
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents
Tiago
Rodrigues
a,
Florian
Sieglitz
a and
Gonçalo J. L.
Bernardes
*ab
aInstituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Av. Prof. Egas Moniz, 1649-028 Lisboa, Portugal
bDepartment of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK. E-mail: gb453@cam.ac.uk; Tel: +44 (0)1223 336305
First published on 18th February 2016
Abstract
Treatment of cancer is a significant challenge in clinical medicine, and its research is a top priority in chemical biology and drug discovery. Consequently, there is an urgent need for identifying innovative chemotypes capable of modulating unexploited drug targets. The transient receptor potential (TRPs) channels persist scarcely explored as targets, despite intervening in a plethora of pathophysiological events in numerous diseases, including cancer. Both agonists and antagonists have proven capable of evoking phenotype changes leading to either cell death or reduced cell migration. Among these, natural products entail biologically pre-validated and privileged architectures for TRP recognition. Furthermore, several natural products have significantly contributed to our current knowledge on TRP biology. In this Tutorial Review we focus on selected natural products, e.g. capsaicinoids, cannabinoids and terpenes, by highlighting challenges and opportunities in their use as starting points for designing natural product-inspired TRP channel modulators. Importantly, the de-orphanization of natural products as TRP channel ligands may leverage their exploration as viable strategy for developing anticancer therapies. Finally, we foresee that TRP channels may be explored for the selective pharmacodelivery of cytotoxic payloads to diseased tissues, providing an innovative platform in chemical biology and molecular medicine.
 Tiago Rodrigues | Dr Tiago Rodrigues received an MSc in Pharmaceutical Sciences (2006) and a PhD in Medicinal Chemistry (2010) from the University of Lisbon, Portugal, under the supervision of Dr Francisca Lopes. He then completed a postdoctoral stay in the group of Prof. Gisbert Schneider at ETH Zürich working on de novo design, microfluidics-assisted syntheses and macromolecular target identification for small molecules. He is currently working with Dr Gonçalo Bernardes on the target deorphanization of natural products and development of drug delivery constructs. |
 Florian Sieglitz | Dr Florian Sieglitz received his MSc in Biomedicine in 2009 and a PhD in developmental neuroscience in 2013 from the University of Münster, Germany, focussing on feedback signalling downstream of receptor tyrosine kinases. Currently, he is working in the lab of Dr Gonçalo Bernardes on hydrogen sulphide mediated posttranslational protein modification during neuroinflammation. |
 Gonçalo J. L. Bernardes | Dr Gonçalo Bernardes is a Group Leader at the Department of Chemistry, University of Cambridge, U.K. He is also the Director of the Chemical Biology and Pharmaceutical Biotechnology Unit at the Instituto de Medicina Molecular, Portugal. After completing his DPhil degree in 2008 at the University of Oxford, U.K., he undertook postdoctoral work at the Max-Planck Institute of Colloids and Interfaces, Germany, and the ETH Zürich, Switzerland, and worked as a Group Leader at Alfama Lda in Portugal. He started his independent research career in 2013, and his research group interests focus on the development of site-selective chemical protein modification for basic biology and drug development. He is a Royal Society University Research Fellow and was recently awarded a Starting Grant from the European Research Council. |
Key learning points(1) Transient receptor potential (TRP) channel modulation as a strategy for cancer cell killing.(2) Natural products are privileged chemical matter for TRP modulation. (3) Natural products may be promiscuous multi-TRP channel modulators. (4) TRP modulators may be used to vectorize cytotoxic payloads. |
1. Introduction
Cancer remains a leading cause of death and morbidity worldwide, despite extensive basic and clinical research efforts dedicated to finding ways to control tumour growth and cure diseased tissues. Typically, anti-cancer small molecules aim to selectively modulate crucial and often up-regulated drug targets and signalling pathways. However, the rapid evolution of resistance in tumour cells to the current therapeutic armamentarium poses a great challenge to the design of effective long-term chemotherapeutic schemes and prototypes for sustained personalized medicines.1,2 Thus, innovative chemotypes capable of modulating unexplored cancer-relevant targets are constantly sought for.Such targets, amongst others, involve signals elicited by the intracellular second messenger calcium (Ca2+). It is well established that cytosolic free Ca2+ acts as a key regulator of fundamental down-stream processes in cancer, including proliferation, differentiation, and gene transcription.3,4 As a result, Ca2+ influx remodelling may disrupt intracellular pathophysiological events and induce cancer cell death.3
Transient receptor potential (TRP) cation channels encompass six different subfamilies that control Mg2+/Ca2+ homeostasis, including the vanilloid (TRPV), melastatin (TRPM), canonical (TRPC), ankyrin (TRPA), mucolipin (TRPML), and polycystic (TRPP).5 While technical challenges on both NMR spectroscopy and X-ray crystallography have generally proven insurmountable for obtaining structural data, information on domain composition has been primarily gained through in silico modelling and structure–function relationship studies. More recently, breakthrough cryo-electron microscopy structures of the mammalian TRPV1 have provided critical insights on a dual gating mechanism for ion transport.6,7 Furthermore, allosteric coupling between the upper and lower gates rationalize the finely controlled channel dynamics,6,7 and reveal opportunities for future drug discovery endeavours. Typically, TRP channels are comprised by six transmembrane-spanning domains (S1–S6), a pore-forming loop between S5 and S6, and assemble as homo- or hetero-tetramers (Fig. 1a–c). TRP channels are widely distributed in diverse tissues.8 Moreover, together with accessory proteins, they mediate a plethora of cellular processes. Surprisingly, TRP channels have seldom been investigated as drug targets, despite their “druggability” and relevance to a multitude of pathologies.5 For example, only 4 of the 28 mammalian TRP counterparts have yielded clinical stage ligands to date, primarily as analgesic agents.9
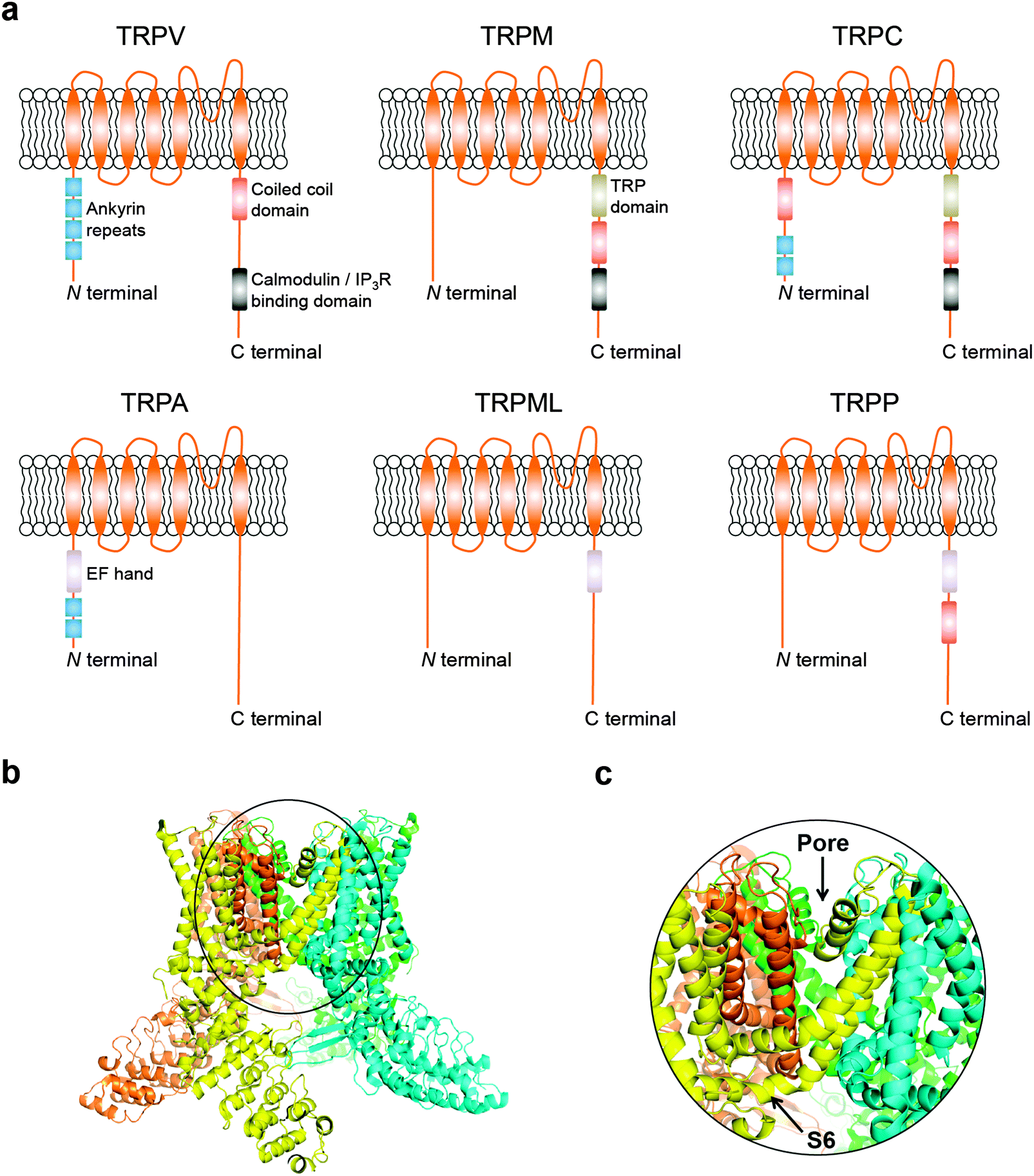 | ||
| Fig. 1 (a) Schematics of TRP channel subfamilies and domain organization. (b) General overview of TRPV channels exemplified by the TRPV1 atomic model (PDB 3J5Q); (c) focus on the Pore-S6 domain of TRPV1. Images generated with PyMOL (Schrödinger LLC). | ||
The (patho)physiology associated to TRP channels, i.e. channelopathies, and the deorphanization of TRP channels with natural products have already been extensively reviewed elsewhere.5,10,11 In this Tutorial Review we focus on how TRP channels are attractive drug targets per se or for the targeted delivery of cytotoxic payloads. We provide a selection of prototypical natural products and derivatives with anti-cancer activity partly or fully related to TRP engagement. Special attention is given to chemical matter modulating the TRPV, TRPM and TRPC channels, including future implications in cancer drug discovery.
2. Transient receptor potential channels in cancer
The role of TRP channels in cancer is highly heterogeneous, ranging from the control of Ca2+ homeostasis to regulation of tumourigenic and metastatic events. Tumour progression can be generally associated with misregulation of either one or more TRP channels.4 Though, as Gkika and co-workers8 point out, a full expression profiling of these channels is still required in order to correlate a cancer-specific pattern to carcinogenesis staging. Furthermore, several members of the TRP channel family are highly expressed in normal tissues while for many others thorough expression profiles, e.g. on a cellular level, are lacking. For example, TRPC4 is found in brain, bone, heart and prostate, whereas TRPM1-3 are highly expressed in brain tissue.5,8 Thus, the advantages of targeting TRP channels in cancer must be carefully traded off against possible adverse reactions in other tissues. Interestingly, the role of TRP channels appears to be dependent on the cell type, i.e. a given channel may be either oncogenic or tumour suppressive in different cancers. TRPV2 presents a perfect example. On one hand it is overexpressed and required for invasiveness of metastatic prostate adenocarcinoma. On the other hand, its overexpression inhibits growth of glioblastoma.5TRPV1 and TRPV6 rank among the most studied channels in cancer. Studies on clinical samples have shown that TRPV6 is overexpressed in most prostate cancers and thus can be generally used as a marker to predict clinical outcomes.12
TRPM8 channels constitute some of the most studied counterparts within the melastatin channel family. Its widespread overexpression in many prostate, breast, colon, lung and skin tumours, in opposition to low or undetectable levels in the corresponding normal tissues, make it an attractive target for cancer modulation.13 Moreover, TRPM8 may be effectively used in prostate cancer diagnosis and staging.14 Among the TRPC subfamily, it has been reported that expression and activation of TRPC1 decreases Ca2+-dependent apoptosis and cancer progression in the neuroblastoma SH-SY5Y cell line.15 Furthermore, together with TRPC6 it mediates Ca2+ signalling in MCF7 breast cancer cells.16 TRPC6 may simultaneously have an important role in most liver and gastric cancers given its up/mis-regulation in tumourigenic tissues.17,18 Finally, activation of TRPC4/5 was recently unveiled to induce death in the renal cell carcinoma A498 cell line via high Ca2+ influx and overload.19,20
Taken together, a deeper understanding of TRP channel biology in diverse cancer cell-lines promises a new window of opportunity for developing innovative anti-cancer agents. While blockage of cancer-relevant drug targets has been a better-established concept for developing therapeutic agents and tackling tumours, promotion of the TRP-mediated Ca2+ influx for intracellular Ca2+ overload is not only toxic to cells, but also attainable and effective19 (Fig. 2). Hence, modulation of TRP channels offers a much-sought opportunity for unravelling drug-relevant chemical space, while exploiting innovative and presumably intellectual property-free chemotypes. Moreover, the evolutionary sequence similarity between these channels5 provides a solid basis to consider promiscuous target engagement profiles by TRP ligands and tight integration of TRP channels in diverse polypharmacology networks. Nevertheless, considering the heterogeneous expression of TRPs in diverse cancers, its ligands may be best employed in personalized medicine schemes.
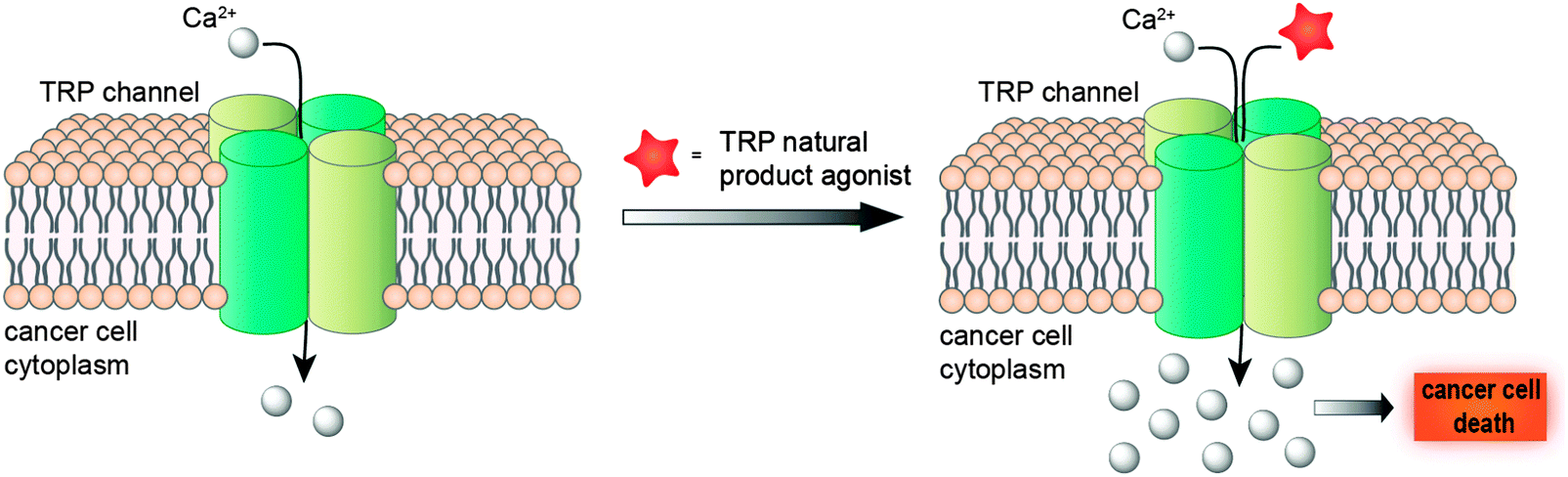 | ||
| Fig. 2 Modulation of TRP channels with agonists induces intracellular Ca2+ overload, with subsequent cell death. | ||
3. Natural product modulators of TRP channels
Natural products have long been a valuable source of chemical matter for interrogating biological systems, while acting as biologically pre-validated prototypes for chemical probe and drug discovery. Such role has been especially prominent in anti-cancer research with ca. 50% of FDA-approved drugs since 1940 being either natural products or their derivatives.21 Thus, it is widely accepted that the intricate architectures generally comprised in natural products offer a multitude of opportunities for exploring drug-relevant chemical space only sparingly covered by synthetic small molecules. In particular, deorphanisation of TRP channels by natural product ligands11 has, together with knock-down/knock-out studies, contributed to the understanding of the gating mechanism and function for this ion channel family.11 Moreover, the discovery of natural product modulators of TRP channels has led to the development of mimics as chemical probes and drug leads.11On- and off-targets remain largely unknown for the majority of naturally occurring chemotypes, hindering the use of rational approaches to design natural product-inspired chemical entities.22 While proteomics is the technology of choice for identifying macromolecular binding counterparts, target prediction software tools have increasingly proven useful for that purpose.22,23 In fact, from a pharmacophore feature pair point of view, the ligand-based software SPiDER24 suggests that a significant proportion of natural products annotated in the Dictionary of Natural Products database display potential pharmacophores for modulation of one or more TRP channels.22 Hence, although experimental confirmation is required for said predictions, the data advocates that natural products present privileged scaffolds for engaging TRP ion channels. Given that several of these compounds theoretically present “pathological” moieties, i.e. substructural motifs commonly flagged for promiscuous unspecific binding, it will be imperative to validate TRP channel screening hits by assessing their potential to form colloidal aggregates, and counter screen them in orthogonal assays and/or against unrelated proteins.
3.1 TRPV modulators
TRPV1 remains the most studied receptor within the TRP channel family and is important for nociception. It can be activated by a wide range of stimuli amongst them heat, low pH, allyl isothiocyanate, N-oleyl-dopamine and vanilloids.5 Its prototypical and best studied ligand capsaicin (Fig. 3) is a natural product isolated from Capsicum spp. (i.e. “hot” chilli pepper), which potently mediates intracellular Ca2+ influx (EC50 = 40 nM). Recent cryo-electron microscopy data,6,7 together with computational modelling and mutagenesis studies,25 provide solid evidence of the molecular mechanisms of TRPV1 activation. Capsaicin sits in an intracellular binding site26 in a “tail-up, head-down” configuration. While the vanillyl and amide moieties establish directed interactions to anchor its binding position, the flexible hydrophobic tail may adopt several bioactive conformations and mediate hydrophobic contacts within the receptor.25 | ||
| Fig. 3 Chemical structure of selected TRPV1 natural product agonists with anticancer activity. | ||
Recognition of the importance of TRPV1 and putative ligands in cancer progression has spurred interest in their exploitation as potential drug lead candidates. The TRPV1-dependent increase of intracellular reactive oxygen species (ROS) endorsed by capsaicin is known to inhibit cancer progression.27 Considering that experimental conditions often include capsaicin at concentrations several orders of magnitude higher than required for TRPV1 activation, it comes at no surprise that several mechanisms and signalling pathways, from off-target engagement, interplay in phenotype changes. In fact, it has been realized that capsaicin-induced cell death is mediated by TRPV1 at low doses in a variety of cancer cell lines, whereas TRPV1-independent effects take over from high dose and prolonged exposure.28 Interestingly, capsaicin equally promotes the production of ROS through TRPV1-independent pathways.29 Without evidence of a clear mechanism of action, 10 μM capsaicin causes G0/G1 cell-cycle arrest in ER-positive and negative breast carcinoma variants, further contributing to confounding effects.30 The anti-proliferative effects of capsaicin against the androgen-sensitive PC-3 prostate cancer cells proceed via a TRPV1-dependent pathway and the increase of intracellular Ca2+ concentration is responsible for apoptosis.31
Malignant gliomas are one of the most aggressive and therapeutically challenging cancer types. Whereas important progresses in the understanding of relevant pathobiology and medicinal chemistry have been made in the past decade, innovative therapies are still urgently required.5 Amantini et al. have shown TRPV1-dependent apoptosis of U373 glioma cells (grade III), involving Ca2+ influx, p38 MAPK activation, mitochondrial transmembrane potential dissipation and Caspase-3 activation at capsaicin concentrations ranging from 1–50 μM.28 Importantly, oral administration of 2.5–5 mg kg−1 capsaicin is able to significantly reduce growth of pancreatic tumour xenografts in mice without any reported side effects.27 However, contradictory studies report capsaicin induced up-regulation of tumour associated NADPH oxidase in colon carcinoma cells promoting proliferation and migration32 as well as enhanced invasiveness of colorectal cancer cells upon capsaicin treatment due to induction of Matrixmetalloprotease-2 and -9.33 Taken this into account as well as the broad expression of TRPV1 in various tissues, carefully planned studies will have to show if capsaicin-inspired small molecules may find applicability in early drug discovery programs.
Natural products structurally related to capsaicin are commonly labelled as capsaicinoids. In general, these share the vanillin head coupled to a hydrophobic tail of varying lengths and topologies. One such example, piperine, activates moderately TRPV1 (EC50 = 38 μM) and produces cell cycle arrest of human prostate cancer PC3 and LNCaP cell lines.34 In similar fashion to capsaicin, it induces production of ROS, loss of mitochondrial membrane integrity, caspase and p38 MAPK activation,35 suggesting the relevance of TRPV1 for the observed functional effects. In particular, 75–150 μM of piperine was shown to selectively inhibit growth of several colon cancer cell lines.35 6-Gingerol is a major component of ginger and a single-digit micromolar agonist of TRPV1 that significantly affects cell viability of the HeLa and LoVo cancer cell lines, by increasing the level of ROS.36 6-Shogaol is a potent agonist of TRPV1 channels with an EC50 value of ca. 770 nM. Intracellular Ca2+ is not only rapidly increased during 6-shogaol-mediated TRPV1 activation, but also reversible upon wash out of the natural product. Hence, data suggests that a lipophilic surrogate, i.e. similar pharmacophore features, may substitute the Michael acceptor in the hydrophobic tail of 6-shogaol without sacrificing bioactivity.37 Interestingly, it was recently reported the anti-cancer activity of 6-shogaol at TRPV1 activation-relevant concentrations against breast carcinoma cell lines. Though, it remains unknown whether the engagement of TRPV1 channels is responsible for the cytotoxicity or not.38
Resiniferatoxin is a diterpene-based natural product with high affinity (KD = 25–480 pM) and potent agonist activity in TRPV1 channels (EC50 = 1 nM).39 Cryo-electron microscopy data shows that resiniferatoxin sits in an overlapping, yet non-identical binding site to capsaicin.7 Like other TRPV1 agonists, it induces apoptosis in several bladder cancer cell lines by modulating mitochondrial function, depolarization and increase of ROS production at ca. 20 μM. However, data also advocates that the observed effects are TRPV1-independent, thus supporting a promiscuous profile by the natural product at high concentrations. Xenograft mouse models of bladder cancer have shown significantly reduced tumour growth upon treatment with 10 μM resiniferatoxin without detectable toxicity.40
In opposition to TRPV1, the osmo and mechano sensory related TRPV2 is one of the least studied vanilloid receptors. The absence of specific TRPV2-modulating chemical tools has been causative for the current lack of knowledge on the underlying pharmacology. The only known ligand for TRPV2 is the natural product cannabidiol (Fig. 4), which is a potent single-digit micromolar agonist of TRPV2, yet targeting TRPA1 and other cellular receptors as well. No selective antagonists have been validated thus far.41
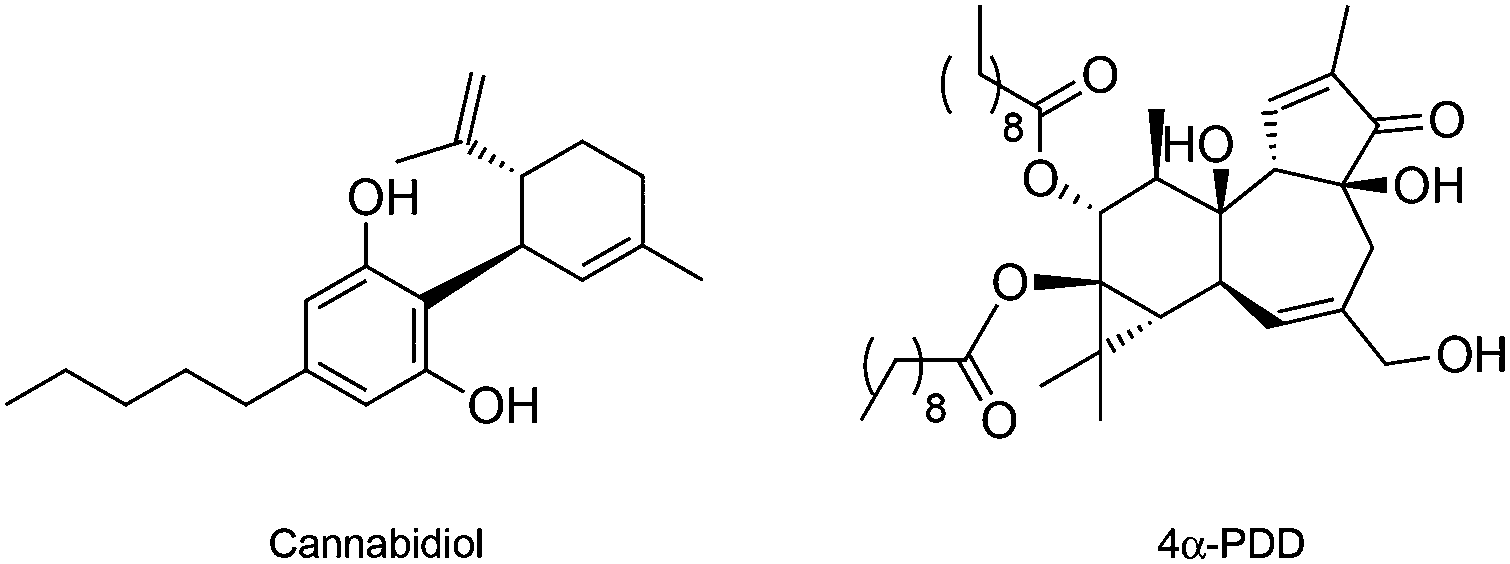 | ||
| Fig. 4 Chemical structure of the TRPV2 and TRPV4 agonists. | ||
TRPV2 has been found overexpressed in a multitude of cancer cell lines – human hepatocarcinoma cells (HepG2), bladder and prostate cancers. Moreover, TRPV2 transcript levels seem to correlate with the metastatic potential of prostate cancers.42 Introduction of TRPV2 in the LNCaP prostate cancer cell line, enhances migration and cancer progression, as it promotes the expression of invasion markers such as Matrix metalloproteinase 9 and Cathepsin B. In line with these observations, silencing of TRPV2 with siRNA reduces tumour growth and invasiveness of TRPV2-expressing prostate cancers.42 Consequently, effective blockage of TRPV2 channels may offer an innovative means of inhibiting cancer metastases.
A large body of evidence supports the anticancer activity of cannabidiol, whose putative targets further include the cannabinoid receptor 2, serotonin 5-HT1A receptor, GPR55 and the peroxisome proliferator-activated receptor gamma.43 In bladder cancer, 30 μM cannabidiol was shown to lead to cell death through TRPV2-mediated Ca2+ influx, suggesting the feasibility of activating TRPV2 channels as therapeutic strategy.44 Interestingly, cannabidiol may be additionally used to sensitize TRPV2-expressing glioblastoma cells to effectively promote the uptake of small cytotoxic molecules (e.g. temozolomide, carmustine or doxorubicin) with no effect in normal astrocytes. Although data from Santoni and co-workers might indicate that cytotoxic drugs such as doxorubicin can directly permeate through the pores of activated TRP channels, such as TRPV1, TRPV2 or TRPM845 a validation of this mechanism is still missing. Namely, it remains to be determined if the small molecules enter the tumour cells directly through the TRP pores or if the drug uptake is indirectly regulated through a yet unknown mechanism that requires TRP-mediated Ca2+ signals to induce sensitization. Indeed, Santoni and co-workers reported that depletion of Ca2+ from the cell culture medium by EGTA blocks small molecule uptake and cytotoxicity despite channel activation, favouring the latter hypothesis. Being subject to industrial property protection (US20100311678 A1), where several TRP channels and respective agonists are foreseen to improve the uptake of doxorubicin to cancer cells, research efforts have to be done to unravel this promising mechanism.
Like for its congeners, the discovery of specific agonist and antagonist small molecules for TRPV4 is bound to clarify its roles in cancer and other (patho)physiological events.14,46 Fusi et al. have provided evidence that TRPV4 mediates the release of IL-8 in keratinocytes to limit cancer progression. Moreover, exposure to the semi-synthetic diterpenoid agonist 4α-PDD (EC50 = 200 nM, Fig. 4) elicits a concentration-dependent release of IL-8,47 which may result in favourable therapeutic outcomes.
3.2 TRPM modulators
Regarding the TRPM subfamily most studies focused on either TRPM7 or TRPM8. For instance, the expression of melastatin TRPM8 in sensory neurons is required for cold sensation and regulation of serum insulin levels.48 Similarly to TRPV1, prolonged agonist exposure to TRPM8 causes Ca2+ currents to adapt, desensitizing stimulation. Here, increased Ca2+ influx subsequently leads to cleavage of a membrane phospholipid – phosphatidylinositol 4,5-bisphosphate – which activates cellular downstream events to inactivate the channel. TRPM8 has been found on several tissues, e.g. bladder, prostate, where it serves unknown functions other than sensory. TRPM8 is dramatically overexpressed in many prostate, breast, colon, lung and skin cancers, suggesting its important roles in cancer.49 Importantly, menthol (Fig. 5) is a prototypical TRPM8 terpene agonist with its (−) isomer being more potent than the (+) counterpart (EC50 = 4–80 μM) in promoting cold sensation.50 TRPM8 ligands may equally find potential application as probes or drug leads against cancerous diseases.11 Exposure of melanoma cells to menthol down-regulates TRPM8 expression dose-dependently and induces cell death (IC50 = 12 μM).51 As such, Dendreon Corp. developed a menthol-inspired benzoimidazole (D-3263; Fig. 5) for the treatment of prostatic hyperplasia and have recently conducted phase I clinical trials on advanced solid tumours (clinicaltrials.gov identifier: NCT00839631), exploring the therapeutic benefits of selective Ca2+ overload in transformed cells. Cannabigerol (Fig. 5) is a cannabinoid that potently blocks TRPM8 (IC50 = 0.11 ± 0.02 μM) while moderately activating TRPV1, TRPV2 and TRPA1 channels. It selectively inhibits growth of colon carcinoma cells both in vitro and in xenograft models. In fact, the cannabigerol-induced apoptosis is associated with ROS generation and is independent from the activation of other TRP channels.52 Camphor (Fig. 5) is a known TRPM8 modulator that has been linked with the arrest of keratinocyte proliferation through activation of TRPV3 (WO/2014/078868).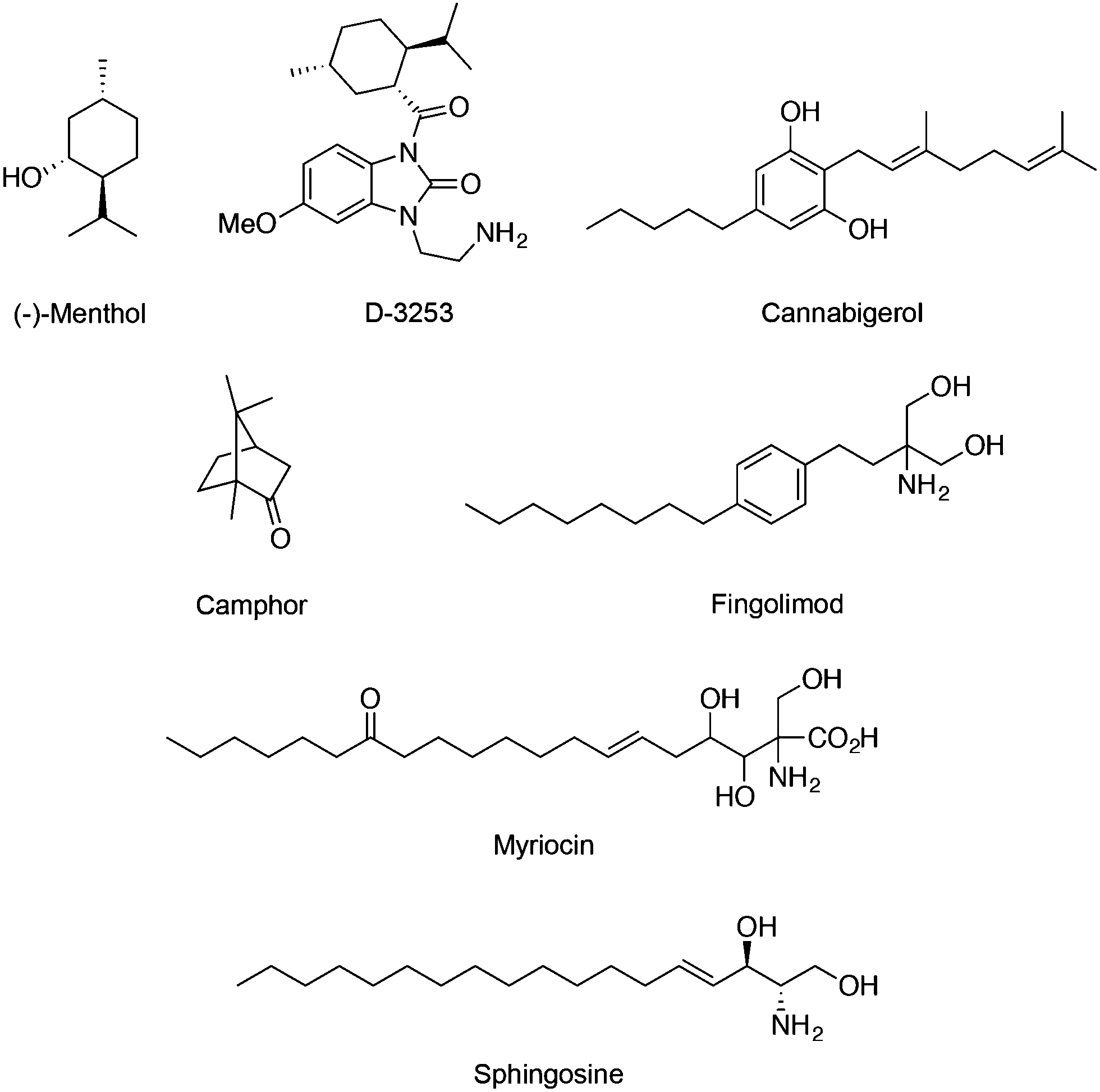 | ||
| Fig. 5 Chemical structure of the TRPM7 and TRPM8 agonists. | ||
Fingolimod (Fig. 5) is a derivative of myriocin, a fungal metabolite of Isaria sinclairii, and an analogue of sphingosine. Both fingolimod and sphingosine potently block TRPM7 with IC50 values of 0.72 and 0.59 μM, respectively. Interestingly, modulation of TRPM7 by fingolimod significantly alters both HEK-293 cell migration and proliferation, and is in line with its anticancer effect in vitro and in vivo. Considering the requirement of TRPM7 activity for breast cancer cell metastasis, treatment with fingolimod may deserve further attention.53
As seen from the cited examples, the confounding effects of fragment-like natural products on TRP biology appear to be commonplace. Hence, the phenotype change induced by such molecules is generally best explained via multi-target engagement.
3.3 TRPC modulators
Although the exact role in cancer is yet to be determined, several members of the TRPC subfamily are crucial for cancer proliferation, as exemplified by experiments that employed siRNA-mediated gene silencing. TRPC expression may be linked with cancer staging of progressive cancers, offering a valid diagnostic marker.54,55 Recently, the sesquiterpene agent (−)-englerin A (Fig. 6) was identified as a discriminative and nanomolar-potent TRPC4/5 agonist in renal cell carcinoma cell lines with moderate effects on other ion channels.19,20 Waldmann, Beech and co-workers determined that (−)-englerin A evoked Ca2+ entry rather than Ca2+ release from intracellular storages with high effectiveness (IC50ca. 10 nM). This Ca2+ overload was subsequently responsible for the potent anti-cancer-selective effects. Data suggests that (−)-englerin A has limited effect against TRPC6/M2/V4 and engages reversibly a binding site of TRPC4 that is either extracellular or only accessible from the external leaflet of the membrane bilayer.19 Despite being a powerful chemical tool to assess TRPC4/5 channels, the development of (−)-englerin A into an efficacious investigational anti-cancer drug candidate may still prove challenging. In fact, pieces of evidence show that oral administration of 5 mg kg−1 to rats is well tolerated, but insufficient blood exposure is achieved. On the other hand, a single 1 mg kg−1 intravenous administration results in immediate animal death, while a 1.5 mg kg−1 subcutaneous injection leads to tolerable yet undetectable (−)-englerin A exposure. Given the narrow therapeutic window in mouse models, and the perceived selectivity of (−)-englerin A for TRPC4/5, it is possible that its associated toxicity is linked to agonizing TRPC4/5 in diverse tissues. Accordingly, further studies on TRPC4/5 biology are required to validate their engagement benefits in anti-cancer therapy.20 Modulation of hitherto unknown off-targets by (−)-englerin A cannot be irrevocably neglected and biochemical validation of these may shed light on complex systems biology networks.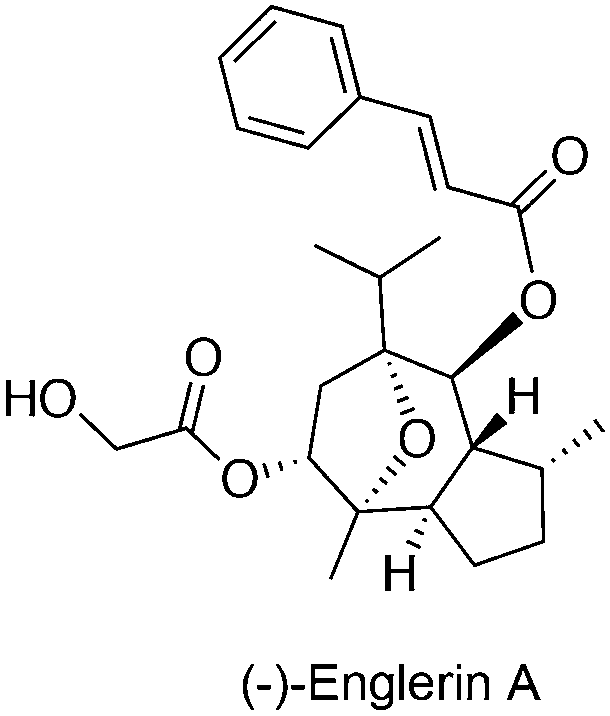 | ||
| Fig. 6 Chemical structure of a TRPC4/5 agonist. | ||
4. Outlook
While natural products offer innovative scaffolds and viable starting points for exploring chemical space relevant to TRP channel biology, the latter continue under-explored in the chemical biology and drug discovery contexts. Growing experimental evidence suggests that TRP channels play a leading role in cancer progression, and that disrupting Ca2+ homeostasis triggers important cancer-cell phenotype changes. Some may be used inclusively as biomarkers for tumour staging. Moreover, unlike many other target families, both agonists and antagonists may present clinical value.With the advent of drug delivery systems and an impending need to selectively deliver cytotoxic payloads to cancer cells, TRP channels may become candidates for directing anti-cancer drugs. To that end, it is relevant to: (i) understand the expression patterns of these channels across all body tissues; (ii) identify high affinity and selective TRP ligands for suitable vectorization of cytotoxic drugs. In general, current TRP ligands identified from the natural product realm display only modest affinity. On the other hand, different expression levels of some TRP channels between healthy and diseased cells may be regarded as an opportunity to develop multiple TRP-centred drug discovery programs. Nonetheless, engagement of TRP channels on healthy tissues will result in toxicity as recently demonstrated with (−)-englerin A. Thus, while modulators may be properly validated as probes, pharmacodynamic/pharmacokinetic issues can preclude their development into efficacious drugs.
It has been realized that TRPV1/2 channels may mediate cell entry of charged small molecules. The outreach of such findings may significantly impact on cancer research and go well beyond simple stoichiometric combinations of TRP ligands and small molecules of interest. Thus, one may envisage taking advantage of TRPV biology and build capsaicin-drug conjugates for the pharmacodelivery of cytotoxic payloads.
In summary, target deorphanisation of natural products as TRP ligands will continuously leverage understanding of this emerging family of ion channels in diseases, while providing innovative scaffolds for groundbreaking chemical biology and drug discovery. Henceforth, natural products, their derivatives and TRP-directed drug delivery constructs may yield an original means of tackling cancers with potential application in translational medicine, e.g. as chemotherapeutics and probes for fluorescence-guided surgery.
Acknowledgements
We thank FCT Portugal (FCT Investigator to G. J. L. B.), the EU (Marie-Curie CIG and Marie-Curie ITN Protein Conjugates to G. J. L. B.), Deutsche Forschungsgemeinschaft (Postdoctoral Fellowship to F. S.), the EPSRC and MRC for funding. G. J. L. B. is a Royal Society University Research Fellow and the recipient of an European Research Council Starting Grant (TagIt).References
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed
.
- L. Chin, J. N. Andersen and P. A. Futreal, Nat. Med., 2011, 17, 297–303 CrossRef CAS PubMed
- G. R. Monteith, D. McAndrew, H. M. Faddy and S. J. Roberts-Thomson, Nat. Rev. Cancer, 2007, 7, 519–530 CrossRef CAS PubMed
- N. Prevarskaya, R. Skryma and Y. Shuba, Nat. Rev. Cancer, 2011, 11, 609–618 CrossRef CAS PubMed
- B. Nilius and A. Szallasi, Pharmacol. Rev., 2014, 66, 676–814 CrossRef PubMed
- M. Liao, E. Cao, D. Julius and Y. Cheng, Nature, 2013, 504, 107–112 CrossRef CAS PubMed
- E. Cao, M. Liao, Y. Cheng and D. Julius, Nature, 2013, 504, 113–118 CrossRef CAS PubMed
- M. Bernardini, A. Fiorio Pla, N. Prevarskaya and D. Gkika, Int. J. Dev. Biol., 2015, 59, 399–406 CrossRef PubMed
- Y. Kaneko and A. Szallasi, Br. J. Pharmacol., 2014, 171, 2474–2507 CrossRef CAS PubMed
- J. Zheng, Compr. Physiol., 2013, 3, 221–242 Search PubMed
- J. B. Calixto, C. A. Kassuya, E. Andre and J. Ferreira, Pharmacol. Ther., 2005, 106, 179–208 CrossRef CAS PubMed
- T. Fixemer, U. Wissenbach, V. Flockerzi and H. Bonkhoff, Oncogene, 2003, 22, 7858–7861 CrossRef CAS PubMed
- L. Tsavaler, M. H. Shapero, S. Morkowski and R. Laus, Cancer Res., 2001, 61, 3760–3769 CAS
- N. Prevarskaya, L. Zhang and G. Barritt, Biochim. Biophys. Acta, 2007, 1772, 937–946 CrossRef CAS PubMed
- S. Bollimuntha, B. B. Singh, S. Shavali, S. K. Sharma and M. Ebadi, J. Biol. Chem., 2005, 280, 2132–2140 CrossRef CAS PubMed
- Y. El Hiani, A. Ahidouch, M. Roudbaraki, S. Guenin, G. Brule and H. Ouadid-Ahidouch, J. Membr. Biol., 2006, 211, 127–137 CrossRef CAS PubMed
- C. El Boustany, G. Bidaux, A. Enfissi, P. Delcourt, N. Prevarskaya and T. Capiod, Hepatology, 2008, 47, 2068–2077 CrossRef CAS PubMed
- R. Cai, X. Ding, K. Zhou, Y. Shi, R. Ge, G. Ren, Y. Jin and Y. Wang, Int. J. Cancer, 2009, 125, 2281–2287 CrossRef CAS PubMed
- Y. Akbulut, H. J. Gaunt, K. Muraki, M. J. Ludlow, M. S. Amer, A. Bruns, N. S. Vasudev, L. Radtke, M. Willot, S. Hahn, T. Seitz, S. Ziegler, M. Christmann, D. J. Beech and H. Waldmann, Angew. Chem., Int. Ed., 2015, 54, 3787–3791 CrossRef CAS PubMed
- C. Carson, P. Raman, J. Tullai, L. Xu, M. Henault, E. Thomas, S. Yeola, J. Lao, M. McPate, J. M. Verkuyl, G. Marsh, J. Sarber, A. Amaral, S. Bailey, D. Lubicka, H. Pham, N. Miranda, J. Ding, H. M. Tang, H. Ju, P. Tranter, N. Ji, P. Krastel, R. K. Jain, A. M. Schumacher, J. J. Loureiro, E. George, G. Berellini, N. T. Ross, S. M. Bushell, G. Erdemli and J. M. Solomon, PLoS One, 2015, 10, e0127498 Search PubMed
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed
- D. Reker, A. M. Perna, T. Rodrigues, P. Schneider, M. Reutlinger, B. Mönch, A. Koeberle, C. Lamers, M. Gabler, H. Steinmetz, R. Müller, M. Schubert-Zsilavecz, O. Werz and G. Schneider, Nat. Chem., 2014, 6, 1072–1078 CrossRef CAS PubMed
- T. Rodrigues, D. Reker, J. Kunze, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2015, 54, 10516–10520 CrossRef CAS PubMed
- D. Reker, T. Rodrigues, P. Schneider and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4067–4072 CrossRef CAS PubMed
- F. Yang, X. Xiao, W. Cheng, W. Yang, P. Yu, Z. Song, V. Yarov-Yarovoy and J. Zheng, Nat. Chem. Biol., 2015, 11, 518–524 CrossRef CAS PubMed
- H. Li, S. Wang, A. Y. Chuang, B. E. Cohen and H. H. Chuang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8497–8502 CrossRef CAS PubMed
- R. Zhang, I. Humphreys, R. P. Sahu, Y. Shi and S. K. Srivastava, Apoptosis, 2008, 13, 1465–1478 CrossRef CAS PubMed
- C. Amantini, M. Mosca, M. Nabissi, R. Lucciarini, S. Caprodossi, A. Arcella, F. Giangaspero and G. Santoni, J. Neurochem., 2007, 102, 977–990 CrossRef CAS PubMed
- C. B. Gonzales, N. B. Kirma, J. J. De La Chapa, R. Chen, M. A. Henry, S. Luo and K. M. Hargreaves, Oral Oncol., 2014, 50, 437–447 CrossRef CAS PubMed
- N. H. Thoennissen, J. O'Kelly, D. Lu, G. B. Iwanski, D. T. La, S. Abbassi, A. Leiter, B. Karlan, R. Mehta and H. P. Koeffler, Oncogene, 2010, 29, 285–296 CrossRef CAS PubMed
- S. Malagarie-Cazenave, N. Olea-Herrero, D. Vara, C. Morell and I. Diaz-Laviada, Cytokine, 2011, 54, 330–337 CrossRef CAS PubMed
- N. C. Liu, P. F. Hsieh, M. K. Hsieh, Z. M. Zeng, H. L. Cheng, J. W. Liao and P. J. Chueh, J. Agric. Food Chem., 2012, 60, 2758–2765 CrossRef CAS PubMed
- J. Yang, T. Z. Li, G. H. Xu, B. B. Luo, Y. X. Chen and T. Zhang, Neoplasma, 2013, 60, 364–372 CrossRef CAS PubMed
- F. N. McNamara, A. Randall and M. J. Gunthorpe, Br. J. Pharmacol., 2005, 144, 781–790 CrossRef CAS PubMed
- P. B. Yaffe, M. R. Power Coombs, C. D. Doucette, M. Walsh and D. W. Hoskin, Mol. Carcinog., 2015, 54, 1070–1085 CrossRef CAS PubMed
- J. Poltronieri, A. B. Becceneri, A. M. Fuzer, J. C. Filho, A. C. Martin, P. C. Vieira, N. Pouliot and M. R. Cominetti, Mini-Rev. Med. Chem., 2014, 14, 313–321 CrossRef CAS PubMed
- P. Rebellato and M. S. Islam, JOP, 2014, 15, 33–37 Search PubMed
- A. Ray, S. Vasudevan and S. Sengupta, PLoS One, 2015, 10, e0137614 Search PubMed
- M. Raisinghani, R. M. Pabbidi and L. S. Premkumar, J. Physiol., 2005, 567, 771–786 CrossRef CAS PubMed
- V. Farfariello, S. Liberati, M. B. Morelli, D. Tomassoni, M. Santoni, M. Nabissi, A. Giannantoni, G. Santoni and C. Amantini, Chem. -Biol. Interact., 2014, 224C, 128–135 CrossRef PubMed
- A. Peralvarez-Marin, P. Donate-Macian and R. Gaudet, FEBS J., 2013, 280, 5471–5487 CrossRef CAS PubMed
- M. Monet, V. Lehen'kyi, F. Gackiere, V. Firlej, M. Vandenberghe, M. Roudbaraki, D. Gkika, A. Pourtier, G. Bidaux, C. Slomianny, P. Delcourt, F. Rassendren, J. P. Bergerat, J. Ceraline, F. Cabon, S. Humez and N. Prevarskaya, Cancer Res., 2010, 70, 1225–1235 CrossRef CAS PubMed
- P. Massi, M. Solinas, V. Cinquina and D. Parolaro, Br. J. Clin. Pharmacol., 2013, 75, 303–312 CrossRef CAS PubMed
- T. Yamada, T. Ueda, Y. Shibata, Y. Ikegami, M. Saito, Y. Ishida, S. Ugawa, K. Kohri and S. Shimada, Urology, 2010, 76(509), e501–e507 Search PubMed
- M. Nabissi, M. B. Morelli, M. Santoni and G. Santoni, Carcinogenesis, 2013, 34, 48–57 CrossRef CAS PubMed
- A. O. Jo, D. A. Ryskamp, T. T. Phuong, A. S. Verkman, O. Yarishkin, N. MacAulay and D. Krizaj, J. Neurosci., 2015, 35, 13525–13537 CrossRef CAS PubMed
- C. Fusi, S. Materazzi, D. Minocci, V. Maio, T. Oranges, D. Massi and R. Nassini, J. Invest. Dermatol., 2014, 134, 2408–2417 CrossRef CAS PubMed
- D. D. McCoy, L. Zhou, A. K. Nguyen, A. G. Watts, C. M. Donovan and D. D. McKemy, Am. J. Physiol.: Endocrinol. Metab., 2013, 305, E78–E88 CrossRef CAS PubMed
- N. S. Yee, Cancers, 2015, 7, 2134–2146 CrossRef PubMed
- D. M. Bautista, J. Siemens, J. M. Glazer, P. R. Tsuruda, A. I. Basbaum, C. L. Stucky, S. E. Jordt and D. Julius, Nature, 2007, 448, 204–208 CrossRef CAS PubMed
- T. Kijpornyongpan, A. Sereemaspun and C. Chanchao, Asian Pac. J. Cancer Prev., 2014, 15, 1551–1556 CrossRef PubMed
- F. Borrelli, E. Pagano, B. Romano, S. Panzera, F. Maiello, D. Coppola, L. De Petrocellis, L. Buono, P. Orlando and A. A. Izzo, Carcinogenesis, 2014, 35, 2787–2797 CrossRef PubMed
- X. Qin, Z. Yue, B. Sun, W. Yang, J. Xie, E. Ni, Y. Feng, R. Mahmood, Y. Zhang and L. Yue, Br. J. Pharmacol., 2013, 168, 1294–1312 CrossRef CAS PubMed
- H. N. Jiang, B. Zeng, Y. Zhang, N. Daskoulidou, H. Fan, J. M. Qu and S. Z. Xu, PLoS One, 2013, 8, e67637 CAS
- B. Zeng, C. Yuan, X. Yang, S. L. Atkin and S. Z. Xu, Curr. Cancer Drug Targets, 2013, 13, 103–116 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |