma, R. J. Chimentao, F. Medina, J. Sueiras, I. Angurell, M. Seco and O. Rossell, J. Catal., 2008, 258, 187–198 CrossRef CAS.
- Y. Tai, W. Yamaguchi, K. Tajiri and H. Kageyama, Appl. Catal., A, 2009, 364, 143–149 CrossRef CAS.
- Y. Tai, W. Yamaguchi, M. Okada, F. Ohashi, K. Shimizu, A. Satsuma, K. Tajiri and H. Kageyama, J. Catal., 2010, 270, 234–241 CrossRef CAS.
- D. J. Gavia and Y. S. Shon, Langmuir, 2012, 28, 14502–14508 CrossRef CAS PubMed.
- D. J. Gavia, J. Koeppen, E. Sadeghmoghaddam and Y. S. Shon, RSC Adv., 2013, 3, 13642–13645 RSC.
- N. G. Hamilton, I. P. Silverwood, R. Warringham, J. Kapitan, L. Hecht, P. B. Webb, R. P. Tooze, S. F. Parker and D. Lennon, Angew. Chem., Int. Ed., 2013, 52, 5608–5611 CrossRef CAS PubMed.
- D. Lennon and S. F. Parker, Acc. Chem. Res., 2014, 47, 1220–1227 CrossRef CAS PubMed.
- I. P. Silverwood, S. M. Rogers, S. K. Callear, S. F. Parker and C. R. A. Catlow, Chem. Commun., 2016, 52, 533–536 RSC.
- S. F. Parker, K. Refson, A. C. Hannon, E. R. Barney, S. J. Robertson and P. Albers, J. Phys. Chem. C, 2010, 114, 14164–14172 CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/ 1997–2015.
- S. F. Parker, F. Fernandez-Alonso, A. J. Ramirez-Cuesta, J. Tomkinson, S. Rudic, R. S. Pinna, G. Gorini and J. Fernández Castañon, J. Phys.: Conf. Ser., 2014, 554, 012003 CrossRef.
- S. F. Parker, D. Lennon and P. W. Albers, Appl. Spectrosc., 2011, 65, 1325–1341 CrossRef CAS.
- http://www.isis.stfc.ac.uk/ .
- M. A. Adams, S. F. Parker, F. Fernandez-Alonso, D. J. Cutler, C. Hodges and A. King, Appl. Spectrosc., 2009, 63, 727–732 CrossRef CAS PubMed.
- http://accelrys.com/products/collaborative-science/biovia-materials-studio/ .
- Schrödinger Release 2014-1: MacroModel, version 10.3, Schrödinger, LLC, New York, NY, 2014.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- K. Refson, P. R. Tulip and S. J. Clark, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- V. Milman, A. Perlov, K. Refson, S. J. Clark, J. Gavartin and B. Winkler, J. Phys.: Condens. Matter, 2009, 21, 485404 CrossRef PubMed.
- K. Refson, Phonons and Related Calculations in CASTEP, http://www.castep.org/.
- A. J. Ramirez-Cuesta, Comput. Phys. Commun., 2004, 157, 226 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, New York, 1991 Search PubMed.
- N. Nakamura, K. Uno and Y. Ogawa, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o508 CAS.
- D. A. Braden, S. F. Parker, J. Tomkinson and B. S. Hudson, J. Chem. Phys., 1999, 111, 429–437 CrossRef CAS.
- J. Tomkinson, S. F. Parker, D. A. Braden and B. S. Hudson, Phys. Chem. Chem. Phys., 2002, 4, 716–721 RSC.
| This journal is © the Owner Societies 2016 |