
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent syntheses of functional chromophores†
Lucilla
Levi
and
Thomas J. J.
Müller
*
Institut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, D-40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@hhu.de
First published on 22nd February 2016
Abstract
Multicomponent reactions are a valuable tool for the synthesis of functional π-electron systems. Two different approaches can be taken into account for accessing the target structures. In the more conventional scaffold approach an already existing chromophore is coupled with other components to give a complex functional π-system. Here, electronically monotonous components can also be introduced, which may exert synergistic electronic effects within the novel compound. The more demanding chromophore concept generates a complete π-electron system and a scaffold concurrently. The latter approach is particularly stimulating for methodologists since π-systems might be accessible from simple starting materials. This review encompasses the advances in the preparation of functional π-electron systems via multicomponent processes during the past few years, based both on the scaffold and chromophore concepts. Besides the synthetic strategies the most important properties, i.e. redox potentials, absorption and emission maxima or fluorescence quantum yields, of the synthesized molecules are highlighted.
 Thomas J. J. Müller (left) and Lucilla Levi (right) | Lucilla Levi (right) was born in Milan, Italy, and studied chemistry at the Heinrich-Heine Universität Düsseldorf in 2007. She obtained her bachelor's degree in 2010 and two years later a master's degree in chemistry. She joined the group of Prof. Dr T. J. J. Müller in 2012 for her PhD thesis on the synthesis and application of phenothiazinyl merocyanines in dye-sensitized solar cells. She received her PhD in 2016. |
Thomas J. J. Müller (left) was born in Würzburg, Germany, and studied chemistry at Ludwig-Maximilians-Universität München (LMU) from 1984 to 1989 (diploma in 1989 and PhD in 1992 with Prof. Dr R. Gompper). After a post-doctoral stay with Prof. Dr B. M. Trost at Stanford University (USA) in 1993 he returned to Germany in 1994 to begin his independent research as a Liebig scholar at Technical University Darmstadt. He moved to LMU as a DFG scholar in 1997, obtained his habilitation and was appointed to Privatdozent in 2000. From 2002 to 2006 he was a professor of organic chemistry at Ruprecht-Karls-Universität Heidelberg. Since 2006 he has been a full professor and has held the chair of organic chemistry at Heinrich-Heine-Universität Düsseldorf. He is the author of more than 200 journal articles and book chapters. |
1 Introduction
Chromophores are functional π-electron systems that lay the molecular foundation of modern organic electronics and solar energy conversion with tailored small molecules.1 For instance they find applications in organic light-emitting diodes (OLEDs),2 dye-sensitized solar cells (DSSCs),3 organic photovoltaics (OPVs),4 organic field effect transistors (OFETs),5 or sensor arrays in bio- or environmental analytics.6 Hence, the efficient and rapid preparation of these novel functional organic molecules with specific photophysical and electrochemical properties remains an ultimate goal and challenge for researchers both in organic chemistry and materials science. Inevitably, prerequisites for productivity and efficacy are represented by selectivity criteria, i.e. chemo-, regio-, and stereoselectivity, in combination with economic and ecological aspects. Therefore, the creation of concise and conceptually novel syntheses that intelligently concatenate fundamental organic reactions in one-pot sequences defines the paramount goal.7In particular, multicomponent processes8 exactly tackle these synthetic challenges and, therefore, they have received considerable interest both in academia and industry, predominantly for synthesizing biologically active molecules.9 By definition a multicomponent process is a reaction of more than two starting materials leading to a product that contains most of the employed atoms.7,8 More specifically, multicomponent reactions (MCRs) as a reactivity based concept10 can be divided into three different categories. In domino-type MCRs all reagents have to be present from the very beginning of the process. A sequential MCR implements the subsequent addition of components in a well-defined order without changing the reaction conditions. Finally, the consecutive MCR implements the subsequent addition of reagents by changing the conditions from step to step. All three types of processes assure high structural and functional diversity and bear an immense explorative potential. At best, MCR will commence with easily accessible and variable starting materials to warrant convenient, interesting and occasionally unusual results.7 The implementation of MCR in diversity-oriented syntheses of chromophores, fluorophores and redox-active molecules has just started in the past one and half decades and represents a fruitful concept for exploring large structural and functional molecular spaces.11,12 By the nature of MCR they can be either employed to introduce a preformed chromophore into a scaffold (scaffold approach) or the MCR acts as a chromogenic event (chromophore approach) (Scheme 1). While the scaffold approach is the more traditional concept for ligating functional chromophores with concomitant formation of an electronically innocent molecular framework, the chromophore approach generates a functional π-system and a scaffold simultaneously.
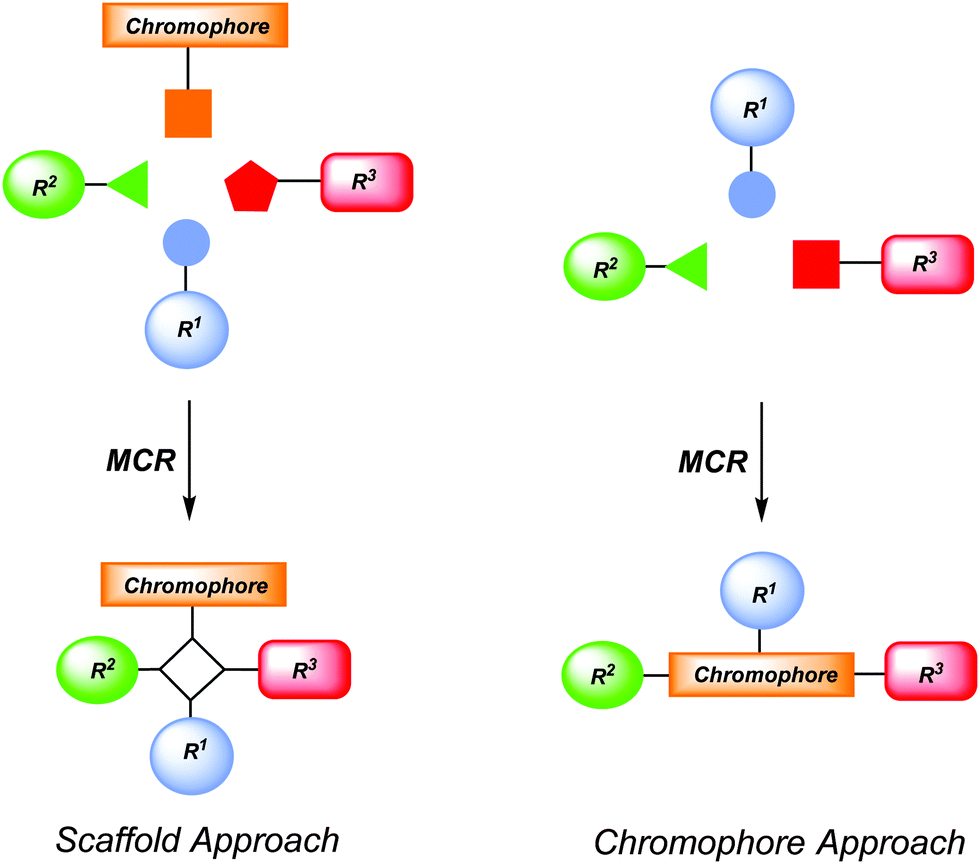 | ||
| Scheme 1 Conceptual MCR formation of functional chromophores by scaffold and chromophore approaches. | ||
Synthetically this review focuses on scaffold and chromophore approaches for the new MCR syntheses of chromophores and functional π-systems that have been developed in the past decade. However the outline is oriented at the functionality of the chromophores and comprises redox systems, including PET (photoinduced electron transfer) systems, and fluorophores, including NLO-(nonlinear optical) chromophores. As clearly defined by MCR methodology only those processes have been considered that are conducted in a one-pot fashion sensu stricto, i.e. without isolation of intermediate products, filtrations, or intermediate evaporation and change of solvents.
2 Redox systems
Redox-active systems can undergo a change in the oxidation state, often in a fully reversible fashion, and they have received attention both for biomedical applications and as functional materials. For the former by oxidation or reduction reactivity can be induced that affects protein action, whereas in the latter the change of the oxidation stage can induce intra- and intermolecular electron transfer that ultimately leads to material (semi)conductivity and macroscopically to current flow.Antioxidants have shown promising results in medicinal chemistry, for instance, in devising Alzheimer's disease therapies.13 The reduced accumulation of certain proteins leads to a slower decline of the brain function in mice. Therefore, the search for powerful antioxidants for disease treatment is an ongoing task both in academia and industry. Barate et al. recently reported on the antioxidant activity of 4-arylquinolines in Alzheimer' treatment.14 In the sense of the chromophore concept, a multicomponent, metal-free approach was developed by reacting substituted anilines with terminal alkynes and ethyl glyoxolate to rapidly furnish ethyl 4-arylquinolinyl-2-carboxylates 1 at room temperature in excellent yields (Scheme 2).
 | ||
| Scheme 2 Domino three-component synthesis of redox active 4-arylquinolines 1. | ||
The efficiency of the molecule to reduce iron, the so called iron-reducing power, and free-radical scavenging activity determine the antioxidant potential of the prepared 4-arylquinolines. In comparison to ascorbic acid (set at 100% as a standard) quinolines 1a and 1b show the highest Fe-reducing power, approximately 45%. The molecules only possess a low free radical scavenging activity.
Ferrocenyl derivatives are organometallic redox systems and exhibit a broad range of biological activities, ranging from antitumor, antimalaria to anti-HIV active compounds.15 By a consecutive three-component synthesis ferrocenyl isoxazoles 2 were readily synthesized according to the scaffold concept (Scheme 3).16 Interestingly, the isoxazole core is obtained by Sonogashira coupling of acyl chlorides with alkynes to give alkynones, which react as dipolarophiles in subsequent (3+2)-cycloadditions with nitrile oxides (in situ generated from hydroxyiminoyl chlorides by 1,3-elimination) in the same reaction vessel. By choice of the alkyne and the acid chlorides ferrocenyl moieties can be placed at two points of diversity in this coupling-1,3-dipolar cycloaddition sequence.
 | ||
| Scheme 3 Consecutive three-component synthesis of ferrocene substituted isoxazoles 2. | ||
The MCR proceeds with excellent regioselectivity always placing the carbonyl group at the 4-position of the isoxazole. Cyclic voltammetry reveals slightly anodically shifted oxidation potentials compared to ferrocene (E0/+11/2 = 450 mV). This can be rationalized by the electron withdrawal of the conjugated carbonyl group. For compounds 2b and 2c second oxidation waves are detected, which can be assigned to the phenothiazinyl moiety (2b) or a second ferrocenyl substituent (2c). Interestingly the strongly electron donating character of phenothiazine attenuates the carbonyl capacity on the metallocene potential, leading to a smaller anodic shift in the first oxidation potential.
Ferrocenyl-substituted pyridines have also been applied in drug research17 and find use as potential bio-receptor ligands.18 The desired ferrocenyl pyridines 3 can be isolated after the multicomponent condensation of malononitrile, 2-cyano-3-ferrocenyl acrylonitrile and an alcohol in the sense of the scaffold concept (Scheme 4). As an inevitable side reaction a twofold addition of the ferrocenyl moiety results in the formation of 3,4-dihydropyridines 4 as byproducts.19
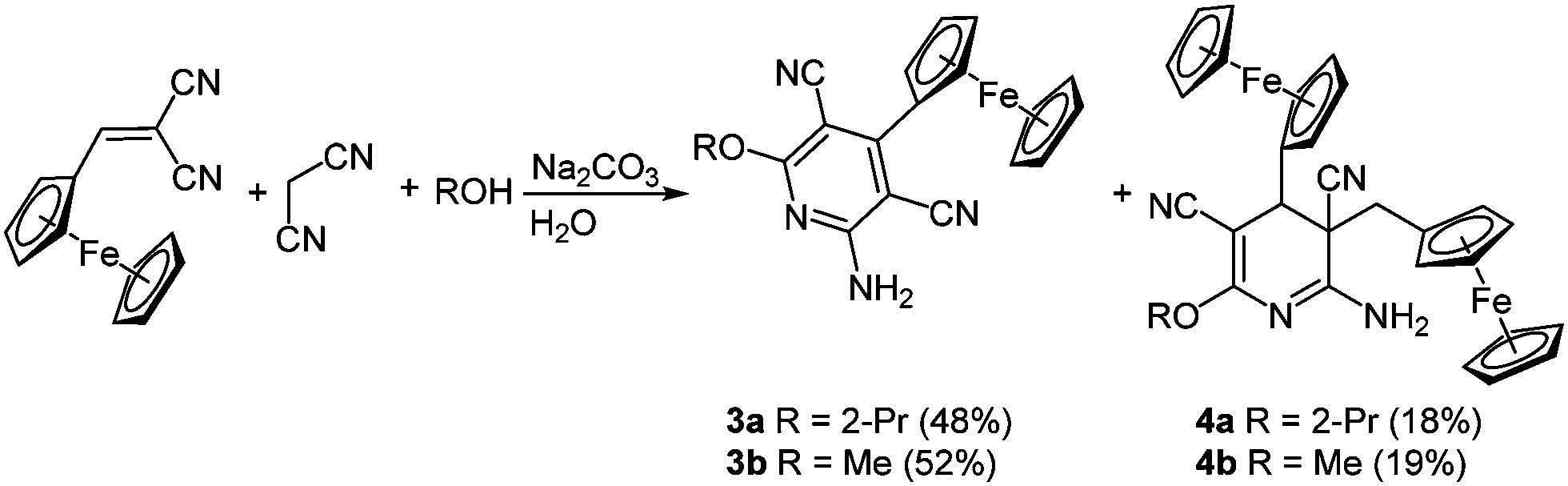 | ||
| Scheme 4 Three-component synthesis of ferrocenyl pyridines 3 and 3,4-dihydropyridines 4. | ||
The cyclic voltammograms of the aromatic compound 3a reveals a single reversible Nernstian oxidation potential at around 215 mV deriving from the ferrocenyl substituent. Expectedly a second oxidation potential can be detected for dihydropyridine 4a, originating from the second ferrocenyl moiety. Surprisingly this second oxidation is irreversible.20
In organic electronics the use of oligothiophenes as organic semiconducting materials is ubiquitous.21 According to the chromophore concept Bäuerle and coworkers presented a domino three-component synthesis of thiophene-1,2,3-triazole co-oligomers 5 as novel donor–acceptor–donor models.22 1,2,3-Triazoles are most conveniently accessed by Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC).23 This regiospecific reaction generally produces the 1,4-disubstituted triazole with high yields, moderate reaction times and tolerates many functional groups. For the synthesis of co-oligomers azidothiophenes are formed in situ from halothiophenes with sodium azide, which complete the CuAAC-based MCR24 with 2-ethynylthiophenes (Scheme 5).
 | ||
| Scheme 5 Domino three-component synthesis of thiophene-1,2,3-triazole co-oligomers 5. | ||
The absorption spectra show several broad bands, which can be assigned to the different constituting subunits of the oligomers. The longest wavelength absorption bands around 280 nm correlate with the triazole core, while the thienyl substituent at the 1-position of the triazole shows a maximum at around 250 nm. Longer oligomer chains display bathochromically shifted longest wavelength absorption bands and symmetrical oligomers give more structured spectra. The length of the chain also affects the oxidation potentials of the compounds, where shorter oligomers possess higher potentials. A reduction potential for the triazole was not detected in the cyclic voltammograms under the given conditions.
Oxygen and nitrogen containing macrocycles with large cavities have a high binding affinity to numerous anion and cation receptors,25 proteins26 and organic guest molecules.27 The use of these ligands as model compounds for biological processes results from the possible complexation with biologically relevant anions.28 The preparation of piperazinophanes 6 and the expanded cyclic dimers 7 was realized via an amine–aldehyde–alkyne (A3) reaction29 of piperazine and formaldehyde with aromatic bispropargylic ethers in the sense of the scaffold approach (Scheme 6).30
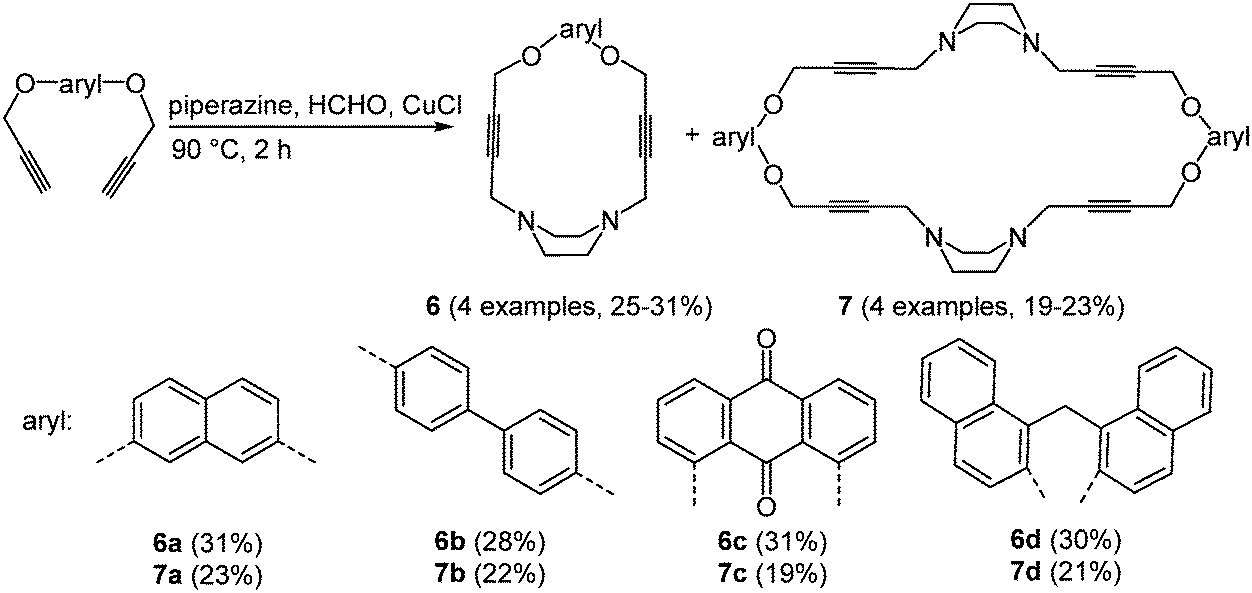 | ||
| Scheme 6 Amine–aldehyde–alkyne (A3) synthesis of piperazinophanes 6 and 7. | ||
The absorption maxima of piperazinophanes 6 and 7 appear between 243 and 356 nm. Systems with anthraquinone as an aromatic moiety also show a second weaker absorption band at 284 nm. In addition all macrocycles display intense luminescence with emission maxima between 432 and 501 nm. In the cyclic voltammograms irreversible reductions can be readily identified. Again the anthraquinone substituted macrocycles 6c and 7c possess several reduction peaks. The UV/Vis spectra of the compounds after complexation with 7,7,8,8-tetracyanoquinodimethane show strongly red-shifted absorption bands at around 730, 750 and 770 nm, which were assigned to charge transfer transitions between the macrocycle and the guest molecule. The complexation ratio was photospectrometrically determined to result in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex.
:1 complex.
3 Photoinduced electron transfer (PET) systems
Effective systems in molecular electronics and optoelectronics are often based upon donor–acceptor (D–A) conjugate motifs.31 These systems can be classified by the nature of ligation of the functional units. Constitutionally, they are distinguished in conjugated, non-conjugated, and non-covalently bound D–A molecules. For non-conjugated topologies, a photoinduced electron transfer (PET) causing a persistent charge separation represents the primordial step in photovoltaics and permits a light-to-current conversion.32 This is also reminiscent of the most important electron transfer reaction in nature, i.e. photosynthesis converting sunlight into electrochemical potential. Here, sunlight is absorbed, funneled by an antenna complex and followed by an electron transfer to the reaction center which leads to charge separation. Inspired by this process many studies were directed towards porphyrin induced charge separation in organic solar cells. Simple porphyrin–quinoline-dyads were first reported by Tabushi et al. in 1979,33 followed by many other research groups.34 Astoundingly, only very few one-pot syntheses of porphyrins were reported to date. The standard procedures deal with the decoration or extension of porphyrin rings in a multicomponent fashion followed by metalation in a one-pot reaction.35In 1959, Ugi presented a new four-component reaction (Ugi-4CR) condensing an acid, an isonitrile, an aldehyde, and an amine.36 This event set the stage for an eager search for novel MCRs.8,9,37 Due to the concomitant formation of two amide bonds in the course of this process the Ugi-4CR was established as a flexible and rapid access to peptides, peptoids and a series of many biologically active compounds.38 Although this powerful process found entry in modular syntheses of bioactive ingredients only in recent years the Ugi-4CR has been explored for the preparation of electronic and photoactive systems.
Just recently the Ugi reaction was applied to access donor–acceptor-systems in a novel and straightforward fashion. Due to their tunable and reversible oxidation potentials phenothiazine derivatives are particularly interesting as electron donor moieties.39 On the other hand 9,10-anthraquinone has a distinct electron deficiency, which makes it an important electron acceptor.40 When employing the donor as an amine, the acceptor as an aldehyde, acetic acid and tert-butyl isocyanide in the Ugi-4CR the desired condensation products 8 are obtained in good to high yields in the sense of a scaffold approach.41 The scope of 14 examples was realized via this method by varying the donor and/or the acceptor moiety (Scheme 7).
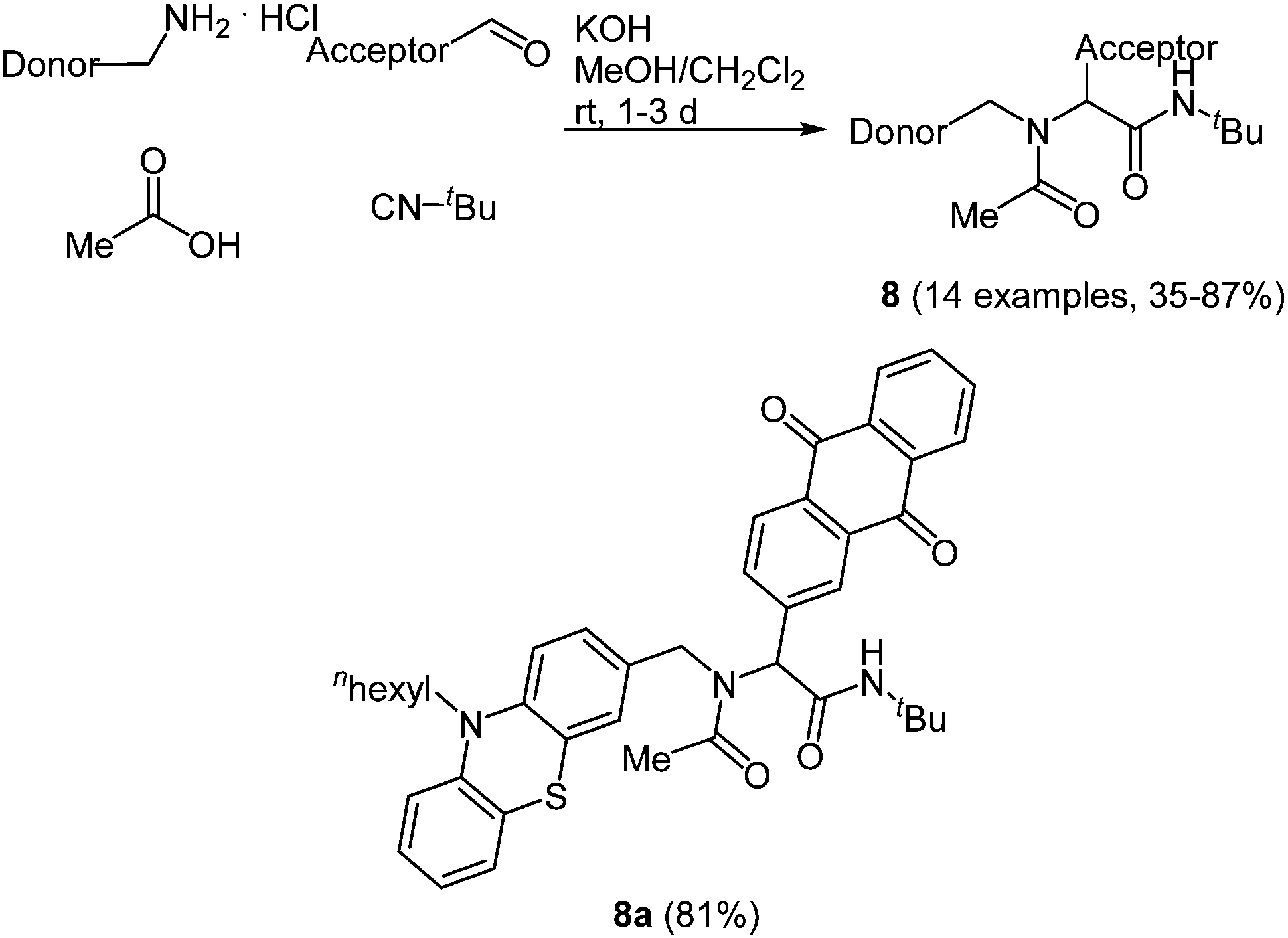 | ||
| Scheme 7 Ugi-4CR synthesis of donor–acceptor dyads 8 and structure of the phenothiazine–anthraquinone system employed in transient absorption studies. | ||
Phenothiazine based dyads 8 show absorption characteristics in solution that support the absence of an electronic communication in the electronic ground state, as expected for non-conjugated D–A systems. Most strikingly, the D–A conjugates display considerable intramolecular fluorescence quenching caused by an excited state communication between phenothiazine and acceptor moieties. Femtosecond spectroscopy unambiguously elucidated that in the excited state an ultrafast electron transfer to the anthraquinone accounts responsible for the observed emission quenching. Thereby, in the dyads rapid electron transfer proceeds with ps rate constants via excited singlet or triplet states to terminate in a short lived singlet and a long lived triplet charge separated state.
Dihydropyridines already found application as dyads in biosensors and photosensitive polymers, due to their ability to modulate vectorial transport of energy or charge transfer upon photoexcitation.42 The synthesis of 2-unsubstituted dihydropyridines with strong UV absorption characteristics was achieved via a Hantzsch-type pseudo four-component reaction with enaminones as starting materials in the sense of the chromophore approach.43 The introduction of suitable electron acceptor moieties in the molecule affects the intramolecular PET characteristics. The reaction gives excellent results when performed with sterically undemanding aromatic aldehydes, for instance with benzaldehyde, furnishing the corresponding dihydropyridines within 2 min under microwave irradiation in 84–95% yield. With sterically more demanding substituents such as naphthalene, conventional heating has to be applied for extended times giving the desired heterocycles 9 only in moderate yields (Scheme 8).
 | ||
| Scheme 8 Pseudo four-component reaction for the synthesis of naphthyl substituted dihydropyridines 9. | ||
The UV/Vis spectra display two distinct absorption bands in the regimes of 277–306 and 383–406 nm, respectively. The higher energy absorption maximum is assigned to the aryl moieties, whereas the longest wavelength absorption maximum is ascribed to dihydropyridine. The emission spectra exhibit maxima between 454 and 486 nm with fluorescence quantum yields of up to 50%.
4 Fluorophores
Luminescent dyes exhibit intense light emission upon light excitation. The underlying chromophore is called the fluorophore.44 Ultimately the electronic properties of the fluorophore determine excitation wavelengths. In addition, fluorophores often display pronounced sensitivity for the environment (solvent polarity, hydrophobic media, pH). Interestingly, due to numerous interactions and processes the spectral properties can often be altered, switched and controlled. Therefore, fluorophores are highly interesting as functional π-electron systems. The emission range lies between UV and NIR with lifetimes varying from nanoseconds (fluorescence in a broader sense) to micro- and milliseconds (generally referred to as phosphorescence), strongly depending on the structures and excited states' characteristics. Fluorescent molecules are often employed for DNA or protein labeling, enabling unique sensitivity and highly selective diagnosis in biomedical analytics.45As a consequence of this manifold of scientific applications and motivated by the demand for stable tailor-made fluorophores, novel and concise syntheses of this class of functional chromophores are currently a hot topic and the synthetic activity is steadily increasing. Various approaches have been followed to date ranging from consecutive over sequential to domino MCR. In many cases the spectral properties of the fluorophores primarily depend on the (hetero)aromatic or polyene core structure. By the choice of the delocalized π-electron system the excitation in the visible region can be achieved and fine-tuned.
4.1 MCR formation of nitrogen heterocycles
Five-membered N-heterocycles like pyrazoles and imidazoles display a multifold chemical reactivity accompanied by multiple types of applications.46 Much interest resides in the broad spectrum of biological activity but also in unique electronic and optical properties of these classes of heterocycles.47 Depending on the substitution pattern different photophysical and electrochemical properties can be addressed. As a consequence they find use as optical brighteners, as UV stabilizers, and as constituents in photoinduced electron transfer systems.48Studies on OLEDs proved that excited states with charge transfer (CT) character are very valuable for increasing the efficiency of the module. Many chromophores, based on both the covalent donor–acceptor structure and complexes, have already been synthesized and their electronic properties were tested. Predominantly red and green emissive fluorophores have been reported.49 CT excited states typically exhibit a narrower band gap than normal locally excited states, while blue, non-CT emitters usually retain a wider band gap.50 A deep-blue electroluminescent chromophore 10 with a current efficiency of >5.0% was recently reported by Ma and coworkers.51 The acceptor moiety 10 was synthesized by a four-component reaction of an aldehyde, an amine, ammonium acetate, and phenanthryl quinone derived from the reaction presented by Sun.52 Suzuki coupling of the donor as a boronic acid ester and the acceptor 10 resulted in the desired push–pull chromophore 11 following the scaffold concept (Scheme 9).
 | ||
| Scheme 9 Synthesis of the deep-blue emitting donor (blue)-acceptor (red) chromophore 11. | ||
An interesting multicomponent approach to imidazo-pyridines under bismuth catalysis was very recently presented by Esmati and coworkers.53 In a Groebke–Blackburn–Bienaymé (GBB)54 multicomponent condensation 4-pyrone carbaldehydes react with isonitriles and 1-amino pyridines under bismuth chloride catalysis furnishing the desired imidazo[1,2-a]pyridines 12 within a reaction time of 3 min and in good to excellent yields in the sense of the chromophore concept (Scheme 10).
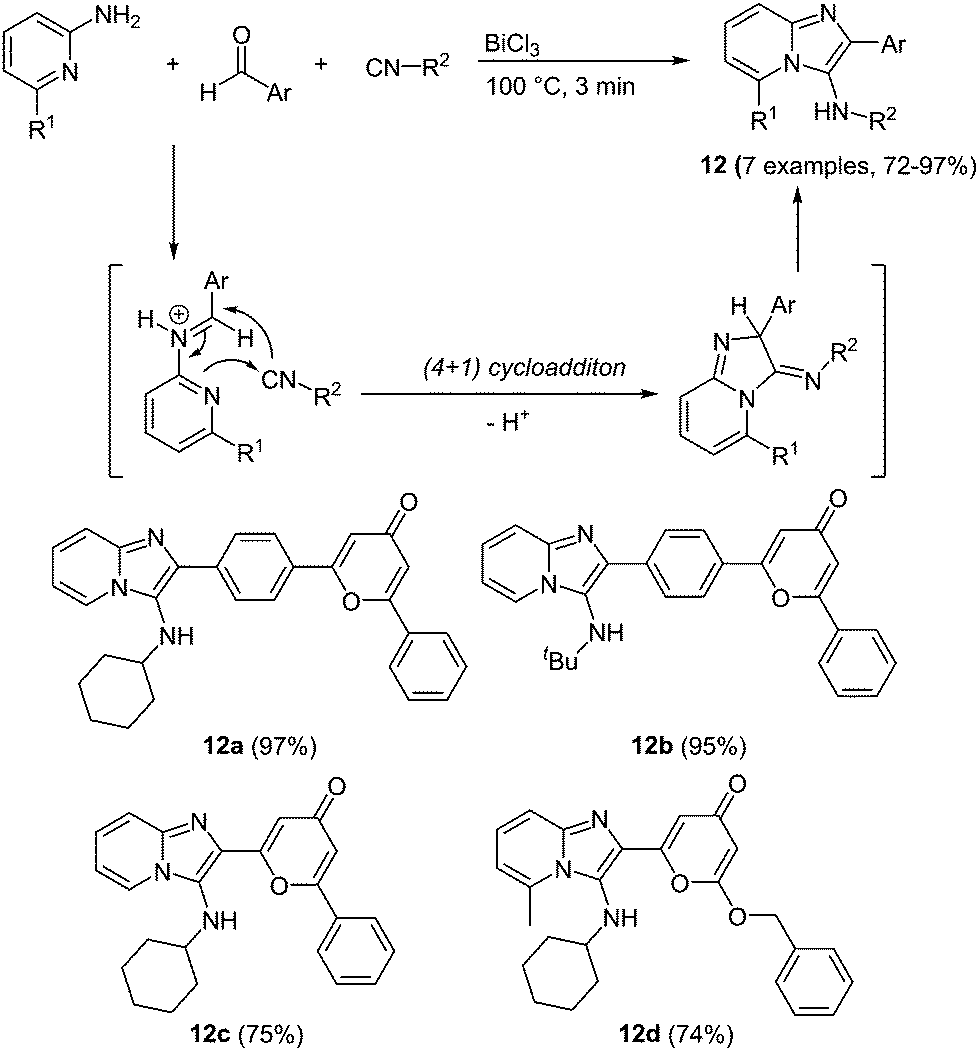 | ||
| Scheme 10 GBB multicomponent synthesis of imidazo[1,2-a]pyridines 12. | ||
A distinct influence of the benzene spacer between the imidazo-pyridine core and 4-pyrone can be observed in the absorption spectra of these chromophores. A blue shift can be detected in the strongest maxima, ascribed to the π–π* transition, of the compounds without a spacer (λmax = 265–273 nm) compared to the benzene substituted systems (λmax = 283 nm), which possess an extended conjugated system. More remarkable is the difference in the shape of the absorption bands. The compounds containing a spacer show very broad, shapeless spectra with an intense maximum at 356 nm, whereas clearly a less intense but more structured longest wavelength absorption band is observed for the spacer-free compounds. Also in the emission spectra a hypsochromic shift can be determined for the compounds without the benzene spacer. The compounds display intense blue to green fluorescence with maxima between 500 and 515 nm for 12a–12b and 475–478 nm for 12c–12d.
Imidazo[1,2-a]-pyridines 13 with a similar structure were prepared under copper and glucose catalysis, resulting in systems with excited state intramolecular proton transfer (ESIPT) characteristics.55 The reaction between phenylacetylene, 2-aminopyridines and benzaldehydes give the desired products in 24 h with yields between 41 and 89% following the chromophore approach (Scheme 11).
 | ||
| Scheme 11 Three-component synthesis of imidazo[1,2-a]-pyridines 13 under copper and D-glucose catalysis and selection of examples showing ESIPT characteristics. | ||
The longest wavelength absorption maxima of compounds 13 lie between 305 and 360 nm, reaching extinction coefficients up to 22000 M−1 cm−1. The substituents do not reveal any systematic influence on the absorption characteristics. With very large Stokes shifts of 12400 to 14700 cm−1 the compounds bearing a 2-(2′-hydroxyphenyl) substituent clearly show excited state intramolecular proton transfer (ESIPT) characteristics with low fluorescence quantum yields (Fig. 1). The emission maxima of all the synthesized molecules are found between 371 and 556 nm.
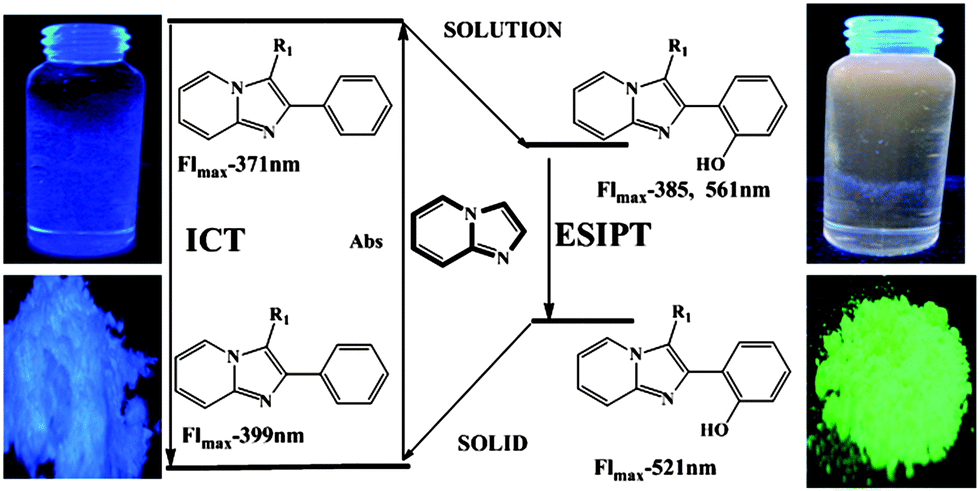 | ||
| Fig. 1 ESIPT characteristics of imidazo[1,2-a]-pyridines 13. Reprinted from Dyes and Pigments, 121, B. Umamahesh, K. I. Sathiyanarayanan, CuO–CuAl2O4 and D-glucose catalyzed synthesis of a family of excited state intramolecular proton transfer imidazo[1,2-a]pyridine analogues and their optical properties, 88–98, Copyright (2015), with permission from Elsevier. | ||
Various one-pot multicomponent syntheses of pyrazoles are found in the literature; however, predominantly for accessing biologically active compounds.56 Highly fluorescent pyrazoles can be synthesized in an efficient and straightforward fashion by consecutive three- or four-component reactions.57 Sonogashira coupling between acyl chlorides and terminal alkynes results in reactive alkynone intermediates, which can readily react with all kinds of binucleophiles in a one-pot fashion.58 Hence, after addition of hydrazines 1,3,5-trisubstituted pyrazoles are formed in a regioselective fashion. A step by step synthesis of these compounds was already known, however only a moderate scope of substituents could be introduced and despite the high regioselectivity the isolation of both isomers was inevitable.59 Therefore, the advantages of the multicomponent approach lie in easier experimental handling as well as in better chemical results. An extension of the sequence allows for the efficient de novo synthesis of even more complex tetrasubstituted pyrazoles. After the formation of pyrazole by coupling–addition–cyclocondensation the 4-position can be halogenated by an N-halosuccinimide, still in the same reaction vessel. A subsequent concluding Suzuki coupling with boronates gives the desired products in good yields in a one-pot fashion in the sense of a chromophore approach (Scheme 12).
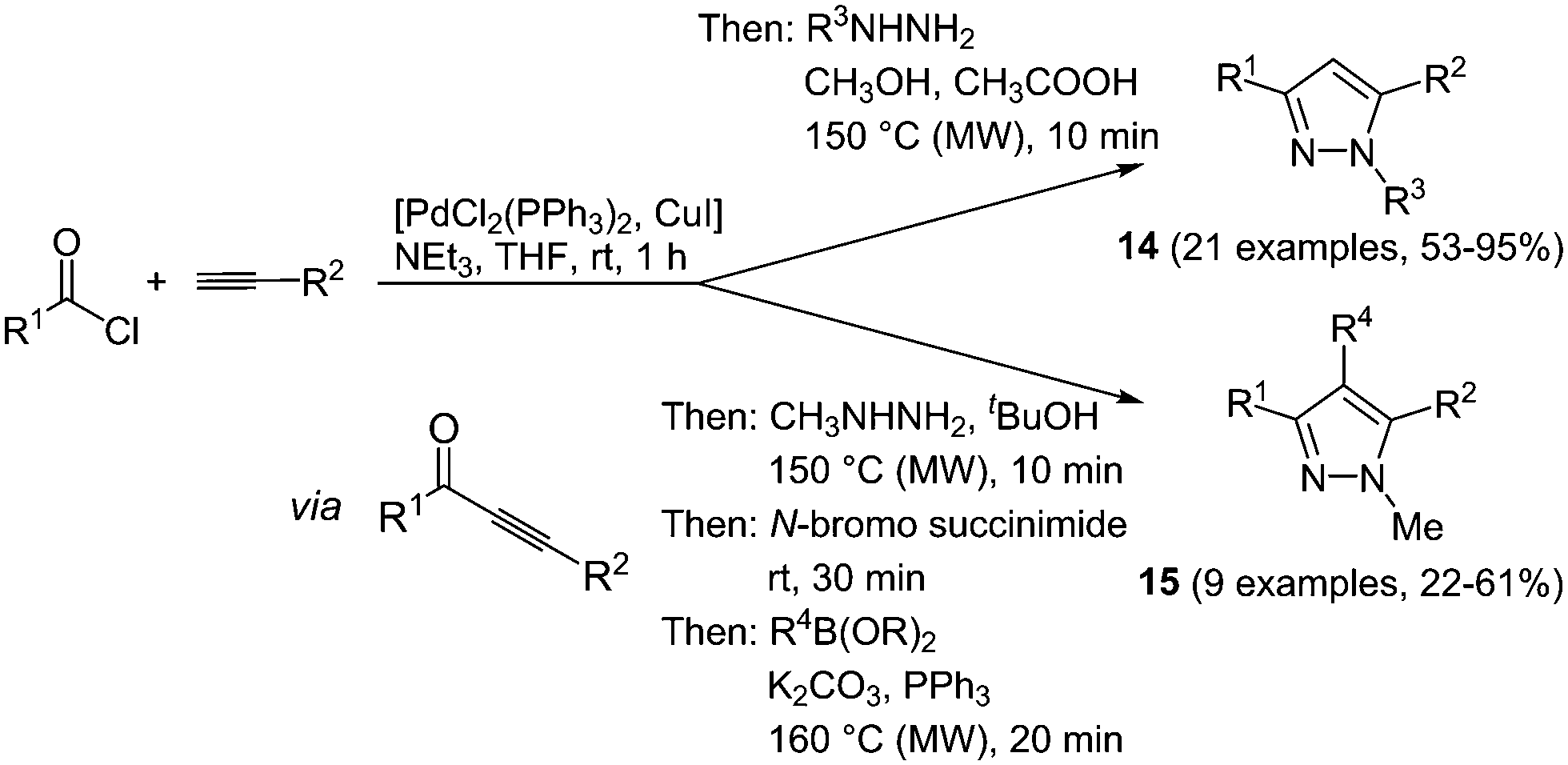 | ||
| Scheme 12 One-pot 3- and 4-component synthesis of tri- and tetrasubstituted pyrazoles 14 and 15. | ||
This diversity-oriented one-pot approach allows for the introduction of many electronically different substituents, generating a broad scope for the investigation of the spectroscopic behavior of pyrazoles. The absorption maxima for the trisubstituted pyrazoles 14 are found between 260 and 385 nm, whereas for tetrasubstituted derivatives 15 values between 240 and 311 nm were determined. It is noteworthy that these molecules display intense fluorescence with emission maxima between 320 and 395 nm and quantum yields of up to 74%. An even higher fluorescence quantum yield of up to 97% could be achieved by the introduction of a biaryl system at one of the substituent positions for the 1,3,5-substituted pyrazoles 14.60 Therefore the Suzuki coupling was concatenated directly after the cyclization reaction. Three different isomers 16–18 were prepared by variation of the arylbromides as substituents in the starting materials giving 19 examples in up to good yields (19–76%). The best results both in reaction and in fluorescence quantum yield were achieved when the biaryl moiety was placed at the position of the alkynyl fragment (Scheme 13).
 | ||
| Scheme 13 Palladium catalyzed four-component synthesis of biaryl-substituted pyrazoles 16–18. | ||
For all substitution patterns large Stokes shifts, between 6000 and 11300 cm−1, were recorded. The longest wavelength absorption maxima of the substances lie between 257 and 311 nm with molar extinction coefficients of up to 44000 M−1 cm−1.
The simultaneous deprotection of three alkyne functionalities on a 1,3,5-triazine core and subsequent CuAAC23 with aromatic azides results in the formation of highly substituted strongly luminescent chromophores 19 with low reduction potentials in a pseudo multicomponent reaction in the sense of a scaffold approach (Fig. 2).61 Absorption spectra in solution and as thin films display almost the same maxima for the longest wavelength absorption bands, however, with considerably higher extinction coefficients for the solution spectra. The compounds are strongly blue fluorescent with quantum yields of up to 43%. Furthermore a liquid crystalline behavior could be observed, showing columnar mesophases at room temperature.
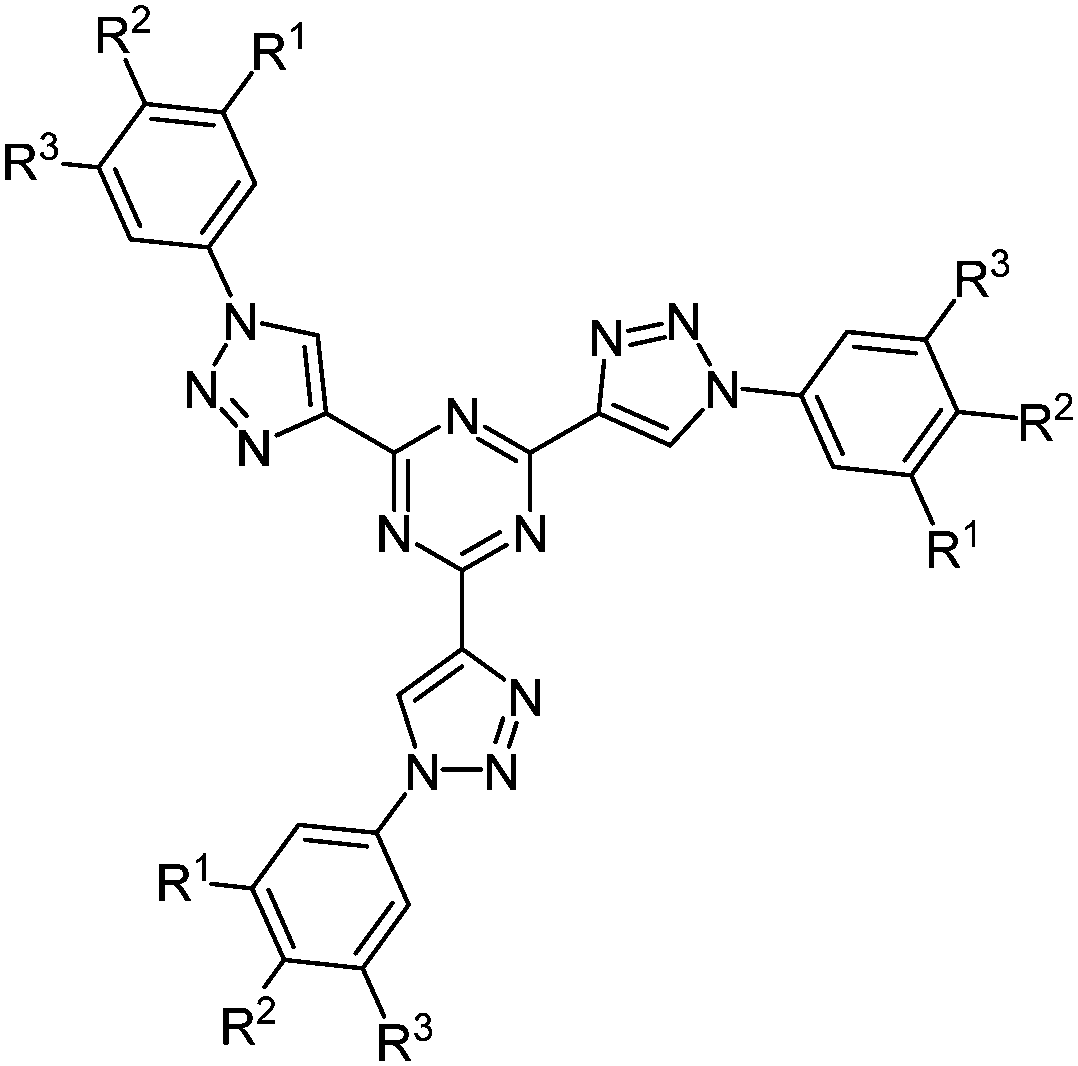 | ||
| Fig. 2 Structure of 2,4,6-tris(triazolyl)-1,3,5-triazine chromophores 19. | ||
Tetrahydropyrimidines are another noteworthy class of biologically active compounds. Additionally some derivatives show interesting magnetic62 and photophysical63 properties. Recently, a novel, efficient five-component synthesis of tetrahydropyrimidines was presented by Liu and coworkers.64 Here, two different amines, formaldehyde, an aromatic aldehyde and acetylene dicarboxylic acid ester are coupled with urea as an organo catalyst (Scheme 14). The reaction proceeds chemoselectively furnishing the desired C-4 unsubstituted product 20 between 21 and 63% yield according to the chromophore concept. The chromophores show aggregation-induced emission (AIE)65 and are almost nonfluorescent in solution. The strong green to blue luminescence in the solid state and in suspension reveals quantum yields of up to 93%.
 | ||
| Scheme 14 Five-component synthesis of C-4 unsubstituted tetrahydropyrimidines 20. | ||
Due to the steadily growing industrialization the detection of toxic heavy metal ions in the environment is an important task.66 Chemosensors with defined optical and mechanistic properties are the most elegant method for the recognition of metal ions.67 Effective fluorescent chemosensors for mercury ions are already well represented in the literature,68 but often with inefficient synthetic routes.69 The Biginelli reaction70 allows for a straightforward preparation of pyrene substituted 1,2,3,4-tetrahydropyrimidine chromophores with sensor characteristics for Hg2+ recognition.71 The zinc catalyzed multicomponent reaction of pyrene-1-carboxaldehyde, methyl (or ethyl) acetoacetate and (thio)urea yields the two desired chromophores 21 in 79 and 89% in the sense of a scaffold approach (Scheme 15).
 | ||
| Scheme 15 Biginelli reaction for the synthesis of tetrahydropyrimidine based fluorescent Hg2+ chemosensors 21. | ||
The characterization of the cation recognition properties of these chromophores was performed by a number of different fluorescence measurements and the ratiometric detection of anions was determined via a counterion displacement assay.72 By screening 13 different metal salts the metal binding ability of sensors 21a and 21b was determined. A possible kinetic influence on the fluorescence spectra was excluded by a second measurement of the probes after one hour. The original emission maximum of 21a at around 395 nm assigned to the pyrene corpus was completely quenched by addition of Hg2+ and a new maximum at 460 nm evolved, while no relevant influence was detected with other cations. The bathochromic shift was ascribed to excimer formation by two pyrene rings that come into close proximity by coordination to the mercury ion.
Singh and coworkers used the above mentioned Biginelli reaction to prepare even more complex and multifunctional pyrene based chemosensors 22 always based on the scaffold concept.73 Thereby zinc ions can be detected in organic media, whereas iron ions are selectively recognized in aqueous medium (Scheme 16).
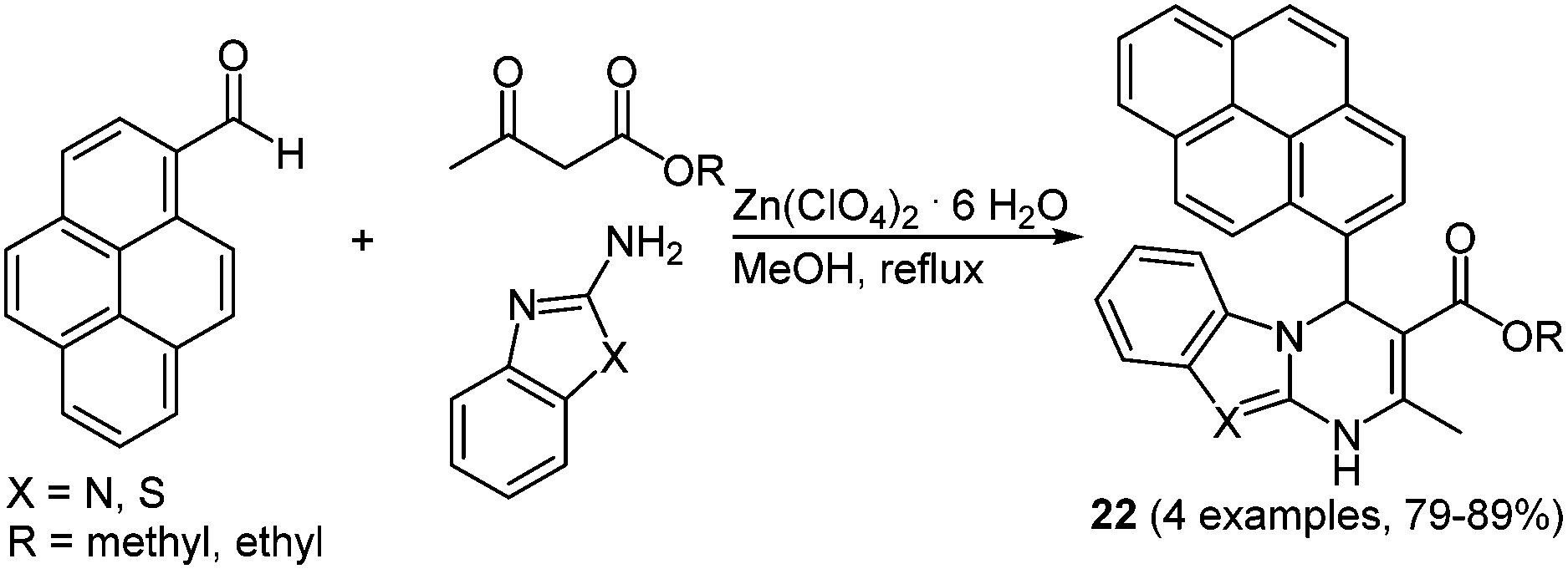 | ||
| Scheme 16 Pyrene based chemosensors 22 for the selective detection of zinc or iron cations. | ||
Similar fluorescence measurements with different metal salts were performed with tetrahydropyrimidines 22. Instead of mercury ions zinc(II) showed the highest variation in the UV/Vis spectra in acetonitrile. Here, the original absorption maximum at around 470 nm was hypsochromically shifted to 370 nm.
Indoles display a high reactivity in the 3-position in electrophilic aromatic substitutions. This aspect has often been exploited to access molecules with remarkable biological activities.74 In addition indoles are particularly interesting as structures and auxochromes in chromophores.
The scaffold of indolyl oxazoles, both with simple but also very complex substitution patterns, is often encountered in naturally occurring alkaloids showing a broad spectrum of biological activity.75 Although many synthetic strategies were developed searching for more biologically active derivatives, the interesting photophysical properties of this class of fluorophores have been completely neglected. A microwave assisted consecutive three-component alkynylation–cycloisomerization–Fischer indole synthesis offers a concise access to 5-(3-indolyl)oxazoles in good yields.76 Starting with a Sonogashira coupling of acid chlorides and 1-(aryl) N-(prop-2-yn-1-yl)acetamides the formed ynones directly undergo an acid catalyzed cycloisomerization to furnish the oxazole core.77 Subsequent addition and reaction with aryl hydrazine hydrochloride conclude the sequence by a Fischer indole synthesis (Scheme 17). The multicomponent strategy with a chromophore approach furnished 25 diversely substituted 5-(3-indolyl)oxazoles 23 with noteworthy electronic properties.
 | ||
| Scheme 17 5-(3-Indolyl)oxazoles 23via the microwave assisted coupling-cycloisomerization Fischer indole sequence. | ||
All indolyl oxazoles 23 display similar absorption and emission behavior. The longest wavelength absorption band is broad and unstructured and appears between 298 and 328 nm. Therefore, only a minor electronic influence of the aryl substituents on the maxima can be deduced. All the emission maxima are found between 426 and 445 nm, resulting in an intense blue luminescence and accompanied by massive Stokes shifts ranging from 7700 to 10600 cm−1. The fluorescence quantum yields lie in the range of 10−25%.
An interesting constitutional isomer of indole is indolizine, where the nitrogen atom is positioned at the bridge head. The fusion of an electron-poor pyridine and an electron-rich pyrrole moiety results in an intriguing scaffold with respect to bioactivity.78 Hence, the synthesis of these heterocycles gained considerable significance and already numerous novel syntheses have evolved.79 However, novel multicomponent reactions have not been explored. An interesting approach presented by Mishra et al. takes advantage of cheap and non-toxic zinc salts as catalysts.80 Zinc iodide has turned out to be the most active catalyst for the sequential three-component coupling-cycloisomerization reaction in the sense of the chromophore concept. The A3-reaction29 of an aromatic aldehyde, a secondary amine, and an alkyne with zinc iodide as a catalyst results in the desired aminoindolizines 24 in good to excellent yields at short reaction time (Scheme 18).
 | ||
| Scheme 18 Zinc catalyzed 3-component synthesis of aminoindolizines and pyrrolo[1,2-a]-quinolines 24. | ||
The introduction of an aliphatic terminal alkyne proceeds also successfully. Likewise employing quinoline-2-carboxaldehydes allows for the preparation of the corresponding pyrrolo[1,2-a]-quinolines 24d in very good yields. Especially these compounds showed interesting photophysical properties, for instance high fluorescence quantum yields of up to 47% and large Stokes shifts, as important characteristics for biological probes. Both absorption and emission solvatochromicity studies were performed with the pyrrolo[1,2-a]-quinoline 24d. While only a minimal shift can be detected in the absorption spectra, the emission spectra show maxima change from 514 and 541 nm with increasing solvent polarity.
Quinoline and quinoxaline derivatives are well known as biologically and pharmaceutically relevant heterocyclic cores,81 and many new efficient syntheses have been reported.82 In addition, these molecules show inherent fluorescence, with a substantial solvent effect on the efficiency and energy of the emission bands, thereby fueling the search for novel emissive chromophores.83 Consequently, many novel push–pull systems were prepared and analyzed pertaining to their spectroscopic behavior.84 However, only very few one-pot multicomponent reactions have been found.
2,4-Diaryl substituted quinolines 25 can be synthesized efficiently by a regiospecific consecutive three-component reaction starting from commercially available acyl chlorides and alkynes.85 First the Sonogashira conditions furnish the corresponding alkynones86 that are subsequently reacted with 2-aminothiophenols. Condensation is accompanied by a concomitant sulfur extrusion upon microwave irradiation giving the desired quinolines in moderate to good yields in the sense of the chromophore concept (Scheme 19).
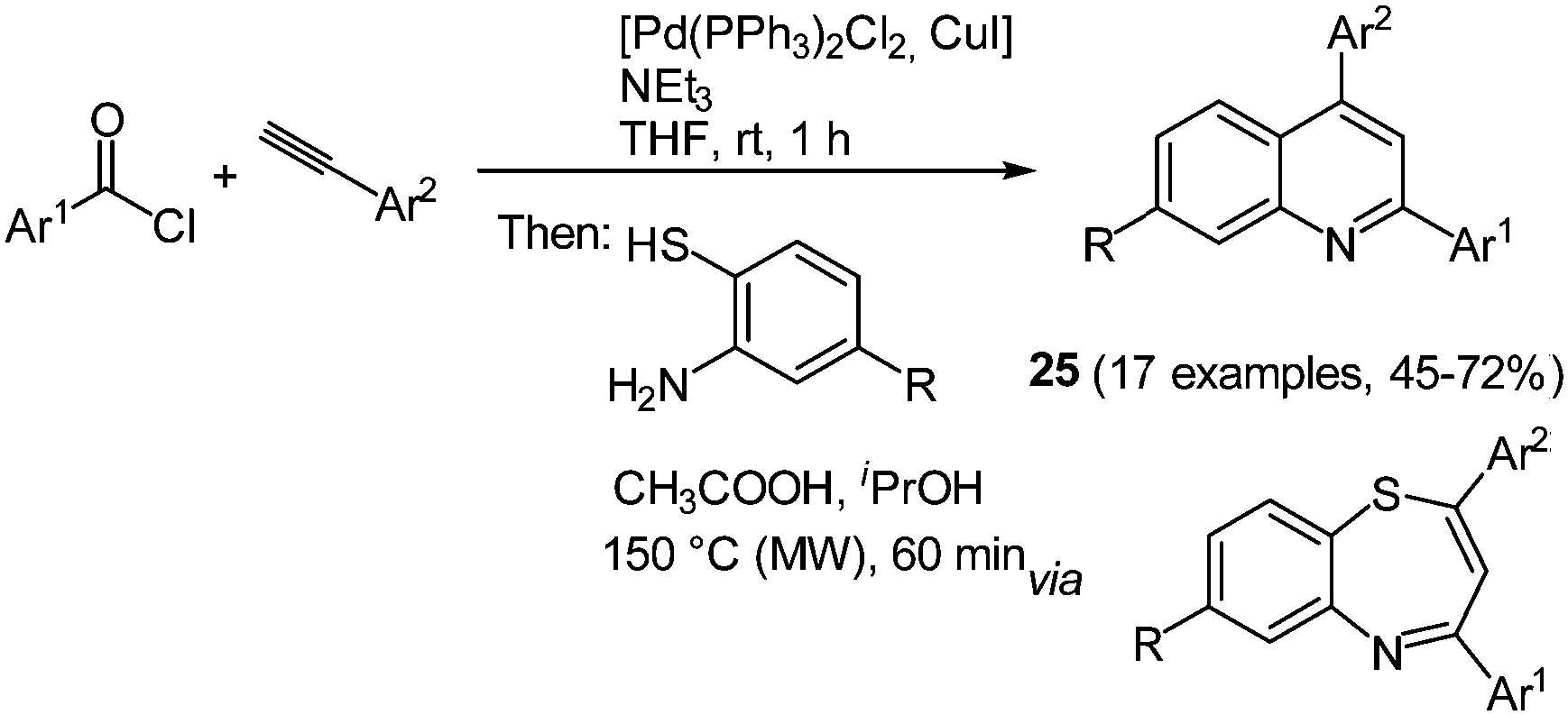 | ||
| Scheme 19 Consecutive 3-component synthesis of 2,4-diaryl substituted quinolines 25. | ||
Comparison of the electronic properties of 2,4-diaryl substituted quinolines 25 evidently shows that 2-substitution on the quinoline core only has a minor influence on the electronic properties. In general, all 2,4-aryl substituted quinolines show similar absorption and emission spectra with just small shifts of the maxima. Only strong donor substituents result in a bathochromic shift in the absorption spectra as a consequence of the push–pull system. The molecules display variable Stokes shifts ranging from 600 to 4000 cm−1. In addition, quantum chemical calculations were performed to qualitatively assign the absorption bands in the electronic spectra.
The implementation of another nitrogen atom in the heterocycle, which leads to the class of quinoxalines, even further intensifies the interesting biological and photophysical properties of the π-system.87–89 Many studies have been conducted with this heterocyclic core, but ethynylquinoxalines are remarkably underexplored. The few reported approaches to ethynylquinoxalines either just furnished intermediates or the obtained substitution pattern was very restricted.88 The novel glyoxylation–alkynylation–cyclocondensation sequence opened an attractive pathway for the preparation of this chromophore in a consecutive four-component reaction.89 As reactive intermediates ynediones are generated by glyoxylation with oxalyl chloride and subsequent Stephens–Castro coupling with a terminal alkyne. The cyclocondensation of a 1,2-diaminoarene concludes the one-pot sequence to give 3-ethynylquinoxalines 26 in moderate to excellent yield in the sense of the chromophore concept (Scheme 20). Interestingly all four components are applied in strictly equimolar ratios, thereby warranting a high degree of process economy.
 | ||
| Scheme 20 Glyoxylation–alkynylation–cyclocondensation sequence for the synthesis of 2-substituted 3-ethynylquinoxalines 26. | ||
A considerable scope becomes apparent by variation of one or more of the components. Glyoxylation is in principle limited to electron-rich π-nucleophiles that ultimately end up in the 2-position. While good results were obtained with aryl- and silyl-substituted alkynes, aliphatic alkynes were no substrates in the Stephens–Castro coupling. Furthermore the photophysical properties of selected derivatives were studied (Scheme 20, 26a–26e). The absorption maxima lie in a narrow range between 393 and 418 nm. The ethynyl substituent only has a minor effect on the spectral properties, while a distinct influence is exerted by the substituents in the 2-position of the quinoxalines. Here, the more electron-rich 1-phenylpyrrol-2-yl substituent causes a blue shift of the longest wavelength absorption band. The emission spectra show a similar behavior, where the shortest wavelength emission band is situated between 474 and 520 nm. Again the electron donating character of the 2-position substituent affects a hypsochromic shift. Furthermore 3-ethynyl quinoxalines display large Stokes shifts of 4400−4800 cm−1 and an intense emission solvatochromicity (Fig. 3).
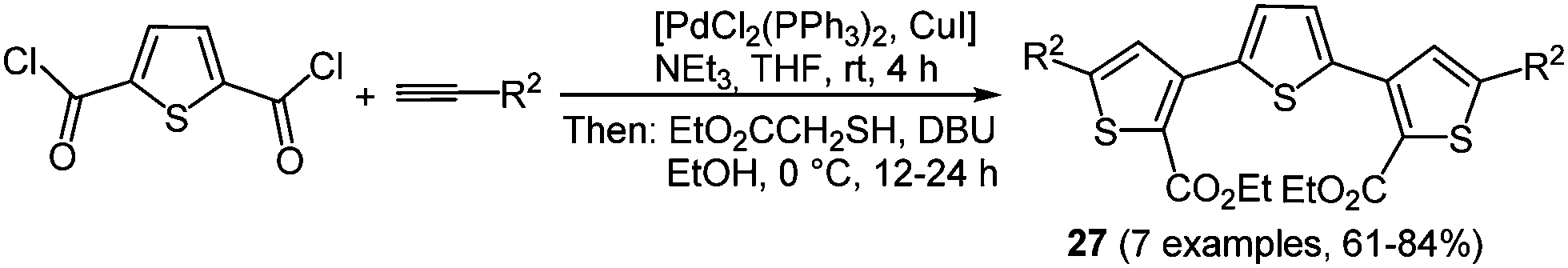 | ||
| Scheme 21 Consecutive multicomponent synthesis of ter- and quinquethiophenes 27. | ||
 | ||
| Fig. 3 Emission solvatochromism of compound 26b (recorded in cyclohexane, toluene, THF, EtOAc, acetone, CH2Cl2, MeCN, iPrOH, EtOH, MeOH (from left to right); T = 293 K; c(26b) = 10−4 M; λexcit = 356 nm, hand-held UV lamp). Reprinted with permission from J. Org. Chem., 2014, 79, 3296–3310. Copyright 2014 American Chemical Society. | ||
4.2 MCR formation of sulfur heterocycles
Thiophenes and oligomers are particularly electron rich heteroarenes, a characteristic giving them peculiar relevance as functional π-electron systems in organic materials science.21 They already find application as hole transport materials in OLED, OFET, and OPV.90 Electropolymerization is one of the well-established methods to prepare 2,5-linked polythiophenes in a resourceful fashion.91 The employment of functionalized monomers to increase the solubility of these systems is also possible. Nevertheless using this method only polydisperse product mixtures are obtained, leaving out the possibility of preparing oligomers with a defined number of chain links. Therefore, a multicomponent approach could allow for variable substitution patterns on thiophenes within an oligomer. This commutability results in different extensions of π-electron conjugation, strongly influencing the photophysical and electronic properties.A simple way for enabling different substitution patterns on the heterocycle core is the de novo formation of the thiophene core from easily accessible starting materials. The Fiesselmann cyclocondensation92 between alkynones and ethyl mercapto acetate was corroborated and implemented in a chromophore approach based MCR strategy.93 Here, simple acid chlorides or a thiophene dicarbonyl chloride first react with terminal alkynes under Sonogashira conditions setting the stage for a terminal Fiesselmann reaction in a one-pot fashion. A large number of (un)symmetrically substituted (oligo)thiophenes 27 were synthesized by these consecutive three- and pseudo-five-component reactions (Scheme 21).
The symmetrically substituted ter- and quinquethiophenes 27 cover a broad range of absorption maxima (305 to 398 nm) in the near UV whereas the shortest wavelength emission bands are located around 445 nm for all compounds. This behavior results in very large Stokes shifts of 10000 cm−1.
An enhancement of the photophysical properties was achieved by building thiophene functionalized macromolecules via the scaffold approach. A tandem polymerization was developed based on this three-component synthesis of oligothiophenes resulting in structurally regioregular polymers 28 with advanced functionality.94 Tetraphenylethene was chosen as a dialkyne component due to its strong aggregation-induced emission characteristics which should enhance the polymer emission.95 High molecular weights are obtained by the polymerization of diyne, diaroyl chloride and 2-mercaptoacetate under mild conditions (Scheme 22).
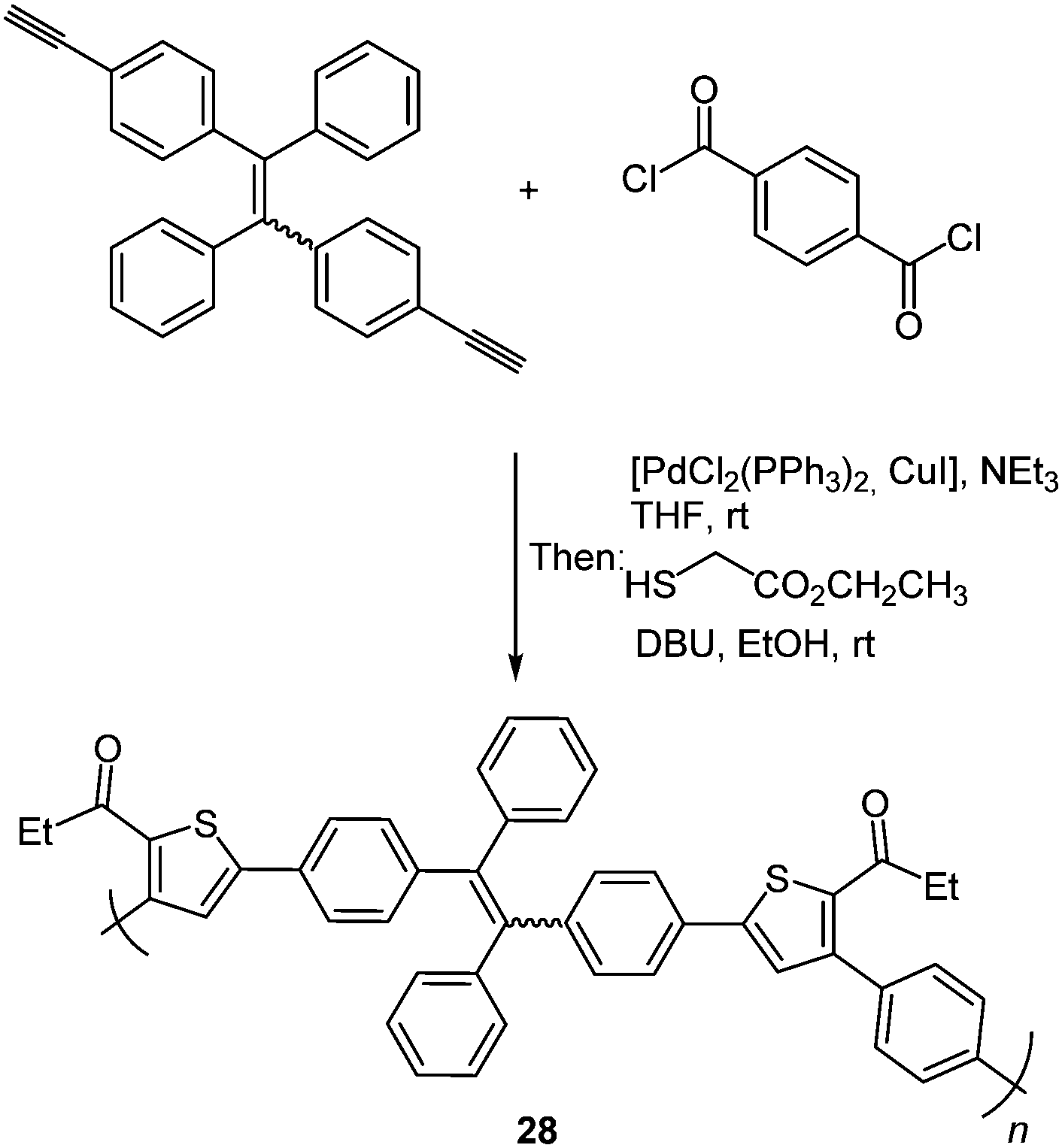 | ||
| Scheme 22 Three-component polymerization synthesis of regioregular AIE polymers 28. | ||
Measurements of the photophysical properties with respect to possible AIE characteristics showed a bright green emission for the nanoaggregates. The emission spectra were recorded in THF/water-mixtures displaying a remarkable enhancement of the intensity in an 80% water solution with a small decrease at higher water concentrations, confirming that the strong emission characteristics originate from aggregates.
Another multicomponent reaction to synthesize various poly(heterocycles) takes advantage of the reactivity of 1,3-dipoles which easily undergo cycloaddition reactions.96 This approach is an important development in polymer chemistry for the construction of highly substituted molecules consisting of a simple variant. Besides overcoming usual tedious multistep syntheses simultaneously rapid and easier tuning of the structures becomes possible.97 An aromatic bis(acid chloride) and a diimine react with CO as a monomer in a palladium-catalyzed polymerization reaction in the sense of the scaffold concept. Upon generating poly(1,3-dipoles) cycloaddition with various dipolarophiles results in a large number of poly(heterocycles) 29. By variation of the monomers avenues to larger scopes of polymers are opened. In Scheme 23 three representative polymers prepared via this multicomponent reaction with interesting photophysical properties are shown.
 | ||
| Scheme 23 Selection of possible reactions for the synthesis of poly(heterocycles) 29. | ||
The longest wavelength absorption maxima of polymers 29 are found between 280 and 368 nm. Expectedly, polymer 29a without a heterocyclic bridge between the monomer units displays the maximum at highest energy, whereas polymers 29b and 29c are red shifted. A similar behavior is found in the emission spectra, where the maxima lie between 413 and 501 nm with fluorescence quantum yields of up to 47%; however, polymer 29a does not fluoresce.
4.3 MCR formation of merocyanines
Merocyanines are categorized as polymethine chromophores and they are typically composed of an auxochrome (donor) and an antiauxochrome (acceptor) at the termini of an even-numbered polymethine chain.98 Due to their tunable photophysical, photochemical and electrochemical properties by modification of the end groups, the substituents and the length of the polymethine chain, merocyanines have found entry as functional dyes in the design of new materials for solar cells, optoelectronics and optical carriers. In the field of organic photovoltaics, both in bulk heterojunction99 and dye sensitized solar cells,3 merocyanines can be employed as visible light absorbing materials. With very few exceptions,100 most syntheses of merocyanines have not been accessed in a multicomponent and a one-pot fashion. Therefore, the search for novel, efficient reaction routes is an ongoing task. A reactivity-based approach takes advantage of the inherent electrophilicity of an intermediary formed Michael system, where an enamine can attack as a nucleophile.101 This consecutive three-component reaction commences with Sonogashira coupling of an aryl acid chloride and a terminal alkyne. The subsequent concluding Michael addition on the ynone intermediate with different enamines gives the desired butadienyl merocyanines 30/32 and 2-styryl enaminones 31 in moderate to good yields in the sense of the chromophore approach (Scheme 24).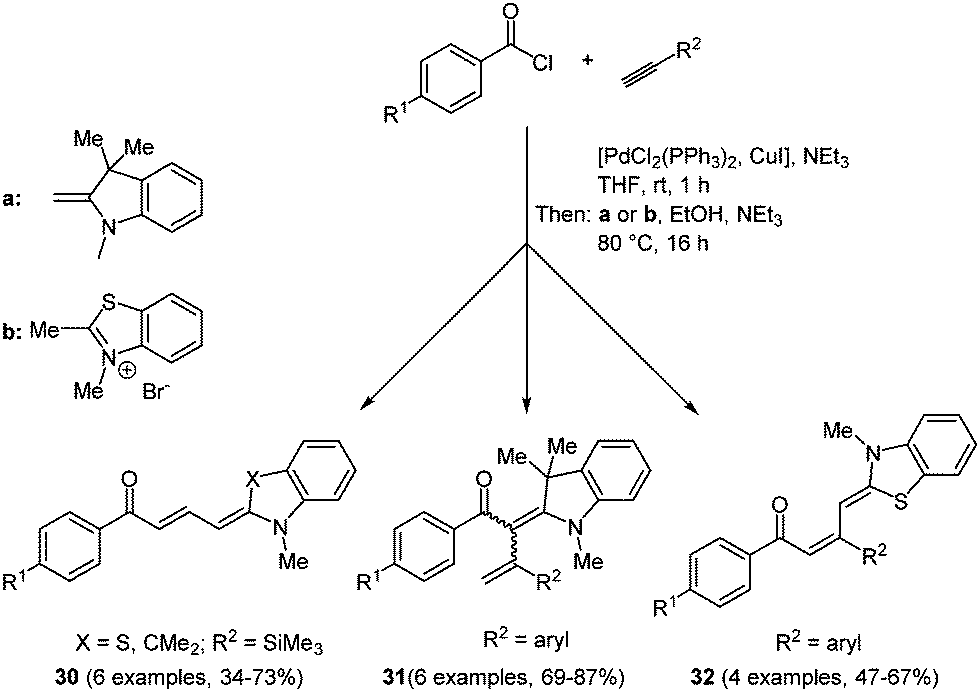 | ||
| Scheme 24 3-Component syntheses of butadienyl merocyanines 30 and 32, and 2-styryl enaminones 31. | ||
Due to the electronic differences of enamines and ketene acetal intermediates 2-styryl enaminones 31 originate from a (2+2)-cycloaddition with subsequent electrocyclic ring opening. The absorption spectra of merocyanines 30/32 underline the strong influence of the electronic substituent effects on the photophysical properties of the chromophores. A bathochromic shift of the longest wavelength absorption band occurs for electron-withdrawing substituents on the aroyl moiety. Thereby, the dipolar character of the π-system can be considerably enhanced. This feature can even be enhanced by introducing a more electron-rich donor on the opposite side of the conjugated π-system. This trend is consistently observed for all three merocyanine series 30, 31, and 32. The solution absorption maxima are found between 395 and 516 nm and just a small bathochromic shift can be detected for the thin films prepared by dropcasting.
Indolone based solid-state luminescent merocyanine chromophores 33 can be readily synthesized in a consecutive three-component reaction, based on the chromophore concept, with excellent yields (60–99%) (Scheme 25).102 By reacting alkynoyl o-iodo anilides with a palladium complex cyclization takes place after insertion of the triple bond in the aryl-metal bond. The following alkynylation under Sonogashira conditions allows for a subsequent Michael-type addition of primary or secondary amines concluding the insertion–coupling–addition sequence. The obtained push–pull butadienes display intense orange to red solid state luminescence (λ = 622–644 nm), while no emission can be detected in solution.
 | ||
| Scheme 25 Insertion–coupling–addition sequence for the synthesis of 4-aminoprop-3-enylidene indolones 33. | ||
Many heterocycle syntheses based on copper catalysis can be found in the literature,103 including the combination of both Cu(I) and Cu(II) salts in the same process.104 Recently, a novel pseudo four-component synthesis of merocyanine analogues by binary Cu(I)/Cu(II) catalysis in the sense of the chromophore approach was reported.105 The coupling of N-methyl isatin with an amine and an alkyne is catalyzed by Cu(I) chloride and Cu(II) triflate, giving rise to merocyanines in 35–80% yield (Scheme 26).
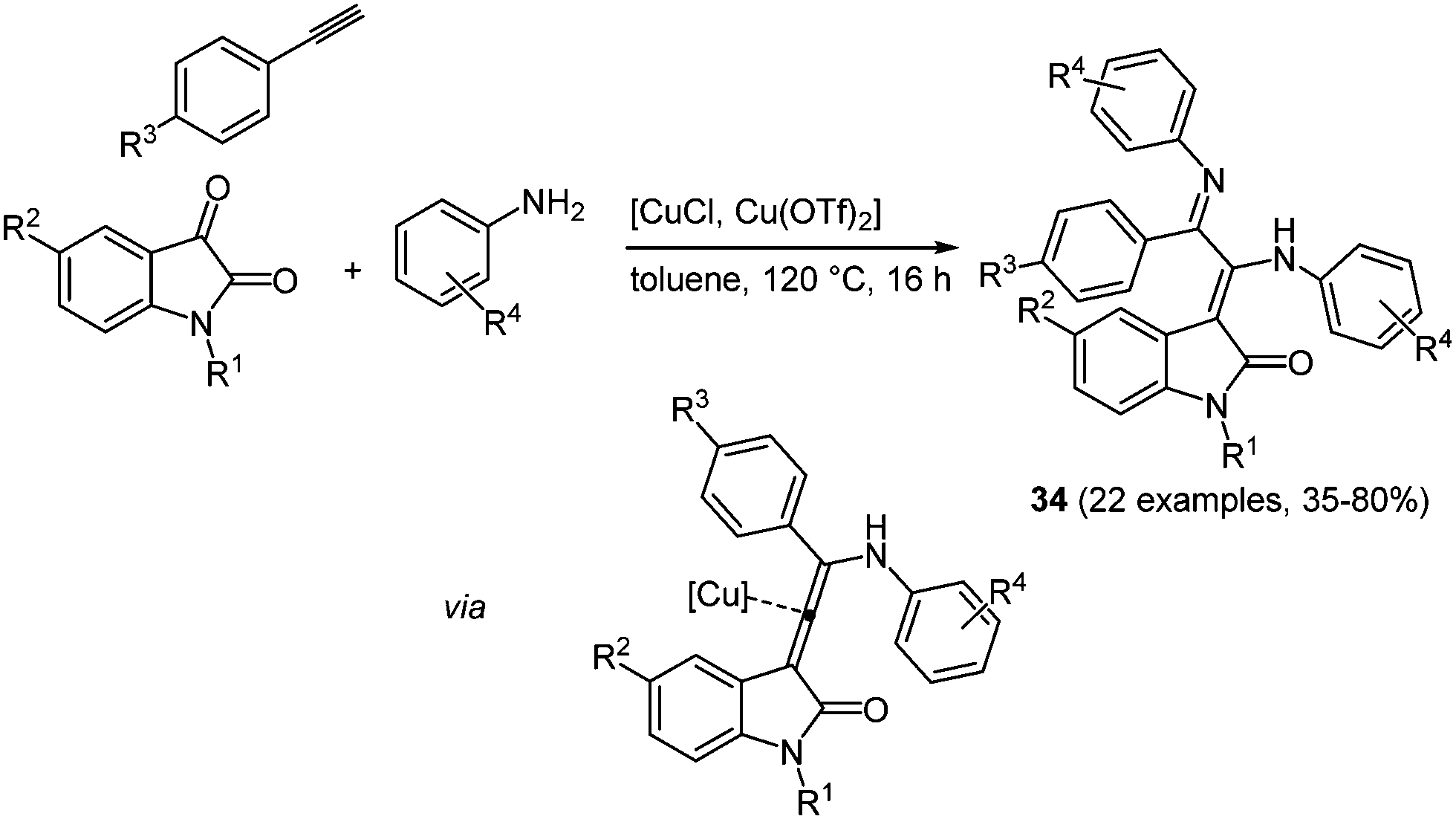 | ||
| Scheme 26 Binary copper catalysis for the pseudo four-component synthesis of merocyanine analogues 34. | ||
Interestingly, these compounds show blue luminescence (438–487 nm) with quantum yields of up to 20%, significantly different in comparison to 4-aminoprop-3-enylidene indolones 34. The absorption spectra possess two major bands at 250–275 and 350–380 nm, resulting in large Stokes shifts of up to 6500 cm−1. The substituent on the aniline rings has the largest influence on the photophysical properties of the merocyanine dyes; for instance, larger Stokes shifts can be observed for electron deficient systems.
4.4 MCR formation of NLO-chromophores
The term NLO property summarizes a variety of different effects.106 Most important for organic NLO materials are photorefractive or multiphoton effects. Although the phenomenon of two-photon absorption has been known for a long time,107 the conceptual design of easily accessible and effective molecules with this characteristic started only in the past two decades.108 The first step beyond normal spectroscopic usage was taken due to a ubiquitous request for novel imaging and data storage technologies.109 For the application in biological imaging most requested molecules are green and red emitters, nevertheless also blue emitting chromophores are interesting, when more than two probes have to be introduced. Blue light emitting small molecules were indeed easily prepared by using various one-pot multicomponent syntheses. The reaction of 9,10-phenanthraquinone or diaryl 1,2-diones with an appropriate aromatic aldehyde and ammonium acetate as a third component in glacial acetic acid under microwave irradiation results in the formation of imidazole (and oxazole) based chromophores 35.52 By replacing the aldehyde with an aryl bis(hydrazide) also 1,2,4-triazine based derivatives 36 can be prepared in the sense of the scaffold approach (Scheme 27). | ||
| Scheme 27 Synthesis of imidazole (35) and 1,2,4-triazine (36) based two-photon absorption chromophores. | ||
The imidazole based chromophores 35 show strong blue emission maxima at around 443–476 nm after two-photon excitation, whereas triazoles 36 clearly display weak non-linear optical characteristics, where fluorescence is essentially absent. Also the single-photon absorption properties reveal a pronounced influence of the nature of the N-heterocycle. In the absorption spectra a considerable bathochromic shift is found by replacing the triazine core (λmax = 275–276 nm) with an imidazole moiety (λmax = 357–376 nm). In contrast, the one-photon excited fluorescence of both series shows similar shortest wavelength maxima in a range between 434 and 483 nm. Hence, huge Stokes shifts for the triazine systems (14600–15500 cm−1) and considerably smaller shifts for the imidazole based chromophores (4200–6400 cm−1) are found.
4.5 MCR formation of complexes
Initially, a wide range of metal complexes was synthesized due to their interesting dye characteristics. However, since the early 1990s these systems have gained increasing importance due to their peculiar electronic and photophysical properties. In particular, the development of DSSCs by Grätzel was decisive.110 Complexes with various metals as core elements were prepared and applied in DSSCs giving overall efficiencies of up to 13%.111Organoboron complexes have been known since the end of the 19th century,112 nevertheless, it was not before the 1950s that the chemistry of these particular compounds has been strongly developed.113 The interest of researchers aroused due to their immense fluorescence, making them important candidates for application in OLEDs as emitting components.114 Due to the increasing significance of these luminescent systems in materials science, the quest for novel straightforward multicomponent reactions became evident.
BODIPY (boron dipyrrin) complexes are the best known and the most investigated boron containing fluorophores. These boron chelates have already found application in a variety of functional materials, for instance as luminescent biomolecular labels, as laser dyes, light harvesters and fluorescent switches.115 Furthermore, dipyrrins can form mono- and dinuclear chelates with many different transition metals, resulting in distinct characteristic properties.116 In 2000 a simple one-pot methodology to prepare these organoboron complexes has been presented by Burgess and coworkers following the chromophore concept. First an aromatic acyl chloride reacts with two equivalents pyrrole derivatives. After addition of triethylamine as a base and boron trifluoride etherate the desired BODIPY complex 37 can be isolated (Scheme 28).117
 | ||
| Scheme 28 One-pot synthesis of a BODIPY complex 37 according to Burgess. | ||
This synthesis became well established and many different complexes were prepared also for other systems than dipyrrin ligands (Fig. 4).118 All boronic systems display strong fluorescence in solution or solid state and they are often accompanied by large Stokes shifts.
 | ||
| Fig. 4 Examples for organoboron complexes by Burgess (left),117 Bröring (middle),118 and Xiao (right).118 | ||
In the past decades investigations on boronic complexes were mostly concerned with fine-tuning of the fluorescence wavelength and with increasing stability.114,119 Another attractive ligand with respect to emerging photochemical properties is the 8-oxyquinolinato ligand. For these compounds the emission color can be tuned by chemical functionalization of the heterocycle corpus. Both homo-120 and heteroleptic diarylboron complexes are known. More interesting are unsymmetrically substituted 8-oxyquinolinato diarylboron derivatives 38 which can be prepared via consecutive three-component synthesis in the sense of the chromophore concept (Scheme 29).121
 | ||
| Scheme 29 Synthesis of heteroleptic 8-oxyquinolinato diarylboron complexes 38. | ||
Most advantageously, a second heterocycle in addition to the 8-oxyquinoline core can be introduced. Here, diethyl 2-fluoro-3-pyridineboronate reacts with aryl lithium to give the diarylborinic acid “ate” complex which after hydrolysis results in the free diarylborinic acid. After addition of the quinolinato ligand the colorful crystalline product can be isolated in good to excellent yields (56–97%). It is also possible to introduce further complexation by addition of zinc chloride. This complexation occurs at the pyridine nitrogen coordinating two boron complexes to the zinc ion. All compounds show absorption maxima between 378 and 407 nm with extinction coefficients of the zinc complexes up to 8900 M−1 cm−1. The shortest wavelength emission band lies in a range of 502–525 nm with fluorescence quantum yields of up to 20%. For the zinc complexes the quantum yields drop to 2 or 3%.
Rhenium(I) complexes with coordinated diimine ligands often show interesting photophysical properties. Therefore, their use as responsive probes for a variety of analytes122 and the local medium,123 or in fluorescence microscopy cell imaging studies has already been established.124 In metal complexes luminescence properties usually originate from metal-to-ligand charge transfer in the excited state.125 Consequently, large Stokes shifts and reasonably long luminescence lifetimes can be reached, which are particularly relevant for bioanalytical application. The synthesis of phenanthroline based chromophore ligands can be performed in a domino four-component reaction following the scaffold approach starting from the corresponding 1,10-phenanthroline-5,6-dione.126 The condensation of the dione with an amine, an aldehyde and ammonium acetate gives rise to imidazo-phenanthroline ligands 39 in moderate to excellent good yields (28–90%) (Scheme 30). Complexation to luminescent [ReBr(CO)3L] complexes 40 was achieved by stirring the ligand with [ReBr(CO)5] overnight. The substituent variation on the ligand core only shows moderate influence on the fluorescence properties.
 | ||
| Scheme 30 Four-component synthesis of chromophoric imidazo-phenanthroline ligands 39 for the complexation with Re(I) to complexes 40. | ||
The photophysical properties of both free ligands and complexes revealed significant differences. While the emission of ligands 39 appears at around 370 nm, complexes 40 fluoresce at around 580 nm. Even more remarkable is the enlargement of the Stokes shifts (up to 7000 cm−1) caused by complexation to rhenium, as a consequence of metal-to-ligand charge transfer.
For modern technology the preparation of white organic light-emitting diodes (WOLED) is an ongoing task. Their potential application in full color displays or backlights for liquid crystals and the prospect of energy saving systems are only some of their advantages.127 Mixing the three primary colors, red, green and blue, with a single layer or the combination of two complementary colors are two strategies developed to realize white light emission. Zinc complexes turned out to be promising candidates for these systems with high efficiency, low cost, easy tunable colors and high thermal stability as advantageous characteristics.128 In the sense of the chromophore concept a pseudo four-component reaction allows for a straightforward synthesis of blue fluorescent zinc-complexes with white-light emitting properties when doped on 4,4′,4′′-tris(N-carbazolyl)triphenylamine in OLEDs.129 Here, a substituted aliphatic diamine reacts with zinc acetate and salicylaldehyde resulting in the desired zinc(II) complexes 41 with chelate ligands in excellent yields (84–92%) (Scheme 31).
 | ||
| Scheme 31 Synthesis of fluorescent zinc(II) complexes 41via a pseudo four-component synthesis. | ||
All complexes 41 display two distinct absorption maxima in the UV/Vis spectra, the first at around 260 nm and the second at around 360 nm, which can be assigned to the metal-to-ligand charge transfer. Furthermore, a strong similarity is observed in the emission characteristics, where the maxima lie in a narrow range between 441 and 447 nm showing a more or less intense blue luminescence. The electroluminescence of the compounds was also studied. Here, the compounds revealed typical diode characteristics emitting a sky-blue to white light in the devices (Fig. 5).
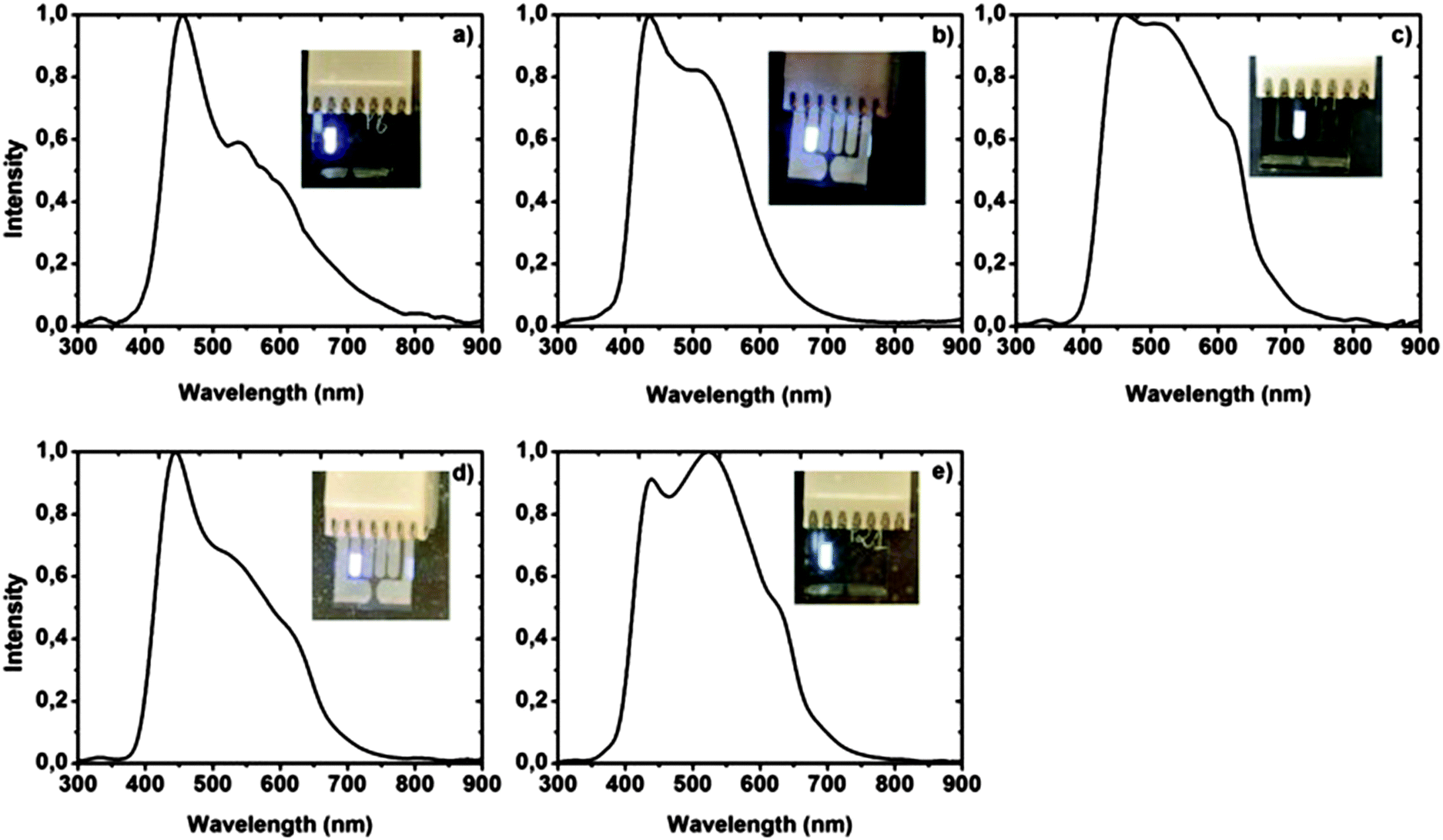 | ||
| Fig. 5 Electroluminescence spectra of sky-blue and white OLEDs of the doped device with zinc complexes 41. Reprinted from F. Dumur, L. Beouch, M.-A. Tehfe, E. Contal, M. Lepeltier, G. Wantz, B. Graff, F. Goubard, C. R. Mayer, J. Lalevée and D. Gigmes, Low-cost zinc complexes for white organic light-emitting devices, Thin Solid Films, 2014, 564, 351–360, Copyright (2014), with permission from Elsevier. | ||
4.6 Miscellaneous
The preparation of chromophore families appended to macrocycles was reported by Main et al. in 2008.130 These novel DOTA (1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid) monoamide ligands were synthesized by Ugi condensation as the key step, starting from an acid functionalized macrocycle. The complexation of DOTA-ligands with lanthanides gives luminescent complexes as agents for the imaging of biological systems (Fig. 6).131 | ||
| Fig. 6 DOTA-lanthanide complexes with an Ugi-4CR product as a side chain (highlighted in blue). | ||
These most intensively luminescent complexes show two emission maxima, the first appearing between 250 and 337 nm and the second between 545 and 980 nm. But also free ligands have received great interest for biological studies. Actually, the isolation, identification, and characterization of metabolites, generally referred to as metabolomics, plays a key role in disease diagnosis.132 The facile mass determination of metabolites can be achieved by chemical derivatization, whereupon the attained novel physicochemical properties simplify both LC-MS and GC-MS analyses.133 For instance, the chemoselective derivatization of aldehydes and ketones as imines, hydrazones and oximes or the reaction of amines with electrophilic reagents is often employed.134 By employing the Ugi-4CR for derivatizing more than a single functional group, different classes of metabolites were derivatized in high yields and with excellent chemoselectivity. Gratifyingly, the MCR can be performed in water giving stable and UV-active products which can be easily identified via LC-MS.135
The investigation of the protein composition is an important issue for understanding complex biological processes, such as protein–protein interactions. The studies are predominantly performed with biomarkers in biomedical sciences.136 For this purpose an activity-based protein profiling (ABPP)137 was developed among other more biological approaches.138 The probes employed in this analysis consist of a reactive group for the attachment to the protein, a group for specific target recognition and a reporter-tag for target detection, for instance, a fluorophore. Westermann and coworkers developed rhodamine based reporter-tags 42 with different bio-orthogonal functional systems as a side chain.139 In the sense of a scaffold approach the Ugi-4CR36 turned out to be the excellent method of choice to prepare these chomophores by reacting rhodamine B, i.e. a carboxylic acid, formaldehyde, an amino ether with tert-butyl isocyanide (Scheme 32).
 | ||
| Scheme 32 Ugi-4CR for the preparation of modified rhodamine dyes 42 for protein detection. | ||
The typical pH-dependency of the rhodamine emission characteristics was absent with this substitution pattern, the compound showing emission both under acidic and basic conditions. The compounds show absorption maxima between 532 and 586 nm with high extinction coefficients of up to 72000 M−1 cm−1 and emission maxima between 550 and 600 nm. The photophysical properties are not altered compared to the starting material rhodamine B, which shows the longest wavelength maximum at 554 nm and the emission maximum at 576 nm.
Pericyclic processes, such as cycloadditions and electrocyclic reactions, are particularly suited for accessing conjugated systems.140 Interestingly, electrocyclic ring opening following a cycloaddition not only lead to the formation of two new carbon–carbon σ-bonds but also to a rearrangement of the π-system. Conjugated arylalkynyl systems can be considered as simple rigid molecular wires that enable fast electron transfer processes between the two redox termini.141 By reaction of the alkynyl moiety with tetracyanoethylene (TCNE) biscyanomethylene moieties can be readily introduced by the above mentioned mechanistic scenario.142 Therefore, the combination of (2+2)-cycloaddition and electrocyclic cyclobutene ring opening represents a powerful methodology for the preparation of tetracyanohexatrienes and -octatetraenes.31,143 The repetitive (2+2)-cycloaddition-cyclobutene sequence has been elaborated into a consecutive (pseudo)-multicomponent reaction with differently substituted alkynes furnishing trienes 43 (3 examples, 30–85%) and tetraene 44 (75%) in the sense of the chromophore concept (Scheme 33).
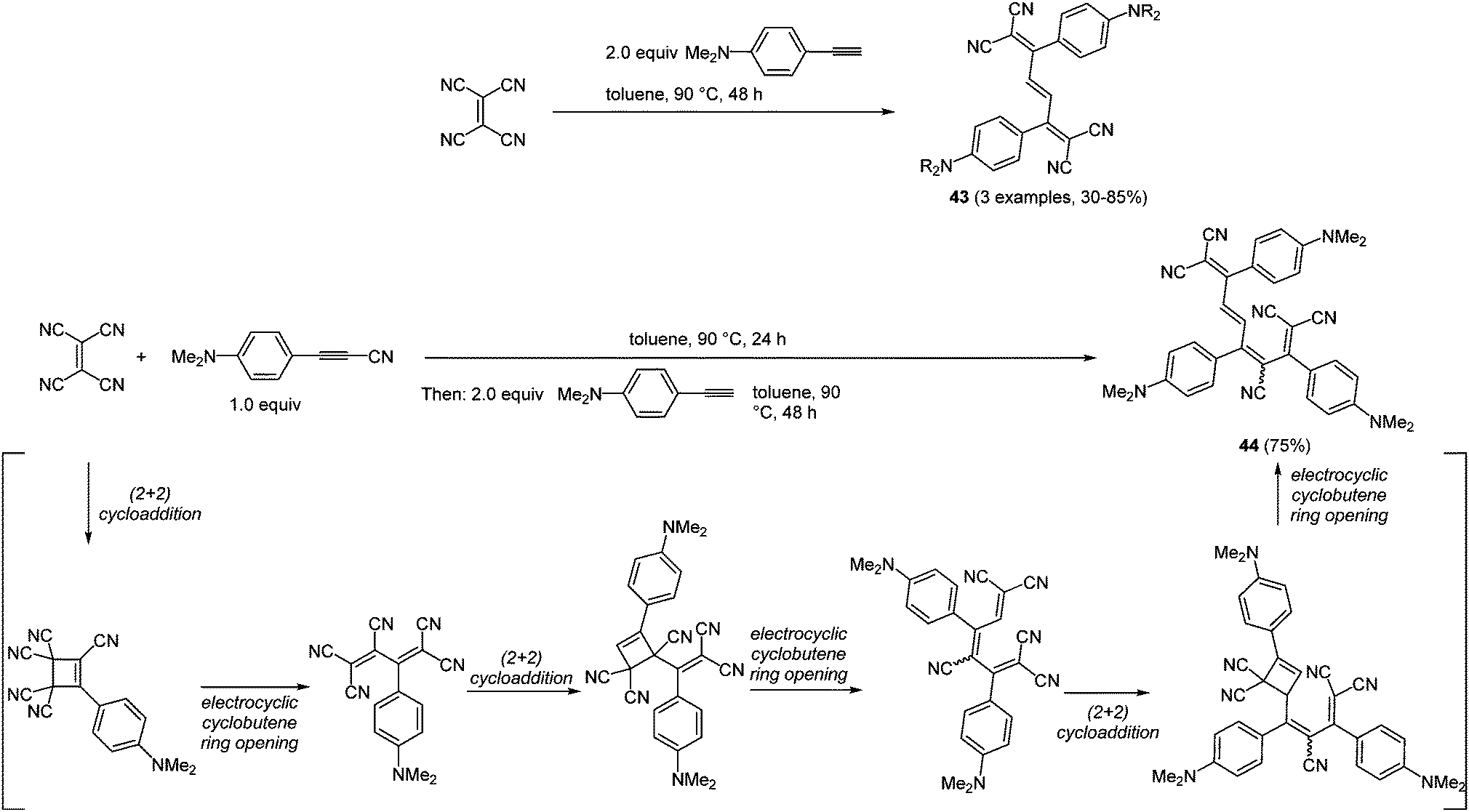 | ||
| Scheme 33 (2+2)-Cycloaddition–electrocyclic cyclobutene opening sequence for the synthesis of tretracyano-trienes 43 and tetracyano-tetraene 44. | ||
Trienes 43 exhibit simple UV/Vis spectra with two major bands at around 350 and 560 nm, where the maxima at the lowest energies correspond to intramolecular charge transfer transitions (ICT). In contrast to PET processes, in ICT the electron transfer instantaneously takes place upon absorption of a photon. Although the two configurational isomers of tetraene 44 display more complex absorption spectra, the positions of the intense ICT-bands do not alter significantly. These results encouraged other research groups to synthesize further donor–acceptor molecules. For instance the introduction of sterically and structurally more demanding alkynes and also dialkynes separated by a spacer were successfully accomplished.144
5 Conclusion
As highlighted in this review, multicomponent reactions as an ongoing task in organic synthetic chemistry have turned out to be a valuable tool in the preparation of functional chromophores. A broad spectrum of domino and consecutive MCR has already found entry to the literature. Besides concatenation of reactions with diverse reactivity patterns, sequentially catalyzed processes are increasingly gaining attention, in particular, due to their process efficiency and catalyst efficacy.Classical MCR, such as the Ugi-4CR and the Biginelli-3CR, are particularly useful for accessing functional materials in the sense of the scaffold concept. This approach is very favorable for applications, where covalent ligation and nonconjugated placement chromophores are important, e.g. for fluorescent probes with molecular recognition tags or in general for photo-induced electron transfer. In turn, the in situ formation of the functional chromophore by an MCR, as outlined in the chromophore concept, requires novel processes with the major challenge to tackle concise syntheses of special functional targets. Fascinating results have been unraveled and molecules with tailor-made electrochemical and photophysical properties can be designed, systematically studied and optimized by skillfully applying this powerful tool of diversity oriented synthesis.
Many synthetic pathways and structures are still unexplored in the world of multicomponent reactions. The unique combination of synthetic creativity, programmed reactivity, and systematic exploration of functional chromophores will stimulate the development and rational design of functional materials by MCR as an intellectual challenge in the topical field of organic materials science in the years to come.
Acknowledgements
The authors gratefully acknowledge the continuous support by the Fonds der Chemischen Industrie.Notes and references
- Functional Organic Materials - Syntheses, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VHC, Weinheim, 2007 Search PubMed.
- Organic Light-Emitting Materials and Devices, ed. Z. R. Li, CRC Press, 2nd edn, 2015 CrossRef; N. Thejo Kalayani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696 CrossRef; Organic Light Emitting Devices: Synthesis, Properties and Applications, ed. K. Müllen and U. Scherf, Wiley VCH, Weinheim, 2006 Search PubMed; For a review on flexible AM-OLED, see e.g. J.-S. Park, H. Chae, H. K. Chung and S. I. Lee, Semicond. Sci. Technol., 2011, 26, 1 Search PubMed.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS PubMed; A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef PubMed.
- Y.-W. Su, S.-C. Lan and K.-H. Wei, Mater. Today, 2012, 15, 554 CrossRef CAS; T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390 Search PubMed; V. W. W. Yam, M. M.-Y. Chan and C.-H. Tao, WOLEDs and Organic Photovoltaics, Springer-Verlag, Berlin, 2010 Search PubMed; Organic Photovoltaics, ed. C. Brabec, V. Dyakonov and U. Scherf, Wiley-VCH, Weinheim, 2008 Search PubMed.
- L. Torsi, M. Magliulo, L. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612 RSC; C.-H. Kim, Y. Bonnassieux and G. Horowitz, IEEE Trans. Electron Devices, 2014, 61, 278 CrossRef CAS; I. Kymissis, Organic Field Effect Transistors Theory, Fabrication and Characterization, Springer, New York, 2009 Search PubMed.
- D. Nilsson, T. Kugler, P.-O. Svensson and M. Berggren, Sens. Actuators, B, 2002, 86, 193 CrossRef CAS; C.-T. Chen, H. Wagner and W. C. Still, Science, 1998, 279, 851 CrossRef PubMed.
- For reviews on one-pot methodologies, see e.g. G. H. Posner, Chem. Rev., 1986, 86, 831 CrossRef CAS; L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed. Engl., 1993, 32, 131 CrossRef; L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef PubMed; B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef PubMed; S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948 RSC.
- For reviews, see e.g. A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3169 CrossRef CAS; H. Bienaymé, C. Hulme, G. Oddon and P. Schmitt, Chem. – Eur. J., 2000, 6, 3321 CrossRef; L. Weber, K. Illgen and M. Almstetter, Synlett, 1999, 366 CrossRef; Multi-component Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VHC, Weinheim, 2005 Search PubMed; Multi-component Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VHC, Weinheim, 2015 Search PubMed.
- L. Weber, Curr. Med. Chem., 2002, 9, 2085 CrossRef CAS PubMed; L. Weber, Drug Discovery Today, 2002, 7, 143 CrossRef PubMed; Y. Huang, A. Yazbak and A. Dömling, in Multicomponent Reactions in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, John Wiley & Sons, Ltd, Chichester, UK, 2012, p. 499 CrossRef; C. Kalinski, H. Lemoine, J. Schmidt, C. Burdack, J. Kolb, M. Umkehrer and G. Ross, Synthesis, 2008, 4007 CrossRef.
- T. J. J. Müller, in Multicomponent Reactions 1. General Discussion and Reactions Involving a Carbonyl Compound as Electrophilic Component, ed. T. J. J. Müller, Science of Synthesis Series, Georg Thieme Verlag KG, Stuttgart, 2014, p. 5 Search PubMed.
- For a review, see T. J. J. Müller, Functional Organic Materials - Syntheses, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VHC, Weinheim, 2007, p. 179 Search PubMed.
- For an account, see T. J. J. Müller and D. M. D'Souza, Pure Appl. Chem., 2008, 80, 609 CrossRef.
- S. Sung, Y. Yao, K. Uryu, H. Yang, V. M. Lee, J. Q. Trojanowski and D. Pratico, FASEB J., 2004, 18, 323 CAS.
- J. B. Bharate, A. Wani, S. Sharma, S. I. Reja, M. Kumar, R. A. Vishwakarma, A. Kumar and S. B. Bharate, Org. Biomol. Chem., 2014, 12, 6267 CAS.
- M. de Champdoré, G. Di Fabio, A. Messere, D. Montesarchio, G. Piccialli, R. Loddo, M. La Colla and P. La Colla, Tetrahedron, 2004, 60, 6555 CrossRef CAS; M. M. Harding and G. Mokdsi, Curr. Med. Chem., 2000, 7, 1289 CrossRef PubMed.
- B. Willy, W. Frank, F. Rominger and T. J. J. Müller, J. Organomet. Chem., 2009, 694, 942 CrossRef CAS.
- R. Epton, G. Marr and G. K. Regers, J. Organomet. Chem., 1976, 110, C42–C44 CrossRef CAS; G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3 CrossRef PubMed.
- T. Yao and G. A. Rechnitz, Biosensors, 1987, 3, 307 CrossRef PubMed.
- E. I. Klimova, M. Flores-Alamo, S. C. Maya, M. E. Martínez, L. Ortiz-Frade and T. Klimova, Molecules, 2012, 17, 10079 CrossRef CAS PubMed.
- J. R. Wright, K. J. Shaffer, C. J. McAdam and J. D. Crowley, Polyhedron, 2012, 36, 73 CrossRef CAS.
- For reviews on thiopenes in electronic materials, see e.g.Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley-VCH, Weinheim, 1999 CrossRef CAS PubMed; A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS PubMed; Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998 Search PubMed.
- S. Potratz, A. Mishra and P. Bäuerle, Beilstein J. Org. Chem., 2012, 8, 683 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS; V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef; M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef PubMed.
- S. Hassan and T. J. J. Müller, Adv. Synth. Catal., 2015, 357, 617 CrossRef CAS.
- R. D. Hancock, S. M. Dobson, A. Evers, P. W. Wade, M. P. Ngwenya, J. C. A. Boeyens and K. P. Wainwright, J. Am. Chem. Soc., 1988, 110, 2788 CrossRef CAS; Y. J. Li, I. Murase, J. Reibenspies and A. E. Martell, Inorg. Chim. Acta, 1996, 246, 89 CrossRef.
- M. Inouye, K. Fujimoto, M. Furusyo and H. Nakazumi, J. Am. Chem. Soc., 1999, 121, 1452 CrossRef CAS; H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495 CrossRef PubMed.
- H. M. Colquhoun, B. W. Greenland, Z. Zhu, J. S. Shaw, C. J. Cardin, S. Burattini, J. M. Elliott, S. Basu, T. B. Gasa and J. F. Stoddart, Org. Lett., 2009, 11, 5238 CrossRef CAS PubMed.
- E. Kimura, Y. Kuramoto, T. Koike, H. Fijioka and M. Kodama, J. Org. Chem., 1990, 55, 42 CrossRef CAS.
- For reviews on the A3 reaction, see e.g. V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790 RSC; W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43 Search PubMed; Y. Liu, ARKIVOC, 2014, 1 Search PubMed.
- A. Thirunarayanan and P. Rajakumar, Synlett, 2014, 2127 CAS.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, VCH Publishers Inc., New York, 1993 RSC; O. S. Wenger, Chem. Soc. Rev., 2011, 40, 3538 RSC; A. B. Ricks, K. E. Brown, M. Wenninger, S. D. Karlen, Y. A. Berlin, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4581 CrossRef CAS PubMed.
- I. Tabushi, N. Koga and M. Yanagita, Tetrahedron Lett., 1979, 20, 257 CrossRef.
- D. Gust and T. A. Moore, Science, 1989, 244, 35 Search PubMed; J. van Gersdorff, M. Huber, H. Schubert, D. Niethammer, B. Kirste, M. Plato, K. Mobius, H. Kurreck, R. Eichberger, R. Kietzmann and F. Willig, Angew. Chem., Int. Ed. Engl., 1990, 29, 670 CrossRef CAS.
- T. Carofiglio, A. Varotto and U. Tonellato, J. Org. Chem., 2004, 69, 8121 CrossRef CAS PubMed; M. H. R. Mahmood, H.-Y. Liu, H.-H. Wang, Y.-Y. Jiang and C.-K. Chang, Tetrahedron Lett., 2013, 54, 5853 CrossRef; F. Mandoj, S. Nardis, R. Pudi, L. Lvova, F. R. Fronczek, K. M. Smith, L. Prodi, D. Genovese and R. Paolesse, Dyes Pigm., 2013, 99, 136 CrossRef PubMed; S. Ajit, S. Palaniappan, P. U. Kumar and P. Madhusudhanachary, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 884 CrossRef.
- I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 386 Search PubMed; I. Ugi and C. Steinbrückner, Angew. Chem., 1960, 72, 267 CrossRef CAS.
- For a recent synthetic advances in isonitrile based MCR, see e.g.R. Riva, L. Banfi and A. Basso, in Science of Synthesis - Multicomponent Reactions 1, ed. T. J. J. Müller, Thieme, Stuttgart, 2014, p. 327 Search PubMed; L. A. Wessjohann, G. N. Kaluđerovic, R. A. W. Neves Filho, M. C. Morejon, G. Lemanski and T. Ziegler, Science of Synthesis - Multicomponent Reactions 1, ed. T. J. J. Müller, Thieme, Stuttgart, 2014, p. 415 Search PubMed.
- For reviews on isonitrile based MCR, see e.g. U. K. Sharma, N. Sharma, D. D. Vachhani and E. V. Van der Eycken, Chem. Soc. Rev., 2015, 44, 1836 RSC; G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544 CrossRef CAS PubMed; J. Zhu, Eur. J. Org. Chem., 2003, 1133 CrossRef; L. Banfi, A. Basso, G. Guanti and R. Riva, Multi-component Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VHC, Weinheim, 2005, p. 1 CrossRef PubMed; A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371 CrossRef PubMed.
- Y. Sasaki, Y. Araki, O. Ito and M. M. Alam, Photochem. Photobiol. Sci., 2007, 6, 560 Search PubMed; T. Miura, R. Carmieli and M. R. Wasielewski, J. Phys. Chem. A, 2010, 114, 5769 CrossRef CAS PubMed; X. Sun, Y. Liu, X. Xu, C. Yang, G. Yu, S. Chen. Z. Zhao, W. Qiu, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 10786 CrossRef PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; Y. K. Kang, P. M. Iovine and M. J. Therien, Coord. Chem. Rev., 2011, 255, 804 CrossRef; J. Hankache and O. S. Wenger, Phys. Chem. Chem. Phys., 2012, 14, 2685 RSC.
- S. Bay and T. J. J. Müller, Z. Naturforsch., 2014, 69b, 541 Search PubMed; S. Bay, G. Makhloufi, C. Janiak and T. J. J. Müller, Beilstein J. Org. Chem., 2014, 10, 1006 CrossRef CAS PubMed; S. Bay, T. Villnow, G. Ryseck, V. Rai-Constapel, P. Gilch and T. J. J. Müller, ChemPlusChem, 2013, 78, 137 CrossRef.
- T. Yamaoka, S. Yamaoka, T. Omote, K. Naitoh and K. Yoshida, J. Photopolym. Sci. Technol., 1996, 9, 293 CrossRef CAS; E. Katz, V. Heleg-Shabtai, A. Bardea, I. Willner, H. K. Rau and W. Haehnel, Biosens. Bioelectron., 1998, 13, 741 CrossRef PubMed; V. K. Ramanujan, J. A. Jo, F. Cantu and B. A. Herman, J. Microsc., 2008, 230, 329 CrossRef PubMed; E. Fasani, M. Fagnoni, D. Dondi and A. Albini, J. Org. Chem., 2006, 71, 2037 CrossRef PubMed; A. J. Jimenez, M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 2009, 74, 6615 CrossRef PubMed.
- N. A. Al-Awadi, M. R. Ibrahim, M. H. Elnagdi, E. John and Y. A. Ibrahim, Beilstein J. Org. Chem., 2012, 8, 441 CrossRef CAS PubMed.
- For a monograph on fluorescence spectroscopy, see J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- For reviews, see e.g.J. R. Lakowicz, Principles of Fluorescence Sectroscopy, 3rd edn, Springer, New York, 2006, p. 63 CAS; R. Alford, H. M. Simpson, J. Duberman, G. C. Hill, M. Ogawa, C. Regino, H. Kobayashi and P. L. Choyke, Mol. Imaging, 2009, 8, 341 CAS.
- For general reviews, see e.g. A. N. Kost and I. I. Grandberg, Adv. Heterocycl. Chem., 1966, 6, 347 CrossRef CAS; Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky and C. W. Rees, Elsevier, Oxford, 2008, vol. 4 Search PubMed.
- D. J. Wustrow, T. Capiris, R. Rubin, J. A. Knobelsdorf, H. Akunne, M. D. Davis, R. MacKenzie, T. A. Pugsley, K. T. Zoski, T. G. Heffner and L. D. Wise, Bioorg. Med. Chem. Lett., 1998, 8, 2067 CrossRef CAS PubMed; F. Bellina, S. Cauteruccio and R. Rossi, Tetrahedron, 2007, 63, 4571 CrossRef; T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef PubMed; D. Raffa, B. Maggio, M. V. Raimondi, S. Cascioferro, F. Plescia, G. Cancemi and G. Daidone, Eur. J. Med. Chem., 2014, 97, 732 CrossRef PubMed.
- See e.g.A. K. Sarkar, British Pat., GB 1052179 19661221, 1966 CrossRef CAS; A. Dolars, C.-W. Schelhammer and J. Schoreder, Angew. Chem., Int. Ed. Engl., 1975, 14, 665 CrossRef CAS; J. Catalan, F. Fabero, R. M. Claramunt, M. D. Santa Maria, M. C. Foces-Foces, F. Hernandez Cano, M. Martinez-ripoll, J. Elguero and R. Sastre, J. Am. Chem. Soc., 1992, 114, 5039 CrossRef; T. Karatsu, N. Shiochi, T. Aono, N. Miyagawa and A. Kitamura, Bull. Chem. Soc. Jpn., 2003, 76, 1227 CrossRef.
- See e.g. Y. S. Yao, J. Xiao, X. S. Wang, Z. B. Deng and B. W. Zhang, Adv. Funct. Mater., 2006, 16, 709 CrossRef CAS; Y. Tao, Q. Wang, C. L. Yang, C. Zhong, K. Zhang, J. G. Qin and D. G. Ma, Adv. Funct. Mater., 2010, 20, 304 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef CAS PubMed; S. M. King, I. I. Perepichka, I. F. Perepichka, F. B. Dias, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 586 CrossRef; J. Herbich and A. Kapturkiewicz, J. Am. Chem. Soc., 1998, 120, 1014 CrossRef.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797 CrossRef CAS.
- Y.-F. Sun, W. Huang, C.-G. Lu and Y.-P. Cui, Dyes Pigm., 2009, 81, 10 CrossRef CAS; Y.-F. Sun, W.-L. Pan, R.-T. Wu and H.-C. Song, Chin. J. Org. Chem., 2006, 26, 1079 Search PubMed.
- A. Shahrisa, K. D. Safa and S. Esmati, Spectrochim. Acta, Part A, 2014, 117, 614 CrossRef CAS PubMed; A. Shahrisa and S. Esmati, Synlett, 2013, 595 CrossRef.
- K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661 CrossRef CAS; C. A. Blackburn, Tetrahedron Lett., 1998, 39, 5469 CrossRef; H. Bienayme and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234 CrossRef.
- B. Umamahesh and K. I. Sathiyanarayanan, Dyes Pigm., 2015, 121, 88 CrossRef.
- For reviews, see e.g. S. Fustero, A. Simón-Fuentes and J. F. Sanz-Cervera, Org. Prep. Proced. Int., 2009, 41, 253 CrossRef CAS; K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 11960 CrossRef PubMed; A. Jamwal, A. Javed and V. Bhardwaj, J. Pharm. BioSci., 2013, 114 Search PubMed.
- B. Willy and T. J. J. Müller, Eur. J. Org. Chem., 2008, 4157 CrossRef CAS; B. Willy and T. J. J. Müller, Org. Lett., 2011, 13, 2082 CrossRef PubMed.
- B. Willy and T. J. J. Müller, ARKIVOC, 2008, 195 Search PubMed; B. Willy and T. J. J. Müller, Curr. Org. Chem., 2009, 13, 1777 CrossRef CAS.
- B. C. Bishop, K. M. J. Brands, A. D. Gibb and D. J. Kennedy, Synthesis, 2004, 43 CrossRef CAS.
- M. Denißen, J. Nordmann, J. Dziambor, B. Mayer, W. Frank and T. J. J. Müller, RSC Adv., 2015, 5, 33838 RSC.
- E. Beltrán, J. S. Serrano, T. Sierra and R. Giménez, Org. Lett., 2010, 12, 1404 CrossRef PubMed.
- P. Brough, J. Pécaut, A. Rassat and P. Rey, Chem. – Eur. J., 2006, 12, 5134 CrossRef CAS PubMed.
- V. Friese, S. Nag, J. Wang, M.-P. Santoni, A. Rodrigue-Witchel, G. S. Hanan and F. Schaper, Eur. J. Inorg. Chem., 2011, 39 CrossRef CAS.
- Q. Zhu, L. Huang, Z. Chen, S. Zheng, L. Lv, Z. Zhu, D. Cao, H. Jiang and S. Liu, Chem. – Eur. J., 2013, 19, 1268 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- Q. Wang, D. Kim, D. D. Dionysiou, G. A. Sorial and D. Timberlake, Environ. Pollut., 2004, 131, 323 CrossRef CAS PubMed; E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443 CrossRef PubMed; G. Guzzi and C. A. M. La Porta, Toxicology, 2008, 244, 1 CrossRef PubMed.
- G. D. Huy, M. Zhang, P. Zuo and B.-C. Ye, Analyst, 2011, 136, 3289 RSC.
- S. Yoon, A. E. Albers, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16030 CrossRef CAS PubMed; D. W Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168 CrossRef PubMed.
- B.-C. Yin, M. You, W. Tan and B.-C. Ye, Chem. – Eur. J., 2012, 18, 1286 CrossRef CAS PubMed; W. Xuan, C. Chen. Y. Cao, W. He, W. Jiang, K. Liu and W. Wang, Chem. Commun., 2012, 48, 7292 RSC.
- P. Biginelli, Ber. Dtsch. Chem. Ges., 1891, 24, 1317 CrossRef; P. Biginelli, Ber. Dtsch. Chem. Ges., 1891, 24, 2962 CrossRef.
- A. Kaur, H. Sharma, S. Kaur, N. Singh and N. Kaur, RSC Adv., 2013, 3, 6160 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185 RSC.
- T. Raj, P. Saluja and N. Singh, Sens. Actuators, B, 2015, 206, 98 CrossRef CAS.
- For reviews, see e.g. A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef PubMed.
- See e.g. G. R. Pettit, J. C. Knight, D. L. Herald, R. Davenport, R. K. Pettit, B. E. Tucker and J. M. Schmidt, J. Nat. Prod., 2002, 65, 1793 CrossRef CAS PubMed; N. Lindquist, W. Fenical, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1991, 113, 2303 CrossRef; Z. Cruz-Monserrate, H. C. Vervoort, R. Bai, D. J. Newman, S. B. Howell, G. Los, J. T. Mullaney, M. D. Williams, G. R. Pettit, W. Fenical and E. Hamel, Mol. Pharmacol., 2003, 63, 1273 CrossRef PubMed.
- O. Grotkopp, A. Ahmad, W. Frank and T. J. J. Müller, Org. Biomol. Chem., 2011, 9, 8130 CAS.
- E. Merkul, O. Grotkopp and T. J. J. Müller, Synthesis, 2009, 502 Search PubMed; E. Merkul and T. J. J. Müller, Chem. Commun., 2006, 4817 RSC.
- For reviews on bioactive indolizines, see e.g. V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593 CrossRef CAS; G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237 CrossRef PubMed; J. P. Michael, Nat. Prod. Rep., 2000, 17, 597 Search PubMed.
- I. V. Seregin, A. W. Schammel and V. Gevorgyan, Org. Lett., 2007, 9, 3433 CrossRef CAS PubMed; D. Chernyak, S. B. Gadamsetty and V. Gevorgyan, Org. Lett., 2008, 10, 2307 CrossRef PubMed; J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200 CrossRef PubMed; D. Chernyak, C. Skontos and V. Gevorgyan, Org. Lett., 2010, 12, 3242 CrossRef PubMed.
- S. Mishra, A. K. Bagdi, M. Ghosh, S. Sinha and A. Hajra, RSC Adv., 2014, 4, 6672 RSC.
- J. P. Michael, Nat. Prod. Rep., 1991, 8, 53 RSC; M. F. Grundon, Nat. Prod. Rep., 1990, 7, 131 RSC; P. M. S. Chauhan and S. K. Srivastava, Curr. Med. Chem., 2001, 8, 1535 CrossRef CAS PubMed; J. P. Michael, Nat. Prod. Rep., 2003, 20, 476–493 RSC.
- K. Kobayashi, K. Yoneda, K. Miyamoto, O. Morikawa and H. Konishi, Tetrahedron, 2004, 60, 11639 CrossRef CAS; R. Martinez, D. J. Ramon and M. Yus, Eur. J. Org. Chem., 2007, 1599 CrossRef; R. P. Korivi and C. H. Cheg, J. Org. Chem., 2006, 71, 7079 CrossRef PubMed.
- N. Mataga and S. Tsuno, Bull. Chem. Soc. Jpn., 1957, 30, 368 CrossRef CAS; R. E. Atkinson and P. R. H. Speakman, J. Chem. Soc. B, 1971, 2077 RSC; G. E. Tumambac, C. M. Rosencrance and C. Wolf, Tetrahedron, 2004, 60, 11293 CrossRef; K. B. Woody, E. M. Henry, S. Jagtap and D. M. Collard, Macromolecules, 2011, 44, 9118 CrossRef; M. Schaffroth, B. D. Lindner, V. Vasilenko, F. Rominger and U. H. F. Bunz, J. Org. Chem., 2013, 78, 3142 CrossRef PubMed.
- F. Mitzel, S. FitzGerald, A. Beeby and R. Faust, Chem. – Eur. J., 2003, 9, 1233 CrossRef CAS PubMed; T. C. Lin, W. Chien, C. Y. Liu, M.-Y. Tsai and Y.-J. Huang, Eur. J. Org. Chem., 2013, 4262 CrossRef.
- S. Rotzoll, B. Willy, J. Schönhaber, F. Rominger and T. J. J. Müller, Eur. J. Org. Chem., 2010, 3516 CrossRef CAS.
- A. S. Karpov and T. J. J. Müller, Org. Lett., 2003, 5, 3451 CrossRef CAS PubMed; D. M. D'Souza and T. J. J. Müller, Nat. Protoc., 2008, 3, 1660 CrossRef PubMed.
- M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694 RSC; D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- D. E. Ames and M. I. Brohi, J. Chem. Soc., Perkin Trans. 1, 1980, 1384 RSC; W. Wang, Y. Shen, X. Meng, M. Zhao, Y. Chen and B. Chen, Org. Lett., 2011, 4514 CrossRef CAS PubMed.
- C. F. Gers, J. Nordmann, C. Kumru, W. Frank and T. J. J. Müller, J. Org. Chem., 2014, 79, 3296 CrossRef CAS PubMed.
- T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390 Search PubMed; Y.-W. Su, S.-C. Lan and K.-H. Wei, Mater. Today, 2012, 15, 554 CrossRef CAS; K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169 CrossRef PubMed; WOLEDs and Organic Photovoltaics, ed. V. W. W. Yam, M. M.-Y. Chan and C.-H. Tao, Springer-Verlag, Berlin, 2010 Search PubMed; B. Kippelen and J.-L. Brédas, Energy Environ. Sci., 2009, 2, 251 Search PubMed; Organic Photovoltaics, ed. C. Brabec, V. Dyakonov and U. Scherf, Wiley-VCH, Weinheim, 2008 CrossRef; K. Schulze, C. Uhrich, R. Schüppel, K. Leo, M. Pfeiffer, E. Brier, E. Reinold and P. Bäuerle, Adv. Mater., 2006, 18, 2872 CrossRef; H. Sirringhaus, T. Kawase, R. Fried, T. Shimoda, M. Inbasekaran, W. Wu and E. Woo, Science, 2000, 290, 2123 CrossRef PubMed; F. Geiger, M. Stoldt, H. Schweizer, P. Bäuerle and E. Umbach, Adv. Mater., 1993, 5, 922 CrossRef.
- E. Nasybulin, I. de Albuquerque and K. Levon, Electrochim. Acta, 2012, 63, 341 CrossRef CAS.
- H. Fiesselmann and P. Schipprak, Chem. Ber., 1954, 87, 835 CrossRef CAS; H. Fiesselmann, P. Schipprak and L. Zeitler, Chem. Ber., 1954, 87, 841 CrossRef; H. Fiesselmann and P. Schipprak, Chem. Ber., 1956, 89, 1879 Search PubMed; H. Fiesselmann and F. Thoma, Chem. Ber., 1956, 89, 1907 CrossRef.
- M. Teiber and T. J. J. Müller, Chem. Commun., 2012, 48, 2080 RSC; M. Teiber, S. Giebeler, T. Lessing and T. J. J. Müller, Org. Biomol. Chem., 2013, 11, 3541 Search PubMed.
- H. Deng, R. Hu, E. Zhao, C. Y. K. Chan, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2014, 47, 4920 CrossRef CAS; C. Zheng, H. Deng, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Macromolecules, 2015, 48, 1941 CrossRef.
- R. Hu, J. I. Maldonado, M. Rodriguez, C. Deng, C. K. W. Jim, J. W. Y. Lam, M. M. F. Yuen, G. Ramos-Ortiz and B. Z. Tang, J. Mater. Chem., 2012, 22, 232 RSC; R. Hu, N. L. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- D. C. Leitch, L. V. Kayser, Z.-Y. Han, A. R. Siamaki, E. N. Keyzer, A. Gefen and B. A. Arndtsen, Nat. Commun., 2015, 6, 1 Search PubMed.
- F. Babudri, G. M. Farinola and F. Naso, J. Mater. Chem., 2004, 14, 11 RSC; L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597 CrossRef CAS PubMed.
- For reviews, see e.g. A. V. Kulinich and A. A. Ischenko, Russ. Chem. Rev., 2009, 78, 141 CrossRef CAS; A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973 CrossRef PubMed; F. M. Hamer, The Chemistry of Heterocyclic Compounds, The Cyanine Dyes and Related Compounds, Interscience Publishers, New York, 1964 Search PubMed.
- See e.g. N. M. Kronenberg, M. Deppisch, F. Würthner, H. W. A. Lademann, K. Deing and K. Meerholz, Chem. Commun., 2008, 6489 RSC; F. Würthner and K. Meerholz, Chem. – Eur. J., 2010, 16, 9366 CrossRef CAS PubMed; H. Bürckstümmer, N. M. Kronenberg, K. Meerholz and F. Würthner, Org. Lett., 2010, 12, 3666 CrossRef PubMed.
- F. Würthner, R. Sens, K.-H. Etzbach and G. Seybold, Angew. Chem., Int. Ed., 1999, 38, 1649 CrossRef; F. Würthner, Synthesis, 1999, 2103 CrossRef.
- C. Muschelknautz, W. Frank and T. J. J. Müller, Org. Lett., 2011, 13, 2556 CrossRef CAS PubMed; C. Muschelknautz, R. Visse, J. Nordmann and T. J. J. Müller, Beilstein J. Org. Chem., 2014, 10, 599 CrossRef PubMed.
- D. M. D'Souza, C. Muschelknautz, F. Rominger and T. J. J. Müller, Org. Lett., 2010, 12, 3364 CrossRef PubMed.
- See e.g. F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef; Y. Ohta, H. Chiba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 7052 CrossRef PubMed.
- N. Sakai, N. Uchida and T. Konakahara, Tetrahedron Lett., 2008, 49, 3437 CrossRef CAS; N. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743 CrossRef PubMed.
- S. Periyaraja, A. B. Mandal and P. Shanmugam, Org. Lett., 2011, 13, 4980 CrossRef CAS PubMed.
- D. S. Chemla and J. Zyss, Nonlinear Optical Properties of Organic Molecules and Crystals, Academic Press Inc., London, 1987 RSC; Nonlinear Optical Materials – The IMA volumes in mathematics and its applications, ed. J. V. Moloney, Springer, New York, 1998 RSC; T. Verbiest, S. Houbrechts, M. Kauranen, K. Clays and A. Persoons, J. Mater. Chem., 1997, 7, 2175 RSC.
- M. Göppert-Mayer, Ann. Phys., 1931, 401, 273 CrossRef.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653 CrossRef CAS PubMed.
- For a review, see M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- M. Grätzel and F. P. Rotzinger, Chem. Phys. Lett., 1985, 118, 474 CrossRef; B. O`Reagan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- See e.g. A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed; Y. Chiba, A. Islam, Y. Watanabe, R. Koiyma, N. Koide and L. Han, Jpn. J. Appl. Phys., 2006, 45, L638 CrossRef; M. K. Nazeeruddin, A. Kay, L. Rodicio, R. Humpry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef; M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, C. Le, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613 CrossRef PubMed.
- A. Michaelis, Ber., 1894, 27, 244 CrossRef; A. Michaelis and E. Richter, Ann. Chem., 1901, 315, 26 Search PubMed.
- For monographs on organoboron compounds, see: H. Braunschweig, I. Krummenacher and J. Wahler, Advances in Organometallic Chemistry, ed. A. F. Hill and M. J. Fink, Academic Press, Elsvier Inc., Oxford, 2013, vol. 61, p. 1 Search PubMed; T. Onak, Organoborane Chemistry, Academic Press Inc., New York, 1975 Search PubMed.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927 CrossRef CAS; C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574 CrossRef; F. Jäkle, Chem. Rev., 2010, 110, 3985 CrossRef PubMed; Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263 CrossRef PubMed.
- T. Chen, J. H. Boyer and M. L. Trudell, Heteroat. Chem., 1997, 8, 51 CrossRef CAS; F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin III, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001 CrossRef; R. K. Lammi, A. Amboise, T. Balasubramanian, R. W. Wgner, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 2000, 122, 7579 CrossRef; T. A. Golovkova, D. V. Kozlov and D. C. Neckers, J. Org. Chem., 2005, 70, 5545 CrossRef PubMed; T. K. Khan, M. Bröring, S. Mathur and M. Ravikanth, Coord. Chem. Rev., 2013, 257, 2348 CrossRef.
- M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chem. – Eur. J., 2008, 14, 2976 CrossRef CAS PubMed; S. A. Baudron, Dalton Trans., 2013, 42, 7498 RSC; A. N. Kursunlu, E. Guler, H. I. Ucan and R. W. Boyle, Dyes Pigm., 2012, 94, 496 CrossRef; S. R. Halper and S. M. Cohen, Chem. – Eur. J., 2003, 9, 4661 CrossRef PubMed.
- J. Chen, A. Burghart, C.-W. Wan, L. Thaim, C. Ortiz, J. Reibenspies and K. Burgess, Tetrahedron Lett., 2000, 41, 2303 CrossRef CAS.
- M. Bröring, R. Krüger and C. Kleeberg, Z. Anorg. Allg. Chem., 2008, 634, 1555 CrossRef CAS; M. Mao, S. Xiao, J. Li, Y. Zou, R. Zhang, J. Pan, F. Dan, K. Zou and T. Yi, Tetrahedron, 2012, 68, 5037 CrossRef.
- H. Li and F. Jäkle, Angew. Chem., Int. Ed., 2009, 48, 2313 CrossRef CAS PubMed; A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef PubMed; Y. Nagata and Y. Chujo, Macromolecules, 2008, 41, 2809 CrossRef; Y. Tokoro, A. Nagai and Y. Chujo, Macromolecules, 2010, 43, 6229 CrossRef.
- G. Wesela-Bauman, L. Jastrzbski, P. Kurach, S. Luliński, J. Serwatowski and K. Woźniak, J. Organomet. Chem., 2012, 711, 1 CrossRef CAS.
- G. Wesela-Bauman, P. Ciećwierz, K. Durka, S. Luliński, J. Serwatowski and K. Woźniak, Inorg. Chem., 2013, 52, 10846 CrossRef CAS PubMed.
- P. D. Beer, V. Timoshenko, M. Maestri, P. Passaniti, V. Balzani and B. Balzani, Chem. Commun., 1999, 1755 RSC; D. Curiel and P. D. Beer, Chem. Commun., 2005, 1909 RSC; M. P. Coogan, V. Fernandez-Moreira, B. M. Kariuki, S. J. A. Pope and F. L. Thorp-Greenwood, Angew. Chem., Int. Ed., 2009, 48, 4965 CrossRef CAS PubMed; S. Bullock, A. J. Hallett, L. P. Harding, J. J. Higginson, S. A. F. Piela, S. J. A. Pope and C. R. Rice, Dalton Trans., 2012, 41, 14690 RSC.
- D. Beck, J. Brewer, J. Lee, D. McGraw, B. A. DeGraff and J. N. Demas, Coord. Chem. Rev., 2007, 251, 546 CrossRef CAS; M. Cattaneo, F. Fagalde, C. D. Borsarelli and N. E. Katz, Inorg. Chem., 2009, 48, 3012 CrossRef PubMed; X.-F. Wang, J. Lumin., 2013, 134, 508 CrossRef.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603 CrossRef CAS; F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12 CrossRef.
- S. M. Fredericks, J. C. Luong and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 7415 CrossRef CAS; D. R. Striplin and G. A. Crosby, Chem. Phys. Lett., 1994, 221, 426 CrossRef.
- R. O. Bonello, I. R. Morgan, B. R. Yeo, L. E. J. Jones, B. M. Kariuki, I. A. Fallis and S. J. A. Pope, J. Organomet. Chem., 2014, 749, 150 CrossRef CAS.
- J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1994, 64, 815 CrossRef CAS; W. Brian, D. Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef.
- X. Chen, D. Qiu, L. Ma, Y. Cheng, Y. Geng, Z. Gie and L. Wang, Transition Met. Chem., 2006, 31, 639 CrossRef CAS; S. Tokito, K. Noda, H. Tanaka, Y. Taga and T. Tsutsui, Synth. Met., 2000, 111–112, 393 CrossRef; S. F. Liu, Q. Wu, H. L. Schemider, H. Aziz, N. X. Hu, Z. Popavic and S. Wang, J. Am. Chem. Soc., 2000, 12, 3671 CrossRef.
- F. Dumur, L. Beouch, M.-A. Tehfe, E. Contal, M. Lepeltier, G. Wantz, B. Graff, F. Goubard, C. R. Mayer, J. Lalevée and D. Gigmes, Thin Solid Films, 2014, 564, 351 CrossRef CAS.
- M. Main, J. S. Snaith, M. M. Meloni, M. Jauregui, D. Sykes, S. Faulkner and A. M. Kenwright, Chem. Commun., 2008, 5212 RSC.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC; P. Caravan, J. J. Ellison and T. J. McMurry, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed.
- W. Lu, B. D. Bennett and J. D. Rabinowitz, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 871, 236 CrossRef CAS PubMed; S. S. Rubakhin, E. V. Romanova, P. Nemes and J. V. Sweedler, Nat. Methods, 2011, 8, S20 CrossRef PubMed; B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593 CrossRef PubMed.
- J. M. Halket, D. Waterman, A. M. Przyborowska, R. K. P. Patel, P. D. Fraser and P. M. Bramley, J. Exp. Bot., 2005, 56, 219 CrossRef CAS PubMed.
- G. Lunn and L. C. Hellwig, Handbook of Derivatization Reactions for HPLC, Wiley, New York, 1998 Search PubMed.
- K. A. Totaro, B. O. Okandeji and J. K. Sello, ChemBioChem, 2012, 13, 987 CrossRef CAS PubMed.
- W. P. Blackstock and M. P. Weir, Trends Biotechnol., 1999, 17, 121 CrossRef CAS PubMed; R. Aebersold, Nature, 2003, 422, 115 CrossRef PubMed; W. S. D. Patterson and R. Aebersold, Nat. Genet., 2003, 33, 311 CrossRef PubMed.
- B. F. Cravatt and E. J. Sorensen, Curr. Opin. Chem. Biol., 2000, 4, 663 CrossRef CAS PubMed; K. T. Barglow and B. F. Cravatt, Nat. Methods, 2007, 4, 822 CrossRef PubMed; B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383 CrossRef PubMed.
- A. Pandey and M. Mann, Nature, 2000, 405, 837 CrossRef CAS PubMed.
- S. Brauch, M. Henze, B. Osswald, K. Naumann, L. A. Wessjohann, S. S. van Berkel and B. Westermann, Org. Biomol. Chem., 2012, 10, 958 CAS.
- S. Sankararaman, Pericyclic Reactions – A Textbook: Reactions, Applications and Theory, Wiley-VCH, Weinheim 2005 Search PubMed.
- Charge and Exciton Transport through Molecular Wires, ed. L. D. A. Siebbeles and F. C. Grozema, Wiley-VCH, Weinheim, 2011, p. 1 CrossRef CAS; S. Etemad, A. J. Heeger and A. G. MacDiarmid, Annu. Rev. Phys. Chem., 1982, 33, 443 CrossRef CAS.
- J. S. Meisner, D. F. Sedbrook, M. Krikorian, J. Chen, A. Sattler, M. E. Carnes, C. B. Murray, M. Steigerwald and C. Nuckolls, Chem. Sci., 2012, 3, 1007 RSC.
- G. Jayamurugan, A. D. Finke, J.-P. Gisselbrecht, C. Boudon, W. B. Schweizer and F. Diederich, J. Org. Chem., 2014, 79, 426 CrossRef CAS PubMed; S.-i. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994 RSC.
- T. Shoji, J. Higashi, S. Ito, T. Okujima, M. Yasunami and N. Morita, Org. Biomol. Chem., 2012, 10, 2431 Search PubMed; T. Shoji, S. Ito, T. Okujima and N. Morita, Eur. J. Chem., 2011, 5134 Search PubMed; T. Shoji, S. Ito, K. Toyota, T. Iwamoto, M. Yasunami and N. Morita, Eur. J. Org. Chem., 2009, 4316 CrossRef CAS; W. Zhou, J. Xu, H. Zheng, H. Liu and D. Zhu, J. Org. Chem., 2008, 73, 7702 CrossRef PubMed; S. Chen, Y. Li, C. Liu, W. Yang and Y. Li, Eur. J. Org. Chem., 2011, 6445 CrossRef.
Footnote |
| † Dedicated to Prof. Dr Peter Bäuerle on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2016 |