
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the flexibility in MOFs by SBU functionalization†
Volodymyr
Bon
a,
Negar
Kavoosi
a,
Irena
Senkovska
a,
Philipp
Müller
a,
Jana
Schaber
b,
Dirk
Wallacher
c,
Daniel M.
Többens
d,
Uwe
Mueller
e and
Stefan
Kaskel
*a
aDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstrasse 66, D-01062 Dresden, Germany. E-mail: Stefan.Kaskel@tu-dresden.de
bDepartment of Bioanalytical Chemistry, Technische Universität Dresden, Bergstrasse 66, D-01062 Dresden, Germany
cDepartment of Sample Environments, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
dDepartment of Crystallography, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
eMX Group, Institute for Soft Matter and Functional Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany
First published on 3rd February 2016
Abstract
A new approach for the fine tuning of flexibility in MOFs, involving functionalization of the secondary building unit, is presented. The “gate pressure” MOF [Zn3(bpydc)2(HCOO)2] was used as a model material and SBU functionalization was performed by using monocarboxylic acids such as acetic, benzoic or cinnamic acids instead of formic acid in the synthesis. The resulting materials are isomorphous to [Zn3(bpydc)2(HCOO)2] in the “as made” form, but show different structural dynamics during the guest removal. The activated materials have entirely different properties in the nitrogen physisorption experiments clearly showing the tunability of the gate pressure, at which the structural transformation occurs, by using monocarboxylic acids with varying backbone structure in the synthesis. Thus, increasing the number of carbon atoms in the backbone leads to the decreasing gate pressure required to initiate the structural transition. Moreover, in situ adsorption/PXRD data suggest differences in the mechanism of the structural transformations: from “gate opening” in the case of formic acid to “breathing” if benzoic acid is used.
Introduction
Flexible Metal–Organic Frameworks (MOFs) or so called Soft Porous Crystals are a unique class of materials capable to switch between the porous and dense (or less porous) states as a response to external stimuli such as temperature, pressure, electromagnetic irradiation or adsorption of guest molecules.1–3 The existence of two different states with different physical and textural properties gives rise to a wide potential application field of such materials. For example, NH2-MIL-53(Al) is able to transform between the narrow and large pore phases and therefore could be used as an optical switch.4 Sensor technology is also one of the prospective application fields for switchable MOFs. Thus a Mg based flexible MOF with the composition [Mg(H2dhtp)(H2O)2]·DMAc, denoted as AEMOF-1·DMAc (AEMOF-1 – alkaline earth MOF-1; H4dhtp – 2,5-dihydroxy-terepthalic acid; DMAc – N,N′-dimethylacetamide) was used as a highly efficient luminescent sensor for the real time detection of water traces in various organic solvents.5 The flexibility was also successfully used to achieve the separation of CO and N2, which have very similar physical properties.6 The selective trapping of NO molecules in the flexible Ru- and Rh-based coordination polymers could find its application in biomedicine.7All envisioned applications based on the flexibility require materials with well-defined switching behavior. However, tuning and designing of flexible MOFs with specific switching properties remain very challenging, which limits the application potential of flexible crystalline materials. The most often applied strategies to influence the flexibility in soft MOFs8 are: (i) introduction of flexible side chains into the ligand;9 (ii) modulation of host–guest interaction strength by linker functionalization;10 (iii) incorporation of different metals into the same framework;11 (iv) adjustment of the crystal size.12
In this contribution, we present a switchability tuning approach based on the systematic substitution of monocarboxylates in the secondary building unit (SBU) of a “gate pressure” MOF [Zn3(bpydc)2(HCOO)2] (bpydc – 2,2′-bipyridine-5,5′-dicarboxylate), also known as JLU-Liu4 (JLU – JiLin University).13 It contains two formic acid anions directly coordinated to the metal cluster. The solvent assisted exchange of the monocarboxylic ligand was already demonstrated for rigid Zr based MOFs.14 We could show that utilizing monocarboxylic acids with different backbones in the synthesis (for instance acetic, benzoic, or cinnamic acid) results in the formation of isomorphous frameworks, showing completely different adsorption behavior. Crystal structures of the large pore and narrow pore phases as well as adsorption behavior of the materials bearing different monocarboxylic ligands were studied to enlighten the structural transformations in quest.
Experimental
Materials
All chemicals were purchased from commercial sources and used without further purification. N,N-Dimethylformamide (DMF) (anhydrous 99.8%), trans-cinnamic acid (97%), acetonitrile (99.8%), and 5,5′-dimethyl-2,2′-dipyridine (98%, dmpby) were purchased from Sigma Aldrich. 2,2′-Bipyridyl-5,5′-dicarboxylic acid (H2bpydc) was synthesised according to the reported procedure15 (for more details see the ESI†). Benzoic acid (99.5%) and formic acid (99%) were purchased from Grüssing GmbH, Zn(NO3)2·4H2O from Merck, acetic acid (100%) from Roth and Company, heptane (99%) and dichloromethane 99.99% from Fisher, anhydrous toluene from VWR, and 1-propanol 99.5% from TCI.Synthesis
Elemental analysis (wt%): calc. C 42.89, N 6.98, H 2.74; found C 41.13, N 7.26, H 1.03.
Elemental analysis (wt%): calc. C 49.23, N 6.04, H 2.8; found C 48.18, N 6.16, H 1.27.
Elemental analysis (wt%): calc. C 51.52, N 5.72, H 3.06; found C 49.77, N 5.87, H 1.96.
Single crystal X-ray diffraction
The crystal structure of compounds 1–4 were determined by single crystal X-ray diffraction. All single crystals were prepared in glass capillaries with some amount of solvent. The capillaries were sealed with melted wax. The datasets were collected at the BESSY MX BL14.2 beamline of Helmholtz-Zentrum Berlin für Materialien und Energie (HZB).16 All diffraction experiments were performed at room temperature using radiation with an energy of 14 keV (λ = 0.88561 Å). The ϕ-scans with an oscillation range of 1° were used for data collection. The datasets were processed using CCP4 software.17 The crystal structures were solved by direct methods and refined by full matrix least-squares on F2 using the SHELXTL program package.18 All non-hydrogen atoms were refined in anisotropic approximation. Hydrogen atoms were refined in the geometrically calculated positions using the “riding model” with Uiso(H) = 1.2Uiso(C) for sp, sp2 hybridized carbon atoms and with Uiso(H) = 1.5Uiso(C) for sp3 hybridized carbon atoms. The atoms of the phenyl rings of the benzoate (compound 3) and cinnamate (compound 4) anions are disordered and were treated using the AFIX 66 instruction. In the case of compound 4, SIMU and DELU instructions have been used to restrain the thermal parameters of disordered fragments. Because of the high symmetry of the crystal system it was not possible to localize the lattice solvent molecules in the pores. Therefore, the SQUEEZE procedure was used to correct reflection intensities, corresponding to disordered solvent molecules.19 CCDC 1422814, 1422816, 1422818 and 1422820 contain the supplementary crystallographic data for compounds 1, 2, 3, and 4, respectively. The extended crystallographic data for the structures 1–4 are given in Table S1 of the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 822/5847, Rint = 0.0194, R1 = 0.0287, wR2 = 0.0852, S = 1.089, largest diff. peak 0.569 e Å−3 and hole −0.543 e Å−3.
800/6337, Rint = 0.0275, R1 = 0.0313, wR2 = 0.0960, S = 1.099, largest diff. peak 0.629 e Å−3 and hole −0.532 e Å−3.
507/6196, Rint = 0.0556, R1 = 0.0515, wR2 = 0.1546, S = 1.079, largest diff. peak 0.656 e Å−3 and hole −0.764 e Å−3.
822/5847, Rint = 0.0194, R1 = 0.0287, wR2 = 0.0852, S = 1.089, largest diff. peak 0.569 e Å−3 and hole −0.543 e Å−3.
800/6337, Rint = 0.0275, R1 = 0.0313, wR2 = 0.0960, S = 1.099, largest diff. peak 0.629 e Å−3 and hole −0.532 e Å−3.
507/6196, Rint = 0.0556, R1 = 0.0515, wR2 = 0.1546, S = 1.079, largest diff. peak 0.656 e Å−3 and hole −0.764 e Å−3.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHCOO)2] (4).
C42H26N4O12Zn3, M = 974.78 g mol−1, tetragonal, P43212 (no. 96), a = 15.270(2) Å, c = 23.050(5) Å, V = 5374.6(18) Å3, Z = 4, ρcalc = 1.205 g cm−3, λ = 0.88561 Å, T = 298 K, θmax = 32.1°, reflections collected/unique 20740/6207, Rint = 0.0556, R1 = 0.0377, wR2 = 0.1195, S = 1.123, largest diff. peak 0.500 e Å−3 and hole −0.756 e Å−3.
CHCOO)2] (4).
C42H26N4O12Zn3, M = 974.78 g mol−1, tetragonal, P43212 (no. 96), a = 15.270(2) Å, c = 23.050(5) Å, V = 5374.6(18) Å3, Z = 4, ρcalc = 1.205 g cm−3, λ = 0.88561 Å, T = 298 K, θmax = 32.1°, reflections collected/unique 20740/6207, Rint = 0.0556, R1 = 0.0377, wR2 = 0.1195, S = 1.123, largest diff. peak 0.500 e Å−3 and hole −0.756 e Å−3.
Crystal structures of 1′–4′ determined from PXRD
The crystal structures for narrow pore phases were solved from the PXRD patterns that were recorded at room temperature on the evacuated samples. Data for 1′, 3′, and 4′ were collected at the BESSY KMC-2 beamline of HZB in transmission geometry using 2 scans in the range of 5 to 50°. Data for 2′ were collected on STOE Stadi P. The patterns were indexed using the X-Cell program implemented in Material Studio 5.0 software.20 The most intensive reflections in the 2θ range between 2 and 20° were used for indexing. The C2221 space group was chosen for 1′, 2′, and 4′ considering the group–subgroup relationships and extinction rules. The starting models for the crystal structure refinement were derived from the “as made” 1, 2, and 4 structures, which were converted into the corresponding space group. After adapting the unit cell parameters to those obtained during indexing, the structural models were optimized using a geometry optimization tool with implementation of a universal force field. Due to the low data/parameter ratio, the rigid-body Rietveld refinement with energy options was used to refine the structures.The crystallographically independent Zn atoms, carboxylate groups, and phenyl rings were treated as rigid bodies.
In the case of compound 2′, the minor impurity of open phase 2 is present after activation. Therefore, reflections at 7.0° and 11.6° 2θ, which belong to the open phase 2 (Fig. 3), were excluded from the Rietveld refinement.
The final Rietveld plots are given in Fig. 1. CCDC 1422815, 1422817, 1422819 and 1422821 contain the supplementary crystallographic data for compounds 1′, 2′, 3′, and 4′, respectively.
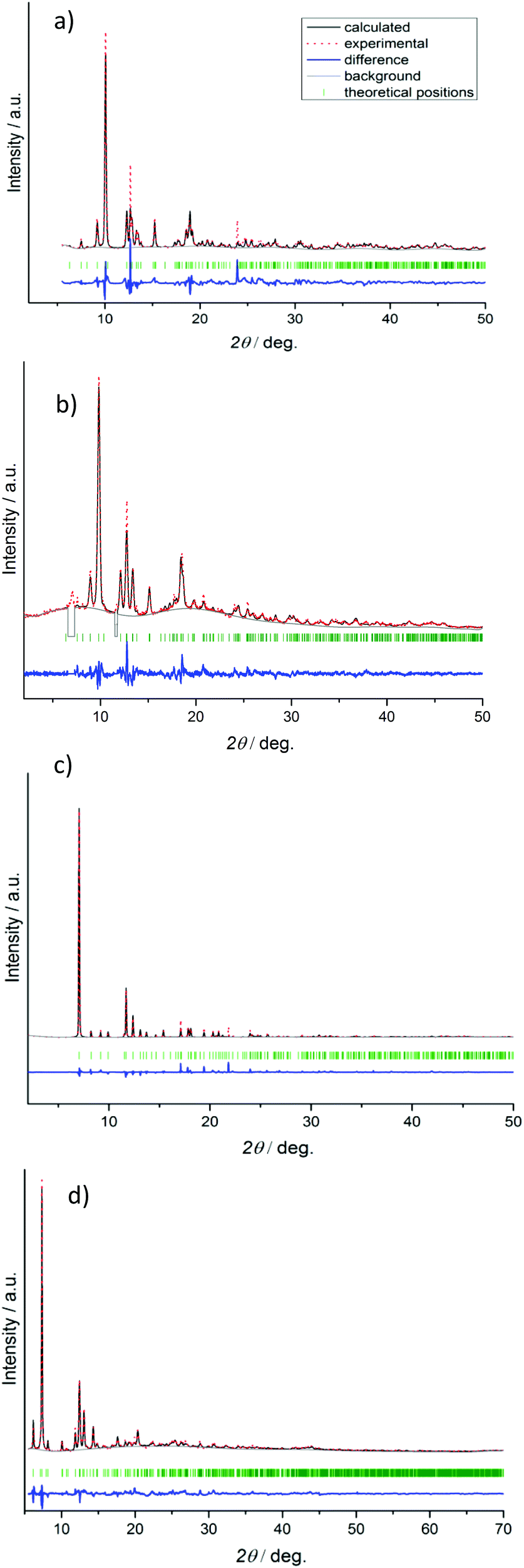 | ||
| Fig. 1 Rietveld plots for compounds 1′ (a), 2′ (b), 3′ (c), and 4′ (d). | ||
In situ monitoring of nitrogen adsorption (77 K) by powder X-ray diffraction
Concerted adsorption of N2 and X-ray powder diffraction experiments on compounds 3 and 4 were performed at the KMC-2 beamline of HZB using specially designed instrumentation.21 All adsorption experiments were performed at 77 K in the relative pressure range between 10−3 and 1 (p0 = 101.3 kPa). All diffraction measurements were performed in the 2θ range from 2 to 50° using monochromatic synchrotron radiation (8048 eV; λ = 1.5406 Å). The diffraction images from the VÅNTEC-2000 2D detector were integrated using Datasqueeze 2.2.9 software.22 Hexagonal boron nitride was used as an external standard.Physical measurements
Thermogravimetric analyses were carried out under air using STA 409 PC from the NETZSCH Company. Mass analysis was performed using Aëolos QMS 403 from the NETZSCH Company. The heating rate was set to 5 K min−1. FT-IR spectra were measured on a VERTEX-70 spectrophotometer (Bruker) using the DRIFT technique. Nitrogen adsorption measurements up to 100 kPa at 77 K were performed on approximately 35 mg of sample using BELSORP-max apparatus. The nitrogen with the purity of 99.999% was used for the experiments. Powder X-ray diffraction patterns were obtained on a STADI P (Stoe) diffractometer at RT using monochromatic Cu–Kα1 radiation (λ = 1.5406 Å) (step size: 0.05, exposure time: 23 s per point). All measurements were performed in transmission geometry. The samples were prepared in the Ar-filled glovebox. Temperature dependent PXRD measurements were performed on an X'Pert Pro diffractometer (PANalytical) in reflection geometry using θ/2θ scans and a MRI (Material Research Instruments) sample stage. The PXRD patterns were obtained under dynamic vacuum every 25 °C in the temperature range between 25 and 450 °C. The heating rate was set to 3 K min−1.Results and discussion
JLU-Liu4 developed by Liu and co-workers consists of trinuclear Zn(II) units coordinated by four carboxylic groups, two 2,2′-bipyridine units, and two formate anions. The structure comprises of a 3D framework (ant topology) and displays channels 4.9 × 5.1 Å in dimensions.13 The 3D framework of JLU-Liu4 undergoes structural transformation upon removal of guest molecules from the pores to give a MOF with gate opening behavior. This is indicated by shifting and intensity changing of the peaks in the PXRD pattern. However, the structure of the desolvated compound has not yet been reported.To study the influence of the monocarboxylic ligand nature on the structure formation of JLU-Liu4 (1), acetic (HAc), benzoic (HBz), or cinnamic (HCn) acid was used in the synthesis instead of the originally reported formic acid.
In all cases crystalline products with the composition Zn3(bpydc)2(Ac)2 (2), Zn3(bpydc)2(Bz)2 (3), and Zn3(bpydc)2(Cn)2 (4) could be obtained. Since all new materials could be obtained as crystals suitable for single crystal X-ray analysis, the crystal structures of all new materials were determined. All compounds crystallize in the same space group P43212, and are isostructural, possessing similar frameworks with the ant topology (Fig. 2a). As expected, the main difference in the structures is the monocarboxylic ligand being included (Fig. 2b–e). According to the PLATON report, the solvent accessible void in the as made crystal structures changes from 59.4% in 1 to 32.6% in 4 (Table 1). The volume of monocarboxylic anions (calculated as the Connolly solvent excluded volume using ChemDraw software) and the change in the solvent accessible void volume of the unit cell during activation are given in Scheme 1.
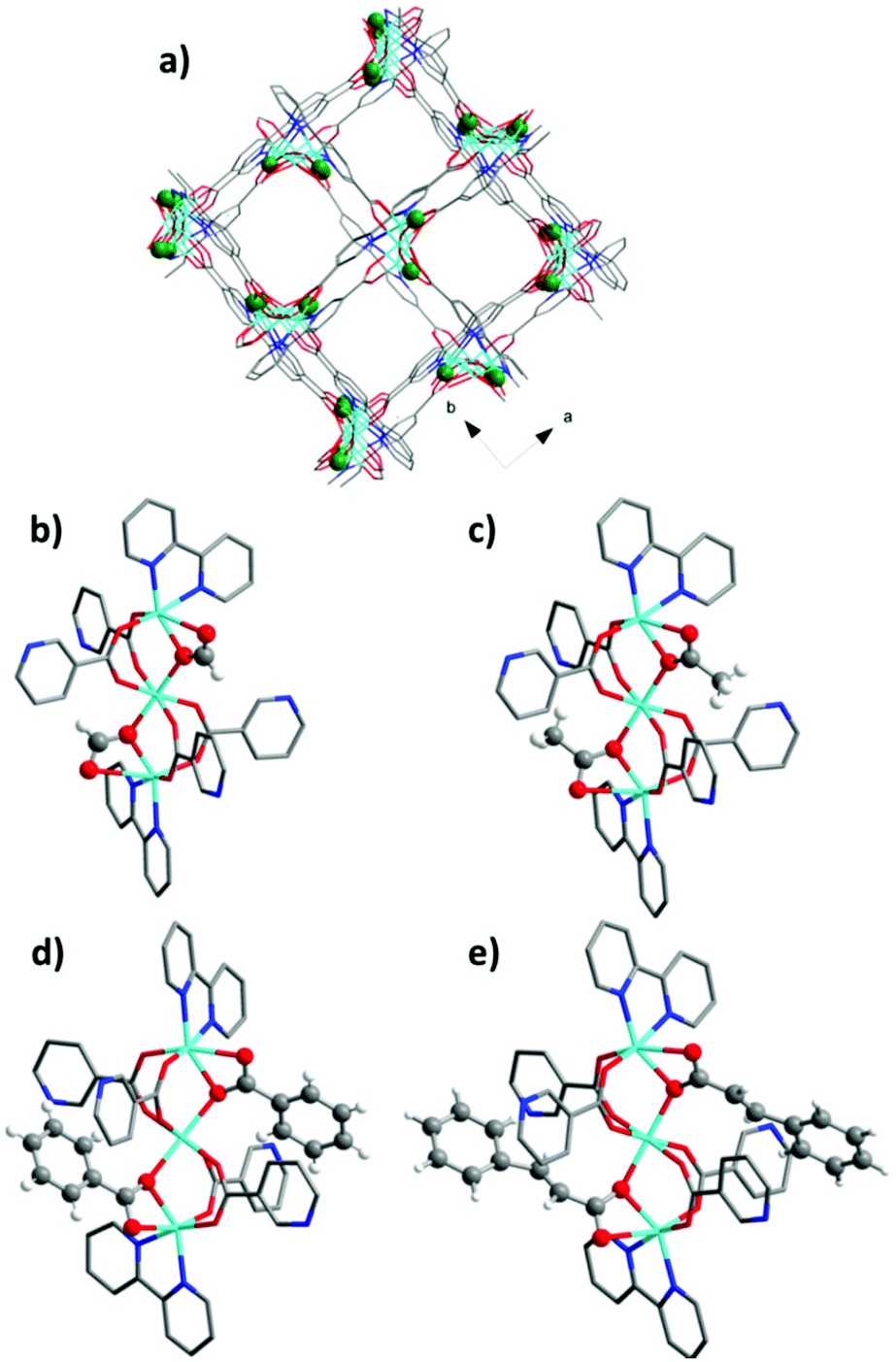 | ||
| Fig. 2 (a) View of the structure of the JLU-LIU4 framework along the c axis (green spheres represent monocarboxylate anions). (b–e) Structure of SBUs in compounds 1–4. Colour code: turquoise – Zn atoms, grey – carbon atoms, red – oxygen atoms, and blue – nitrogen atoms. | ||
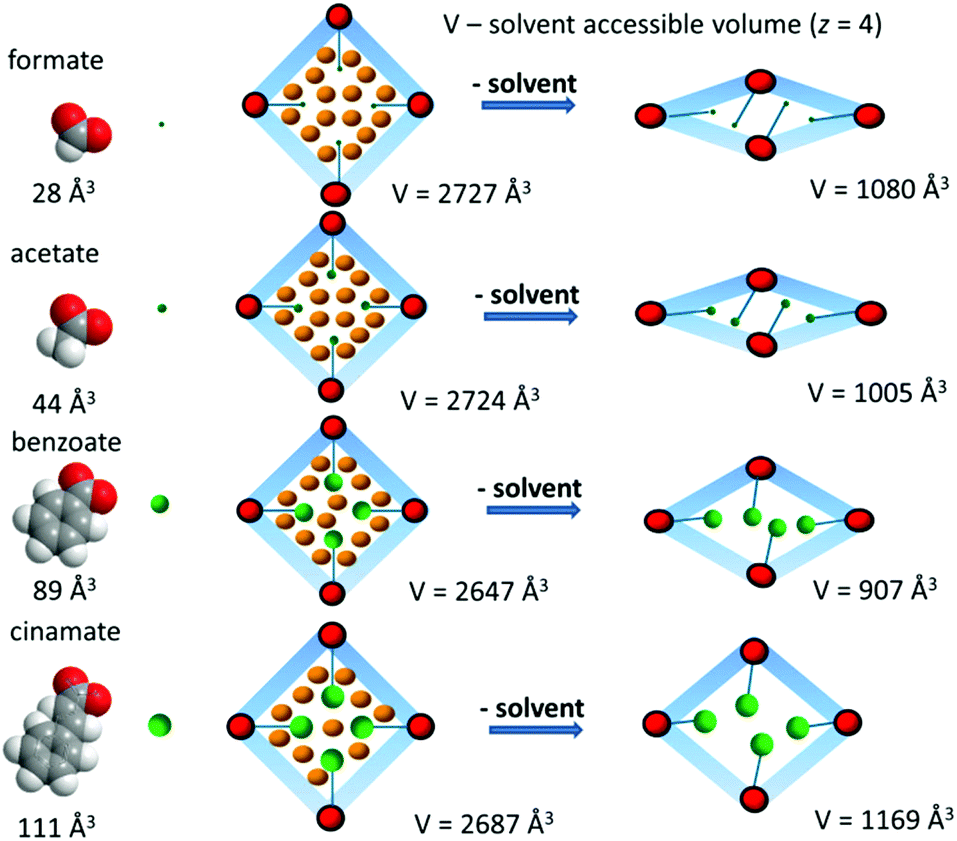 | ||
| Scheme 1 Schematic representation of the monocarboxylic ligand influence on the activation of the LiU-4 series. The volume of monocarboxylic anions was calculated as the Connolly solvent excluded volume using ChemDraw software. The specified volume represents the solvent accessible volume of the unit cell for Z = 4. | ||
| Compound | Solvent accessible void volume of unit cell/% | Calculated pore volume (Vp)/cm3 g−1 | Calculated geometrical surface area/m2 g−1 |
|---|---|---|---|
| 1 | 59.4 | 0.70 | 2425 |
| 1′ | 17.6 | 0.09 | 76 |
| 2 | 55.7 | 0.63 | 2142 |
| 2′ | 15.8 | 0.09 | 66 |
| 3 | 40.5 | 0.36 | 1047 |
| 3′ | 36.7 | 0.30 | 690 |
| 4 | 32.6 | 0.30 | 747 |
| 4′ | 24.8 | 0.17 | 188 |
After removal of the solvent molecules, crystallographic transformation takes place in all compounds, according to the changes in the PXRD patterns (Fig. 3). The patterns of all activated compounds (further referred to as 1′, 2′, 3′, and 4′, respectively) were successfully indexed and the crystal structures were solved using molecular simulations combined with the Rietveld refinement.
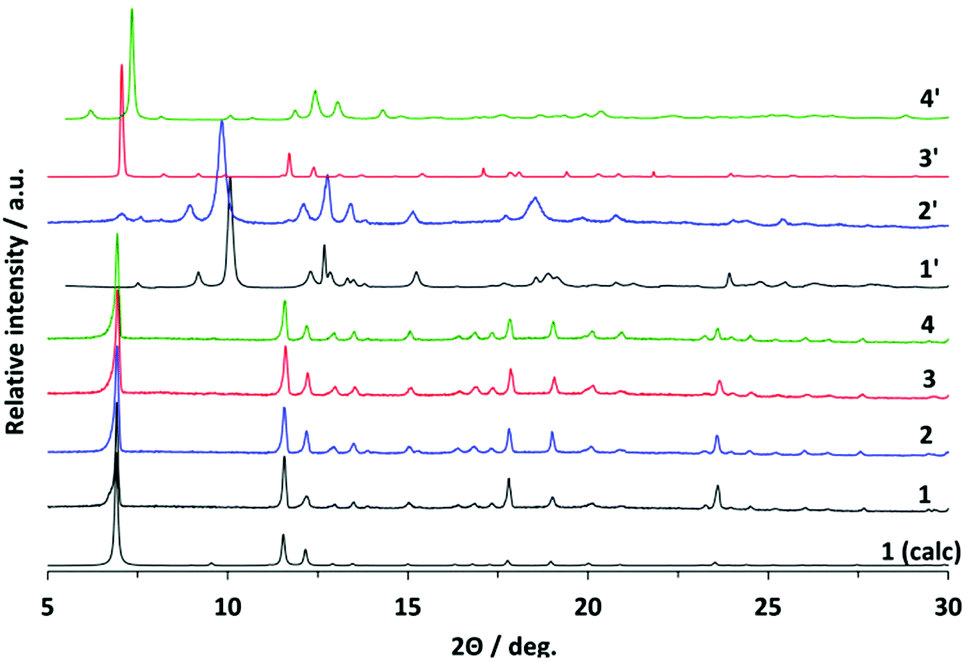 | ||
| Fig. 3 PXRD patterns for compounds 1–4 and 1′–4′. | ||
Activated phases 1′ and 2′ have very similar structures: both crystallize in the C2221 space group with similar unit cell parameters (due to this similarity, only the structure of 1′ is described in detail below). The C2221 space group is a sub-group of the P43212 (the space group of the parent compounds 1 and 2).
Due to the rigidity of the linker, the main contribution to the structural transformation comes from the cluster distortion. The reduction of the symmetry creates two independent SBUs (SBU 1 and SBU 2) in 1′ showing differences in distortion with respect to the linearity of the Zn3 array and coordination environment of the ligand molecules. In SBU 1, the angle between three Zn atoms decreases from 178.65(1)° in 1 to 160.48(1)° in 1′. The angle between the planes involving two 2,2′-bipyridine moieties coordinated to the same cluster changes from 80.31(5)° in 1 to 57.53(5)° in 1′ (Fig. 4a and b). A significant contribution to the structural contraction originates from the bending of carboxylate hinges (dihedral angles between the plane of the carboxylate group and the O–Zn–Zn–O plane). These change from 5.75(3)° and 6.07(3)° in 1 to 19.42(3)° and 20.63(3)° in 1′. This is also often the source of flexibility and structural transitions in other switchable MOFs.23,24 The Zn atoms in SBU 2 are arranged in a nearly linear fashion with a Zn⋯Zn⋯Zn angle of 176.67(1)°. The formate anions, which are coordinated on the opposite sides of SBU in 1 (Fig. 4a and b) change the position in SBU 1 of 1′ and move to the same side of the cluster (Fig. 4b).
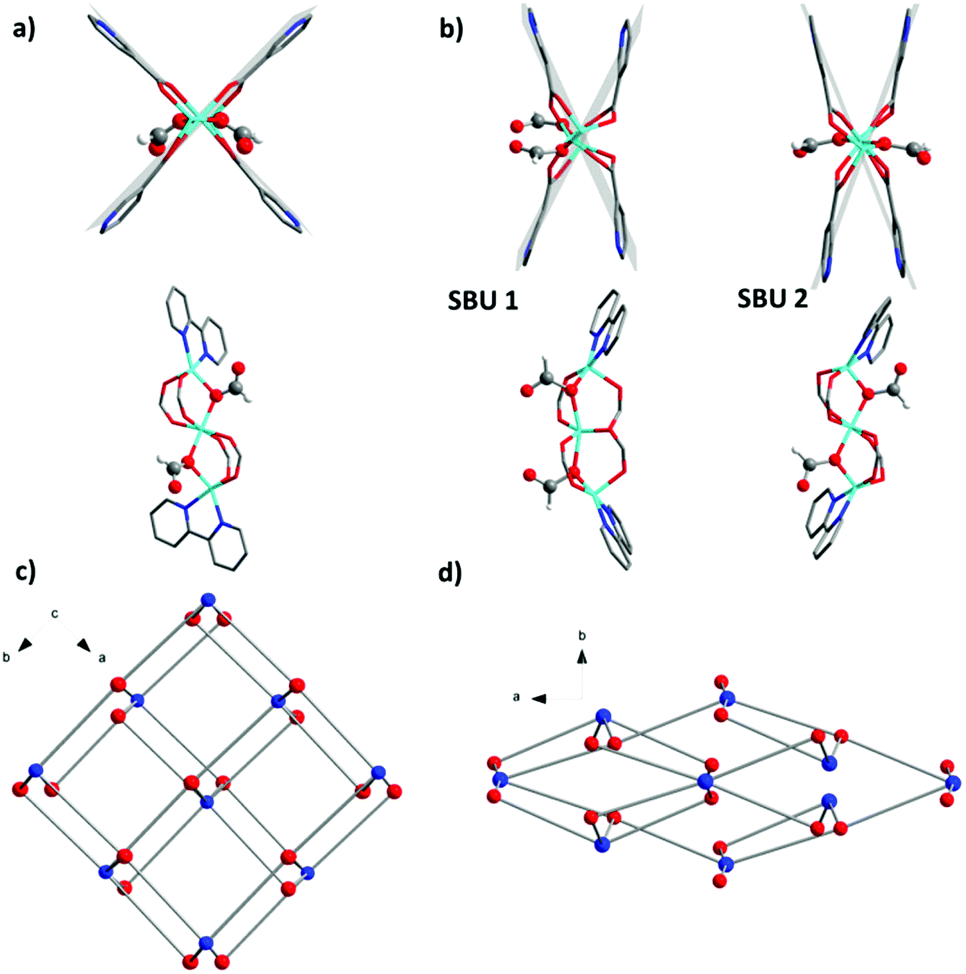 | ||
| Fig. 4 (a) SBU 1 of 1; (b) SBU 1 and SBU 2 of 1′; (c) crystal structure of 1; (d) crystal structure of 1′. (Colour code in (c) and (d): blue spheres – SBU units, and red spheres – bpydc2− linkers). | ||
On the framework level, the structural contraction in 1 occurs along the [110] direction (Fig. 4c), corresponding to the [010] direction in 1′ (Fig. 4d).
The textural properties of 1′ calculated using Poreblazer 3.0.2 software25 show that the compound has nearly no accessible pore volume (0.09 cm3 g−1) as well as a very small pore limiting diameter of 2.11 Å. This completely supports the experimental results, since 1′ shows no uptake of nitrogen up to reaching the “gate opening pressure” of 0.47.
The crystal structure of compound 3′ was refined in the same space group as the structure of 3 (Fig. 1) with different lattice cell constants. Obviously, compound 3 cannot undergo complete transformation because of the bulky phenyl rings of the benzoate. They hinder the cluster transformation as it occurs in 1. Namely, two coordinated benzoate anions, located on the opposite sides of the Zn cluster in 3 cannot regroup in the same way as in 1, and are located on the same side of the SBU plane, because of the small aperture between the ligand molecules, coordinated to the cluster.
The comparison of the calculated pore volumes for 3 and 3′ shows only minor changes from 0.36 cm3 g−1 to 0.30 cm3 g−1.
The thermal stability of the investigated materials was studied by temperature dependent PXRD and thermogravimetric (TG) analysis (Fig. S2–S8 ESI†). Investigation of 1′ by thermo-XRD under an argon atmosphere confirms the retention of crystallinity up to 300 °C (see ESI, Fig. S1†) confirming the TG data reported earlier.13 According to the TG analysis data, materials 2′, 3′, and 4′ are stable up to 300 °C, 330 °C, and 320 °C, respectively (see ESI, Fig. S6–S8†). Therefore, the samples were activated at 150 °C in a vacuum prior to the adsorption experiments.
Compound 1′ shows classical gate pressure behaviour during physisorption of nitrogen at 77 K, with almost no uptake up to p/p0 = 0.47. The total pore volume calculated at p/p0 = 0.95 amounts to 0.55 cm3 g−1 which is slightly lower than the theoretical value (Table 1). As expected, the nitrogen adsorption isotherm of 2′ shows a slightly different trend, reaching the plateau after adsorbing nearly 25 cm3 g−1 nitrogen at very low relative pressures. After reaching p/p0 = 0.3, a very steep increase in uptake takes place. The isotherm reaches a second plateau at p/p0 = 0.95. The adsorbed amount in saturation is 295 cm3 g−1 that corresponds to the pore volume of 0.456 cm3 g−1. Materials 3′ and 4′ show completely different behaviour during the adsorption of the nitrogen at 77 K (Fig. 5). Thus, compound 3′ adsorbs nearly 100 cm3 g−1 of nitrogen already at p/p0 = 10−4 confirming the incompleteness of the crystal structure closing (narrow pore state) of 3′ discussed above. The second step in the isotherm, most likely connected to the structural transformation, occurs in the relative pressure range between 0.03 and 0.07. After that the course of the isotherm is characterized by the continuous increase of the uptake reaching 227 cm3 g−1 at p/p0 = 0.97.
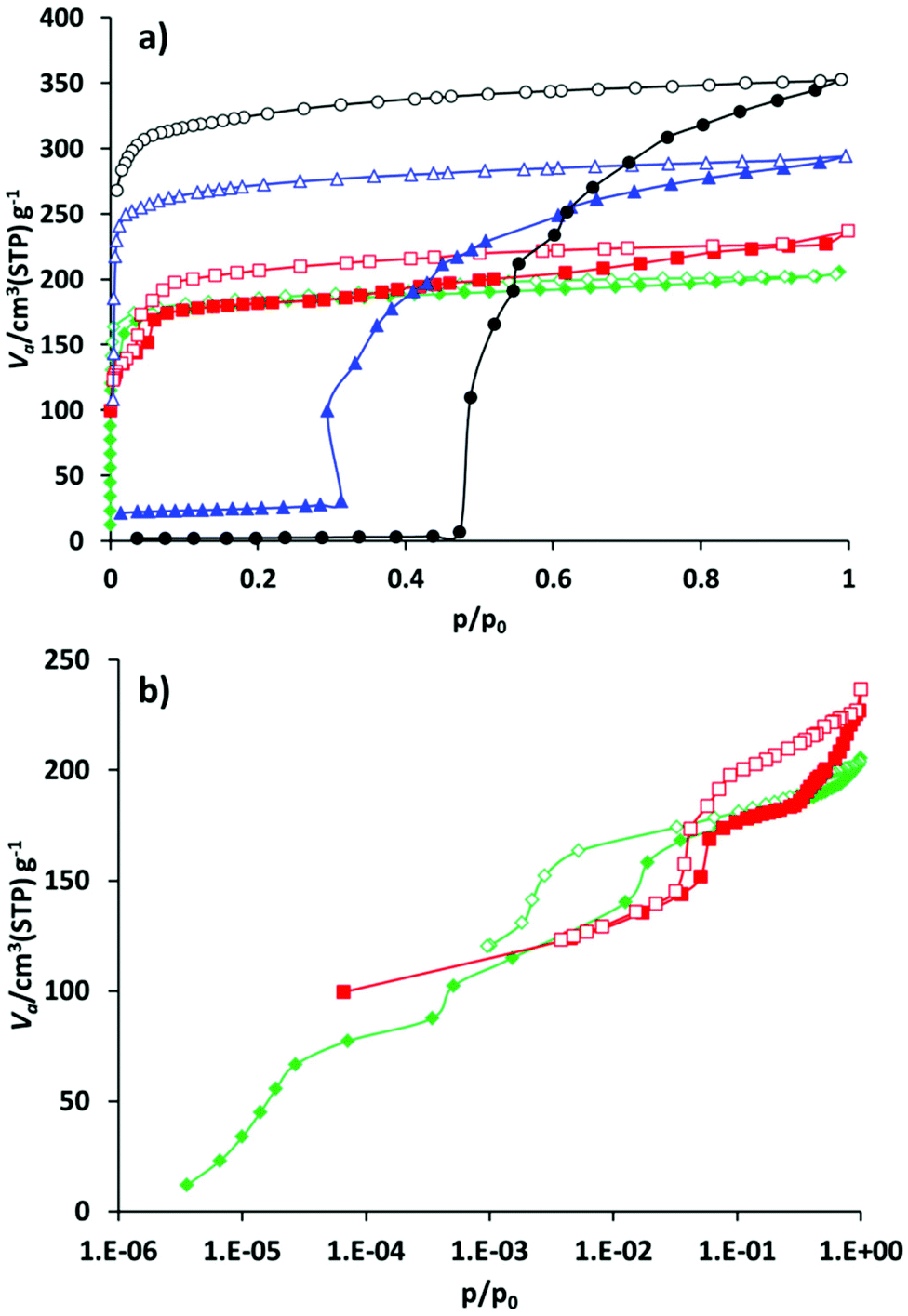 | ||
| Fig. 5 Nitrogen physisorption isotherms for compounds 1′–4′ at 77 K: (a) linear representation of the isotherms; (b) isotherms of compounds 3′ and 4′ on a semilogarithmic scale. Black circles – 1′, blue triangles – 2′, red squares – 3′, and green diamonds – 4′. Close symbols – adsorption, and open symbols – desorption. | ||
Implementation of the larger cinnamate anion into JLU-Liu4 has further influence on the adsorption properties of the solid. As in the case of 3′, the hysteresis related to the guest induced structural changes is shifted to a lower pressure range, and is located between p/p0 = 5 × 10−3 and p/p0 = 5 × 10−2. The isotherm reaches saturation at p/p0 = 0.95 showing the nitrogen uptake of 203 cm3 g−1.
While the structural transformations during the adsorption of nitrogen on 1′ and 2′ are supposed to be one step “gate opening” (Scheme 2), the adsorption behaviour of 3′ and 4′ was studied by parallelized adsorption and PXRD experiments. The PXRD patterns recorded on the evacuated material 3′ and at the first adsorption point (Fig. 6, PXRD no. 1) show the presence of a narrow pore phase. Surprisingly, further five PXRD patterns, recorded in the relative pressure range from 5 × 10−4 up to 10−1, show a shift of the main (101) reflection from 2θ = 7.19° to 7.44°, indicating the formation of an intermediate phase (ip) and contraction of the unit cell (Fig. 4, PXRD patterns no. 2–6). Unfortunately, the patterns could not be indexed unambiguously because of the presence of the reflections of more than one phase. Further increase of the gas pressure leads to the gradual shift of the (101) reflection back to lower angles, but the initial position is not completely reached (Fig. 4, PXRDs no. 2 and 7). The PXRD patterns, obtained in the p/p0 range of 0.1 to 1 are characterized by the intensity increase of the reflection at 2θ = 6.98° characteristic for the as made phase 3. Although the PXRD patterns from 2 to 6 could not be indexed, the shape of the isotherm as well as the reflection shift suggest that the transition is similar to the breathing phenomena, observed in MIL-53(Cr) and MIL-53(Al) materials.26 Desorption of nitrogen proceeds without phase transitions showing the presence of a mixture of compounds 3′ and 3 up to low pressures. Only evacuation of the sample after the experiment at room temperature leads to structural transformation to the initial narrow pore state.
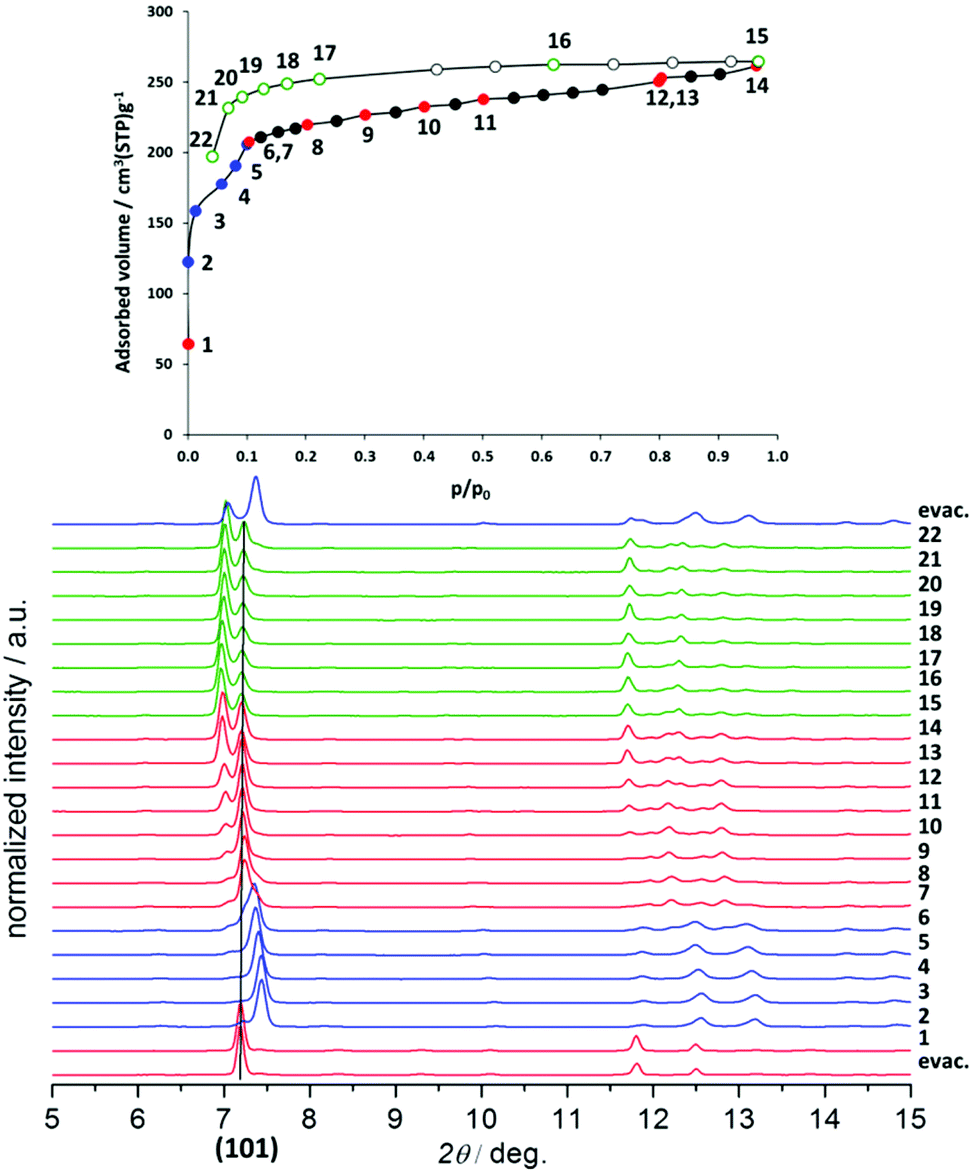 | ||
| Fig. 6 Adsorption of N2 at 77 K on 3′ monitored in situ by PXRD. | ||
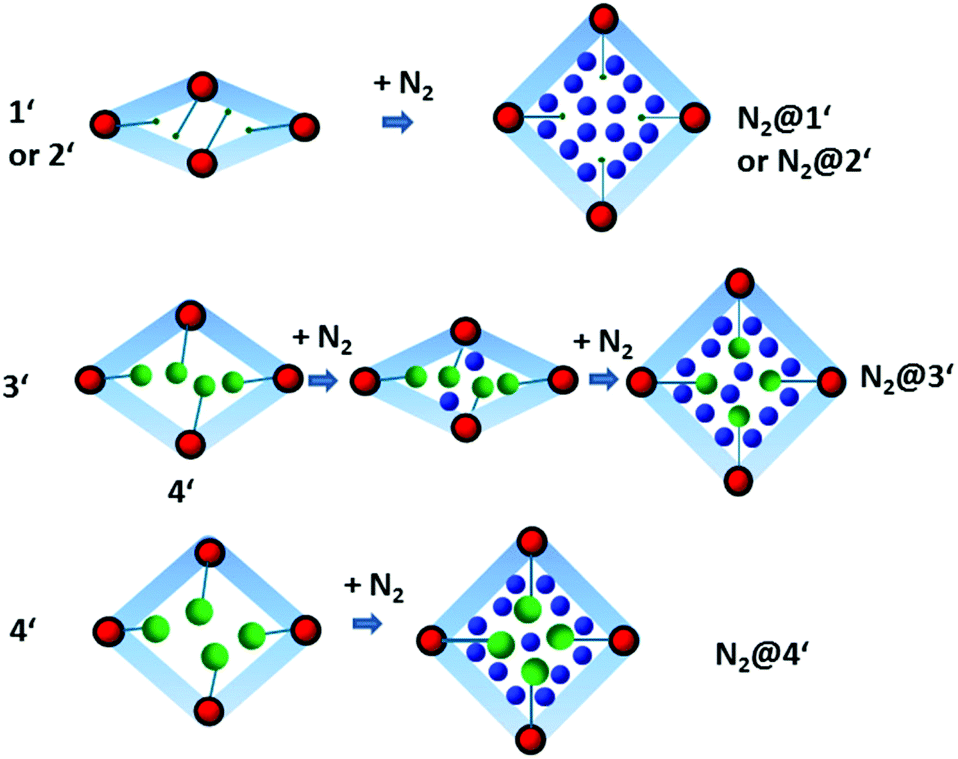 | ||
| Scheme 2 Mechanisms of structural transformation during the adsorption of N2 at 77 K for 1–4. | ||
Nitrogen physisorption at 77 K combined with PXRD analysis of 4′ (Fig. 7) reveals a completely different structural transformation induced by the cinnamate anion (Scheme 2). The PXRD pattern of completely evacuated 4′ shows a reflection at 2θ = 7.50° matching the intermediate phase, obtained during the adsorption of nitrogen on 3′. Indeed, the direct comparison of the above mentioned PXRD patterns shows good match (Fig. S11, ESI†). The indexing of the PXRD patterns obtained from evacuated 4′ gives several possible unit cells, one of which is an orthorhombic C-centered unit cell. Taking into account that 1′ crystallizes in the same crystal system, this unit cell was used for the Rietveld refinement. The pore volume calculated from the final structural model matches with the value estimated from the adsorption isotherm of the compound.
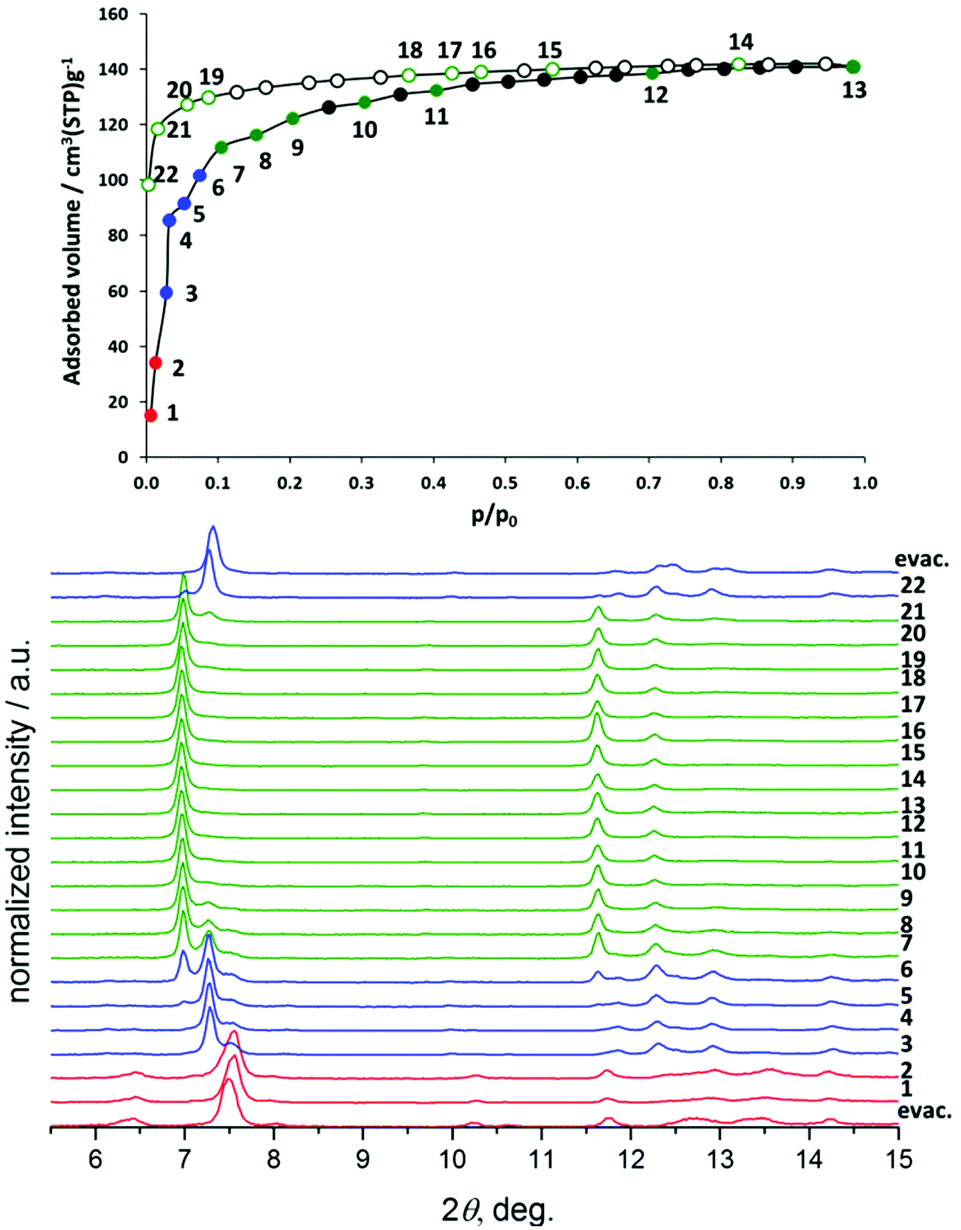 | ||
| Fig. 7 Adsorption of N2 at 77 K on 4′ monitored in situ by PXRD. | ||
Although the adsorption isotherms of 3′ and 4′ are similar to some extent, in situ adsorption/PXRD experiments suggest a distinct pathway for the structural evolution in 4′. In the case of 4′, the most intense reflection shifts towards lower 2θ angles with increasing pressure, probably indicating the stepwise opening of the crystal structure. The phase 4′ exists up to the relative pressure of 0.013. Further adsorption of nitrogen leads to phase transition, accompanied by the shift of the most intense reflection from 7.50° to 7.29° 2θ. Unfortunately, the pure intermediate phase could not be obtained and therefore the PXRD patterns could not be indexed unambiguously. In the relative pressure range between 0.2 and 1 during the adsorption and desorption almost pure phase 4 is presented. Only at relative pressures lower than 10−2, the reversible transformation from 4 to the intermediate phase is observed.
Conclusions
In summary, the fine tuning of the gate pressure and switchability in JLU-Liu-4 was possible by modification of the SBU. Compounds containing coordinated formate, acetate, benzoate or cinnamate anions on the cluster have the same frameworks with the ant topology, but show variations in structural transformation during the adsorption of nitrogen at 77 K. While the utilisation of small carboxylic acids during the synthesis (compounds 1 and 2) results in complete closing of the framework after solvent removal and therefore “gate opening” behaviour during the adsorption, bulkier carboxylates lead to incomplete closing of the frameworks after desolvation. Compounds containing benzoate (3′) and cinnamate (4′) anions show nearly the same adsorption behaviour but undergo completely different structural transformation during the adsorption. While the structural changes during adsorption on 3′ could be rather described as classical breathing,27 the changes in 4′ lead to the step-wise increase of the unit cell volume. We believe that this approach can be further generalized and applied for other flexible MOFs with coordination vacancies on the metal cluster (typically Zn3 and Co3 clusters) in order to obtain materials with targeted adsorption properties.Acknowledgements
N. K. thanks the “excellence initiative by the German federal and state government” (Institutional strategy, measure “support the best”). V. B. thanks the German Federal Ministry for education and research (Project BMBF No. 05K13OD3). The HZB is gratefully acknowledged for the allocation of the synchrotron radiation beamtime on KMC-2 and MX BL14.2 beamlines and for travel grants.References
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- P. Serra-Crespo, M. A. van der Veen, E. Gobechiya, K. Houthoofd, Y. Filinchuk, C. E. A. Kirschhock, J. A. Martens, B. F. Sels, D. E. De Vos, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2012, 134, 8314–8317 CrossRef CAS PubMed.
- A. Douvali, A. C. Tsipis, S. V. Eliseeva, S. Petoud, G. S. Papaefstathiou, C. D. Malliakas, I. Papadas, G. S. Armatas, I. Margiolaki, M. G. Kanatzidis, T. Lazarides and M. J. Manos, Angew. Chem., Int. Ed., 2015, 54, 1651–1656 CrossRef CAS PubMed.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167–170 CrossRef CAS PubMed.
- W. Kosaka, K. Yamagishi, A. Hori, H. Sato, R. Matsuda, S. Kitagawa, M. Takata and H. Miyasaka, J. Am. Chem. Soc., 2013, 135, 18469–18480 CrossRef CAS PubMed.
- Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed.
- S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464–9474 CrossRef CAS PubMed.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- O. Kozachuk, M. Meilikhov, K. Yusenko, A. Schneemann, B. Jee, A. V. Kuttatheyil, M. Bertmer, C. Sternemann, A. Pöppl and R. A. Fischer, Eur. J. Inorg. Chem., 2013, 2013, 4546–4557 CrossRef CAS.
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193–196 CrossRef CAS PubMed.
- J. Wang, J. Luo, J. Zhao, D.-S. Li, G. Li, Q. Huo and Y. Liu, Cryst. Growth Des., 2014, 14, 2375–2380 CAS.
- P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC.
- I. Gamba, I. Salvadó, G. Rama, M. Bertazzon, M. I. Sánchez, V. M. Sánchez-Pedregal, J. Martínez-Costas, R. F. Brissos, P. Gamez, J. L. Mascareñas, M. Vázquez López and M. E. Vázquez, Chem. – Eur. J., 2013, 19, 13369–13375 CrossRef CAS PubMed.
- U. Mueller, N. Darowski, M. R. Fuchs, R. Forster, M. Hellmig, K. S. Paithankar, S. Puhringer, M. Steffien, G. Zocher and M. S. Weiss, J. Synchrotron Radiat., 2012, 19, 442–449 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Material Studio 5.0, Accelrys Software Inc., San Diego, USA, 2009 Search PubMed.
- V. Bon, I. Senkovska, D. Wallacher, A. Heerwig, N. Klein, I. Zizak, R. Feyerherm, E. Dudzik and S. Kaskel, Microporous Mesoporous Mater., 2014, 188, 190–195 CrossRef CAS.
- P. A. Heiney, Datasqueeze 2.2.9 Graphical Tool for X-ray Data Analysis, 2012 Search PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Férey, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS.
- V. Bon, N. Klein, I. Senkovska, A. Heerwig, J. Getzschmann, D. Wallacher, I. Zizak, M. Brzhezinskaya, U. Mueller and S. Kaskel, Phys. Chem. Chem. Phys., 2015, 17, 17471–17479 RSC.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Temperature dependent PXRD measurement for 1′, TG curves for 1–4 and 2′–4′, FT-IR spectra for 1′–4′, and PXRD patterns for 3′ and 4′. CCDC 1422814–1422821. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03504j |
| This journal is © The Royal Society of Chemistry 2016 |