
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and catalytic applications of 1,2,3-triazolylidene gold(I) complexes in silver-free oxazoline syntheses and C–H bond activation†
René
Pretorius
ab,
Manuel R.
Fructos
c,
Helge
Müller-Bunz
b,
Robert A.
Gossage
ad,
Pedro J.
Pérez
*c and
Martin
Albrecht
*ab
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: perez@dqcm.uhu.es; martin.albrecht@dcb.unibe.ch
bSchool of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland
cLaboratorio de Catálisis Homogénea, Unidad Asociada al CSIC, CIQSO-Centro de Investigación en Química Sostenible and Departamento de Química, Universidad de Huelva, Campus de El Carmen, 21007 Huelva, Spain
dDepartment of Chemistry & Biology, Ryerson University, 350 Victoria Street, Toronto, ON M5B 2K3, Canada
First published on 24th June 2016
Abstract
A series of novel 1,2,3-triazolylidene gold(I) chloride complexes have been synthesised and fully characterised. Silver-free methodologies for chloride ion abstraction of these complexes were evaluated for their potential as Au-based catalyst precursors. Using simple potassium salts or MeOTf as chloride scavengers produced metal complexes that catalyse both the regioselective synthesis of oxazolines and the C–H activation of benzene or styrene for carbene transfer from ethyl diazoacetate. These results indicate that Ag-free activation of 1,2,3-triazolylidene gold(I) chloride complexes is feasible for the generation of catalytically active Au triazolylidene species. However, silver-mediated activation imparts substantially higher catalytic activity in oxazoline synthesis.
Introduction
Homogeneous catalysts comprised of gold as the active centre have enjoyed increased attention in recent years as more and more unique reactivity patterns have been revealed.1 In particular, ligation by N-heterocyclic carbenes (NHCs) as for example in AuCl(IPr) (IPr = N,N′-bis(2,6-diisopropylphenyl)imidazolylidene) imparted exceptionally high catalytic activity,1k,2 which spurred widespread efforts in this burgeoning area of catalysis. In comparison to classical, imidazole-derived NHCs, structurally related 1,2,3-triazolylidene (trz) ligands3 have been significantly less studied as ligands in gold chemistry.4 The triazole framework offers several benefits when compared with NHCs, including, for example, the ease of functional group incorporation,3a,5 increased σ-donation compared to classical NHCs,3,6 and a mesoionic resonance form which is able to potentially stabilise different metal oxidation states.7 Catalytic activity in carbene gold complexes has typically been instigated by using a silver(I) salt as halide scavenger for catalyst activation.8 However, Ag+ can be a non-innocent activator in many Au-catalysed transformations, and often plays an active (and not always beneficial) role in the catalytic cycle.1d,9 Current trends in Au catalysis are therefore focusing on using Ag-free protocols to avoid any undesired side-reactions. For example, pre-activation and subsequent silver salt removal from a stable cationic gold(I) complex as catalyst precursor has been described by Gagosz10 Echavarren,11 and others.12 Also, the use of a gold(I) hydroxide13 or acetate14 pre-catalyst in lieu of gold(I) chloride complexes has also been shown to be an effective method to avoid the necessity of silver salts for catalyst activation. Several approaches towards Ag-free activation have been probed, though almost none are based on simple and inexpensive (commercial) salts. Thus, the in situ generation of active gold(I) catalysts has been explored by using Lewis acids as activators such as metal triflates,15 and alkali metal salts of borates,4a,16 to name a few.17 However, with the exception of Cu(OTf)2 (OTf = CF3SO3), the activators are typically non-commercial and customised salts.We have recently demonstrated the non-innocent behaviour of silver(I) salts when activating 1,2,3-triazolylidene gold complexes.18 Specifically, silver mediated carbene transfer reactions have been demonstrated, likely via a reverse carbene transfer from gold(I) to silver(I). Considering the significant complications arising from such ligand transfer, we endeavoured to establish alternative procedures for the activation of AuCl(trz) complexes for catalytic bond activation purposes, which do not rely on Ag+ for chloride abstraction. Herein we demonstrate that simple potassium salts can be used to form catalytically active triazolylidene gold complexes. These methodologies are of further use for exploring the specific impact of triazolylidene ligands in gold(I)-mediated catalytic activity.
Results and discussion
Synthesis of triazolylidene gold complexes
A set of novel triazolylidene gold complexes AuCl(trz) were synthesised as representative examples intending to probe ligand induced catalytic effects. The ligand precursor triazolium salts 1a–e (Scheme 1) were readily available through copper(I)-catalysed [2 + 3] dipolar cycloaddition reactions and subsequent alkylation with either (Me3O)BF4, MeI, or MeOTf (OTf− = SO3CF3−). Of note, triazolium 1b, which contains a pendant acetate group was highly sensitive to hydrolysis and hence the alkylation of the triazole with MeOTf was performed at low temperature (below 0 °C). Subsequent storage of the triazolium salt 1b below −20 °C is paramount to avoid spontaneous degradation over time. Salt 1c was obtained in relatively low yields (about 30%) when compared to the other triazolium salts (≥79%), presumably because the electron withdrawing nature of the perfluorinated tail decreases the nucleophilicity of the triazole unit. The influence of the fluorocarbon unit is supported by the 1H NMR chemical shifts for the NCH2 signals, which are shifted downfield by 0.72 and 0.38 ppm, for N1 and N3 respectively, in comparison with the analogous protons of 1a. Moreover, the N-bound methylene group appears in the 13C NMR spectrum as a triplet (3JCF = 21 Hz), indicating an interaction between the perfluorinated tail and the methylene group. In contrast, the 13C NMR chemical shifts do not show any significant variation due to the more electron withdrawing nature of the fluoro-chain.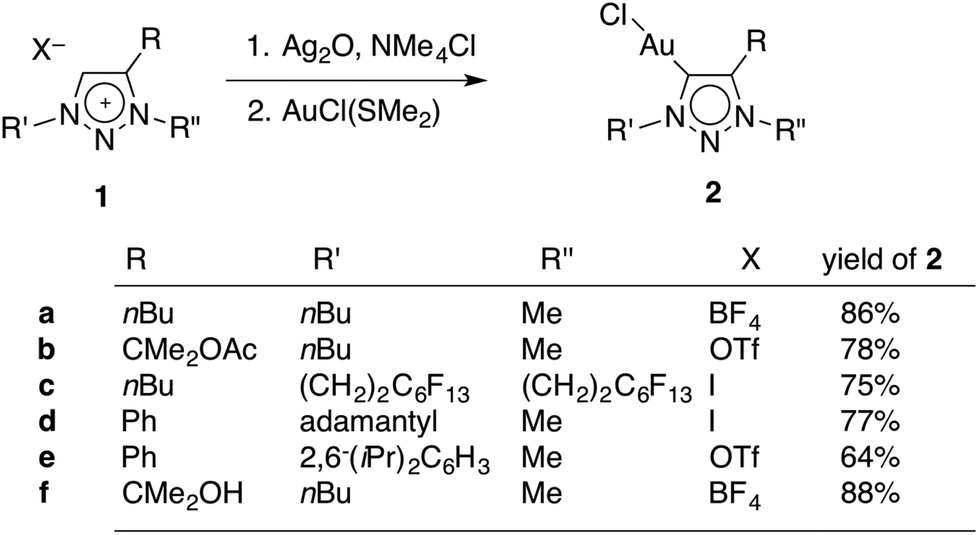 | ||
| Scheme 1 Synthesis of triazolylidene gold(I) complexes 2. | ||
The AuCl(trz) complexes were synthesised from the triazolium salts 1a–f using well established methodologies (Scheme 1).4g,18,19 Accordingly, treatment of the appropriate triazolium salt with Ag2O yielded the corresponding carbene silver intermediate, which was not isolated and used in situ for transmetallation with AuCl(SMe2). To ensure that chlorido complexes were formed exclusively, all novel AuCl(trz) complexes were prepared from triazolium salts containing either a BF4− or a OTf− counterions, or alternatively by counterion exchange with AgBF4 directly before the reaction with Ag2O (for 1c). Only traces of triazolylidene silver intermediate were formed in the absence of a chloride source according to 1H NMR spectroscopic monitoring of the reaction, while clean and essentially quantitative formation of the intermediate was observed in the presence of Me4NCl. The subsequent transmetalation was straightforward and good yields ranging from 64–88% were obtained for all gold complexes 2a–f (see Experimental section).
Metallation of the hydroxyl-functionalised triazolium salt 1f gave a mixture of products, with ratios that were dependent on the reaction conditions. In the presence of an excess Ag2O and in moist solvents, complex 2f was obtained in nearly 90% yield. Metallation of 1f under an atmosphere of dry nitrogen and substoichiometric amounts of Ag2O afforded significant amounts (up to 46%) of complexes 2g and 2g′ as the major products (Scheme 2). We have not been able to separate these two products preparatively, though fractional crystallisation afforded single crystals of pure 2g that were suitable for X-ray diffraction analysis (see below). Dissociation of the C-bound substituent in 2g and 2g′ with formal loss of acetone was indicated by NMR spectroscopy. Two sets of resonances were observed, both featuring an additional resonance in the aromatic region and the disappearance of the two methyl groups of the wingtip unit of 1f. Integration of the 1H NMR resonances of the NCH2 and NCH3 signals suggest a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of 2g and 2g′. The spectroscopic assignment was based on long range 1H–13C correlation spectroscopy (HMBC). Accordingly, the NCH3 group of the C4-metallated complex 2g resonantes at 4.20 ppm (cf. δH = 4.17 ppm in the C5-metallated homologue 2g′), whereas the NCH2 group in 2g′ appears at lower field than in 2g (δH = 4.50 vs. 4.38 ppm) and provides the clearest spectroscopic distinction between the two isomers. The Ctrz–H resonance shift is essentially identical in both complexes and appears at 7.58 and 7.57 ppm, respectively. Similarly, the 13C NMR shifts of the two isomers are only marginally disparate. In agreement with the 1H NMR data, the largest difference again pertains to the NCH3 group (δC = 42.1 and 38.8) and the NCH2 group (δC = 52.5 and 55.4 for 2g and 2g′, respectively). The resonances due to the CtrzH carbons were significantly upfield shifted (δC = 134 ± 0.4) when compared to the normal 145–150 ppm range observed for C5-alkylated or -arylated triazolylidene gold complexes.
:4 ratio of 2g and 2g′. The spectroscopic assignment was based on long range 1H–13C correlation spectroscopy (HMBC). Accordingly, the NCH3 group of the C4-metallated complex 2g resonantes at 4.20 ppm (cf. δH = 4.17 ppm in the C5-metallated homologue 2g′), whereas the NCH2 group in 2g′ appears at lower field than in 2g (δH = 4.50 vs. 4.38 ppm) and provides the clearest spectroscopic distinction between the two isomers. The Ctrz–H resonance shift is essentially identical in both complexes and appears at 7.58 and 7.57 ppm, respectively. Similarly, the 13C NMR shifts of the two isomers are only marginally disparate. In agreement with the 1H NMR data, the largest difference again pertains to the NCH3 group (δC = 42.1 and 38.8) and the NCH2 group (δC = 52.5 and 55.4 for 2g and 2g′, respectively). The resonances due to the CtrzH carbons were significantly upfield shifted (δC = 134 ± 0.4) when compared to the normal 145–150 ppm range observed for C5-alkylated or -arylated triazolylidene gold complexes.
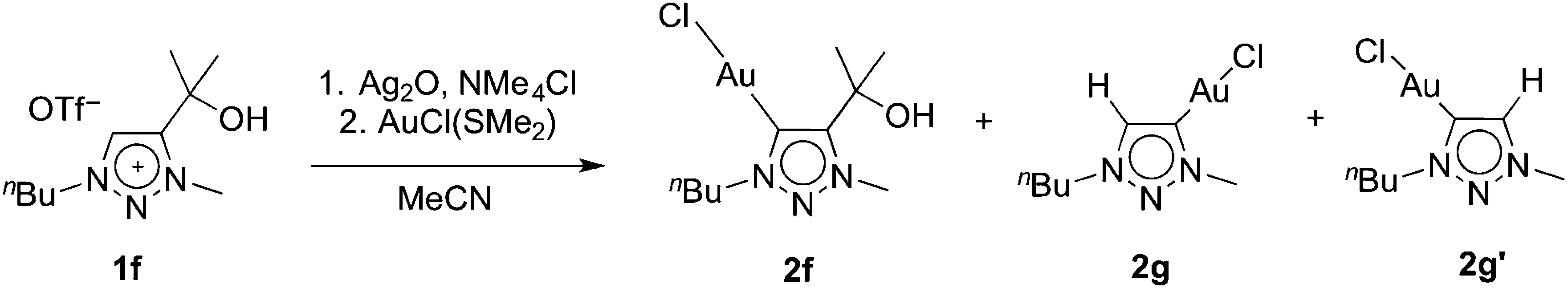 | ||
| Scheme 2 Transmetallation of 1f and concurrent partial formal loss of acetone resulting in the formation of 2f, 2g, and 2g′. | ||
Formation of complexes 2g and 2g′ may be rationalised by deprotonation of the hydroxyl group, which is followed by elimination of acetone from the ligand. The observation of isomers suggests that the ligand degraded before the triazolylidene silver intermediate is formed. Metallation of a C4,C5-diprotonated triazolium salt is unselective and may thus account for the formation of a mixture of 2g and 2g′. The influence of Ag2O was therefore investigated in more detail. Using various 1f/Ag2O ratios (0.5, 1, 2) and monitoring of the triazolylidene silver complex formation indicated that higher equivalents of Ag2O promoted the preservation of the wingtip group and hence promoted the formation of the silver intermediate that eventually leads to complex 2f, while sub-stoichiometric quantities of Ag2O increased substituent degradation. The isomeric ratio of the unsubstituted triazolylidene silver complexes is independent of the quantity of silver salt (cf. Table S1†), remains unchanged over time, and is in good agreement with the final product distribution observed for formation of complexes 2g and 2g′. These data therefore suggest that wingtip degradation of the ligand occurs prior to transmetallation and that acetone elimination is a competitive process to triazolium Ctrz–H bond activation. Moreover, these results indicate that wingtip degradation only takes place from the triazolium salt, yet it is suppressed once the carbene silver precursor is formed. Otherwise, larger quantities of Ag2O should shift the isomeric ratio due to degradation of the silver precursor of 2f.
Characterisation of triazolylidene gold complexes
The 13C NMR spectra of all triazolylidene gold complexes feature a resonance in the 156–162 ppm range for the Au-bound Ctrz nucleus, consistent with related triazolylidene gold complexes.4,18–20 The carbenic carbon is most deshielded in complex 2e (δC = 162.1 ppm), and resonates at higher field in the alkyl-substituted triazolylidene complexes (δC = 157.5 ± 2.3 ppm) than in aryl-substituted triazolylidene complexes, suggesting that the electronic configuration of this carbon is affected by the triazolylidene substitution pattern. The polyfluorinated tail had no significant influence on the NMR frequency of the carbenic carbon. However, the NCH2 group in complex 2c is deshielded by 0.4 ppm (1H NMR) when compared to the butyl homolog 2a, and likewise shielded by about 8 ppm in the 13C NMR spectrum.Single crystal X-ray diffraction analysis of complexes 2b, 2f, and 2g confirmed the bonding situation in these complexes and in particular demonstrated the wingtip dissociation in 2g (Fig. 1). Bond lengths and angles of these complexes (Table 1) do not significantly deviate from related carbene gold complexes characterised previously.4,18–20 However, some packing trends are worth noting. Complex 2g displays a head-to-tail type packing, attributed to presumably weak aurophilic interactions (Au⋯Au separation of 3.46 Å) which may add to the electrostatic interaction between the chlorido ligand and the heterocyclic ring.1f,21 This arrangement may be preferred due to the low steric demand of this ligand system, and hence allows for much closer packing than that of, for example, complex 2b.
 | ||
| Fig. 1 ORTEP representations of complex 2b (a), 2f (b), and 2g (c; all 50% probability ellipsoids, H-atoms and second independent molecule of 2g in the unit cell omitted for clarity). | ||
| Complex | Au–C1 | Au–Cl | C1–Au–Cl |
|---|---|---|---|
| a Only for one of the two non-symmetry related molecules in the unit cell of 2g. | |||
| 2b | 1.995(2) | 2.2896(6) | 178.08(7) |
| 2f | 1.976(6) | 2.2873(19) | 178.1(2) |
| 2g | 1.984(2) | 2.2978(6) | 175.90(7) |
Catalysis results and activation studies
Previous work has clearly shown that Ag(I) can be a non-innocent activator for Au-catalysed transformations.1d,9c,d,18,22 In particular when using AuCl(trz) system, silver has multiple roles beyond solely abstracting the gold-bound chlorido ligand and likely induces a retro-transmetallation to (transiently) yield catalytically active triazolylidene silver complexes.9c,18,23 Because of these complications and the associated uncertainty whether the true catalytic species is (are) based on silver or gold, silver-free catalyst activation methodologies are of paramount importance.16 We considered two specific routes: (i) halide abstraction with a potassium salt, and (ii) the use of CH3+ as an electrophile. Even though methyl cations are only rarely used to scavenge chloride, chloride to halide exchange reactions at the metal centre have been performed successfully with methyl iodide.24 Moreover, C–C bond formation by electrophilic activation of (NHC)Au(Aryl) complexes has been reported.25 To probe this methodology, complex 2a was reacted with stoichiometric equivalents of MeOTf in MeCN. Initial attempts to isolate the putative cationic solvent complex [(trz)Au(solv)]OTf from the reaction mixture only afforded Au(0), indicated by the gold mirror that formed upon evaporation of all volatiles. Monitoring the reaction by 19F NMR spectroscopy (CD2Cl2) indicated a new resonance that is shifted 5 ppm upfield from MeOTf, attributed to the gradual formation of a new ion pair in solution (see ESI, Fig. S1†). The corresponding 1H NMR data showed only minor changes in the ligand, and a new resonance around 3 ppm which has been assigned to CH3Cl. In order to overcome the apparent instability of the cationic species resulting from 2a, it was trapped by the addition of an exogenous ligand. Addition of isonitrile (methyl isocyanoacetate) gave the corresponding cationic complex (see ESI†).18 Likewise, addition of PPh3 before evaporation of the solvent afforded the cationic gold species 3a. This complex was unambiguously identified by NMR spectroscopy and X-ray diffraction (Fig. 2). However, it should be noted that the phosphine carbene gold complex cation is also formed upon treatment of 2a with PPh3 in the absence of any electrophilic chloride scavenger (albeit with a chloride counterion), which limits the relevance of phosphines as trapping reactions. In contrast, isonitrile binding does not occur with the [AuCl(trz)] complexes 2 spontaneously and was only observed in the presence of MeOTf, suggesting that isonitriles are more diagnostic trapping agents. | ||
| Fig. 2 Synthesis and ORTEP representation of complex 3a (50% probability, hydrogens and OTf− anion omitted for clarity); Au1–C1 2.046(3) Å, Au1–P1 2.2817(8), Cl1–Au–P1 172.57(9)°. | ||
Analysis of complex 3a by 13C and 31P NMR spectroscopy reveals similar features when compared to related [(trz)Au(PPh3)]+ and [(NHC)Au(PPh3)]+ complexes (e.g. δP = 39.4 for 3a).18,26 The carbenic nucleus is deshielded by about 15 ppm compared to the chlorido analogue 2a and appears at δC = 172, in agreement with a more electron-deficient metal fragment. X-ray diffraction analysis of single crystals of 3a indicates an elongation of the gold carbene distance compared to 2a (Au–C1 2.046(3) Å in 3avs. 1.98 Å in complexes 2), a consequence of the higher trans influence of PPh3 compared to the chlorido ligand. The coordination geometry slightly deviates from linearity, P–Au–C1 172.57(9)°, presumably due to packing constraints. Alternating layers of triflate ions and triazolium units with close contacts of 3.13 and 3.53 Å suggest electrostatic interactions in the solid state between the formally negatively charged triflate oxygen sites and the partially positively charged heterocycle.
Catalytic activity in oxazoline synthesis
Halide abstraction under catalytically relevant conditions was probed in the presence of isonitriles and bromobenzaldehyde to promote the catalytic formation of oxazolines.12d–g,18,27 Complex 2a was chosen as a representative AuCl(trz) complex because of the simple ligand design and the established benefit of the triazolylidene substitution pattern for monitoring catalytic reactions.7d Activation of this complex for initiating catalytic activity was investigated using MeOTf as well as different potassium salts (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Au] | Additive | Base | Conversion (%) | cis/trans |
| a General conditions: 4-bromobenzylaldehyde (1.6 mmol), methyl isocyanoacetate (1.4 mmol), NiPr2Et (0.14 mmol), AuCl(trz) (0.014 mmol), additive (0.028 mmol), trimethoxybenzene (0.36 mmol, internal standard), CH2Cl2 (4 mL), 40 °C, 8 h; yields and cis/trans ratio determined by 1H NMR spectroscopy, averaged of at least 2 runs. | |||||
| 1 | — | — | — | 6 | — |
| 2 | — | KPF6 | NiPr2Et | 25 | — |
| 3 | 2a | — | NiPr2Et | 24 | 23/77 |
| 4 | 2a | KPF6 | — | <2 | — |
| 5 | 2a | KPF6 | NiPr2Et | 93 | 26/74 |
| 6 | 2a | KBF4 | NiPr2Et | 21 | 18/82 |
| 7 | 2a | KOTf | NiPr2Et | 61 | 28/72 |
| 8 | 2a | MeOTf | NiPr2Et | 62 | 27/73 |
| 9 | 2b | KPF6 | NiPr2Et | 88 | 27/73 |
| 10 | 2c | KPF6 | NiPr2Et | 75 | 29/71 |
| 11 | 2d | KPF6 | NiPr2Et | 87 | 29/71 |
A systematic variation of the different reaction parameters revealed that the gold complex, amine, and a chloride scavenger are needed for generating an active species (entries 1–5).28 Comparison of different chloride scavenging additives revealed that KPF6 (entry 5) is superior to other potassium salts (entries 6 and 7). The addition of NiPr2Et is essential for catalytic turnover, as no catalytic activity was observed in the absence of base (entry 4).18 Likewise the reaction is very slow when no scavenger was used (entry 3). Interestingly, KBF4 has no activating effect (cf. entries 3 and 6). The utilisation of MeOTf enhanced catalytic activity (entry 8), suggesting that chloride methylation is a viable strategy to generate the labile coordination site at the gold centre. It is remarkable that KOTf and MeOTf result in the same catalytic activity (entries 7 and 8) and give identical conversions, suggesting a distinct role for the OTf− anion in levelling catalyst activity.1e Tentatively, weak triflate coordination may be surmised to slow down the turnover frequency when compared to the analogous species containing a PF6− counterion. The relevance of the counterion is also indicated by the variable catalytic activity in dependence of different anions in the potassium salts (entries 5–7).
Further studies on using MeOTf to activate the AuCl(trz) catalyst precursor included the variation of the equivalents of this activator, a modification which affected the conversion rates markedly. Using only 1.1 instead of 2 equiv. MeOTf with respect to 2a reduced the activity to about half (e.g. 21% vs. 38% conversion after 4 h, Fig. 3 and Table S2†). In the presence of 5 equiv. MeOTf, the catalytic activity is doubled and almost full conversion was accomplished after 8 h (45% already after 2 h), which matches the conversion rates achieved upon activation of the gold complex with KPF6. The need for higher relative ratios of additive may be because MeOTf may interact with NiPr2Et or undergo partial hydrolysis.
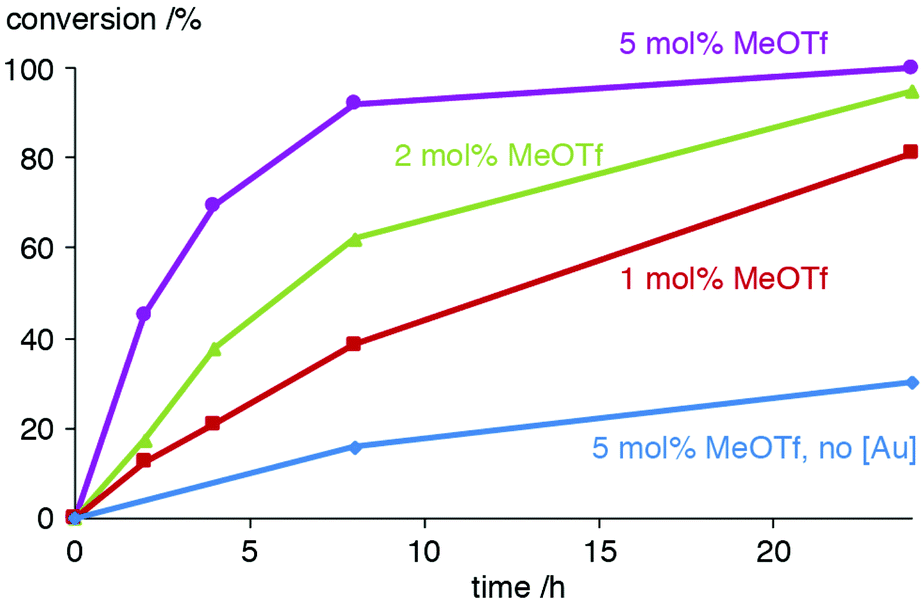 | ||
| Fig. 3 Comparison of the conversion for catalytic oxazoline synthesis using 2a in combination with varying amounts of MeOTf (red, green, and purple traces), and using MeOTf in the absence of a gold complex (blue trace). | ||
The enhancement of catalytic activity in the presence of KOTf, MeOTf and in particular KPF6 suggests the successful formation of a [(trz)Au]+ species for substrate conversion. Therefore, KPF6, was utilised as activator for a catalytic screening of the AuCl(trz) complexes 2a–d (entries 5, 9–11). In this series, the catalytic activity of 2a, 2b, and 2d is identical within the measurement errors (±5%). Complex 2c containing perfluorinated substituents on the triazolylidene induced slightly lower catalytic activity. This effect is particularly pronounced at early conversion stages. Thus after 2 h and 4 h, runs with complex 2c achieved 25% and 43% conversion, respectively, while the other complexes are considerably more active (average 38% and 65% conversion after the same reaction time). The lower performance of complex 2c may be associated with the specific solubility and aggregation properties imparted by the perfluorinated substituents. Variation of the hydrocarbon substituents on the triazolylidene ligand from linear or branched alkyl groups to aryl substituents does not affect the catalytic performance (cf.2avs.2b and 2d). These results therefore suggest that the catalytic rate-determining step is not significantly governed by steric or electronic effects induced by the triazolylidene ligand. This conclusion is further supported by the essentially unaltered cis/trans ratio, even when a sterically demanding adamantyl group was included in the ligand scaffold (2d, entry 11).
Importantly, activation of complex 2a with AgBF4 affords a species that is two to three orders of magnitude more active than the species generated with KPF6 or triflates, reaching quantitative oxazoline formation within 15 min at 0.5 mol% loading.18 Similarly high activities have been recorded when related triazolylidene silver complexes (trz)AgCl were used for catalysis in the absence of gold.29 Thus, the gold-based active species are considerably less competitive than their triazolylidene silver analogues in this catalytic heterocycle synthesis. These data further suggest that the catalytically active component in the AuCl(trz)/AgBF4 system may be a [(trz)Ag]+ species resulting from a retro-transmetallation or the formation of Au nanoparticles.15a,18,29
C–H bond activation in metal-mediated carbene transfer from ethyl diazoacetate
The application of Ag-free [(trz)Au]+ catalysis was further probed in carbene transfer chemistry by the metal-mediated decomposition of diazo compounds. This catalytic system is attractive for several reasons. Firstly, NaBArF4 (BArF4 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) has been shown to be an excellent activator for AuCl(NHC) catalysts in this area of catalysis and hence allows for Ag-free activation.30 In particular the AuCl(IPr)/NaBArF4 system has given promising results with arenes as substrates. With previously described catalytic systems,31 the reaction of benzene and ethyl diazoacetate (EDA) led to cycloheptatriene (4 in Table 3), as the result of consecutive double bond cyclopropanation and ring expansion in the so-called Buchner reaction. However, the gold-based system is selective towards C–H bond functionalisation of benzene and other arenes by formal carbene insertion affording an alkyl-arene (5 in Table 3).30a–c Furthermore, it was shown that AuCl(trz)/AgSbF6 systems are useful carbene transfer reagents from EDA to both reactive N- or O-containing substrates.4g Since the activity of the AuCl(trz) systems for C–H insertion has not been studied, the catalytic C–H activation with EDA seems an attractive expansion to investigate Ag-free activation of AuCl(trz). Benzene was chosen as a representative substrate for preliminary screening, and the activity of complexes 2a–2e in catalytic carbene transfer was monitored using NaBArF4 as additive (Table 3). While a mixture of the aforementioned cycloheptatriene 4 and benzyl acetate 5 is expected, these two products are easily distinguishable by their diagnostic NMR signals.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Au] | Additive | Product ratiob | ||
| 4 | 5 | DEF/DEMc | |||
| a General conditions: [Au] (0.012 mmol), benzene (3 mL), EDA (0.44 mmol), NaBArF4 (0.015 mmol), ratios in % of product mixture. b Distribution of products according to 1H NMR spectroscopy, ca. 15% of EDA were remaining after 24 h as quantified with benzaldehyde as internal standard. c DEF/DEM: diethyl fumarate/diethyl maleate, by products of the carbene coupling reaction from two molecules of EDA. | |||||
| 1 | 2a | NaBArF4 | 68 | 17 | 15 |
| 2 | 2b | NaBArF4 | 65 | 18 | 17 |
| 3 | 2c | NaBArF4 | 61 | 19 | 21 |
| 4 | 2d | NaBArF4 | 57 | 17 | 26 |
| 5 | 2e | NaBArF4 | 67 | 16 | 17 |
| 6 | 2a | AgBF4 | 77 | 9 | 14 |
| 7 | 2a | AgSbF6 | 64 | 18 | 18 |
| 8 | 2e | AgSbF6 | 62 | 22 | 16 |
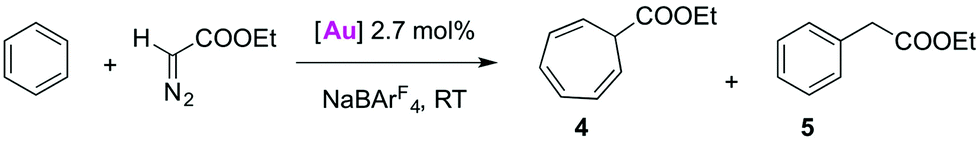
All AuCl(trz) complexes 2a–e favoured the formation of the cycloheptatriene product 4 over the product from C–H insertion in approximately a 3:1 ratio (Table 3, entries 1–5). The catalytic activity is essentially insensitive to the substitution pattern on the triazolylidene heterocycle with overall product ratios, i.e.4 + 5, all in the 75–85% range. The influence of silver salts as halide scavengers was also considered to determine potential interactions of silver in the catalytic cycle (entries 6–8). While the use of AgBF4 further promoted cyclopropanation and yielded a 8:1 selectivity towards 4 (entry 6), AgSbF6 did not alter the product distribution when compared to the activation of AuCl(trz) with NaBArF4 (entries 7 and 8). We also noted that the consumption of EDA was not complete (ca. 85%, see Table 3) even after 24 h. Furthermore, the species generated from AuCl(trz) and NaBArF4 in CH2Cl2 was unstable in the absence of coordinating substrates as displayed by the immediate formation of gold(0) and gold nanoparticles observed as purple particles in solution. This instability required addition of EDA before introducing the additive for generating the active catalyst. This sequence of addition resulted in the formation of fumarate and maleate as minor by-products. Such side-product formation is typically suppressed by slow addition of EDA,32 though this procedure is prohibited here because of the instability of the [Au(trz)]+/BArF4 species.
Similar reactivity patterns were observed when using selected AuCl(trz) complexes for carbene transfer to styrene (Table 4). These runs were performed in CH2Cl2 solution, in contrast to the neat benzene catalysis. The results indicate that the active species is solvent tolerant and apparently does not react with CH2Cl2 but instead selectively activates the aromatic C–H bonds. As in the case of benzene, full conversion of EDA was not achieved within 24 h and about 10% EDA remained unconverted after that time, which resulted in a complex reaction mixture for NMR analysis. Nonetheless, the ratio of products confirmed the same trend noted for diazocarbene transfer to benzene. Thus, carbene addition to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and formation of the phenyl cyclopropane 6 is the major outcome of the reaction, while carbene insertion into a Caryl–H bond and formation of styryl acetate 7 is less favoured. The ratio between cyclopropanation and C–H bond activation is about 3:1 for complexes 2b,d,e and hence very similar to the ratios obtained when using benzene as substrate (entries 2–4). In contrast, complex 2a is considerably more selective and reaches a 6.5:1 product ratio (entry 1).
C double bond and formation of the phenyl cyclopropane 6 is the major outcome of the reaction, while carbene insertion into a Caryl–H bond and formation of styryl acetate 7 is less favoured. The ratio between cyclopropanation and C–H bond activation is about 3:1 for complexes 2b,d,e and hence very similar to the ratios obtained when using benzene as substrate (entries 2–4). In contrast, complex 2a is considerably more selective and reaches a 6.5:1 product ratio (entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Au] | Additive | Product ratiob | ||
| 6 | 7 | DEF/DEMc | |||
| a General conditions: [Au] (0.012 mmol), styrene (2.3 mmol), EDA (0.44 mmol), NaBArF4 (0.015 mmol), CH2Cl2 (2 mL); average of 2 runs. b Distribution of products according to 1H NMR spectroscopy, ca. 10% of EDA were remaining after 24 h as quantified with benzaldehyde as internal standard. c DEF/DEM: diethyl fumarate/diethyl maleate, by products of the carbene coupling reaction from two molecules of EDA. | |||||
| 1 | 2a | NaBArF4 | 77 | 12 | 11 |
| 2 | 2b | NaBArF4 | 65 | 20 | 15 |
| 3 | 2d | NaBArF4 | 59 | 17 | 24 |
| 4 | 2e | NaBArF4 | 66 | 21 | 13 |
| 5 | 2a | AgSbF6 | 74 | 13 | 12 |
| 6 | 2d | NaBArF4, Hg drop | 67 | 20 | 13 |

No significant change in activity or selectivity was observed upon substituting the chloride scavenger from NaBArF4 to a silver salt (entry 5). The similar performance suggests that unlike in the oxazoline syntheses (vide supra), the catalytically active species in the carbene transfer reaction remains the [(trz)Au]+ species. Finally, a Hg-drop test was performed using catalyst 2d. No significant deviation was noted when compared to the parent run (cf. entries 3 and 6), which is indicative of a homogeneous catalytic cycle rather than one involving nanoparticles as an active species.
It is worthwhile to compare complexes AuCl(trz) and AuCl(IPr) as catalyst precursors to give some insight into the ligand effects. Firstly, the activity of the [(trz)Au]+ species is substantially lower than that of [(IPr)Au]+, requiring more than 24 h rather than just 6 h to reach complete conversion under identical conditions.30c Secondly, and perhaps more importantly, the active species resulting from AuCl(trz) strongly favours cyclopropanation over C–H bond activation and carbene insertion, while the use of AuCl(IPr) favours C–H bond activation. However, the product ratio for the AuCl(trz) systems closely matches those found for functionalised AuCl(NHC) systems.30d As the reactivity pattern was essentially identical for all the evaluated triazolylidene complexes, this facet likely reflects the different properties induced by triazolylidenes vs. classical NHC ligands. Previous work has suggested that decreasing electron density on the metal centre favours C–H insertion. In agreement with such a model, the increased σ-donor properties of mesoionic triazolylidenes indeed results in preferential cyclopropanation as compared to carbene insertion via C–H activation.30a,33 Accordingly, an increased electron density at the metal centre favours a carbene type resonance structure for the diazo fragment (A, Scheme 3) while electron-poorer metal centres induce a carbocationic structure (B, Scheme 3).34 It is very likely that the relative weight of each limiting structure in the real structure influences the reaction outcome toward the addition (Buchner) or insertion (alkyl-carbene) products.
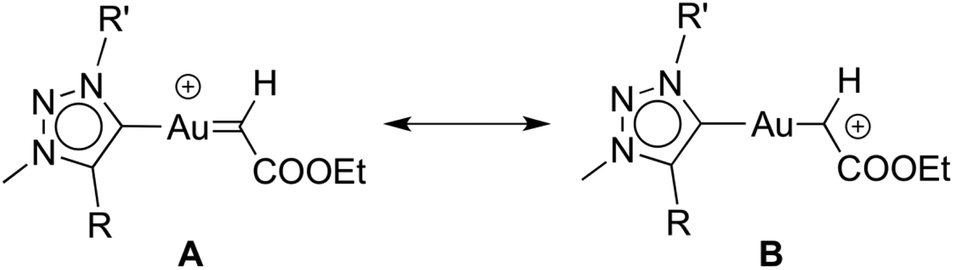 | ||
| Scheme 3 Resonance forms for the gold–carbene intermediate. | ||
Conclusions
A range of Ag-free chloride scavenger reagents for application with triazolylidene gold(I) complexes has been tested and shown to be useful for the activation of these complexes prior to catalysis. The results indicate that the Au complexes are less active catalysts for oxazoline synthesis than the analogous silver complexes such as AgCl(trz).29 Also, due to the linear coordination sphere around the metal centre, variation of the ligand substituents only moderately modulated catalytic activity and did not alter the diastereoselectivity of the resulting product profile. The AuCl(trz)/NaBArF4 system also catalysed the carbene transfer from EDA via benzene and styrene C–H bond activation. This activity demonstrated further applications of Ag-free [(trz)Au]+ catalysis. In this case, cyclopropanation products were favoured over C–H insertions, in contrast to the AuCl(IPr) system, indicating that the strong σ-donor properties of triazolylidene can be used to modulate the selectivity of gold in catalytic processes. Considering these results in combination with previous studies, it can be concluded that the choice of chloride scavenger is important to effectively identify the activity of gold-based catalysts and to assess the impact of the metal, as well as that of the ligand framework.Experimental
General
The precursor 4-butyl-1-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-1H-1,2,3-triazole for the triazolium salts 1c,35 the triazolium salts 1a,36 and 1e,3d and the triazolylidene gold complex 2d were synthesised using literature procedures.18 The synthesis of all novel triazoles and triazolium salts is described in the ESI.† Ag2O was used after regeneration by heating >160 °C under vacuum. Dry, degassed solvents were purified using an alumina/catalyst column (Thermovac Co.). All other reagents are commercially available and were used as received. All microwave experiments were carried out using a Biotage Initiator 2.5 instrument, operating at 0–400 W irradiation power. Unless specified otherwise NMR were recorded at 25 °C, on Varian and Bruker spectrometers operating at 300, 400, or 500 MHz (1H NMR) and 75, 100, 125 MHz (13C{1H} NMR) respectively. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to residual solvents. Assignments are based on homo- and heteronuclear shift correlation spectroscopy. Elemental analyses were performed by the Microanalytical Laboratory at University College Dublin, Ireland using an Exeter Analytical CE-440 Elemental Analyzer, with the exception of complex 2d which was performed in Unidad de Análisis Elemental of the Universidad de Huelva. Residual solvents were identified by NMR spectroscopy GC analysis were recorded on a Varian CP-3800 instrument. High-resolution mass spectrometry was carried out with a Micromass/Waters Corp. USA liquid chromatography time-of-flight spectrometer equipped with an electrospray source.:1 v/v) were added Ag2O (134 mg, 0.58 mmol), and NMe4Cl (140 mg, 1.3 mmol), and the suspension was stirred for 16 h under the exclusion of light under a N2 atmosphere. AuCl(SMe2) (309 mg, 1.1 mmol) was added and stirring continued for 2 h. The reaction mixture was filtered through Celite and volatiles were removed under reduced pressure. The residue was redissolved in CH2Cl2 and filtered through Celite a second time to remove residual salts. Volatiles were removed under reduced pressure yielding the product as a brown oil, which became crystalline after several days of drying in vacuo (0.376 g, 0.88 mmol, 86%). 1H NMR (500 MHz, CDCl3): δ 0.95 (m, 6H, 2 × CH2CH3), 1.32–1.44 (m, 4H, 2 × 2 CH2CH3), 1.68–1.74 (m, 2H, CtrzCH2CH2), 1.98–2.04 (m, 2H, NCH2CH2), 2.76 (t, 3JHH = 7.8 Hz, 2H, CtrzCH2), 4.02 (s, 3H, NCH3), 4.42 (t, JHH = 7.3 Hz, 2H, NCH2) 13C{1H} (126 MHz, CDCl3): δ 13.4 (NCH2CH2CH2CH3), 13.9 (CtrzCH2CH2CH2CH3), 19.8 (NCH2CH2CH2), 22.5 (CtrzCH2CH2CH2), 24.9 (CtrzCH2), 31.4 (CtrzCH2CH2), 32.3 (NCH2CH2), 36.4 (NCH3), 55.1 (NCH2), 147.5 (CtrzCH2), 157.0 (Ctrz–Au) HRMS (ESI+): m/z Found 450.0987 [M + Na]+ (calcd for C11H21N3AuClNa, 450.1028) Anal. Calcd for C11H21N3AuCl: C, 30.89; H, 4.97; N, 9.82%. Found: C, 31.04; H, 4.72; N, 9.69%.
OCH3), 4.14 (s, 3H, NCH3), 4.49 (t, 3JHH = 7.1 Hz, 2H, NCH2) 13C{1H} (126 MHz, CDCl3): δ 13.6 (CH2CH3), 19.8 (CH2CH3), 22.0 (COCH3), 29.1 (C(CH3)2), 32.3 (NCH2CH2), 39.6 (NCH3), 56.1 (NCH2), 76.7 (CCtrz), 149.4 (CCtrz), 156.6 (Ctrz–Au), 169.5 (CO) HRMS (ESI+): m/z Found 494.0886 [M + Na+] (calcd for C12H21N3O2ClAuNa, 494.0886) Anal. Calc. for C12H21N3AuClO2: C, 30.55; H, 4.49; N, 8.91%. Found: C, 30.28; H, 4.31; N, 8.63%.
:1 v/v), an stirred with the exclusion of light under a N2 atmosphere for 5 min. This mixture was treated with Ag2O (27 mg, 0.12 mmol) and NMe4Cl (26 mg, 0.24 mmol) and the reaction was stirred for 16 h in the dark. Then AuCl(SMe2) (56 mg, 0.19 mmol) was added and the reaction stirred for a further 2 h. The reaction mixture was exposed to air and the product was purified as described for 2a by repeated filtering through Celite, affording complex 2c as a pale yellow solid (150 mg, 0.14 mmol, 75%). 1H (500 MHz, CDCl3): δ 0.97 (t, 3JHH = 7.3 Hz, 3H, CH2CH3), 1.39–1.47 (m, 2H, CH2CH3), 1.73–1.79 (m, 2H, CCH2CH2), 2.8–3.01 (m, CCH2, 6H, 2 × NCH2CH2), 4.62 (t,3JHH = 7.1 Hz, 2H, N3CH2), 4.82 (t, 3JHH = 6.8 Hz, 2H, N1CH2) 13C{1H}(126 MHz, CDCl3): δ 13.8 (CH2CH3), 22.4 (CH2CH3), 24.9 (CtrzCH2), 30.9 (t, 3JCF = 22.2 Hz, N3CH2CH2), 31.3 (t, 3JCF = 20.1 Hz, N1CH2CH2), 31.6 (CtrzCH2CH2), 42.2 (N3CH2), 47.7 (N1CH2), 146.7 (CtrzCH2), 158.5 (Ctrz–Au) 19F{1H}(470 MHz, CDCl3): δ −126.27–(−126.12) (m, 4F), −123.4, −123.2 (2 × br, 2 × 2F), −122.9 (br, 4F), −121.97-(−121.79), −114.23–(−14.05) (2 × m, 2 × 4F), −80.88–(−80.80) (m, 6F) Anal. Calcd for C22H17AuClF26N3: C, 25.17; H, 1.63; N, 4.00%. Found: C, 25.18: H, 1.30; N, 3.80%.
:1 v/v) under a N2 atmosphere, for 17 h with the exclusion of light. Volatiles were removed in vacuo, and the residue was suspended in dry CH2Cl2 (15 mL), and stirred with AuCl(SMe2) (54.9 mg, 0.19 mmol) in the dark for 2 h. The reaction mixture was filtered through Celite, eluted with CH2Cl2 and all volatiles were removed. The product was obtained as a white solid (65 mg, 0.12 mmol, 64% yield). Crystals were obtained by slow diffusion of dry Et2O into a CH2Cl2 solution at −20 °C. 1H (500 MHz, CDCl3): δ 1.14 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.34 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 2.31 (sept, 3JHH = 6.9 Hz, 2H, 2 × CH(CH3)2), 4.25 (s, 3H, NCH3), 7.31 (d, 3JHH = 7.8 Hz, 2H, CDiIPrH), 7.53–7.58 (m, 4H, CArH), 7.80–7.82 (m, 2H, CArH) 13C{1H}(126 MHz, CDCl3): δ 24.2, 24.5 (2 × CHCH3), 28.9 (CDiIPrH), 38.4 (NCH3), 124.4 (CArH), 126.0 (CPh), 129.4, 129.8, 130.7 (3 × CPhH), 131.6 (CArH), 135.2, 145.2 (2 × CAr), 146.8 (CtrzC), 162.1 (Ctrz–Au) Anal. Calcd for C21H25AuClN3·½CH2Cl2: C, 43.45; H, 4.41; N, 7.07%. Found: C, 43.28; H, 4.50; N, 6.79%.
:1 v/v) for 16 h protected from light. To this mixture was added AuCl(SMe2) (79.5 mg, 0.27 mmol) and stirring was continued for 2 h. The suspension was filtered through Celite, eluted with CH2Cl2 and the volatiles were removed under reduced pressure yielding the crude product as an off-white solid (98 mg 23 mmol, 88%). Crystals were grown by slow evaporation of a CH2Cl2/Et2O solution of 2f. 1H (400 MHz, CD2Cl2): δ 0.97 (t, 3JHH = 7.4 Hz, 3H, CH2CH3), 1.33–1.43 (m, 2H, CH2CH3), 1.89 (s, 6H, C(CH3)2), 1.99–2.06 (m, 2H, NCH2CH2), 4.29 (s, 3H, NCH3), 4.47 (t, 3JHH = 7.3 Hz) 13C{1H}(101 MHz, CD2Cl2): δ 13.8 (CH2CH3), 21.2 (CH2CH3), 32.6, 32.7 (NCH2CH2 and C(CH3)2), 41.0 (NCH3), 56.2 (NCH2), 151.7 (CtrzC), 155.2 (Ctrz–Au).
Catalytic procedures
For reactions involving 1.1 mol% and 2 mol% MeOTf, a solution of MeOTf (0.265 M in dry, degassed CH2Cl2) was used and added before NiPr2Et. The volume of CH2Cl2 was adjusted to a total reaction volume of 4 mL.
For experiments probing the carbene transfer to styrene, the experimental procedure was identical to that described above,30c,39 except benzene was replaced by a mixture of CH2Cl2 (2 mL) and styrene (267 μL, 2.3 mmol).
Acknowledgements
This work was financially supported by the European Research Council (CoG 615653), the European Science Foundation (COST Action CM 1205), MINECO (CTQ2014-52769-CO3-1-R), the Junta de Andalucía (P10-FQM-06292), Ryerson University (R. A. G.), and the Irish Research Council through a fellowship to. R. P. We thank Dr D. Conseco-Gonzales for the synthesis of complex 2e. We thank the group of Chemical Crystallography of the University of Bern (PD Dr P. Macchi) for the X-ray structure solution of 2f and the Swiss National Science Foundation (R'equip project 206021_128724) for co-funding the single crystal X-ray diffractometer at the department of Chemistry and Biochemistry of the University of Bern.References
- (a) D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC; (b) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016 10.1039/C5CS00887E; (c) M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed; (d) B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103–7118 RSC; (e) M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638–1652 CrossRef CAS; (f) H. G. Raubenheimer and H. Schmidbaur, J. Chem. Educ., 2014, 91, 2024–2036 CrossRef CAS; (g) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC; (h) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed; (i) R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed; (j) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS PubMed; (k) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91–100 CrossRef CAS PubMed; (l) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS PubMed; (m) N. Shapiro and F. Toste, Synlett, 2010, 675–691 CAS; (n) C. Nevado, Chimia, 2010, 64, 247–251 CrossRef CAS PubMed; (o) R. A. Widenhoefer, Chem. – Eur. J., 2008, 14, 5382–5391 CrossRef CAS PubMed; (p) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (q) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed; (r) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (s) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed.
- (a) D. Gatineau, J. Goddard, V. Mouriès-Mansuy and L. Fensterbank, Isr. J. Chem., 2013, 53, 892–900 CrossRef CAS; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (c) H. G. Raubenheimer and S. Cronje, Chem. Soc. Rev., 2008, 37, 1998–2011 RSC. For applications of Au(NHC) complexes in other areas, see for example: (d) M. Baron, S. Bellemin-Laponnaz, C. Tubaro, M. Basato, S. Bogialli and A. Dolmella, J. Inorg. Biochem., 2014, 141, 94–102 CrossRef CAS PubMed; (e) C. Bazzicalupi, M. Ferraroni, F. Papi, L. Massai, B. Bertrand, L. Messori, P. Gratteri and A. Casini, Angew. Chem., Int. Ed., 2016, 55, 4256–4259 CrossRef CAS PubMed.
- (a) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC; (b) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC; (c) J. D. Crowley, A.-L. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118–1132 CrossRef CAS; (d) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed; (e) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed.
- For examples, see: (a) D. R. Tolentino, L. Jin, M. Melaimi and G. Bertrand, Chem. – Asian J., 2015, 10, 2139–2142 CrossRef CAS PubMed; (b) D. Mendoza-Espinosa, R. González-Olvera, G. E. Negrón-Silva, D. Angeles-Beltrán, O. R. Suárez-Castillo, A. Álvarez-Hernández and R. Santillan, Organometallics, 2015, 34, 4529–4542 CrossRef CAS; (c) D. Mendoza-Espinosa, R. González-Olvera, C. Osornio, G. E. Negrón-Silva and R. Santillan, New J. Chem., 2015, 39, 1587–1591 RSC; (d) C. Mejuto, G. Guisado-Barrios, D. Gusev and E. Peris, Chem. Commun., 2015, 51, 13914–13917 RSC; (e) L.-A. Schaper, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 3376–3384 CrossRef CAS; (f) L.-A. Schaper, K. Öfele, R. Kadyrov, B. Bechlars, M. Drees, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2012, 48, 3857–3859 RSC; (g) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC.
- (a) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC; (c) V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS; (d) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- V. Leigh, W. Ghattas, R. Lalrempuia, H. Müller-Bunz, M. T. Pryce and M. Albrecht, Inorg. Chem., 2013, 52, 5395–5402 CrossRef CAS PubMed.
- (a) I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 5, 2714–2718 CrossRef CAS; (b) C. Cesari, R. Mazzoni, H. Müller-Bunz and M. Albrecht, J. Organomet. Chem., 2015, 793, 256–262 CrossRef CAS; (c) A. Petronilho, A. Llobet and M. Albrecht, Inorg. Chem., 2014, 53, 12896–12901 CrossRef CAS PubMed; (d) D. Canseco-Gonzalez and M. Albrecht, Dalton Trans., 2013, 42, 7424–7432 RSC.
- (a) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC; (b) I. J. Lin and C. S. Vasam, Can. J. Chem., 2005, 83, 812–825 CrossRef CAS.
- (a) Z. Lu, J. Han, G. B. Hammond and B. Xu, Org. Lett., 2015, 17, 4534–4537 CrossRef CAS PubMed; (b) A. Zhdanko and M. E. Maier, ACS Catal., 2014, 4, 2770–2775 CrossRef CAS; (c) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed; (d) H. Schmidbaur and A. Schier, Z. Naturforsch., B: Chem. Sci., 2011, 66, 329–350 CrossRef CAS; (e) S. R. Patrick, I. I. F. Boogaerts, S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2011, 7, 892–896 CrossRef CAS PubMed; (f) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733–5736 CrossRef CAS PubMed.
- (a) L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS; (b) A. Buzas and F. Gagosz, J. Am. Chem. Soc., 2006, 128, 12614–12615 CrossRef CAS PubMed; (c) N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed.
- (a) A. Homs, C. Obradors, D. Lebœuf and A. M. Echavarren, Adv. Synth. Catal., 2014, 356, 221–228 CrossRef CAS PubMed; (b) C. Obradors, D. Leboeuf, J. Aydin and A. M. Echavarren, Org. Lett., 2013, 15, 1576–1579 CrossRef CAS PubMed; (c) P. R. McGonigal, C. de León, Y. Wang, A. Homs, C. R. Solorio-Alvarado and A. M. Echavarren, Angew. Chem., Int. Ed., 2012, 51, 13093–13096 CrossRef CAS PubMed; (d) C. R. Solorio-Alvarado, Y. Wang and A. M. Echavarren, J. Am. Chem. Soc., 2011, 133, 11952–11955 CrossRef CAS PubMed; (e) V. López-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292–9294 CrossRef PubMed; (f) C. H. M. Amijs, V. López-Carrillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef CAS PubMed.
- (a) P. García-García, A. Martínez, A. M. Sanjuán, M. A. Fernández-Rodríguez and R. Sanz, Org. Lett., 2011, 13, 4970–4973 CrossRef PubMed; (b) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2011, 50, 5560–5563 CrossRef CAS PubMed; (c) P. de Frémont, E. D. Stevens, M. R. Fructos, M. Mar Díaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045–2047 RSC; (d) M. Sawamura, Y. Nakayama, T. Kato and Y. Ito, J. Org. Chem., 1995, 60, 1727–1732 CrossRef CAS; (e) A. Togni and S. D. Pastor, J. Org. Chem., 1990, 55, 1649–1664 CrossRef CAS; (f) S. D. Pastor and A. Togni, J. Am. Chem. Soc., 1989, 111, 2333–2334 CrossRef CAS; (g) Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405–6406 CrossRef CAS.
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed.
- (a) S. K. Schneider, W. A. Herrmann and E. Herdtweck, Z. Anorg. Allg. Chem., 2003, 629, 2363–2370 CrossRef CAS; (b) P. García-Domínguez and C. Nevado, J. Am. Chem. Soc., 2016, 138, 3266–3269 CrossRef PubMed.
- (a) A. Guérinot, W. Fang, M. Sircoglou, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Angew. Chem., Int. Ed., 2013, 52, 5848–5852 CrossRef PubMed; (b) W. Fang, M. Presset, A. Guérinot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Chem. – Eur. J., 2014, 20, 5439–5446 CrossRef CAS PubMed.
- (a) M. Wegener, F. Huber, C. Bolli, C. Jenne and S. F. Kirsch, Chem. – Eur. J., 2015, 21, 1328–1336 CrossRef CAS PubMed; (b) R. Manzano, T. Wurm, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 6844–6848 CrossRef CAS PubMed; (c) M. J. López-Gómez, D. Martin and G. Bertrand, Chem. Commun., 2013, 49, 4483–4485 RSC; (d) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed; (e) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412–17415 CrossRef CAS PubMed; (f) X. Zeng, M. Soleilhavoup and G. Bertrand, Org. Lett., 2009, 11, 3166–3169 CrossRef CAS PubMed.
- (a) J. Serra, C. J. Whiteoak, F. Acuña-Parés, M. Font, J. M. Luis, J. Lloret-Fillol and X. Ribas, J. Am. Chem. Soc., 2015, 137, 13389–13397 CrossRef CAS PubMed; (b) S. Bastin, C. Barthes, N. Lugan, G. Lavigne and V. César, Eur. J. Inorg. Chem., 2015, 2216–2221 CrossRef CAS.
- D. Canseco-Gonzalez, A. Petronilho, H. Mueller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193–13203 CrossRef CAS PubMed.
- J. R. Wright, P. C. Young, N. T. Lucas, A. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065–7076 CrossRef CAS PubMed.
- L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949–10952 RSC.
- (a) J. Turek, I. Panov, P. Švec, Z. Růžičková and A. Růžička, Dalton Trans., 2014, 43, 15465–15474 RSC; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC; (c) S. Guo, J. C. Bernhammer and H. V. Huynh, Dalton Trans., 2015, 44, 15157–15165 RSC.
- D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962–4965 CrossRef CAS PubMed.
- It has also been postulated that the reverse ligand transfer leads to a slow release of cationic Au(I) species which are underligated, and therefore potentially catalytically highly active, see ref. 15.
- D. M. Gove, G. van Koten, J. N. Louwen, J. G. Noltes, A. L. Spek and H. J. C. Ubbels, J. Am. Chem. Soc., 1982, 104, 6609–6616 CrossRef.
- M. T. Johnson, J. Marthinus Janse van Rensburg, M. Axelsson, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2011, 2, 2373–2377 RSC.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 CrossRef CAS.
- (a) V. A. Soloshonok, A. D. Kacharov, D. V. Avilov, K. Ishikawa, N. Nagashima and T. Hayashi, J. Org. Chem., 1997, 62, 3470–3479 CrossRef CAS; (b) A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235–5331 CrossRef CAS PubMed; (c) C. Hubbert and A. S. K. Hashmi, in Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, Germany, 2012, pp. 237–261 Search PubMed.
- The potassium salts on their own and in the absence of complex 2a reach only medicore conversion (entry 2). When using 5% potassium salt instead of the 2% as in runs containing 2a, a maximum of 20% conversion was observed after 8 h, see also Table S1.†.
- R. Heath, H. Müller-Bunz and M. Albrecht, Chem. Commun., 2015, 51, 8699–8701 RSC.
- (a) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (b) M. M. Díaz-Requejo and P. J. Pérez, J. Organomet. Chem., 2005, 690, 5441–5450 CrossRef; (c) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed; (d) M. Delgado-Rebollo, Á. Beltrán, A. Prieto, M. Mar Díaz-Requejo, A. M. Echavarren and P. J. Pérez, Eur. J. Inorg. Chem., 2012, 1380–1386 CrossRef CAS; (e) A. Prieto, M. R. Fructos, M. Mar Díaz-Requejo, P. J. Pérez, P. Pérez-Galán, N. Delpont and A. M. Echavarren, Tetrahedron, 2009, 65, 1790–1793 CrossRef CAS.
- A. Caballero, M. M. Díaz-Requejo, M. R. Fructos, A. Olmos, J. Urbano and P. J. Pérez, Dalton Trans., 2015, 44, 20295–20307 RSC.
- (a) R. Gava, A. Olmos, B. Noverges, T. Varea, E. Álvarez, T. R. Belderrain, A. Caballero, G. Asensio and P. J. Pérez, ACS Catal., 2015, 5, 3726–3730 CrossRef CAS; (b) M. R. Fructos, T. R. Belderrain, M. C. Nicasio, S. P. Nolan, H. Kaur, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2004, 126, 10846–10847 CrossRef CAS PubMed.
- Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817–9821 CrossRef CAS PubMed.
- (a) Y. Wang, M. E. Muratore and A. M. Echavarren, Chem. – Eur. J., 2015, 21, 7332–7339 CrossRef CAS PubMed; (b) D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard and F. D. Toste, Nat. Chem., 2009, 1, 482–486 CrossRef CAS PubMed; (c) A. M. Echavarren, Nat. Chem., 2009, 1, 431–433 CrossRef CAS PubMed; (d) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2008, 47, 6754–6756 CrossRef CAS PubMed.
- R. W. Read and X. B. Wang, Chiang Mai J. Sci., 2009, 36, 247–257 CAS.
- (a) A. Poulain, D. Canseco-Gonzalez, R. Hynes-Roche, H. Müller-Bunz, O. Schuster, H. Stoeckli-Evans, A. Neels and M. Albrecht, Organometallics, 2011, 30, 1021–1029 CrossRef CAS; (b) S. S. Khan, S. Hanelt and J. Liebscher, ARKIVOC, 2009, xii, 193–208 Search PubMed.
- H. Ohta, Y. Uozumi and Y. M. A. Yamada, Chem. – Asian J., 2011, 6, 2545–2549 CrossRef CAS PubMed.
- (a) W. D. Mackay and J. S. Johnson, Org. Lett., 2016, 18, 536–539 CrossRef CAS PubMed; (b) N. Komine, J. A. Flores, K. Pal, K. G. Caulton and D. J. Mindiola, Organometallics, 2013, 32, 3185–3191 CrossRef CAS; (c) A. K. Padala, V. Saikam, A. Ali and Q. N. Ahmed, Tetrahedron, 2015, 71, 9388–9395 CrossRef CAS.
- (a) J. S. Yadav, B. V. S. Reddy and P. N. Reddy, Adv. Synth. Catal., 2004, 346, 53–56 CrossRef CAS; (b) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726–728 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of all novel triazoles and triazolium salts, NMR spectra of 2c, tables of catalytic runs and crystallographic details. CCDC 1481151–1481154. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02181f |
| This journal is © The Royal Society of Chemistry 2016 |