
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inorganic sulfur–nitrogen compounds: from gunpowder chemistry to the forefront of biological signaling
Miriam M.
Cortese-Krott
a,
Anthony R.
Butler
b,
J. Derek
Woollins
c and
Martin
Feelisch
*d
aCardiovascular Research Laboratory, Department of Cardiology, Pneumology and Angiology, Medical Faculty, Heinrich Heine University of Düsseldorf, Düsseldorf, Germany
bMedical School, University of St-Andrews, St-Andrews, Fife, Scotland, UK
cEaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, Scotland, UK
dClinical and Experimental Sciences, Faculty of Medicine; Institute for Life Sciences, University of Southampton and NIHR Biomedical Research Center, Southampton General Hospital, Tremona Road, Southampton, UK. E-mail: m.feelisch@soton.ac.uk
First published on 15th February 2016
Abstract
The reactions between inorganic sulfur and nitrogen-bearing compounds to form S–N containing species have a long history and, besides assuming importance in industrial synthetic processes, are of relevance to microbial metabolism; waste water treatment; aquatic, soil and atmospheric chemistry; and combustion processes. The recent discovery that hydrogen sulfide and nitric oxide exert often similar, sometimes mutually dependent effects in a variety of biological systems, and that the chemical interaction of these two species leads to formation of S–N compounds brought this chemistry to the attention of physiologists, biochemists and physicians. We here provide a perspective about the potential role of S–N compounds in biological signaling and briefly review their chemical properties and bioactivities in the context of the chronology of their discovery. Studies of the biological role of NO revealed why its chemistry is ideally suited for the tasks Nature has chosen for it; realising how the distinctive properties of sulfur can enrich this bioactivity does much to revive ‘die Freude am experimentellen Spiel’ of the pioneers in this field.
 M. M. Cortese-Krott | Miriam Cortese-Krott was born in Milan (Italy) and obtained her Doctoral degree (Laurea) with honors in Pharmaceutical Biotechnology from the University of Milan in 2002. In 2006, she obtained a PhD in Pharmacology with honors from the Heinrich Heine University of Düsseldorf, working on nitric oxide biochemistry and biology under the supervision of Prof. Victoria Kolb-Bachofen. Later she worked on the chemistry and physiology of NO in erythrocytes with Prof. Malte Kelm. She won 4 Young Investigators’ awards, and was an invited speaker at the NO-Gordon Research Conference. Since her Habilitation in Molecular Medicine, she is an independent Group Leader working on the NO/sulfide cross-talk in biology and cardiovascular medicine. |
 A. R. Butler | Anthony Butler is a graduate of King's College, University of London. His PhD work was supervised by Victor Gold. After his graduation he worked at Northampton Technical College and then, on a post-doctoral fellowship, at Cornell University with Thomas Bruice. After a brief spell at the University of Sussex he moved to the University of St Andrews, where he worked in both the Chemistry Department and the Medical School. His main research interests included the chemistry of urea, iron–sulfur clusters, nitrosothiols and iron nitrosyls, the mechanism of nitration and nitrosylation reactions, and the physiology and medical use of nitric oxide. He is now retired. |
 J. D. Woollins | Derek Woollins was born in Cleethorpes and educated at the University of East Anglia (Norwich), where he went on to carry out his PhD. He was appointed as a lecturer at Imperial College London in 1982. After 12 years at Imperial College, he moved to Loughborough as the Chair in Inorganic Chemistry, where he stayed for 5 years before moving to St Andrews as the Chair in Synthetic Chemistry in 1999. He is currently Vice Principal (Research) and Provost at St Andrews. Woollins has published over 500 research papers in main group chemistry and three books. His research interests center around group 15/16 chemistry including the use of P–Se compounds (such as Woollins’ Reagent) in synthesis and structural science. |
 M. Feelisch | Martin Feelisch studied pharmacy and obtained his PhD degree from the Heinrich Heine University in 1988, working on mechanisms of guanylate cyclase stimulation by nitrovasodilators under the supervision of Professor Eike Noack. As Director of Pharmacology at Schwarz Pharma he led a drug discovery program on novel NO-donors before moving back to academia in 1997. After working with Salvador Moncada (University College London) he spent eight years in the US (LSU Shreveport and Boston University), returning to the UK in 2007 (Warwick University). In 2012 he was appointed as Professor of Experimental Medicine & Integrative Biology at the University of Southampton. His research interests are wide-ranging and include human adaptation to hypoxia, systems biology, and the redox chemistry of NO/HNO and sulfur species. |
Introduction
The chemical reactions of sulfur-centered nucleophiles with a range of nitrogen species have been studied for preparative synthetic and industrial purposes, to better understand fundamental processes in geochemical and atmospheric chemistry and to shed new light on the origins of life.1 The earliest reports, dating back almost a century now,2 along with the related more recent literature3–9 established that the reaction of either hydrogen sulfide (H2S),† alkali sulfide (e.g. NaHS, Na2S) or polysulfide (K2Sx) salts or colloidal sulfur (S8) with nitric oxide (NO), nitrite (NO2−) or nitrous acid (HNO2) gives rise to nitrogen and gaseous nitrogen oxides (NO, N2, N2O) and sulfur-bearing anionic solutes such as polysulfides10,11 and S–N hybrid species, i.e. combined sulfur and nitrogen containing compounds.‡ However, none of this was considered to be of any relevance to contemporary biology.As aptly pointed out by Chivers in the Preface to his authoritative monograph on chalcogen–nitrogen chemistry,12 much of the chemistry of these sulfur–nitrogen (S–N) compounds was explored by German chemists working in the second half of the 20th century and published, as was proper then, in German. Interest in the subject at the time was sparked through a book by Margot Goehring entitled ‘Ergebnisse und Probleme der Chemie der Schwefelstickstoff–verbindungen’ (results and concerns pertinent to the chemistry of sulfur–nitrogen compounds).13 Fritz Raschig, Peter W. Schenk and Fritz Seel also contributed substantially to our knowledge of these compounds. Seel's work in particular described the reaction mechanisms between either sulfide or polysulfide and NO or nitrite, and identified as well as chemically characterized the reaction intermediates to comprise short-chain polysulfide (S32−; S42−) and the S–N compounds, nitrosopersulfide ([SSNO]−) and dinitrososulfite ([ONN(O)–SO3]2−);11 in addition, Seel hypothesized that the same mechanism could be involved in the chain of reactions taking place during the explosion of gun powder.14
The recent surge of interest in this chemistry by the biological community7,15–23 was triggered by the growing appreciation that both, NO and H2S§ are synthesized by discrete, highly conserved enzymatic machineries in cells and tissues18,22,23 and often exert similar and, in part, interdependent biological actions within the same pharmacological or biological model system. This so-called NO/H2S “cross-talk” has been shown to result in either mutual attenuation or potentiation of responses.23–25 The interaction can occur at multiple levels and include the modulation of enzymes belonging to the same signaling pathways,26 reciprocal functional interaction (NO inhibiting H2S production after binding to the heme prosthetic group of cystationine β-synthase,27,28 one of the H2S generating enzymes, and sulfide modulating nitric oxide synthase expression and/or activity,29,30 for example), and direct chemical reactions;31 all of these processes can modulate direction or magnitude of the biological response, or both. While the different elements of the NO signaling cascade are well characterized, those underpinning the biological/pharmacological effects of sulfide are currently less well understood. Some recent studies32,33 suggest that polysulfide or other sulfane sulfur species,34,35 rather than dissolved H2S or hydrosulfide anions (HS−), may be responsible for “sulfide signaling”.36
We recently reported9 that the same S–N intermediates and products originally described by Goehring, Schenk and Seel in the reactions of sulfide/polysulfide with NO or nitrite, and later by Williams37,38 for reaction of S-nitrosothiols and sulfide, are also formed under biologically relevant conditions (pH 7.4, air) and display potent bioactivity in cultured cells in vitro and in rats in vivo, bringing S–N compounds and their interesting chemistry to the attention of contemporary biochemists and to the fore in relation to biological signaling. In recent years, this area of research has occasioned a lively debate over controversial assignments of some of the more labile reaction intermediates/products.8,39 However, this article will not focus on perceived differences in stability under specific reaction conditions. Instead, the current perspective aims at providing a synopsis of bioactive S–N compounds and their potential role in biological signaling. Specifically, we will give a brief overview about the discovery and basic chemical properties of HSNO/SNO−, SSNO− and [ONN(O)–SO3]2− in comparison to nitrosothiols, review what is currently known about their bioactivity, and discuss how some of those fundamental properties relate to the emerging biology and pharmacology. Paraphrasing Margot Goehring, we hope to herewith revive “die Freude am experimentellen Spiel”40 (the joy of the experimental game) in the fledgling research field of the ‘NO/sulfide cross talk’.
Stability of the S–NO bond of nitrosothiols (RSNOs) and their chemical reactivity
Nitrosothiols (RSNO; monothionitrites) in which an alkyl or aryl substituent is attached to the sulfur atom of a nitroso group were, for many years, considered chemical curiosities because of the peculiar properties of the –SNO grouping, sequentially linking sulfur to nitrogen and oxygen. It is now generally accepted that RSNOs occur naturally in bacteria, plants and mammalian tissues (e.g. S-nitrosoglutathione (GSNO), S-nitrosoalbumin, S-nitrosohemoglobin) where they can either modulate protein function or act as ‘nitric oxide stores’, carrying NO bioactivity and releasing NO on demand.41–46 Although the pathways of formation and degradation/bioactivation of endogenous RSNOs are far from clear at present, the involvement of specific enzymes (nitrosylases/denitrosylases) has been proposed (see for example literature by Stamler and co-workers47,48). In addition to these enzymatically regulated processes direct chemical reactions leading to cleavage of the S–N bond and release of the NO group in the form of either NO+, NO˙ or NO− occur under biologically relevant conditions,41,49 and are likely to occur also in cells and tissues.The earliest (1909) solid, but unstable, S-nitrosothiol to be isolated was probably PhSNO50 but some years later a completely stable compound of this type, S-nitroso-N-acetyl-penicillamine (SNAP), was prepared and a crystal structure obtained, followed by the preparation of crystalline S-nitrosoglutathione and S-nitrosocysteine51 as well as S-nitrosocaptopril.52 Efforts to prepare other nitrosothiols were less successful and it soon became apparent that, in general, they are unstable compounds.
The Pauling electronegativities for N, O and S are 3.04, 3.44, and 2.58, respectively. As a result, in binary systems the N–O bond will be polarized with a partially negative charge on oxygen, whereas the S–N bond will be polarized in the other direction.12 The preference for σ-bonding over π-bonding is a common feature of the heavier p-block elements, which include all three elements. Therefore, sulfur-nitrogen π-bonds are weaker than oxygen–nitrogen π-bonds, and there is an increased reluctance for sulfur to form multiple bonds to nitrogen as compared to oxygen.12 For these reasons the S–N bond in RSNOs is predicted to be thermally more labile than the O–N bond in RONOs.12
A probable mechanism of reaction (in the absence of another reaction partner) is light or temperature catalyzed homolysis of the S–N bond to give a thiyl radical and NO (eqn (1)), followed by radical dimerization (eqn (2)).53
| 2RSNO → 2RS˙ + NO˙ | (1) |
| 2RS˙ → 2RSSR | (2) |
The apparent weakness of the S–N bond in nitrosothiols, leading to facile homolysis in solution, was initially something of a conceptual problem. Crystallographic data reveal that in RSNOs there is an elongated S–N bond of 1.76–1.85 Å15,51,54 and that there are syn and anti-conformers, although the energy difference between the two is very small.54,55
The dissociation energy of the S–N bond is reported56,57 to range from 21 to 31 kcal mol−1, an unexpectedly large variation. There have been a number of computational studies on this matter, and the results depend on the computational methods used. One high-level study58 gives a bond dissociation energy for HSNO of 29.2 kcal mol−1, and kinetic data for alkyl-substituted nitrosothiols produce estimates in the order of 28–31 kcal mol,57 whereas calorimetric results59 typically yield lower values (25 kcal mol−1). The situation became clearer when it was observed by Williams and Butler et al.60 that the rate of RSNO homolysis is very sensitive to the presence of copper ions. Even the amount of copper present in normal distilled water is sufficient to bring about a massive increase in the rate of homolysis. A further, more detailed examination61 of the reaction kinetics showed that it was Cu(I) ions, rather than the Cu(II) ions, that were responsible for catalysis. Cu(I) ions are readily formed by reduction of Cu(II) by thiolate (either produced by the slow hydrolysis of the RSNO itself or already present in the form of reduced thiol). In the presence of EDTA or neocuproine (a selective complexing agent for Cu(I) ions) even S-nitrosocysteine is quite stable.
In understanding the catalytic effects of Cu(I) ions it was first assumed that Cu(I) complexes with the sulfur of the S–N bond in a nitrosothiol. That this would have the required effect of weakening the S–N bond has been shown by theoretical calculations.62 A more detailed set of calculations showed also that while complexation to the sulfur of the S–N bond would weaken the bond, complexation to the nitrogen would have the opposite effect.63 So, steric crowding around the sulfur atom of a nitrosothiol could have a profound effect upon its stability. This could be part of the explanation of the stability of SNAP and S-nitroso-t-dodecanethiol (Fig. 1). In addition, recent density functional theory calculations revealed that substituent effects are significant in governing the stability and reactivity of nitrosothiols, as demonstrated for CH3SNO vs. CF3SNO.54
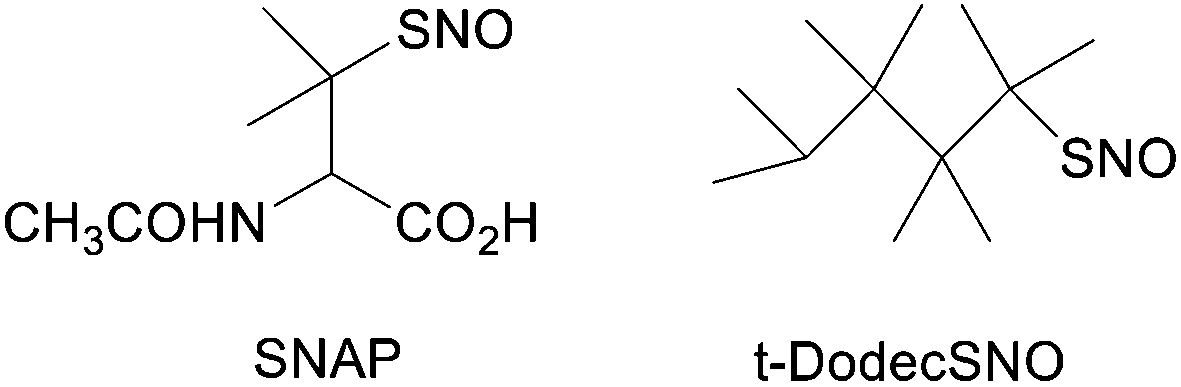 | ||
| Fig. 1 Structures of S-nitroso-N-acetylpenicillamine (SNAP) and S-nitroso-t-dodecanethiol (t-DodecSNO). | ||
A second reaction leading to decomposition of RSNOs is nucleophilic attack of a thiolate at the sulfur of the nitrosothiol with nitroxyl anion (NO−) as a leaving group (eqn (3)). This is a possibility as thiolate is a very powerful nucleophile, more powerful according to Ritchie's scale than hydroxide.64 This pathway is discussed because it is a possible source of nitroxyl in the physiological situation. Which thiols are the most relevant in this context is presently unclear and will depend on where the reaction will take place (e.g. inside cells or in blood), abundance of reduced thiols in this compartment and pKa of the sulfhydryl groups involved. Thiol concentrations may be less important here than actual fluxes, but little is known about this for most thiols including its smallest representative, H2S.
Third, a nitrosothiol may undergo transnitrosation with another thiol following a nucleophilic attack of a thiolate on nitrogen (eqn (4))65
| R′S− + RSNO → RSSR′ + NO− | (3) |
| R′S− + RSNO → R′SNO + RS− | (4) |
In view of the abundance of reduced thiols present in the physiological milieu (many mammalian cells contain millimolar concentrations of glutathione), the reactions according to eqn (3) and (4) are highly likely to occur and very fast.
Fourth, under favorable conditions, there can be the formal transfer of [NO+] from the sulfur atom of a nitrosothiol to the nitrogen of an amine (S→N transnitrosation). The most detailed study of this process is the intramolecular transfer of [NO+] from sulfur to the amine group in the decomposition of S-nitrosocysteine (eqn (5))66
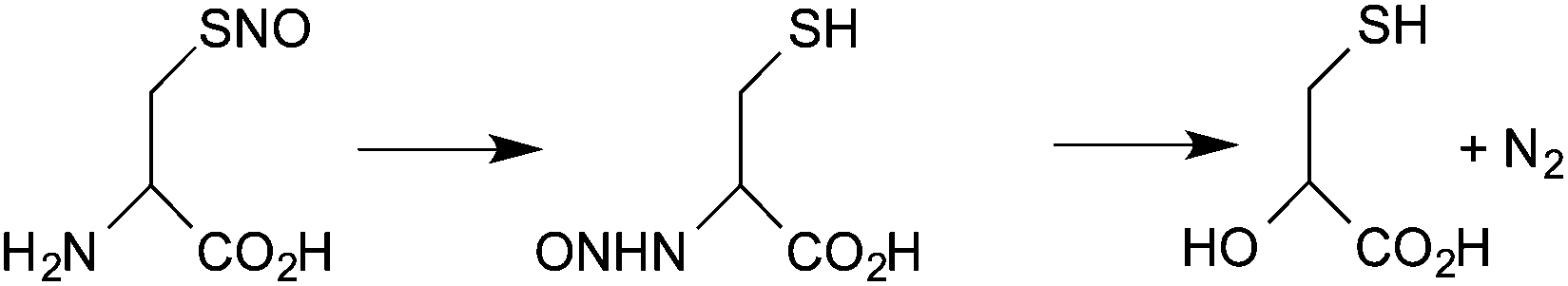 | (5) |
As far as we know, there has been no kinetic study of intermolecular reactions of this type and so it is difficult to assess its possible physiological role. However, the formation of reactive intermediates (such as the diazonium ion and acrylic acid) as a consequence of such reaction has been proposed to contribute to the toxicological properties of this class of NO donors.67
Stability and reactivity of HSNO: a unique nitrosothiol
Although generally named rather differently, thionitrous acid (HSNO) is also a nitrosothiol. Some authors refer to it as the “smallest” and even the “simplest” member of this class of molecules. That it is the smallest is undoubtedly true, although this does not necessarily make its chemistry or its chemical biology special, as its charge and stability under physiological conditions might do. That it is the simplest must be questioned: its very peculiar and rather complex chemical properties urge a re-appraisal of these definitions.We suggest that HSNO is unique amongst nitrosothiols because it possesses a mobile and ionisable proton (eqn (6) and (7)) instead of the alkyl group of other RSNOs. The mobile hydrogen promotes isomerization by intramolecular hydrogen shift (eqn (6) and (7))
 | (6) |
 | (7) |
This gives it an inherent instability and reactivity not found with other nitrosothiols. As a result, there are four compounds of the same formula which, in order of increasing ground state energy, are HNSO > HOSN > HSNO > HONS. These species are in equilibrium with each other (eqn (8)).
| HNSO ⇌ HOSN ⇌ HSNO ⇌ HONS | (8) |
The isomerization of these compounds was proposed by Margot Goehring back in 1952 in a seminal piece of work.68 She used the synthetic procedure reported by Schenk69 to obtain SONH (yellow), and observed that the product rapidly isomerizes to the red SNOH and forms insoluble brown material at temperatures greater that −60 °C.68 Later on, Huber and co-workers characterized all four isomers of OSNH, including the unstable HSNO, by IR spectroscopy with the material trapped in an Argon matrix at −261 °C.69–71 Interconversion between isomers was effected by photolysis, since these molecules polymerize on condensation,70,71 as reported previously by Schenk and Goehring.68,69,72 They proposed that the most stable isomer is HNSO (thionylimide)70,71 again in accordance with the conclusions of Goehring.68 More recent work by Méndez et al.73 showed that the energy barrier to isomerisation between the four compounds is indeed very low.
In addition to isomerization, the S–N bond of HSNO can undergo the same reactions as described for nitrosothiols above. There is spontaneous homolysis of the S–N bond, catalysed by light, temperature and transition metals. Cu(I) should be at its most effective in HSNO as the latter is the least sterically hindered representative. If formed by nitrosation of sulfide, as proposed by Williams,38 there is the possibility of reaction of HSNO with HS− (or RS−) via nucleophilic attack at sulfur. This parallels exactly what happens with an alkyl nitrosothiol (e.g. S-nitrosocysteine) but the disulfide formed (HSSH; disulfane) is a much more reactive species than cystine; HSSH can ionise to give persulfide (HSS−), which could react further, as will be discussed later. Finally, HSNO can also undergo transnitrosation (nucleophilic attack at nitrogen) by reaction with thiols, amines or alcohols present in solution. Possible reactions of thionitrous acid are displayed in Fig. 2.
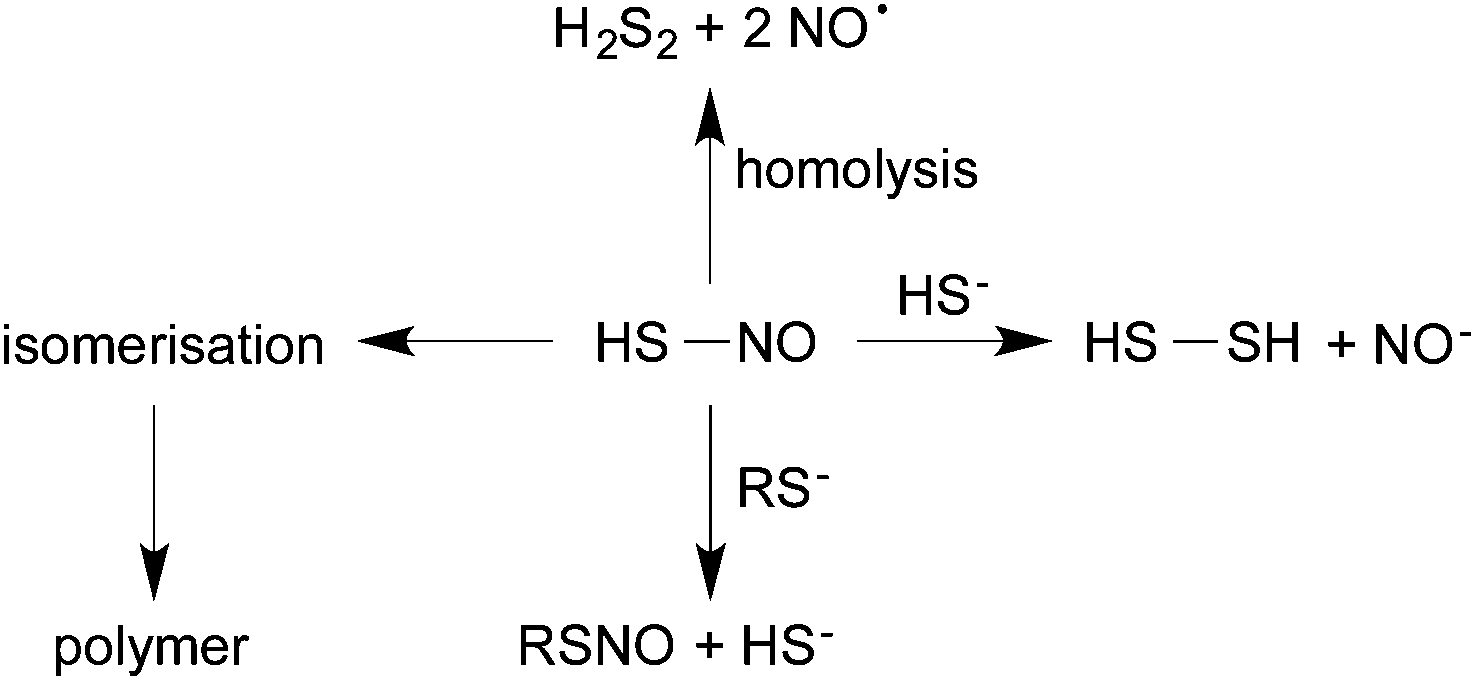 | ||
| Fig. 2 Reactivity of HSNO formed by the reaction of RSNO and HS−: isomerization, homolysis, nucleophilic attack and transnitrosation. | ||
All these reactions suggest either instability or high reactivity. Its instability is also reflected by the need for very low temperatures (<−60 °C) and extreme conditions (frozen Ar matrix) to characterize the physicochemical properties of HSNO and its isomers.68,70–72,74 Importantly, all isomers have a different chemical reactivity.
The pKa of thionitrous acid has not been experimentally determined but we can make an informed estimate. The pKa of nitrous acid (HONO) is 3.25, and the replacement of oxygen by sulfur should lower the pKa (as is the case with cyanic acid and thiocyanic acid); thus at physiological pH, any thionitrous acid formed would be largely present in the form of the anion, apart from the equilibrium concentration of undissociated acid. Therefore, it is important to note that, at physiological pH, HSNO will be ionised and exists largely as the SNO− anion (thionitrite), and except for isomerization of SNO− into NSO− (thiazate)75 (Fig. 4), none of the reaction pathways shown in Fig. 2 apply. This, of course, does not give the species stability as the anion will always be in equilibrium with the undissociated molecule and so reactivity and instability will be reduced, to an extent determined by the pH, but not eliminated.
Much to our surprise, HSNO was reported to be formed in the reaction of acidified nitrite and sulfide, or S-nitrosoglutathione and sulfide and claimed to being “stable for 30 min at physiological pH at room conditions”.76 HSNO formation at pH 7.4 was seemingly confirmed by an ultrahigh-resolution electrospray ionisation time-of-flight mass spectrometer operating in positive ionisation mode. A peak at m/z of 63.9902 was observed and assigned to [HSNO + H]+ (a disturbingly large deviation of 78 ppm from the theoretical m/z 63.9852, in particular considering the instrumentation employed in that study). When GS15NO was used there was an increase of one m/z unit. Further evidence for the presence of HSNO/[SNO]− came from the observation of a peak in the 15N NMR spectrum of the reaction mixture at 322 ppm,76 the same region as that observed for other nitrosothiols.77 We were unable to reproduce the mass spectroscopic identification of [HSNO + H]+ (in positive ionisation mode) or detect [SNO]− (using negative ionization), in spite of strenuous efforts including the use of cryospray ionisation.9 We tried equally hard to obtain a signal from a 15N-NMR spectrum of a reaction mixture containing sulfide and 15N-labelled sodium nitrite at low pH without success (Botting, Feelisch and Butler, unpublished results).
A detailed study of our own mass spectral results tells a rather different story. Studies using time-resolved chemiluminescence and UV-visible spectrophotometry suggested that the initial intermediate formed from the reaction of sulfide with GSNO, probably SNO−, had only a fleeting existence (as also observed by Seel and by Williams11,37 and was replaced by a more stable species with λmax of 412 nm, and it is this species that could be responsible for the bioactivity of the NO/sulfide duo.7–9 We used a different detection technique (a hybrid Linear Iontrap/Orbitrap mass spectrometer) to identify it.9 Because of the pH, we assumed many key intermediates would be present as anions, therefore negative ionisation mode was used throughout. A signal corresponding to perthionitrite SSNO− at m/z of 93.9427 (theoretical 93.94268) was observed. In addition to this signal there were others in the low molecular weight range corresponding to dinitrososulfite [ONN(O)–SO3]2− – called in the paper by the trivial name it is known by in the nitric oxide research field, SULFI/NO9 – observed 140.9612, (theoretical 140.96061), as well as polysulfide species (S32−, S42− and S52−). For both, SSNO− and [ONN(O)–SO3]2−, the assigned structures were confirmed by the fragmentation patterns and stable isotope labelling.
Although we feel that the chemical literature, including but not limited to references58,69–71,78 and our own experimental work7–9 provided overwhelming evidence for the notion that, contrary to other claims,76 the stability of thionitrous acid (HSNO) is incompatible with biologically relevant conditions characterized by neutral pH and ambient temperatures; instead, it is very likely that its deprotonated form, SNO− (thionitrite) is the first product of the reaction of sulfide with a nitrosating agent as this parallels what happens with other thiols.
The discovery of SSNO−
The perthionitrite or nitrosopersulfide (R-SSNO) class of molecules, of which the SSNO− anion is the first member, was discovered and characterized by Fritz Seel in the 1980s,10,11,79,80 shortly before his death in 1987. A life-long interest of Fritz Seel was the speciation of sulfur and polysulfides in polar non-aqueous solvents.81 He was particularly interested in the – at that time – quite mysterious chemistry of the blue trisulfide radical anion (S3˙−),82 which is responsible for the blue color of the gemstone lapis lazuli.83,84 For a tutorial review about the identification, analysis and chemical properties of S3˙−, please refer to ref. 85. Solutions of sulfur in DMF, DMSO and hexamethylphosphoramide result in the formation of the blue radical anion S3˙− (λmax 630 nm) and the red radical anion S4˙− (λmax 510 nm). Schmidt and Wägerle86,87 reported that on addition of nitrite to a solution of sulfur in DMF blue and red intermediates were formed, and proposed that the red intermediates were perthionitrates (Sx–NO2−) obtained by the nucleophilic attack of nitrite on cyclooctane sulfur (S8). The perthionitrates were a new class of molecules the existence of which had been predicted but never been demonstrated before. This together with the observed formation of a blue color indicative of intermediate formation of S3˙− induced Seel to investigate this reaction in greater detail.10,88Seel et al. observed that the same orange/red reaction product (λmax 425 nm in ethanol/methanol; λmax 448 nm in acetone, acetonitrile and electron pair donor solvents; it is characterized by 15N-NMR shift of 339 ppm) formed in the reaction of nitrite with sulfur or polysulfide was also formed by the reaction of polysulfides with NO gas.10 The latter made it highly unlikely that the orange/red product could be a perthionitrate (SSNO2−), but suggested it was a perthionitrite(SSNO−) instead. Intriguingly, Seel observed that SSNO− was in equilibrium with NO and short-chain polysulfides (including S3˙− and S4˙−), presumably originating via thermal homolyis of SSNO− into NO˙ and the highly unstable SS˙− (eqn (9) and (10)), as confirmed by EPR analysis.80
| SSNO− ⇌ S2˙− + NO˙ | (9) |
| 2SS˙− ⇌ S42− ⇌ S˙− + S3˙− | (10) |
According to Steudel, in aqueous solution S2˙− will rapidly dimerize to tetrasulfide or form S˙− radicals and trisulfide.89 As later confirmed by us,7,8 Seel et al. also observed that non-aqueous solutions of SSNO− are relatively stable in air for 1–2 hours,80 but if water is added rapid sulfur precipitation ensues. This is due to the disturbance of the equilibrium between SSNO− and S2˙−/S4˙− + S˙−/S3˙− (eqn (9) and (10)), in the course of which polysulfides are rapidly transformed to water-insoluble S8. Addition of acid will lead to protonation of SSNO− to HSSNO, which undergoes homolysis with formation of NO, longer-chain polysulfides and sulfur precipitation.
| SSNO− + H+ → HSSNO → HSS˙ + NO˙ | (11) |
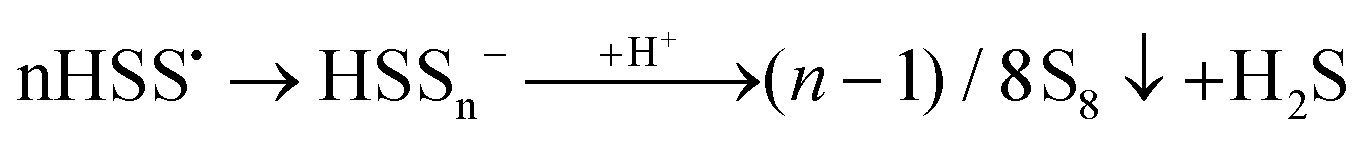 | (12) |
However, SSNO− can also be prepared (with lower yield) in basic non-buffered aqueous solution from the reaction of NO and sulfide or polysulfides,11 or RSNO and sulfide37 (λmax 410 nm in water pH > 9; 412 nm pH = 7.4); importantly, these reactions occur also under aerated conditions at pH 7.4.7–9 The lower yield of formation of SSNO− (max 30%)9 obtained under these conditions prevented Seel from characterizing this product by 15N-NMR.11 Similar to the metastable nature of short-chain polysulfides and persulfide in water,90–92 the chemistry of SSNO− in aqueous solution can only be studied if generated directly in this medium. This is also the reason why solutions of SSNO− (or short chain polysulfides) once prepared in organic solvents cannot be further diluted by addition to aqueous solutions, for example to test their reactivity in water or the bioactivity of SSNO−, as described recently.39
Very recently, the reaction of nitroprusside with aqueous sulfide was shown to result in formation of the blue intermediate [Fe(CN)5N(O)SS]4− with SSNO− serving as the sixth ligand to the transition metal as assessed by a combination of multinuclear (17O, 15N, 13C) NMR, UV/Vis, IR spectroscopic techniques and quantum chemical computation.93
Stability and reactivity of SNO− and SSNO−
Although we have made what we think is a convincing case for the reactivity and instability of thionitrous acid, the thionitrite anion is of course a different matter. The ease with which perthionitrite, rather than thionitrite, can be prepared in solution suggests that the former is an energy minimum in the complex series of reactions between sulfur or polysulfide and a number of nitrosating agents. It may be because perthionitrite is stabilised by charge delocalisation in a way that is not possible for the thionitrite.The way the two anions are drawn by Chivers12 is shown in Fig. 3. The syn configuration is based on the crystallographic structure obtained by Seel et al.79
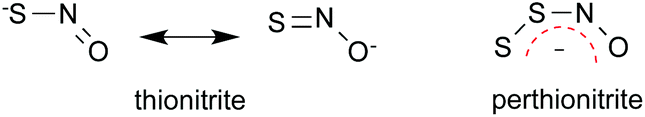 | ||
| Fig. 3 Structures of SNO− and SSNO−, and charge localization in SSNO−. | ||
The canonical form of thionitrite with the negative charge on oxygen rather than sulfur, is unfavourable as sulfur tends to form single bonds to itself rather than double bonds because of the poor overlap of its orbitals, which are held apart by the bulky atomic cores of neighbouring atoms. Thus there is less possibility for charge delocalisation in thionitrite than in perthionitrite. Interestingly, a cyclic intermediate was proposed to account for the rapid isomerisation of SNO− and NSO− (Fig. 4;75), and – similar to the protonated forms described above – the latter appears to be the more stable species.94,95
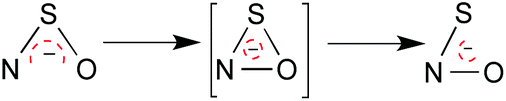 | ||
| Fig. 4 Isomerization of SNO− into NSO−via formation of a cyclic intermediate as described by Chivers.75 | ||
The middle form is unique and most likely a transition state. Although theory in ref. 94 suggests a stable ground state structure, it has not been isolated yet, and bimolecular or even catalyzed processes are more likely (given the calculated barrier). Metal porphyrines may be excellent catalysts of this reaction.96
Nevertheless, SNO− can be isolated as a salt with the aid of a large counter cation like [Ph4As]+ or [N(PPh3)2]+,12,75,79 while its isomer NSO− can even be isolated as a potassium salt.12,75 The fact that perthionitrite appears to be more stable, in the crystalline state, than thionitrite is, of course, not the same as its stability in solution.
Seel et al. were the first to isolate crystals containing [SNO]−, but its mode of preparation is telling. As described previously, reaction of [N(PPh3)2]+[NO2]− with elemental sulfur in DMF gives [N(PPh3)2]+ [SSNO]−. When this was treated with triphenylphosphine, the thionitrite [N(PPh3)2]+[SNO]− was obtained. The structure of both salts was elucidated by X-ray crystallography and NMR spectroscopy.79 From this it would appear that SSNO− is the preferred product of the reaction between sulfide or polysulfide with NO because it is an energy minimum on the reaction pathway, but SNO− can be obtained by reversing the process and removing one sulfur. This is consistent with observations8,9,37,97 that SSNO− rather than SNO− accumulates at reactant concentrations high enough for detection.
The reactivity of SSNO− and SNO− in non-aqueous solutions was described recently in an elegant study conducted by E. Victor in the Lippard laboratory at the Massachusetts Institute of Technology.98 Victor prepared the [N(PPh3)2]+[SSNO]− and [N(PPh3)2]+[SNO]− salts using a glove box adaptation of the original procedures described by Seel79 instead of the classical Schlenk technique. The crystallization of SNO− was reportedly more difficult than that of SSNO− and only successful when conducted in the dark indicative of thionitrite's inherent light sensitivity. The lengths of the S–N bond in SSNO− and SNO− were not very different from each other (1.689 Å and 1.707 Å, respectively), but much shorter than in other crystallographically characterized nitrosothiols. These findings are in full agreement with those of Seel,79 but clearly at variance with another recent publication, which claims that the longer distance between sulfur and nitrogen in SSNO− as compared to nitrosothiols accounts for the “intrinsic instability” of SSNO− as compared to HSNO/SNO−.39 Importantly, on protonation of the SNO− salt by an organic acid (HBF4) Victor found that the UV spectrum of thionitrite disappeared within the mixing time without appearance of a new peak, suggesting HSNO was highly unstable. In accordance to Seel's and our own findings described for dissolved SSNO−,8 Victor also documented that addition of acid to solutions of both [N(PPh3)2]+[SSNO]− and [N(PPh3)2]+[SNO]− leads to immediate release of NO into the headspace, a reaction accompanied by sulfur precipitation. This observation is consistent with homolytic cleavage of protonated SSNO− and SNO− and formation of colloidal sulfur secondary to decomposition of the formed polysulfide (Fig. 2 and eqn (11) and (12)). Thus, both HSSNO and HSNO release NO, and not HNO on decomposition in acid (again at variance with ref. 39 and 76). Victor also compared the reactivity of the two species towards biomimetic iron complexes and found not much difference between SNO− and SSNO−, although both were clearly different from nitrosothiols. It is worth mentioning that [N(PPh3)2]+ salts in general have an extremely low solublity in water,99 and are thus unsuitable to test its reactivity in water (as claimed to have been performed in ref. 39) or its bioactivity in biological experiments. Thus, as with short chain polysulfides,90–92 the reactivity and bioactivity of SSNO− in aqueous solution should be tested by generating the molecule in situ.
In summary, all available data demonstrates the instability and high reactivity of HSNO making the detection as [HSNO + H]+ by mass spectrometry at pH 7.4 and room temperature76 difficult to understand. The formation of SNO− as intermediate in the reaction of nitrosothiols (and the organic nitrite, isopentylnitrite) with sulfide, with transient formation of a UV/Vis peak at λmax 325 nm in DMSO79 has been described by us and others,7–10,37 but was never characterised further. Instead, the formation of SSNO− from NO or RSNO with sulfide and polysulfide under both non-aqueous and aqueous conditions have been demonstrated experimentally and confirmed by us and others.7–11,79,80,93,98,100 Moreover, the SNO− crystals can only be obtained by extraction of the sulphur from SSNO−.12,75,79,98 From all this evidence it would appear that SSNO− is the preferred product of the NO/sulfide/polysulfide interaction because it is an energy minimum along the reaction pathway, and SNO− is an important yet transient reaction intermediate. Both molecules, in their protonated form (HSSNO and HSNO) are unstable and undergo homolysis, releasing NO and persufide radical anion, which finally leads to sulfur precipitation and formation of longer-chain polysulfides via rapid intertwined catenation reactions.
Dinitrososulfite [ONN(O)–SO3]2−: a redox switch for NO
Another potentially important product of the reaction between NO or RSNOs and sulfide/polysulfide under biologically relevant conditions is dinitrososulfite ([ONN(O)–SO3]2−), also known as SULFI/NO,9,101 a different class of S–N compound with very peculiar properties not previously considered in the NO/sulfide cross talk. [ONN(O)–SO3]2− arises as a result of the trapping of two molecules of NO by sulfite (SO32−),101–103 a reaction originally described by Davy in 1802. See also.104 Compounds of this type are known as ‘Drago complexes’ or ‘diazeniumdiolates’ (NONOates; Keefer et al.101) and have found widespread use as nitric oxide donors.105Since the structure of [ONN(O)–SO3]2− remained a mystery for more than a century,6,106 it is perhaps no wonder that it assumed different names including ‘dinitrososulfite’,106 ‘Pelouze's salt’, N-nitrosohydroxylaminesulfonate or ‘Stickoxid-sulfite’ in the German literature (see ref. 6 and references therein). Seel contributed to the clarification of its structural characteristics6 and found SULFI/NO is formed both under non-aqueous and aqueous anaerobic conditions in the reaction between sulfide/polysulfide and sulfur with NO or nitrite.10,11,80 He proposed that SSNO− and [ONN(O)–SO3]2− are the main intermediates of two independent branches of the reaction, occurring in parallel, and that the direction of the reaction is determined by the reaction conditions and the molar ratio of the reactants.80 The formation and decomposition of SULFI/NO leads to release of sulfoxy species and N2O, while formation and decomposition of SSNO− leads to formation of NO (and its oxidation products NO2− and NO3−) along with polysulfide. Seel also proposed that the same reactions participate in the exothermic branched reaction cascade leading to the explosion of gunpowder.14
Consistent with these reports, we found that [ONN(O)-SO3]2− is formed in the reaction of sulfide with NO or nitrosothiols in buffered aerated conditions at physiological pH.9 Under these conditions SULFI/NO has similar stability characteristics as other diazeniumdiolates,101–103,107 co-generates small amounts of NO and HNO at pH 7.4 and releases N2O in high yields. Its formation in the NO/sulfide system suggests that prior production of sulfite contributes to the scavenging of NO, with effective redox conversion of part of this NO to HNO. HNO is a potent signaling molecule in its own right with properties (e.g. thiol oxidation) and biological effects (effects on the contractility of the heart) distinct from NO.
This unexpected chemistry explains why the pharmacological effects of NO may be potentiated, inhibited or fully transformed by the presence of sulfide. Formation of SSNO− and [ONN(O)–SO3]2− may be responsible for NO scavenging, NO release or redox switching of NO into HNO and N2O, propelling an almost forgotten body of inorganic chemistry to the forefront of biological signaling.
Is there a role for S–N compounds in biological signaling?
There is no straightforward answer to the question as to whether or not the intermediates and products of a chemical reaction are relevant to biology. This is particularly true for the reaction between NO and sulfide, which is characterized by a complex cascade of parallel reactions, the outcome of which strongly depends on concentration ratios or fluxes of the reactants. The mechanistic details of the reactions are not fully understood at this juncture, and it is conceivable that several different paths lead to the same final products yet at different yields.7,9 To address this question, it may be instructive to break this general question down into a subset of more specific questions, some of which have already been dealt with in the literature.7–9,108Feasibility: Can NO react with sulfide under biologically relevant conditions (i.e. near-neutral pH, presence of oxygen, high thiol concentrations/reducing conditions)? As alluded to above, we find that sulfide indeed reacts with NO and nitrosothiols, giving rise to three main reaction intermediates namely SSNO−, [ONN(O)–SO3]2−, and polysulfides. In addition to our own experiments7–9 a rich chemical literature attests to the fact that SSNO− releases NO on decomposition.10,11,14,79,80,98,109,110 In particular, release of NO into the headspace during the reaction of sulfide with nitrosothiols has been described many times,7–9,98,109,110 yet was questioned by ref. 39. Irrespective of the recent emergence of controversy, the addition of an excess of sulfide to a biological sample109 and measurement of the NO released by gas phase chemiluminescence110 was proposed a decade ago as a way to quantify nitrosothiol concentrations, although without knowledge of specific reaction mechanisms or intermediates.
So, after having established that NO and sulfide react with one another and give rise to several new products it will be important to know whether any of these are biologically active in cellular systems or in vivo, and therefore have the potential to act as biological messengers. In order to serve as a “biological messenger” reaction intermediates must exhibit a delicate balance between the molecule's reactivity that allows ‘delivering the message’ and the stability that allows ‘making the journey’. We find that SSNO− accumulates at pH 7.4, its decomposition is not affected by the presence of millimolar concentrations of cysteine and glutathione, and releases bioactive NO as evidenced by activation of NO-dependent synthesis of cyclic guanosine monophosphate (cGMP; the so-called “second messenger” of NO) in cultured cells, by vasodilation of aortic rings in organ baths and by decrease of blood pressure in rats.8,9,108 [ONN(O)–SO3]2− releases low levels of bioactive NO along with HNO in cells; for it to markedly activate cellular soluble guanylate cyclase (the enzyme responsible for cGMP production) it requires the presence of high concentrations of superoxide dismutase allowing conversion of NO into HNO; in vivo it not only mildly lowers blood pressure but also potently affects cardiac contractility (which is a hallmark of nitroxyl donors111,112). In addition, polysulfides appear to have a bioactivity that is orders of magnitude greater than that of sulfide itself.21,33 Thus all three classes of compounds formed in the reaction of NO with sulfide are clearly bioactive.9
An obvious but currently unaddressed question is whether SSNO−, [ONN(O)–SO3]2− and polysulfides can actually be formed in blood and tissues, and if so, by what mechanisms. Concerns have been raised about the likelihood of formation of SSNO− based on estimates of reaction rates observed in vitro. We recognize that because it can be formed from NO and sulfide under physiological pH in a test tube this does neither necessarily mean that it is actually formed or that this very same reaction will be its preferred path of production in real life. There may well be enzymatically catalyzed reaction channels giving rise to radical precursors that react with NO almost instantaneously as soon as they are formed – we do not know. In fact, similar doubts were raised two decades ago when peroxynitrite (OONO−) was discussed as an entity of possible biological relevance. Peroxynitrite is the oxygen analogue of SSNO− and formed in the reaction of superoxide with NO (eqn (13)).
| O2˙− + NO˙ ⇌ −OONO | (13) |
This radical–radical reaction occurs at a rate close to the diffusion-controlled limit. When this reaction was first proposed to occur in a biological environment by Beckman, Freeman et al.113 some members of the NO community considered OONO− to be too unstable to be of much biological relevance and, because of the presence of superoxide dismutase, whose speed of removing O2˙− was one of the fastest biological reaction known then, its formation in tissues was questioned.114–116 Since many enzymatic sources of O2˙− (including mitochondria und NADPH oxidases) have since been discovered to occur in close proximity to enzymatic NO sources, OONO− is now an established cellular mediator that contributes to “the dark side” of NO, in particular under inflammatory conditions.116
According to eqn (9), SSNO− is in equilibrium with its products of homolysis, S2˙− and NO˙. We here suggest that in biological environments SSNO− may be formed by a radical–radical reaction between S2˙− and NO according to the following equilibrium (eqn (14)):
| S2˙− + NO˙ ⇌ −SSNO | (14) |
The disulfide radical ion S2˙− is a species well-known to sulfur chemists (please refer to the rich literature by Steudel, e.g.89,117–119). Interestingly, organic persulfides and polysulfides (CySSH and GSSH) have been found in astonishingly high (micromolar) concentrations in tissues,120 and proposed to be formed from cystine or GSSG by enzymes involved in sulfur amino acid metabolism, including CBS, CSE and MST; in addition, mitochondrial enzymes (SQR) or the hemoglobin-mediated oxidation of sulfur-centered radicals, including S2˙− as well as thiosulfate and sulfite may be involved in their formation.121 Sulfite may in turn be enzymatically converted to sulfoxy radicals such as SO2˙, SO3˙−, O3SOO˙− or, SO4˙− by peroxidases.121,122 Interestingly, all these are precursor of formation of both SSNO− and [ONN(O)–SO3]2−, or similar intermediates.9 Since the kinetics of those radical reactions are all expected to be extremely fast these intermediates are likely to be formed in cells and tissues.
We have made the case that SSNO− and [ONN(O)–SO3]2−, along with polysufildes, are products of the reaction of sulfide with NO and nitrosothiols, and we have demonstrated that these products are bioactive in cells and in experimental animals in vivo.9 To answer the question, whether there is a role for S–N compounds in biological signaling, the next efforts should be focused on identifying the biochemical sources of precursors of these intermediates, and developing analytical techniques suitable to analyze their formation in complex biological matrices. This last fundamental, yet still unanswered, question demands finding detection methods capable of determining concentrations and flux rates of S–N compounds and their precursors at low physiological levels in cells and tissues. This is a call for inorganic and analytical sulfur chemists to join the search team.
Summary and conclusions
NO was the first small signaling molecule that was demonstrated to be produced endogenously by mammalian cells and shown to exert an enormous breadth of physiological responses. Fundamental to the understanding of NO's molecular modes of action was (and still is) a sound understanding of its chemical properties, which dictate the interaction with its biological targets including the chemical interaction with other small molecules such as oxygen, superoxide and other free radicals.123,124 Similarly, sulfide and/or sulfane sulfur compounds were shown to be synthesized by conserved enzymatic machineries and exert a plethora of biological signaling roles; unexpectedly, it turned out it cooperates with NO to do so – or is it rather the other way round and sulfide was there long before NO appeared to play a role in biology? Regardless, it is likely that the mutual interaction of these two species is due to their unique chemical reactivity which leads to formation of S–N compounds including SNO− and SSNO− as well as [ONN(O)–SO3]2−, each with their own peculiar chemical biology and distinct bioactivity supporting and/or enabling NO scavenging, NO release and redox switching to HNO. Interestingly, the precursors of these molecules (reactive sulfur species) have been proposed to be formed in cells by a variety of enzymatic and non-enzymatic mechanisms both in the cytoplasm and in the mitochondria and to fulfill signaling functions in their own right.121,125 Considerable analytical development will have to be invested until we will be in a position to determine the sources of those reactive sulfur species, their localization and possibly their co-localization with NO sources as well as the concentrations of the products and their fluxes in cells. Studies of the biological role of NO revealed why its chemistry is ideally suited for the tasks Nature has chosen for it; realizing how the distinctive properties of sulfur can enrich this bioactivity does much to revive ‘die Freude am experimentellen Spiel’ of the pioneers in this field.Acknowledgements
We are indebted to Catherine Botting for constructive discussion of the mass spectrometric results, to Christian M. Kramer for preparing the figures displayed in this manuscript, and Malte Kelm for continuous support. The authors gratefully acknowledge support from the German Research Council (DFG CO 1305/2-1 and SFB1116 TP B06 to MCK), the Susanne-Bunnenberg-Stiftung of the Düsseldorf Heart Center (to MK, and MCK) and the Forschungskommission, Faculty of Medicine, Heinrich Heine University of Düsseldorf (to MCK), the UK Medical Research Council (G1001536 to MF), and the Faculty of Medicine, University of Southampton (to MF).Notes and references
- H. C. Urey, Proc. Natl. Acad. Sci. U. S. A., 1952, 38, 351–363 CrossRef CAS.
- F. Raschig, Schwefel- und Stickstoffstudien, Verlag Chemie, Leipzig-Berlin, 1924 Search PubMed.
- H. Dunnicliff, S. Mohammad and J. Kishen, J. Phys. Chem., 1931, 35, 1721–1734 CrossRef CAS.
- A. Kurtenacker and H. Löschner, Z. Anorg. Allg. Chem., 1938, 238, 335–349 CrossRef CAS.
- D. M. Yost and H. Russell, Systematic inorganic chemistry, Prentice-Hall, 1946 Search PubMed.
- E. Degener and F. Seel, Z. Anorg. Allg. Chem., 1956, 285, 129–133 CrossRef CAS.
- M. M. Cortese-Krott, B. O. Fernandez, M. Kelm, A. R. Butler and M. Feelisch, Nitric Oxide, 2015, 46, 14–24 CrossRef CAS PubMed.
- M. M. Cortese-Krott, B. O. Fernandez, J. L. Santos, E. Mergia, M. Grman, P. Nagy, M. Kelm, A. Butler and M. Feelisch, Redox Biol., 2014, 2, 234–244 CrossRef CAS PubMed.
- M. M. Cortese-Krott, G. G. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS PubMed.
- F. Seel and M. Wagner, Z. Naturforsch., B: Chem. Sci., 1985, 40, 762 Search PubMed.
- F. Seel and M. Wagner, Z. Anorg. Allg. Chem., 1988, 558, 189–192 CrossRef CAS.
- T. Chivers, A guide to chalcogen-nitrogen chemistry, World Scientific, Singapore, 2005 Search PubMed.
- M. Goehring, Ergebnisse und Probleme der Chemie der Schwefelstickstoffverbindungen, Akademie-Verl., Berlin, 1957 Search PubMed.
- F. Seel, Chem. Unserer Zeit, 1988, 22, 9–16 CrossRef CAS.
- B. S. King, Free Radicals Biol. Med., 2013, 55, 1–7 CrossRef PubMed.
- Q. Li and J. R. Lancaster Jr., Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- G. K. Kolluru, S. Yuan, X. Shen and C. G. Kevil, Methods Enzymol., 2015, 554, 271–297 Search PubMed.
- R. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8801–8802 CrossRef CAS PubMed.
- K. R. Olson, Antioxid. Redox Signaling, 2012, 17, 32–44 CrossRef CAS PubMed.
- P. Nagy, Methods Enzymol., 2015, 554, 3–29 Search PubMed.
- H. Kimura, Antioxid. Redox Signaling, 2014, 20, 783–793 CrossRef CAS PubMed.
- B. D. Paul and S. H. Snyder, Antioxid. Redox Signaling, 2015, 22(5), 411–423 CrossRef CAS PubMed.
- M. R. Hellmich, C. Coletta, C. Chao and C. Szabo, Antioxid. Redox Signaling, 2014, 22, 424–448 CrossRef PubMed.
- H. Kimura, Nitric Oxide, 2014, 41, 4–10 CrossRef CAS PubMed.
- K. Shatalin, E. Shatalina, A. Mironov and E. Nudler, Science, 2011, 334, 986–990 CrossRef CAS PubMed.
- C. Coletta, A. Papapetropoulos, K. Erdelyi, G. Olah, K. Modis, P. Panopoulos, A. Asimakopoulou, D. Gero, I. Sharina, E. Martin and C. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9161–9166 CrossRef CAS PubMed.
- J. B. Vicente, H. G. Colaco, M. I. Mendes, P. Sarti, P. Leandro and A. Giuffre, J. Biol. Chem., 2014, 289, 8579–8587 CrossRef CAS PubMed.
- C. Gherasim, P. K. Yadav, O. Kabil, W. N. Niu and R. Banerjee, PLoS One, 2014, 9, e85544 Search PubMed.
- A. L. King, D. J. Polhemus, S. Bhushan, H. Otsuka, K. Kondo, C. K. Nicholson, J. M. Bradley, K. N. Islam, J. W. Calvert, Y. X. Tao, T. R. Dugas, E. E. Kelley, J. W. Elrod, P. L. Huang, R. Wang and D. J. Lefer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3182–3187 CrossRef CAS PubMed.
- Z. Altaany, Y. Ju, G. Yang and R. Wang, Science Signaling, 2014, 7, ra87–ra87 CrossRef PubMed.
- M. Whiteman, L. Li, I. Kostetski, S. H. Chu, J. L. Siau, M. Bhatia and P. K. Moore, Biochem. Biophys. Res. Commun., 2006, 343, 303–310 CrossRef CAS PubMed.
- S. Koike, Y. Ogasawara, N. Shibuya, H. Kimura and K. Ishii, FEBS Lett., 2013, 587, 3548–3555 CrossRef CAS PubMed.
- Y. Kimura, Y. Mikami, K. Osumi, M. Tsugane, J. Oka and H. Kimura, FASEB J., 2013, 27, 2451–2457 CrossRef CAS PubMed.
- J. I. Toohey, Biochem. J., 1989, 264, 625–632 CrossRef CAS PubMed.
- J. I. Toohey and A. J. Cooper, Molecules, 2014, 19, 12789–12813 CrossRef CAS PubMed.
- H. Kimura, Antioxid. Redox Signaling, 2014, 22, 347–349 CrossRef PubMed.
- A. P. Munro and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 2000, 1794–1797 RSC.
- D. L. H. Williams, Nitrosation reactions and the chemistry of nitric oxide, Elsevier, Amsterdam, The Nederlands, 2004 Search PubMed.
- R. Wedmann, A. Zahl, T. E. Shubina, M. Dürr, F. W. Heinemann, B. E. C. Bugenhagen, P. Burger, I. Ivanovic-Burmazovic and M. R. Filipovic, Inorg. Chem., 2015, 54, 9367–9380 CrossRef CAS PubMed.
- M. Becke-Goehring, Jahrbuch der Heidelberger Akademie der Wissenschaften, Heidelberger Akademie der Wissenschaften, Heidelberg, 1978 Search PubMed.
- J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674–7677 CrossRef CAS.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- J. Rodriguez, R. E. Maloney, T. Rassaf, N. S. Bryan and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 336–341 CrossRef CAS PubMed.
- N. S. Bryan, T. Rassaf, R. E. Maloney, C. M. Rodriguez, F. Saijo, J. R. Rodriguez and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4308–4313 CrossRef CAS PubMed.
- K. A. Broniowska and N. Hogg, Antioxid. Redox Signaling, 2012, 17, 969–980 CrossRef CAS PubMed.
- J. Stamler and M. Feelisch, in Methods in nitric oxide research, 1996, pp. 521–539 Search PubMed.
- L. Liu, A. Hausladen, M. Zeng, L. Que, J. Heitman and J. S. Stamler, Nature, 2001, 410, 490–494 CrossRef CAS PubMed.
- M. Benhar, M. T. Forrester and J. S. Stamler, Nat. Rev. Mol. Cell Biol., 2009, 10, 721–732 CAS.
- D. R. Arnelle and J. S. Stamler, Arch. Biochem. Biophys., 1995, 318, 279–285 CrossRef CAS PubMed.
- H. O. Jones and H. S. Tasker, J. Chem. Soc. Trans., 1909, 95, 1904–1909 RSC.
- L. Field, R. V. Dilts, R. Ravichandran, P. G. Lenhert and G. E. Carnahan, J. Chem. Soc., Chem. Commun., 1978, 249–250 RSC.
- L. Jia, X. Young and W. Guo, J. Pharm. Sci., 1999, 88, 981–986 CrossRef CAS PubMed.
- N. Bainbrigge, A. R. Butler and C. Görbitz, J. Chem. Soc., Perkin Trans. 2, 1997, 351–354 RSC.
- J. Yi, P. Coppens, D. R. Powell and G. B. Richter-Addo, Comments Inorg. Chem., 2016, 36, 81–91 CrossRef CAS.
- N. Arulsamy, D. Bohle, J. Butt, G. Irvine, P. Jordan and E. Sagan, J. Am. Chem. Soc., 1999, 121, 7115–7123 CrossRef CAS.
- L. Grossi and P. C. Montevecchi, Chem. – Eur. J., 2002, 8, 380–387 CrossRef CAS.
- M. D. Bartberger, J. D. Mannion, S. C. Powell, J. S. Stamler, K. Houk and E. J. Toone, J. Am. Chem. Soc., 2001, 123, 8868–8869 CrossRef CAS PubMed.
- Q. K. Timerghazin, G. H. Peslherbe and A. M. English, Phys. Chem. Chem. Phys., 2008, 10, 1532–1539 RSC.
- J.-M. Lü, J. M. Wittbrodt, K. Wang, Z. Wen, H. B. Schlegel, P. G. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2001, 123, 2903–2904 CrossRef.
- J. McAninly, D. L. H. Williams, S. C. Askew, A. R. Butler and C. Russell, J. Chem. Soc., Chem. Commun., 1993, 1758 RSC.
- A. P. Dicks, E. Li, A. P. Munro, H. R. Swift and D. L. H. Williams, Can. J. Chem., 1998, 76, 789–794 CrossRef CAS.
- C. Toubin, D. Y. Yeung, A. M. English and G. H. Peslherbe, J. Am. Chem. Soc., 2002, 124, 14816–14817 CrossRef CAS PubMed.
- C. Baciu, K.-B. Cho and J. W. Gauld, Eur. J. Mass Spectrom., 2004, 10, 941–948 CrossRef CAS PubMed.
- C. D. Ritchie and P. Virtanen, J. Am. Chem. Soc., 1972, 94, 4966–4971 CrossRef CAS.
- D. J. Barnett, J. McAninly and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1994, 1131–1133 RSC.
- C. Adam, L. García-Río, J. R. Leis and L. Ribeiro, J. Org. Chem., 2005, 70, 6353–6361 CrossRef CAS PubMed.
- L. A. Peterson, T. Wagener, H. Sies and W. Stahl, Chem. Res. Toxicol., 2007, 20, 721–723 CrossRef CAS PubMed.
- M. Goehring and J. Messner, Z. Anorg. Allg. Chem., 1952, 268, 47–56 CrossRef CAS.
- P. W. Schenk, Ber. dtsch. Chem. Ges. A/B, 1942, 75, 94–99 CrossRef.
- R. P. Müller, M. Nonella and J. R. Huber, Chem. Phys., 1984, 87, 351–361 CrossRef.
- M. Nonella, J. R. Huber and T. K. Ha, J. Phys. Chem., 1987, 91, 5203–5209 CrossRef CAS.
- M. Goehring, H. W. Kaloumenos and J. Messner, Z. Anorg. Allg. Chem., 1951, 264, 48–53 CrossRef CAS.
- M. Méndez, J. S. Francisco and D. A. Dixon, Chem. – Eur. J., 2014, 20, 10231–10235 CrossRef PubMed.
- C.-H. Lai, E. Y. Li and P.-T. Chou, Theor. Chem. Acc., 2007, 117, 145–152 CrossRef CAS.
- T. Chivers, A. B. Da Silva, O. Treu and M. Trsic, J. Mol. Struct., 1987, 162, 351–357 CrossRef CAS.
- M. R. Filipovic, J. Miljkovic, T. Nauser, M. Royzen, K. Klos, T. Shubina, W. H. Koppenol, S. J. Lippard and I. Ivanovic-Burmazovic, J. Am. Chem. Soc., 2012, 134, 12016–12027 CrossRef CAS PubMed.
- B. Roy, A. du Moulinet d'Hardemare and M. Fontecave, J. Org. Chem., 1994, 59, 7019–7026 CrossRef CAS.
- P. O. Tchir and R. D. Spratley, Can. J. Chem., 1975, 53, 2318–2330 CrossRef CAS.
- F. Seel, R. Kuhn, G. Simon, M. Wagner, B. Krebs and M. Dartmann, Z. Naturforsch., B: Chem. Sci., 1985, 40, 1607–1617 Search PubMed.
- F. Seel, G. Simon, J. Schuh, M. Wagner, B. Wolf, I. Ruppert and A. B. Wieckowski, Z. Anorg. Allg. Chem., 1986, 538, 177–190 CrossRef CAS.
- F. Seel and H. J. Güttler, Angew. Chem., 1973, 12, 420–421 Search PubMed.
- T. Chivers and I. Drummond, Inorg. Chem., 1972, 11, 2525–2527 CrossRef CAS.
- F. SeeI, H.-J. GiittIer, A. B. Wieckowski and B. WoIf, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1979, 34, 1671–1677 Search PubMed.
- T. Chivers, Nature, 1974, 252, 32–33 CrossRef CAS.
- T. Chivers and P. J. Elder, Chem. Soc. Rev., 2013, 42, 5996–6005 RSC.
- M. Schmidt and R. Wägerle, Angew. Chem., 1958, 70, 594–595 CrossRef CAS.
- M. Schmidt and R. R. Wägerle, Z. Anorg. Allg. Chem., 1964, 330, 48–58 CrossRef CAS.
- F. Seel, R. Kuhn and G. Simon, Z. Naturforsch., B: Chem. Sci., 1984, 39, 1622–1623 Search PubMed.
- R. Steudel, Ind. Eng. Chem. Res., 1996, 35, 1417–1423 CrossRef CAS.
- I. Artaud and E. Galardon, ChemBioChem, 2014, 15, 2361–2364 CrossRef CAS PubMed.
- T. S. Bailey, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2014, 136, 10573–10576 CrossRef CAS PubMed.
- N. E. Francoleon, S. J. Carrington and J. M. Fukuto, Arch. Biochem. Biophys., 2011, 516, 146–153 CrossRef CAS PubMed.
- Y. Gao, A. Toubaei, X. Kong and G. Wu, Chem. – Eur. J., 2015, 21, 17172–17177 CrossRef CAS PubMed.
- D. S. Bohle, C.-H. Hung and B. D. Smith, Can. J. Chem., 2005, 83, 2021–2031 CrossRef CAS.
- T. Trabelsi, O. Yazidi, J. Francisco, R. Linguerri and M. Hochlaf, J. Chem. Phys., 2015, 143, 164301 CrossRef CAS PubMed.
- D. S. Bohle, C.-H. Hung, A. K. Powell, B. D. Smith and S. Wocadlo, Inorg. Chem., 1997, 36, 1992–1993 CrossRef CAS PubMed.
- A. P. Munro, S-nitrosothiols: novel decomposition pathways including reactions with sulfur and nitrogen nucleophiles, Doctoral thesis, Durham University, 1999 Search PubMed.
- E. Victor, Reactions of S-nitrosothiols with biomimetic iron complexes and other transition metals, Doctoral thesis, Massachusetts Institute of Technology, 2014 Search PubMed.
- A. Martinsen and J. Songstad, Acta Chem. Scand., Ser. A, 1977, 31, 645–650 CrossRef CAS.
- A. Berenyiova, M. Grman, A. Mijuskovic, A. Stasko, A. Misak, P. Nagy, E. Ondriasova, S. Cacanyiova, V. Brezova, M. Feelisch and K. Ondrias, Nitric Oxide, 2015, 46, 123–130 CrossRef CAS PubMed.
- L. K. Keefer, R. W. Nims, K. M. Davies and D. A. Wink, Methods Enzymol., 1996, 268, 281–293 CAS.
- B. J. De Witt, J. R. Marrone, A. D. Kaye, L. K. Keefer and P. J. Kadowitz, Eur. J. Pharmacol., 2001, 430, 311–315 CrossRef CAS PubMed.
- J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135–1154 CrossRef CAS PubMed.
- G. Brauer, Handbuch der präparativen anorganischen Chemie, Ferdinand Enke Verlag, Stuttgart, 3rd edn, 1975 Search PubMed.
- M. Feelisch, Naunyn-Schmiedeberg's Arch. Pharmacol., 1998, 358, 113–122 CrossRef CAS PubMed.
- E. Cox, G. Jeffrey and H. Stadler, Nature, 1948, 162, 770 CrossRef CAS.
- J. Reglinski, D. R. Armstrong, K. Sealey and M. D. Spicer, Inorg. Chem., 1999, 38, 733–737 CrossRef CAS PubMed.
- A. Berenyiova, M. Grman, A. Mijuskovic, A. Stasko, A. Misak, P. Nagy, E. Ondriasova, S. Cacanyiova, V. Brezova, M. Feelisch and K. Ondrias, Nitric Oxide, 2015, 46, 123–130 CrossRef CAS PubMed.
- K. Ondrias, A. Stasko, S. Cacanyiova, Z. Sulova, O. Krizanova, F. Kristek, L. Malekova, V. Knezl and A. Breier, Pflugers Arch., 2008, 457, 271–279 CrossRef CAS PubMed.
- X. Teng, T. Scott Isbell, J. H. Crawford, C. A. Bosworth, G. I. Giles, J. R. Koenitzer, J. R. Lancaster, J. E. Doeller, D. W. Kraus and R. P. Patel, Methods Enzymol., 2008, 441, 161–172 CAS.
- K. M. Miranda, N. Paolocci, T. Katori, D. D. Thomas, E. Ford, M. D. Bartberger, M. G. Espey, D. A. Kass, M. Feelisch, J. M. Fukuto and D. A. Wink, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9196–9201 CrossRef CAS PubMed.
- N. Paolocci, T. Katori, H. C. Champion, M. E. St John, K. M. Miranda, J. M. Fukuto, D. A. Wink and D. A. Kass, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5537–5542 CrossRef CAS PubMed.
- J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1620–1624 CrossRef CAS.
- J. M. Fukuto and L. J. Ignarro, Acc. Chem. Res., 1997, 30, 149–152 CrossRef CAS.
- G. L. Squadrito and W. A. Pryor, Chem.-Biol. Interact., 1995, 96, 203–206 CrossRef CAS PubMed.
- R. Radi, J. Biol. Chem., 2013, 288, 26464–26472 CrossRef CAS PubMed.
- R. Steudel, in Elemental Sulfur und Sulfur-Rich Compounds II, Springer, 2003, pp. 99–126 Search PubMed.
- R. Steudel, in Elemental Sulfur und Sulfur-Rich Compounds II, Springer, 2003, pp. 127–152 Search PubMed.
- C. Dahl, A. Prange and R. Steudel, in Biopolymers Online, 2005, p. 9 Search PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457–464 CrossRef CAS PubMed.
- K. Ranguelova, A. B. Rice, O. M. Lardinois, M. Triquigneaux, N. Steinckwich, L. J. Deterding, S. Garantziotis and R. P. Mason, Free Radicals Biol. Med., 2013, 60, 98–106 CrossRef CAS PubMed.
- B. G. Hill, B. P. Dranka, S. M. Bailey, J. R. Lancaster and V. M. Darley-Usmar, J. Biol. Chem., 2010, 285, 19699–19704 CrossRef CAS PubMed.
- J. M. Fukuto, S. J. Carrington, D. J. Tantillo, J. G. Harrison, L. J. Ignarro, B. A. Freeman, A. Chen and D. A. Wink, Chem. Res. Toxicol., 2012, 25, 769–793 CrossRef CAS PubMed.
- G. I. Giles and C. Jacob, Biol. Chem., 2002, 383, 375–388 CrossRef CAS PubMed.
Footnotes |
| † Hydrogen sulfide is a weak diprotic acid. In aqueous solution at neutral pH it will be mainly present as hydrosulfide anion (HS−), followed by dissolved dihydrogen sulfide (H2S) and negligible amounts of the sulfide dianion (S2−), according to its pKa1 = 6.8 and pKa2 > 12. We use the term ‘sulfide’ to refer collectively to all those species in equilibrium. |
| ‡ Most of the species we will discuss in this context also contain one or more oxygens and are therefore S–N–O species; however, we prefer to stay with the original designation in order to avoid misunderstandings with S-nitrosothiol chemistry as bonding orders can change, and not all species are in fact nitroso compounds. |
| § In the biological literature NO, H2S and CO are often referred to as “gasotransmitters”. Clearly, in biological environments these species are not normally present in gaseous form but as either uncharged (NO, CO, H2S) or charged (HS−) solutes. |
| This journal is © The Royal Society of Chemistry 2016 |