
A modular family of phosphine-phosphoramidite ligands and their hydroformylation catalysts: steric tuning impacts upon the coordination geometry of trigonal bipyramidal complexes of type [Rh(H)(CO)2(P^P*)]†
Rebecca C.
How
a,
Robert
Hembre
b,
James A.
Ponasik
b,
Ginette S.
Tolleson
b and
Matthew L.
Clarke
*a
aSchool of Chemistry, University of St Andrews, EaSTCHEM, St Andrews, Fife KY16 9ST, UK. E-mail: mc28@st-andrews.ac.uk
bEastman Chemical Company, Kingsport, Tennessee 37662, USA
First published on 2nd September 2015
Abstract
Four new phosphine-phosphoramidite bidentate ligands have been synthesised and studied in rhodium-catalysed hydroformylation. Variable temperature NMR studies have been used along with HPIR to investigate the coordination mode of the trigonal bipyramidal complexes formed from [Rh(acac)(CO)2], ligand and syngas. It was found that small changes to the ligand structure have a large effect on the geometry of the active catalytic species. The rhodium catalysts of these new ligands were found to give unusually high iso-selectivity in the hydroformylation of propene and 1-octene.
Introduction
Ligands that respond to subtle structural modification by changing coordination mode or metal geometry are of significant interest to coordination chemists and catalysis chemists. The ability to control geometry and coordination chemistry of a ligand in a predictable manner can help understand structure-performance relationships in catalysis. This can lead to improved ligand design and subsequently developments in applied catalysis. One area where such studies are of particular significance is the study of Rh(I) complexes of phosphine ligands, which are used as pre-catalysts or catalysts for alkene hydroformylation, one of the most important applications of transition metal catalysis in the world. With one or two notable exceptions,1,2 most hydroformylation catalysts show at least some selectivity towards the linear aldehyde isomers.3 In the last thirty years, large bite angle diphosphine ligands have been shown to give excellent linear selectivity in this respect (high n/iso ratios). These large bite angle ligands, such as BISBI4,5 and XantPhos,6 (BISBI = 2,2′-bis(diphenylphosphinomethyl)-1,1′-biphenyl; xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) always form trigonal bipyramidal Rh(I) complexes where the diphosphine is bis-equatorial. Some ligands with smaller bite angles form complexes with the diphosphine occupying axial-equatorial positions. There is not enough data to make any correlations between axial-equatorial coordination mode and selectivity, although catalysts with axial-equatorial coordinating ligands are rarely or never highly linear selective in the hydroformylation of terminal alkyl alkenes. This led us to study a family of ligands that might be finely balanced between bis-equatorial and axial-equatorial coordination modes. Our objective was to understand how ligand structure controls the selectivity for bis-equatorial/axial-equatorial isomers and whether the isomer formed has any implications on rate, selectivity, stability or activation times in hydroformylation catalysis. Our ultimate long-term aim is to gain sufficient understanding that we would be able to design a series of catalysts that could access all possible linear to branched ratios in the industrially important hydroformylation of propene. Here we show a readily accessed family of phosphine-phosphoramidite ligands, use low temperature NMR to characterise their Rh(I) hydroformylation catalysts, and demonstrate their unusually high iso-selectivity in the hydroformylation of propene and 1-octene.Non-symmetric bis-phosphorus ligands with a ferrocenyl amine backbone make up a successful series of asymmetric hydrogenation catalysts invented by researchers at Eastman Chemical Company.7,8 In this case, we were not particularly interested in exploiting the ligands in enantioselective catalysis, but were attracted to the backbone, since it tends to give catalysts that are rigid and stable and a 7 membered chelate could easily form both bis-equatorial and axial-equatorial isomers of catalyst. In addition, ligands with electronically quite different donor sets such as phosphine-phosphite2,9,10 and phosphine-phosphoramidite11–13 have yielded important catalysts for hydroformylation,14 which led us to consider phosphine-phosphoramidite 1 to 4 as targets (Scheme 1).
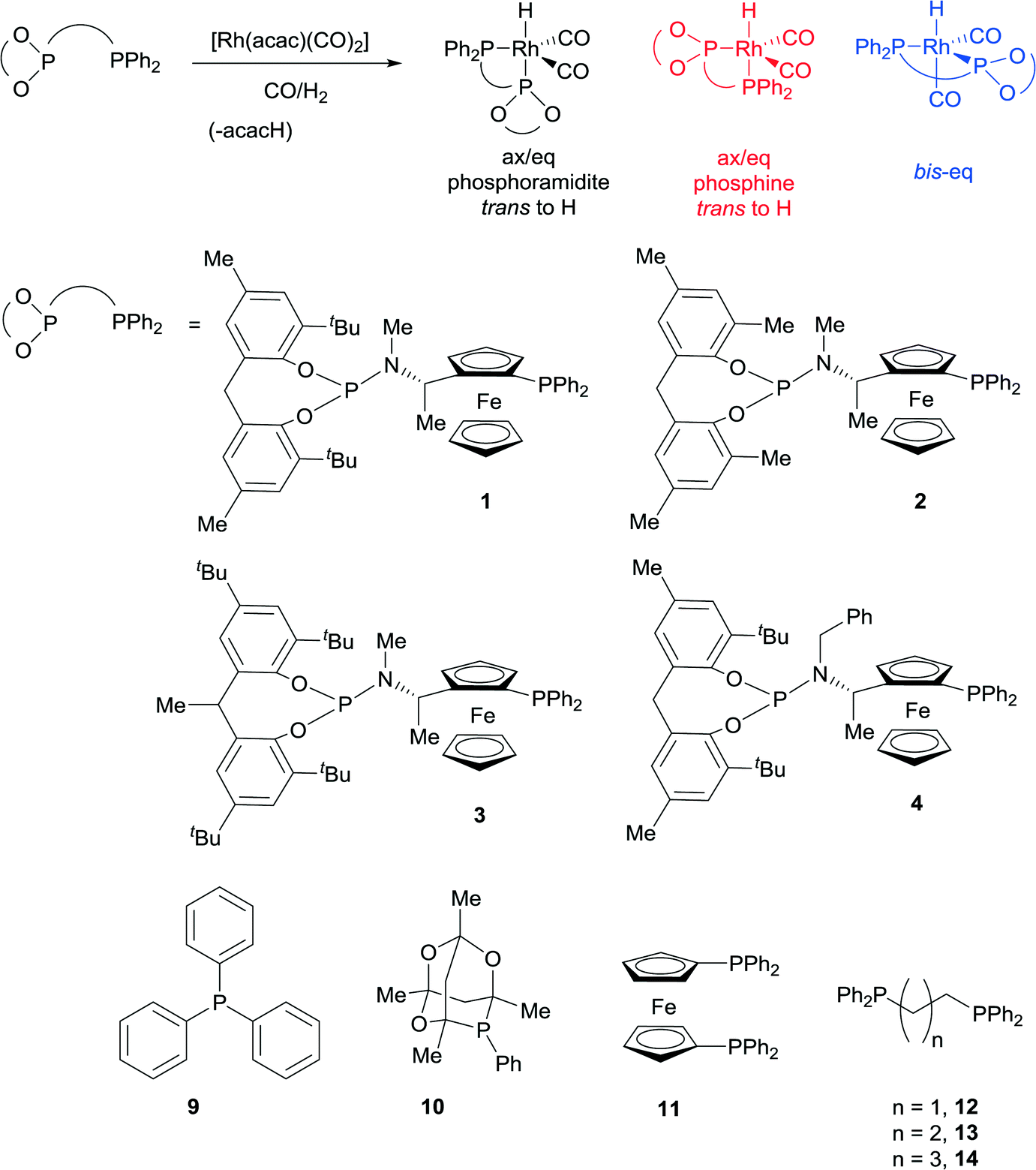 | ||
| Scheme 1 Catalyst structures for non-symmetric ligands (1–4). | ||
Results and discussion
The novel ligands 1–4 were accessed readily from Ugi's amine,155, in 4 steps (Scheme 2).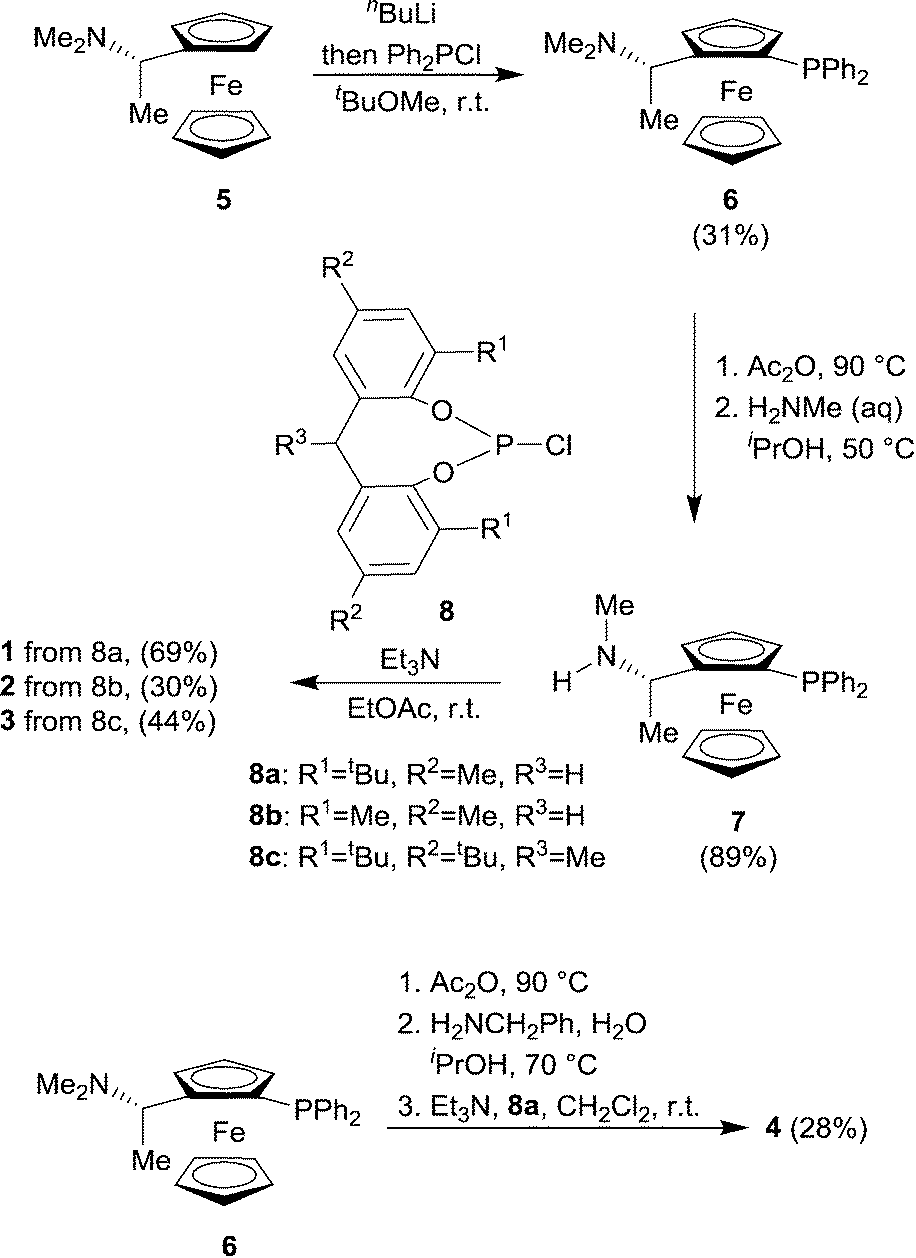 | ||
| Scheme 2 Ligand synthesis. | ||
The ligands react with [Rh(acac)(CO)2] under a syngas atmosphere to form the expected complexes of type [Rh(H)(CO)2(P^P*)] as monitored by low temperature NMR and HPIR (Scheme 3). Since the active pre-catalysts are fairly stable for many hours under a nitrogen atmosphere, we generated the active hydroformylation catalysts by treating either [Rh(acac)(CO)2] and ligand or isolated complex 15 with syngas (Scheme 3), and then subjecting the newly formed complex to NMR spectroscopy under a N2 atmosphere. In a control experiment, the NMR spectrum with ligand 3 was acquired under a pressurised syngas atmosphere; this gave spectra that were the same as those run under a N2 atmosphere.
 | ||
| Scheme 3 Activation of the catalyst. | ||
Comparison of room temperature NMR with low temperature NMR spectra for the activated catalyst formed from ligand 1 (see ESI†) shows the peaks are much sharper at −70 °C in both the 1H NMR and 31P{1H} NMR spectra. In the low temperature 31P{1H} NMR spectrum there were two distinct species, each with a peak in the phosphine [δ 16–20 range] and phosphoramidite [δ 155–180 range] regions. There was also a third species of low intensity in the 31P{1H} NMR, but this was too small to characterise. In the low temperature 1H NMR of the Rh complex of ligand 1, the major Rh-hydride signal has a 2JH–P of 119 Hz. This is very similar to the 117 Hz coupling that Nozaki and co-workers9,16 observed for a phosphine group trans to a rhodium–hydride bond. The minor Rh-hydride signal features a 2JH–P of ≤13 Hz; which is suggestive of a bis-equatorial species. There is a ~9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the size of the peaks assigned to axial-equatorial (phosphine axial, in red) and bis-equatorial species (in blue, see Scheme 3 and Table 1).
:1 ratio between the size of the peaks assigned to axial-equatorial (phosphine axial, in red) and bis-equatorial species (in blue, see Scheme 3 and Table 1).
| Catalyst | Ligand | Phosphorus | 1 J P–Rh (Hz) | 2 J P–P (Hz) | 2 J P–H (Hz) | Ax/eq:bis-eq |
|---|---|---|---|---|---|---|
| a NMR spectra recorded in toluene-d8 at −70 °C. 1H NMR at 500 MHz, 31P{1H} and 31P NMR at 202 MHz. | ||||||
| 1a | 1 | Major phosphoramidite | 215 | 45 | <10 | 89:11 |
| 1a | Major phosphine | 92 | 45 | 119 | ||
| 1b | Minor phosphoramidite | 223 | 167 | 10 | ||
| 1b | Minor phosphine | 140 | 167 | 10 | ||
| 2a | 2 | Only phosphoramidite | 216 | 27 | <10 | 100:0 |
| 2a | Only phosphine | 100 | 27 | 113 | ||
| 3a | 3 | Only phosphoramidite | 219 | 164 | <10 | 0:100 |
| 3a | Only phosphine | 140 | 164 | <10 | ||
| 4a | 4 | 1st major phosphoramidite | 223 | 169 | <10 | 25:75 |
| 4a | 1st major phosphine | 140 | 169 | <10 | ||
| 4b | 2nd major phosphoramidite | 199 | 35 | <10 | ||
| 4b | 2nd major phosphine | 116 | 35 | <10 | ||
| 4c | Minor phosphoramidite | 218 | 49 | 10–20 | ||
| 4c | Minor phosphine | 94 | 49 | 120 | ||
2 J P–H could be easily measured in the 1H NMR, but not the 31P NMR. However, the 31P{1H} NMR and 31P NMR spectra were compared for all peaks. In three of the signals there were no major changes in the appearance of the signal. However, for the major phosphine signal, the peak shape changes due to a large JP–H coupling (119 Hz). This confirms that the phosphine is in the axial position and trans to the rhodium hydride. The ESI† contains all the relevant NMR spectra.
The activated catalysts were then studied for other ligands. Ligand 2 gave only an axial-equatorial species where the phosphine was axial. This was found to give sharp peaks in the NMR at both room temperature and low temperature. Remarkably, when ligand 3 reacted with [Rh(acac)(CO)2] and syngas, only a bis-equatorial isomer is formed. At room temperature the phosphoramidite peak was very broad, but the peak sharpens so that coupling constants could be resolved at −70 °C. For ligand 4 there is a ~3:1 ratio between bis-equatorial and axial-equatorial isomers (phosphine axial). Of the bis-equatorial species, it appears that both diastereomers are present in almost equal levels (see ESI†). These NMR studies show profound changes to the active catalyst geometry are possible just from altering alkyl substituents in the periphery of the ligand structure. These seemingly quite remote substituents might be located close to the coordination sphere of the Rh catalysts. Other workers have seen significant electronic changes to bis-eq/eq-axial ratio's from 1:1 to 9:1.17a
We also studied the formation of the active hydroformylation catalyst of type [Rh(H)(CO)2(P^P*)] using HPIR. Hexane was used as solvent since it has no absorbance in the carbonyl region of the spectrum. The sample was heated to 70 °C, and a spectrum was recorded every 15 minutes to measure the formation of the active catalytic species. The formation of the catalyst requires coordination of the ligand to [Rh(acac)(CO)2] and reaction with CO/H2, or formation of RhH(CO)4 from [Rh(acac)(CO)2] and coordination of the ligand. RhH(CO)4 can be formed rapidly from [Rh(acac)(CO)2] and syngas if ligand coordination is slow (carbonyl stretching adsorptions at 2082 and 2015 cm−1). The catalyst was deemed to have fully formed when the spectrum was constant. Bulky ligands 3 and 4 were found to take longer to activate than ligands 1 and 2 (Fig. 1).
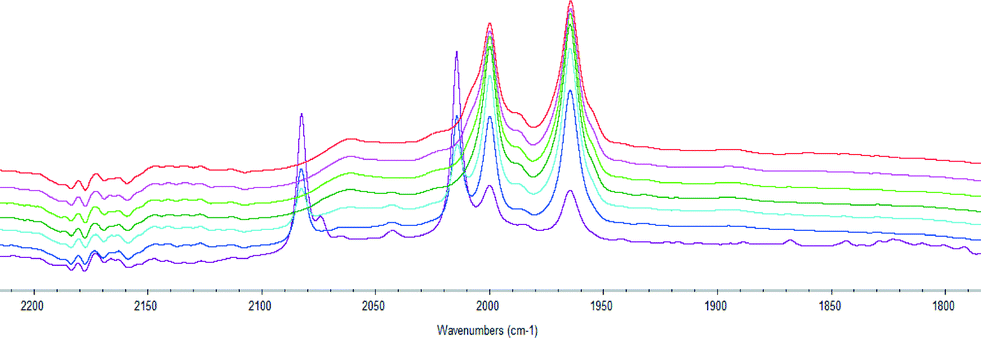 | ||
| Fig. 1 Catalyst activation with ligand 4. One spectra recorded every 15 minutes, RhH(CO)4 was detected in early spectra (purple: 0 minutes, dark blue: 15 min, light blue: 30 min, dark green: 45 min, light green: 60 min, pink: 75 min, red: 90 min); spectra recorded from 45–90 minutes did not change. | ||
Ligand 2 gave only axial-equatorial species by NMR and was found to have only two carbonyl stretches by HPIR. These stretching adsorptions appeared at 2002 and 1954 cm−1 and can be unequivocally assigned as an axial-equatorial species with a phosphine trans to the rhodium–hydride bond.17 No unmodified catalyst was noted after 15 minutes.
Ligand 3 gave only a single bis-equatorial species detectable by NMR spectroscopy. Three carbonyl stretching adsorptions were observed by HPIR. However, there may have been a fourth peak hidden under one of the others. The three bands appeared at 2061, 2000 and 1965 cm−1. Some unmodified catalyst was noted until 60 minutes. Kamer and co-workers17b reported carbonyl stretching adsorptions at 2049 and 1970 cm−1 for a bis-equatorial catalyst with DPEphos; and bands at 2052 and 1980 cm−1 for a bis-equatorial catalyst with a phosphine-phosphite bidentate ligand. The signals at 2061 and 1965 cm−1 were tentatively assigned as the bis-equatorial species on this basis. We also propose that at 70 °C, the axial equatorial isomer with CO stretching adsorptions at 2000 and 1965 (coincident with bis-equatorial) is also present.
The HPIR spectra for ligands 1 and 4 showed a number of peaks: both axial-equatorial and bis-equatorial species, as expected from the NMR studies. For ligand 1 no unmodified catalyst was noted after 15 minutes, but for ligand 4, it was only after 45 minutes that all the unmodified catalyst is converted to the [Rh(H)(L)(CO)2] species. Overall, the HPIR data show there is a strong steric impact on the speed of coordination and that it is likely that broadly similar species are formed at +70 °C under pressure as those detected at −70 °C under an N2 atmosphere. The conditions of the IR experiment are fairly close to those used in hydroformylation (and the same carbonyl stretching adsorptions are observed for ligand 1 at 90 °C and 70 °C). The HPIR data also confirms that the hydroformylation results stem from purely ligand-modified catalysts.
The hydroformylation of propene is one of the most important examples of industrial hydroformylation catalysis. Propene can be considered somewhat distinct from most other terminal alkenes in that the regioselectivity is not influenced by isomerisation in the same way as, for example, 1-octene. Much effort has been expended into optimising the ratio towards linear aldehyde formation to around 95% or higher. However, the branched aldehyde has many markets and uses, and there is significant interest in increasing the range of n/iso ratios that are available (currently around 50–95%). This needs to be accomplished using catalysts that are stable and selective at the typical temperatures needed for industrial application. The unusually high branched selectivities for alkyl alkene hydroformylation recently reported2,18 required low temperatures for good results to be achieved. The hydroformylation of propene with high iso-selectivity under industrially realistic reaction conditions is a formidable challenge. It appears the highest ever branched aldehyde ratio at high temperatures is around 55% iso-selective (n/iso 0.8) using dppp.19
Ligands 1–4 were compared against a range of ligands (Table 2); particularly those that have been previously noted to give significant branched regioselectivity in the hydroformylation of either propene or another alkyl-alkene. Rh catalysts were prepared in situ and the reactions were carried out in a sealed vessel pressurised with a CO/H2 and C3H6 mixture with one hour reaction time. The average TOF are therefore a TON after one hour reaction, not initial rates. The activity data served our purpose of showing that there were no huge differences in activity between the ligands and that their rates are of the same magnitude as those using Rh/PPh3 under these conditions. Dppp ligand 13 (entry 5) gave lower iso-selectivity than literature results, under the reaction conditions tested here. As expected, dppe, 12, and dppb, 14, (entry 4 and 6) gave lower iso-selectivity than dppp with the dppf (entry 3) ligand, 11, giving the lowest iso-selectivity. The cage phosphine, 10, (entry 2) also gave low iso/n ratio, though as expected,20 with high activity.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Average TOF | Iso% | n/i |
|
a Catalyst prepared from [Rh(acac)(CO)2] (5.12 × 10−3 mmol) and ligand by stirring at 20 bar CO/H2 (1:1) at 90 °C for 1 hour in toluene (20 mL) prior to running reactions for 1 hour at 20 bar using propene/H2/CO mixture in a 1:4.5:4.5 ratio. For monodentate ligands, 0.02 mmol ligand (Rh:L 1:4) was used; for bidentate ligands, 6.30 × 10−3 mmol ligand (Rh:L 1:1.25) was used. Products determined by GC using 1-methylnaphthalene as an internal standard. TOFs are average TOF over a 1 hour reaction time (mol mol−1 h−1) in a sealed batch reaction.
|
||||
| 1 | 9 | 903 | 37.3 | 1.7 |
| 2 | 10 | 1013 | 41.4 | 1.4 |
| 3 | 11 | 1082 | 38.8 | 1.6 |
| 4 | 12 | 433 | 41.4 | 1.4 |
| 5 | 13 | 720 | 48.4 | 1.1 |
| 6 | 14 | 513 | 45.9 | 1.2 |
| 7 | 1 | 671 | 50.8 | 1.0 |
| 8 | 2 | 872 | 45.3 | 1.2 |
| 9 | 3 | 602 | 50.6 | 1.0 |
| 10 | 4 | 550 | 55.0 | 0.8 |
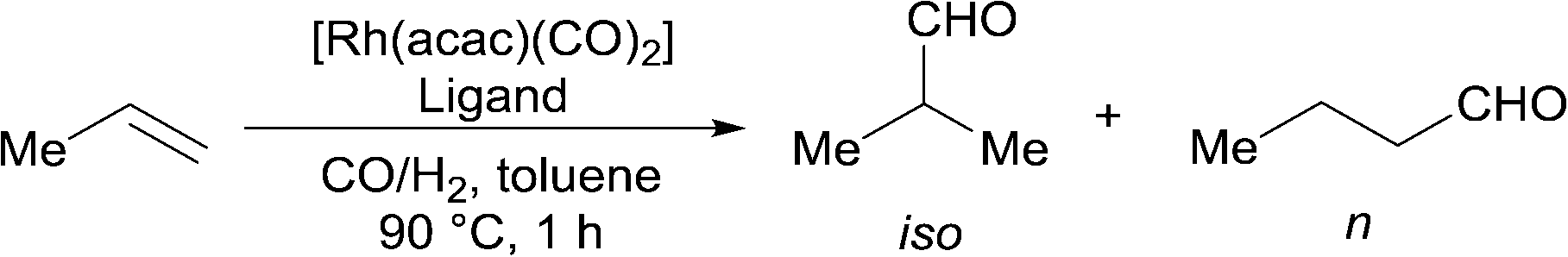
The new ligands 1, 3, and 4 were found to favour the formation of the desired iso-product. Ligand 4 performed the best, with an iso-selectivity of 55/45 (Table 2, entry 10) which is comparable to the best current literature reports. Less bulky ligand, 2, shows significantly less iso-selectivity. This loss of selectivity may be due to direct steric effects between the phosphoramidite part of the ligand and the substrate and not the equatorial-axial arrangement of the ligand on the metal; both ligands 1 and 2 primarily or solely form the equatorial-axial isomer.
The parent ligand, 1, was also tested using a variety of different solvents (Table 3). It was found that the selectivity for the branched product could be improved with a number of solvents, though this was often met with a decrease in average TOF. However, octafluorotoluene was found to give a small increase in activity as well as an increase in iso-selectivity. The increase in activity is proposed to be at least partly due to the known increased solubility of gases in fluorinated solvents.
| Entry | Solvent A | Average TOF | Iso% | n/i |
|---|---|---|---|---|
|
a Catalyst prepared from [Rh(acac)(CO)2] (5.12 × 10−3 mmol) and ligand 1 (6.30 × 10−3 mmol (Rh:L 1:1.25)) by stirring at 20 bar CO/H2 (1:1) at 90 °C for 1 hour in desired solvent mix (19.35 mL solvent A, 0.65 ml toluene) prior to running reactions for 1 hour at 20 bar using propene/H2/CO mixture in a 1:4.5:4.5 ratio. Products determined by GC using 1-methylnaphthalene as an internal standard. TOFs are average TOF over a 1 hour reaction time in mol mol−1 h−1.
|
||||
| 1 | Toluene | 671 | 50.8 | 1.0 |
| 2 | Hexane | 743 | 52.4 | 0.9 |
| 3 | Ethyl acetate | 260 | 54.1 | 0.8 |
| 4 | CF3C6H5 | 585 | 54.9 | 0.8 |
| 5 | Octafluorotoluene | 911 | 55.9 | 0.8 |
The four new ligands were also tested in 1-octene hydroformylation (Table 4). Catalysts derived from both ligands 1 and 2 proceeded rapidly to complete conversion in 17 hours, while catalysts derived from ligands 3 and 4 were found to be slower for the hydroformylation of 1-octene (entries 1–4).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ligand | Time (h) | Conversion (%) | 2-PH (%) | 2-EH (%) | 2-MO (%) | N (%) | n/i |
|
a Catalyst prepared from [Rh(acac)(CO)2] (0.010 mmol) and ligand by stirring at 20 bar CO/H2 (1:1) at 90 °C for 1 hour in toluene (20 mL) prior to running reactions with 1-octene (16 mmol) at 20 bar using CO/H2 (1:1) until at or near full conversion. For monodentate ligands, 0.04 mmol ligand (Rh:L 1:4) was used; for bidentate ligands, 0.013 mmol ligand (Rh:L 1:1.25) was used. Products determined by GC using 1-methylnaphthalene as an internal standard.
|
||||||||
| 1 | 1 | 17 | 99 | 11 | 14 | 35 | 40 | 0.7 |
| 2 | 2 | 17 | 99 | 9 | 14 | 34 | 43 | 0.8 |
| 3 | 3 | 42 | 97 | 12 | 14 | 35 | 39 | 0.6 |
| 4 | 4 | 44 | 97 | 12 | 14 | 35 | 39 | 0.6 |
| 5 | 9 | 4 | 96 | <1 | 2 | 28 | 69 | 2.3 |
| 6 | 1 | 0.5 | 18 | 0 | 2 | 30 | 68 | 2.1 |
| 7 | 1 | 2 | 35 | 4 | 7 | 35 | 54 | 1.2 |
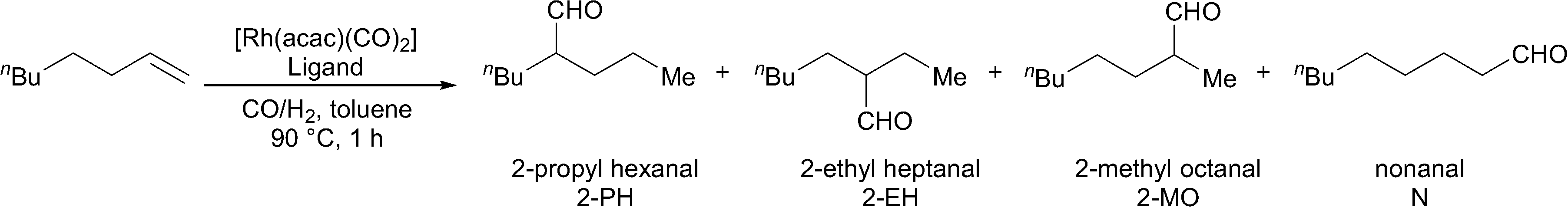
The catalysts derived from all four ligands were found to give high iso-selectivity (Table 4, entries 1–4). However, this does not represent especially high selectivity for 2-aldehydes, since a range of iso-aldehydes are formed. This is in contrast to the results obtained using PPh3 under the same reaction conditions, which lead to almost no apparent isomerisation of the 1-octene (entry 5). The selectivity observed using Rh/ligands 1–4 is due to the isomerisation of 1-octene to internal olefins. This is indeed a very fast reaction, since when the reaction using the Rh/ligand 1 catalyst was repeated with a reaction time of just 30 minutes, almost no 1-octene remains. Isomers of octene were detected, and the higher n:i ratio indicates that the hydroformylation of the terminal alkene favours the formation of nonanal. In a separate experiment, the gas uptake at constant pressure was measured in the reaction catalysed by Rh/ligand 1; the reaction was found to be first order with respect to substrate (see ESI†). The initial turnover frequency measured at 20% conversion was 951 h−1.
Conclusions
Four novel phosphine-phosphoramidites have been prepared and fully characterised. The active Rh hydroformylation catalysts derived from these ligands have been studied using NMR and IR. While we envisaged that this ligand structure may well be finely poised between bis-equatorial and axial-equatorial coordination geometries, it was found that making minor changes to ligand structure completely switched the coordination mode from 100% bis-equatorial to 100% axial-equatorial. Of the ligands synthesised and tested; it was found that the least bulky ligand 2 leads to only an axial-equatorial species. Increasing steric bulk leads to an increase in the bis-equatorial species until only the bis-equatorial isomer was detected by NMR with the more bulky ligand 3. These catalysts were then tested in the hydroformylation of propene and 1-octene and were found to be amongst the most successful catalysts for maximising the branched product at industrially applicable temperatures. The differences in selectivity within the ligand family is very small, limiting the scope of the conclusions that can be drawn. Differences in selectivity between ligand 2, which forms less iso-aldehyde and ligand 3 are more likely ascribed to steric interactions in the transition states for Rh-alkene > Rh-alkyl or Rh-alkyl > Rh-acyl species. Ligands 1 and 3 show very different catalyst geometries but very similar selectivities in hydroformylation, which means there is no strong correlation between coordination mode (alone) and iso-selectivity in propene hydroformylation. While this is not entirely unexpected, confirming that there is no isomeric preference towards iso-selectivity focuses attention on the design of a coordination sphere that can promote the formation of branched intermediates. Highly iso-selective hydroformylation of propene is clearly a formidable challenge and our studies continue. It is hoped that the knowledge gained here will lead us to the rational design of catalysts that form a single isomer and can lead to even higher iso-selective catalysts in the future.Experimental
General information
Full experimental details are available in ESI.†All manipulations were carried out under an inert atmosphere of nitrogen or argon using standard Schlenk techniques. Solvents were dried and degassed before use, with the exception of octafluorotoluene which was degassed only.
General procedure for propene hydroformylation
Ligand (6.40 μmol (Rh:L 1:1.25)) was added to a schlenk tube, which was then purged with N2. [Rh(acac)(CO)2] (5.12 μmol) was added in a toluene stock solution (2 mg mL−1). Toluene was then added to make up to 20 mL total volume, followed by the addition of internal standard 1-methylnaphthalene (0.2 mL). The solution was transferred via syringe to the pressure vessel (which had been purged with CO/H2) through the injection port. CO/H2 (1:1) (20 bar) was added and the heating jacket set to 90 °C while stirring. Once the temperature reached 90 °C, the reaction was stirred for 1 hour to fully activate the catalyst. Then pressure was then slowly released and replaced with propene/CO/H2 (20 bar). The reaction was then run for 1 hour before immediate analysis by GC.
General procedure for ligand synthesis
Synthesis and characterisation of ferrocenyl-ligand precursors, chlorophosphites and phosphine-phosphoramidite ligands is available in ESI.† General synthesis of ligand 1 is given from amine precursor 7 and chlorophosphite 8a.:5 toluene:Et3N) using 30:1 hexane:ethyl acetate as eluent under N2 to give phosphoramidite 1 as an orange solid (0.38 g, 0.48 mmol, 69%).
Acknowledgements
We thank the Eastman Chemical Company for funding, the EPSRC for the use of the national mass spectrometry service, and all the technical staff in the School of Chemistry for their assistance.Notes and references
- (a) T. Besset, D. W. Norman and J. N. H. Reek, Adv. Synth. Catal., 2013, 355, 348–352 CAS; (b) V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef CAS; (c) V. F. Slagt, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526–1536 CrossRef CAS PubMed; (d) M. Kuil, T. Solder, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344 CrossRef CAS PubMed.
- (a) G. M. Noonan, J. A. Fuentes, C. J. Cobley and M. L. Clarke, Angew. Chem., Int. Ed., 2012, 51, 2477–2480 CrossRef CAS PubMed; (b) G. M. Noonan, C. J. Cobley, T. Mahoney and M. L. Clarke, Chem. Commun., 2014, 50, 1475–1478 RSC.
- (a) M. L. Clarke, Curr. Org. Chem., 2005, 9, 701–718 CrossRef; (b) Rhodium Catalysed Hydroformylation, ed. P. W. N. van Leeuwen and C. Claver, Kluwer Academic Publishers, Netherlands, 2000 Search PubMed.
- T. J. Devon, G. W. Phillips, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, US Pat., 4694109, 1987 Search PubMed.
- C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535–5543 CrossRef CAS.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- N. W. Boaz, S. D. Debenham, E. B. Mackenzie and S. E. Large, Org. Lett., 2002, 4, 2421–2424 CrossRef CAS PubMed.
- (a) N. W. Boaz, E. B. Mackenzie, S. D. Debenham, S. E. Large and J. A. Ponasik, J. Org. Chem., 2005, 70, 1872–1880 CrossRef PubMed; (b) See also: X. P. Hu and Z. Zheng, Org. Lett., 2004, 6, 3585–3588 CrossRef CAS PubMed.
- K. Nozaki, N. Sakai, T. Nanno, T. Higashijima, S. Mano, T. Horiuchi and H. Takaya, J. Am. Chem. Soc., 1997, 119, 4413–4423 CrossRef CAS.
- M. Rubio, A. Suárez, E. Álvarez, C. Bianchini, W. Oberhauser, M. Peruzzini and A. Pizzano, Organometallics, 2007, 26, 6428–6436 CrossRef CAS.
- J. Wassenaar and J. N. H. Reek, Dalton Trans., 2007, 3750–3753 RSC.
- R. Bellini and J. N. H. Reek, Chem. – Eur. J., 2012, 18, 13510–13519 CrossRef CAS PubMed.
- Y. Yan and X. Zhang, J. Am. Chem. Soc., 2006, 128, 7198–7202 CrossRef CAS PubMed.
- S. H. Chikkali, J. I. van der Vlugt and J. N. H. Reek, Coord. Chem. Rev., 2014, 262, 1–15 CrossRef CAS.
- D. Marquarding, H. Klusacek, G. Gokel, P. Hoffmann and I. Ugi, J. Am. Chem. Soc., 1970, 92, 5389–5393 CrossRef CAS.
- D. A. Castillo Molina, C. P. Casey, I. Mueller, K. Nozaki and C. Jaekel, Organometallics, 2010, 29, 3362–3367 CrossRef CAS.
- (a) L. A. van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616–11626 CrossRef CAS; (b) C. F. Czauderna, D. B. Cordes, A. M. Z. Slawin, C. Müller, J. I. van der Vlugt, D. Vogt and P. C. J. Kamer, Eur. J. Inorg. Chem., 2014, 1797–1810 CrossRef CAS; (c) S. Schmist, G. Abkai, T. Rosendahl, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 1044 CrossRef; (d) I. del Río, W. G. J. de Lange, P. W. N. M. van Leeuwen and C. Claver, J. Chem. Soc., Dalton Trans., 2001, 1293 RSC; (e) I. del Río, O. Pàmies, P. W. N. M. van Leeuwen and C. Claver, J. Organomet. Chem., 2000, 608, 115 CrossRef.
- D. W. Norman, J. N. H. Reek and T. R. M. L. Besset, US Pat., 8710275B2, 2014 Search PubMed.
- G. W. Phillips, T. J. Devon, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, US Pat., 4760194, 1988 Search PubMed.
- R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079–1085 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A range of control/additional hydroformylation experiments including unselective attempts at asymmetric hydroformylation, HPIR, analytical data and spectra, please consult the supporting information. See DOI: 10.1039/c5cy00886g |
| This journal is © The Royal Society of Chemistry 2016 |