
Silica scale formation and effect of sodium and aluminium ions -29Si NMR study
L.
Lunevich
*,
P.
Sanciolo
,
A.
Smallridge
and
S. R.
Gray
Institute for Sustainability and Innovation, College of Engineering and Science, Victoria University, Melbourne, Victoria 8001, Australia. E-mail: llunevich@yahoo.co.nz
First published on 10th November 2015
Abstract
Silica scale formation on reverse osmosis (RO) membrane surface is a significant problem for operation of high recovery RO desalination plant. In this study we report the effects of sodium ions derived from sodium chloride and aluminium ions derived from aluminium chloride on dissolved silica species obtained from commercial sodium silicate solutions. Five dissolved silica species have been positively identified in sodium silica solutions. 29Si NMR spectroscopy has proven to be particular well suited to obtain in situ information on the connectivity of silicon atoms in the solution and the impact of sodium and aluminium ions on connectivity between monomeric silica acid groups. Such information extends the understanding of how polysilicate ion mixtures change under different chemical conditions. Implications for RO desalination and silica scale formation on the membrane surface were discussed.
Water impactSilica removal processes are critical in desalination of coal seam gas (CSG) water by reverse osmosis (RO) technology as silica can deposit on the membrane surface of RO systems. As a result of silica deposition, the quantity and quality of portable water produced by RO system will be significantly reduced. |
1. Introduction
Given that RO scaling involves precipitation–dissolution (condensation–hydrolysis) reactions of dissolved silica species, metals and inorganic constituents in water, 29Si NMR spectroscopy was used to study dissolved silica species. The technique is useful to gain an understanding of the behaviour of dissolved silica species identified in sodium silicate solutions, under the effects of sodium and aluminium ions commonly influencing silicate precipitation on the membrane surface. The sodium silicate environment provides a homogeneous distribution of colloidal silica. Silica in the sodium silicate environment presents in two domains – colloidal and dissolved groups. Moreover, silicon spectra of dissolved silica species in the sodium silicate system can be extracted without shielding of them by other chemical elements. Table 1 shows formulas and chemical shifts of each dissolved silica species studied, namely Q0, Q1, Q2, Q3 and Q4. Q0 type refers to a silicon atom with zero connections to another silicon species, Q1 type refers to a silicon atom connected to one silicon O–Si species, Q2 type refers to a central silicon atom connected to two silicon species, Q3 refers to a central silicon atom connected to three silicon species and Q4 is a central silicon atom connected to four silicon species. The main information derived from 29Si NMR spectra is the chemical shift and the intensity (peak area) or proportion of each species. The 29Si chemical shift is determined by the number and type of atoms connected to tetrahedral silicon atoms, Table 1. The NMR spectra, thus, allows the detection of the number of structurally equivalent kinds of silicon atoms of various Si–O units in silicates. Meaningful trends of different silica polymerisation paths are constructed as a result of these works.| Silica species (type surrounding) | Q0 | Q1 | Q2 | Q3 | Q4 |
|---|---|---|---|---|---|
| Formula & name group | SiO4 monomer | (SiO4)2O dimer | (SiO4)3 dimer | (SiO4)4 trimer | (SiO4)5 tetramer |
| Chemical shift (ppm) | −67.37 | −80.12 | −88.32 | −97.31 | −108.21 |
2. Concentration polarisation (CP)
Under certain conditions, silicic acid may be deposited on the membrane surface in the form of silicates or it may serve as cementing material (glass-like structures) for setting particles of different composition. Deposition of silica or silicate on the RO membrane surface could occur when the dissolved silicate exceeds its solubility limit. The solubility limit, however, is moderated by cations and anions in the specific water matrix and the pH condition of the RO feed due to their effects on silicate solubility.1,11,16The composition of the water matrix, especially the concentration and types of cations present in the CP zone on the membrane surface, may be seen as the main source of variation in the structure of silicate deposited on membranes. As such, knowledge of the deposition and dissolution of dissolved silicate species under the effect of different cations may thus lead to new insights in silicate deposition. Especially in the field of osmotic membrane separation, it has been noted that only small amounts of cations impurities may impose different deposition structures.11 Moreover, there has been considerable speculation on the role of aluminium in aluminosilicate deposition.1,2,9
Super-saturation conditions arising on RO membrane surfaces can lead silicate to aggregate into colloidal structures and possibly deposit on the surface via coupling points (–OH groups presented in abundance on RO membrane surface as a result of dehydration of water molecules, discussed in detail by El-Manharawy.6,7 Typical fully polymerised silicate structure (homogeneous nucleation) is demonstrated in Fig. 1, which consists of eight silicon atoms surrounded by oxygen atoms. Aggregation of these structures in bulk solution potentially could lead to amorphous silicate deposits on the RO membrane surface.
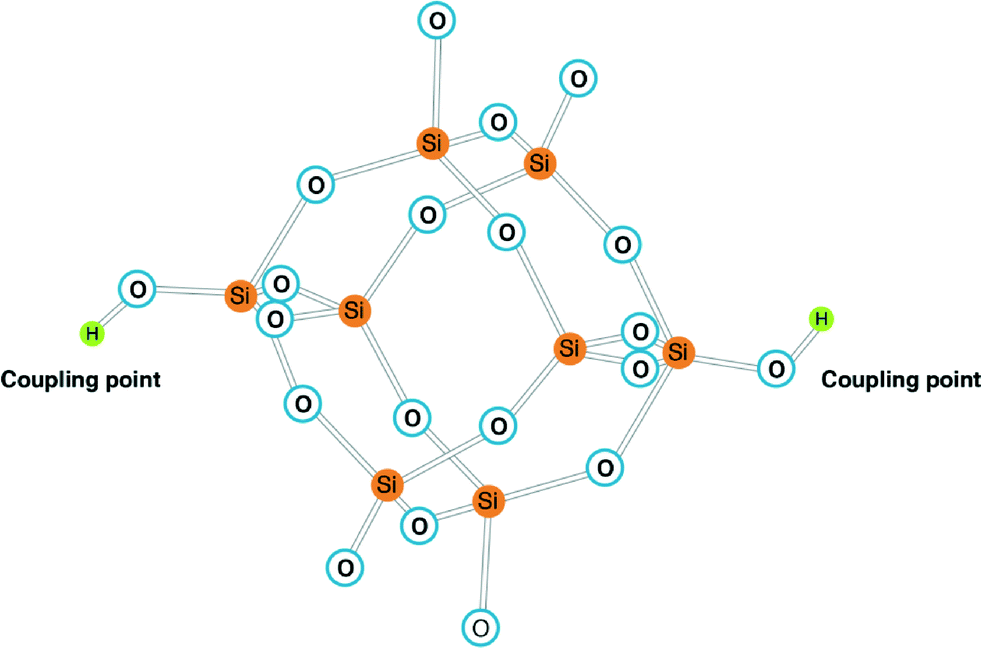 | ||
| Fig. 1 Polymerised silicate anion [Si18O18(OH)−62] adopted from Smolin.12 Coupling points (–OH groups) may link the silicate to RO membrane surfaces. | ||
In order to study the effects of sodium and aluminium ions on dissolved silica species, the species were studied in alkaline sodium silicate solutions.
3. Experimental
3.1 29Si NMR equipment
In this work 29Si NMR experiments were performed using a Bruker DPX300 Spectrometer at Victoria University, Footscray, Melbourne, operating with a 4.7 T magnetic field (29Si Larmor frequency 39.7 MHz). Chemical shifts were externally referenced (calibrated) to tetramethylsilane (TMS) 29Si at 0 ppm. 29Si NMR shift observed for the initial concentrated sodium silicate solution (baseline sample) and corresponding relative quantities of silicon in each Qn type surrounding are summarised in Table 1.3.2 Acquisition parameters
The 29Si NMR aqueous silica species acquisition parameters were designed based on dissolved silica concentrations and references available from the literature.1,2,13–15Table 2 shows 29Si NMR acquisition parameters by a Bruker DPX300 Spectrometer.| Conditions | Selected value |
|---|---|
| Measuring time of each sample | ∼1.5 h |
| Pulse length | 8 μs |
| Acquisition time | 0.8 s |
| Waiting period | 20 s |
| Number of scans | 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 678 678 |
3.3 Experimental solutions and sampling tube
Firstly, the experimental solutions were prepared by adding a relevant volume of deionised water into the mother sodium silicate solution (1.7 Si/M molar ratio). For the effect of sodium ions on silicate species, sodium chloride solution (as NaCl = 1000 mg L−1) was added to the mother sodium silicate solution. Similarly for the effect of aluminium ions, aluminium chloride solution (as AlCl3 = 130 mg L−1) was added to the mother sodium silicate solution. The proportions of the experimental solutions used are described in Table 3. The ratio of silicon to water is expressed as Si/M in molar ratio, where M is the molarity of water (for the reference sodium silicate solution the Si/M ratio was 1.7). The mixed experimental solutions were prepared 8–6 hours prior to each 29Si NMR experiment. The pH of each solution is listed in Table 3 and was measured at the time of preparation of the relevant solution. Approximately 5 mL of the experimental solution was injected into a ∼5 mm diameter Si NMR tube, then ∼0.2 mL of D2O solution was injected into the tube. Then the sample was inserted into the NMR sample chamber for 29Si NMR analysis for approximately 1.5 hours. Acquisition of NMR data was undertaken for 1.5 hours. The sodium silicate experimental solutions are shown in Table 3. No precipitation was observed during the first 48 hours after sample preparation as all samples were stored following the 29Si NMR experiments. Deionised water was used for dilution. Concentrations of total and dissolved Ca, Mg, Ba, Fe, Al, Mn were less than 0.01 mg L−1.| Effect of dilution SiO2:Na2O:H2O × H2O |
Effect of sodium chloride SiO2:Na2O:H2O × NaCl (1000 mg L−1) |
Effect of aluminium SiO2:Na2O:H2O × AlCl3 (130 mg L−1) |
|||
|---|---|---|---|---|---|
| Experimental solutions silica concentration (Si/M molar ratio) & pH of the solution | |||||
| Si/M molar ratio | pH | Si/M molar ratio | pH | Si/M molar ratio | pH |
| 1.7 | 11.56 | 1.7 | 11.48 | 1.7 | 11.48 |
| — | — | — | 1.61 | 10.67 | |
| 1.55 | 11.55 | — | — | 1.55 | 10.63 |
| 1.41 | 11.54 | — | — | 1.41 | 10.23 |
| 1.31 | 11.53 | — | — | — | — |
| 1.21 | 11.53 | — | — | — | — |
| 1.14 | 11.52 | 1.14 | 11.53 | 1.14 | 10.14 |
| 0.85 | 11.50 | 0.85 | 11.51 | 0.85 | 10.16 |
| — | — | — | — | 0.77 | 10.13 |
| 0.67 | 11.49 | 0.67 | 11.49 | 0.67 | 10.13 |
| — | — | 0.49 | 11.48 | — | — |
| — | — | 0.43 | 11.48 | — | — |
| 0.29 | 11.39 | — | — | — | — |
| 0.24 | 11.38 | — | — | — | — |
| 0.19 | 11.37 | — | — | — | — |
3.4 Limitations
One of the limitations of the technique is that the large number of structurally different silicate species observed by 29Si NMR spectroscopy contributes to the observed resonance lines being rather broad. Table 4 shows a number of subspecies recorded, for instance, for mono-substituted cyclic trimer (−86.2, −86.4, −94.7, −97.3, −97.4). However, all these species have a similar number of silicon atoms, overlapping and small difference in resonance frequency of the different lines cause substantial line broadening. The second important limitation of the technique is that the five millimetre diameter Pyrex tubes used by 29Si NMR Spectroscopy were made of borosilicate glass containing approximately 80% silicon dioxide and 12% boron oxide. The resonance of the tube material contributes mainly to Q4 spectra. Because silica species Q4 is very much a gel, for accuracy of interpretation of the results Q4 species were disregarded in all 29Si NMR experiments discussed in this paper. Quartz glass is essentially 100% silicon dioxide. Because quality NMR sample tubes are made from Pyrex, they often contribute substantial and broad signals in 29Si NMR Spectroscopy. The insert of most broad band probes is made from quartz, which can also contribute background signals to 29Si spectra. These background signals can make important spectral features of the sample impossible to discern. Overcoming them adds another challenge to 29Si NMR, because of the low abundance and relative sensitivity of this spin 1/2 nucleus. To overcome these, all spectra obtained during the 29Si NMR work were calibrated with background spectra obtained from the original silica species (five species present in Table 1).| Silica species | Qn site | −δ (ppm) |
|---|---|---|
| Monomer | Q0 | 49.9 |
| 51.9 | ||
| 55.7 | ||
| 58.4 | ||
| 65.1 | ||
| 66.4 | ||
| 67.6 | ||
| Q0 | 69.2 | |
| 69.4 | ||
| Q0 | 71.9 | |
| Dimer | Q1 | 73.6 |
| 78.0 | ||
| Q1 | 76.1 | |
| 76.9 | ||
| Liner trimer | Q1 | 79.9 |
| Q2 | 80.1 | |
| Q2 | 88.1 | |
| Q2 | 90.6 | |
| Cyclic tetramer | Q1 | 77.4 |
| Linear tetramer | Q1 | 88.3 |
| Q2 | 88.4 | |
| Cyclic tetramer | Q2 | 88.7 |
| Mono-substituted cyclic trimer | Q1 | 86.2 |
| Q2 | 86.4 | |
| Q3 | 94.7 | |
| 97.3 | ||
| 97.4 | ||
| Bridge cyclic tetramer | Q2 | 93.8 |
| Q3 | 96.3 | |
| 97.3 | ||
| Mono-substituted cyclic tetramer | Q1 | 95.6 |
| Q2 | ||
| Q2 | ||
| Q3 | 95.3 | |
| Q3 | 97.1 | |
| Tricyclic hexamer 1 | Q3 | 95.6 |
| Tricyclic hexamer II (cisoid) | Q3 | 101.1 |
Table 4 summarises all silica species and sub-species recorded during the experimental works with 29Si NMR technique. For accuracy of the results all subspecies recorded were integrated into relevant silica species.
4. Results and discussion
4.1 Effect of dilution with H2O
Fig. 2 shows consolidated results of 29Si NMR spectra recorded for dilution with deionised water. The results are arranged such that the front diagram presents the chemical shifts for the baseline sample (1.7 Si/M molar ratio) and the last diagram shows the chemical shift for the most diluted sample (0.11 Si/M molar ratio). Fig. 2 illustrates the chemical shift for each silica species for the range of experimental solutions listed in Table 3.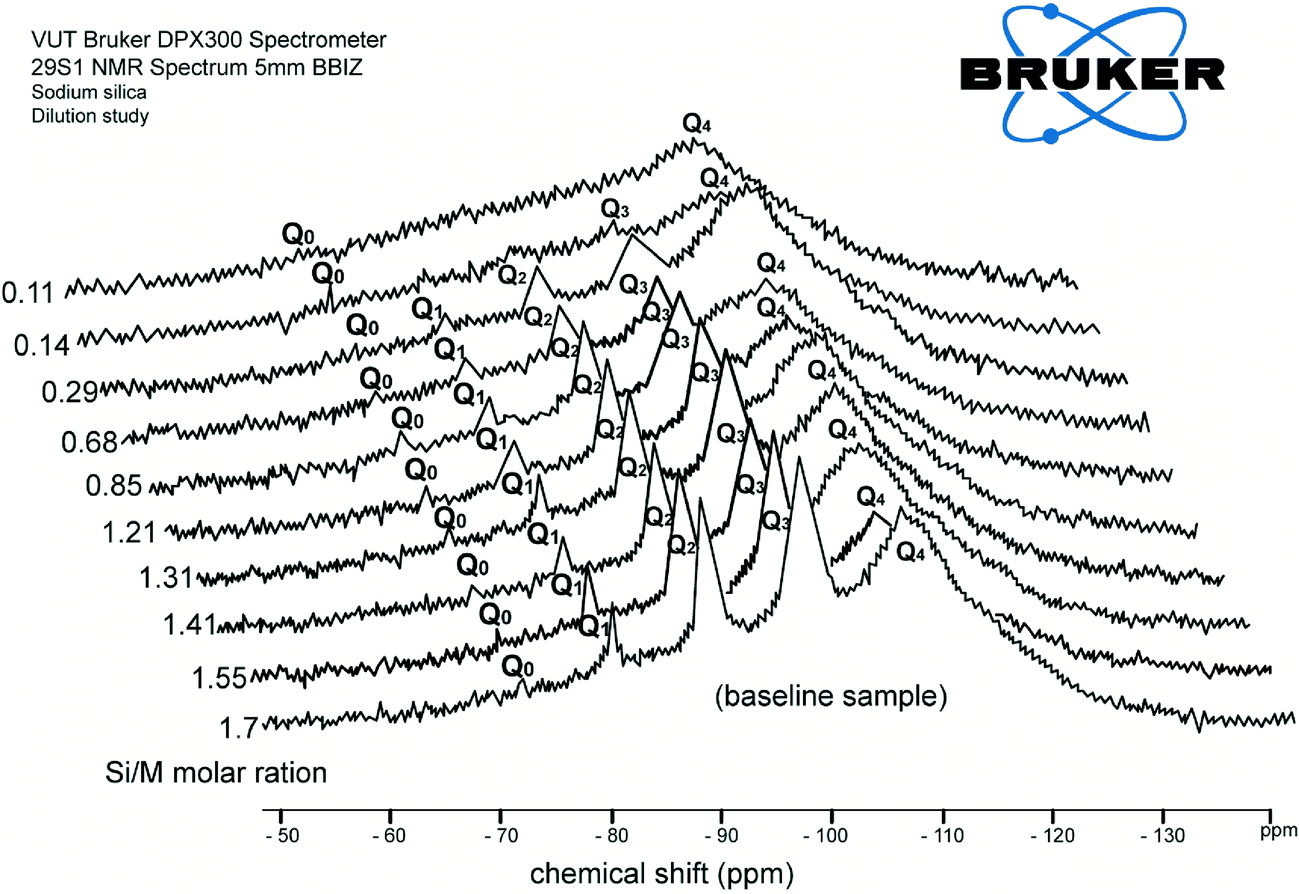 | ||
| Fig. 2 Consolidated results of 29Si NMR spectrum (Q0, Q1, Q2, Q3 type surroundings) of the samples for the range of diluted silicate solutions with H2O (at Si/M molar ratio 1.7, 1.55, 1.41, 1.31, 1.21, 0.85, 0.68, 0.29, 0.14, 0.11). | ||
As can be seen from Fig. 2, each further dilution slightly reduces the peaks for Q1, Q2, Q3 type surroundings until these three silicate species disappeared at the 0.11 Si/M molar ratio. The last sample shows that only Q0 type surroundings present at this silicate concentration. It was observed that each further dilution generates slightly higher concentrations of monomer silicic acid Q0 silicate species, probably due to relatively fast hydrolysis processes affecting this type of silicate species.
For instance, the relative proportion of Q0 type surrounding was 2.5% at the 1.7 Si/M molar ratio and gradually increased with dilutions up to ∼8% at the 0.11 Si/M molar ratio. As can be seen, Q0 (the smallest building block of silicate species or called monomeric silicic acid (Si(OH)4)) can be relatively easily hydrolysed from the more complex species perhaps due to the lower activation energy required to break the –O–Si–O– bonds, compare to formation (hydrolysis and condensation) of the Q2 and Q3 type species.4,5,11 A slight increase of Q0 type was expected as each dilution results in the release of Q0 type as a consequence weak –O–Si–O– bonds.
Fig. 2 also illustrates that Q1 type surroundings were obtainable across all dilution ratios at about 10 to 12% until they disappeared for the 0.11 Si/M molar ratio. Similarly, Q2 type surroundings presented across all dilution ratios at ∼30% for low dilutions until they disappeared at high dilutions. Q3 type surroundings can also be seen across all dilution ratios at approximately 50–60% at low dilution and disappeared at high dilution. The dilution results suggest that Q1, Q2, Q3 type surroundings might be less mobile, are less ionised species.3–5,11
Fig. 2 shows that relative proportions of all four silica species was relatively constant until the disappearance of Q1, Q2 and Q3 at high dilution ratios, possibly indicating equilibrium between species. The experiment was terminated at the 0.11 Si/M molar ratio when Q1, Q2, Q3 species disappeared as complete hydrolysis was likely to have occurred. It is also possible that the designed 29Si NMR acquisition parameters were not suitable to study lower concentrations of silica species.
These results were consistent with the hypothesis developed by Iler, who pointed out that while each solution has some equilibrium for silica species present, the tendency of silanol groups of a silicate molecule to ionise increases in the order: (Q0 > Q1 > Q2 > Q3).1,2,9 The polymerization of silicic acid is a condensation reaction, and requires ionized SiO(OH)3 groups and unionised silica Si(OH)4.6,7,11 This requirement implies that the rate of silicate polymerisation depends on the extent of silicate ionization. The position of the Q0 peak over all range of dilutions in Fig. 2 shows a gradual increase of Q0 surrounding, while the other silicate species (Q1, Q2, Q3) decrease.
Q0 type surrounding silica species plays a key reaction in hydrolysis and condensation (oligomerisation) of other silicate species such as Q1, Q2, Q3 and silicate precipitation reactions.11,12,14 Smolin (1968) points out that during dilution oxygen–silicon bonds weaken as a result of attack by water molecules, with the extent of weakening related to the number of siloxane bridges (Si–O–Si).11,12 For instance, during dilution, colloidal sodium silicate (Si2O18H4Na4) will release Na+ ions and replace it with –OH. OH groups will weaken Si–O bonds leading to potential hydrolysis or dissolution of monosilicic acid (Q0) from polymerised species.
Using 29Si NMR it was also possible to observe dissolved silica species present as monomeric silicate (Q0) as isolated molecules and as linked molecules (Q1, Q2, Q3), referred to as polysilicic acid. The influence of hydrolysis on the structural evolution of silicate polymers was studied in the past.6,7,12,13 It was demonstrated that higher amounts of water enhance hydrolysis and under acid conditions nearly complete hydrolysis occurs.11,13,14,16 The simplified study of the reaction involving additional water molecules, as was undertaken in this study, is very useful to gain a basic insight into the Si–O–Si bridging mechanisms of the hydrolysis process. Similar processes can occur in the CP zone on the membrane surface, where hydrolysis and condensation of silica species occur simultaneously, particularly if there is a shift of super-saturation occurs towards lower concentrations for instance.
The effect of dilution with water demonstrated that higher dilution ratios enhance hydrolysis rates, mainly due to hydrolysis of Si–O–Si bonds, resulting in liberation of monomeric silica groups – Q0 type surroundings. Nevertheless, an understanding of the reaction variables affecting hydrolysis and condensation processes is needed to compare other relevant impacts on silicate precipitation, such as the effect of sodium and aluminium ions to explain why in some cases silicate deposits on membranes and in other cases silicate does not deposit on the membrane surface.11
4.2 Effect of sodium
Limited references are available on the impact of sodium chloride on dissolved silicate species. Generally Na+ acts as a stabilising agent, as it decelerates the condensation of hydrolysed monomer and dimer species.8,13 Sodium ions in solution concentrations >8 g L−1 (as NaCl) tend to create a binding layer around silicate species preventing access of water molecules into polymeric silicate structures.8 However, dissociated sodium ions attract water molecules according to their charge, z to ionic radius, r.6,7 Low (z/r) ions (such as Na+ and Cl−) surrounded with a single water shell of 4 to 6 water molecules. These 4 to 6 water molecules could weaken Na–O–Si–O– bonds so Q0 type could redissolved into the solution unless Na–O–Si–O–Si–O–Si–O–Si–O– bonds (sometime double ring structure exist) will prevent Q0 type from dissolution from the silica polymeric structure, Fig. 1.A detail not found in any previous work, the results described in this paper show that addition of sodium chloride substantially increases the release of monomeric silicic acid (Q0) and impacts hydrolysis and condensation processes of Q1, Q2, Q3 type surroundings silicate species compared to similar dilutions with deionised waters, Fig. 3 and 4.
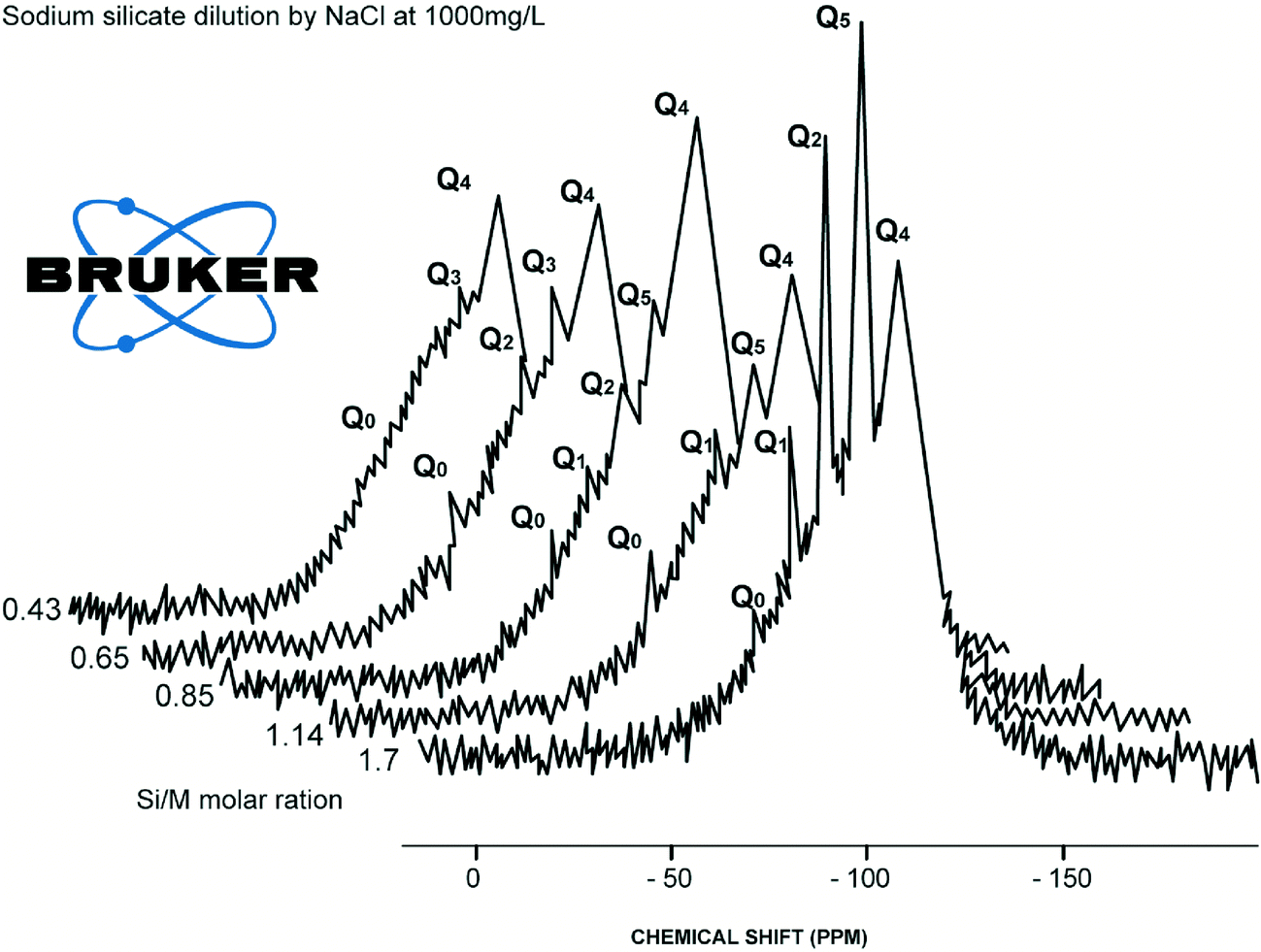 | ||
| Fig. 3 Consolidated results of 29Si NMR spectrum (Q0, Q1, Q2, Q3 type surroundings) from the range of diluted silicate solutions with NaCl (1000 mg L−1 (at Si/M molar ratio 1.7, 1.14, 0.85, 0.68, 0.43)). | ||
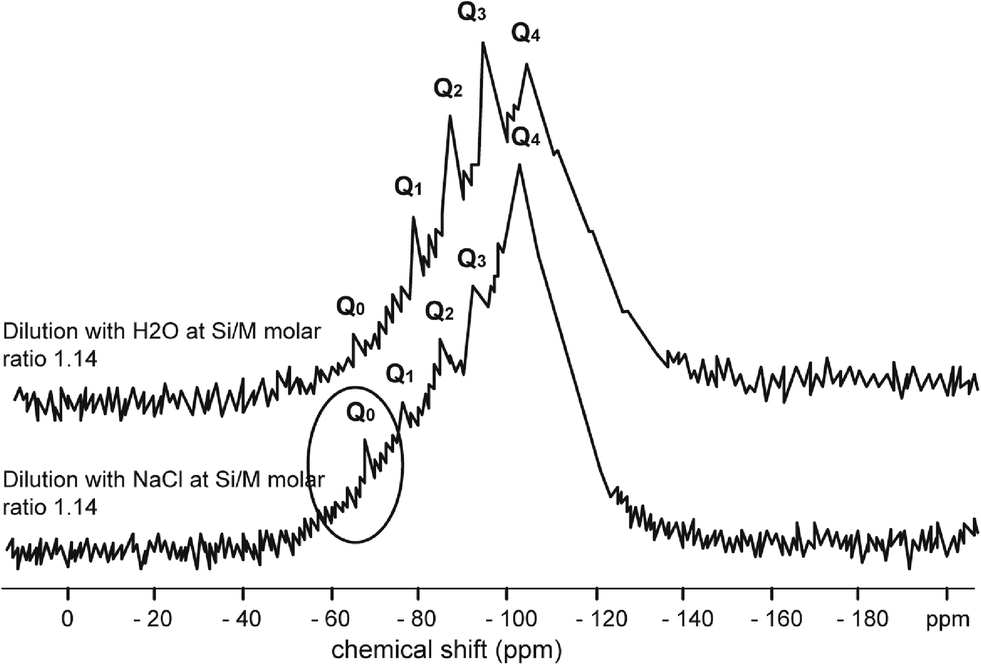 | ||
| Fig. 4 29Si NMR spectrum of sodium silicate diluted with H2O at Si/M molar ratio 1.14 (upper) and 29Si NMR spectrum of sodium silicate diluted with NaCl (1000 mg L−1) at Si/M molar ratio 1.14 (lower). | ||
This effect was observed for Q1, Q2 and Q3 type surroundings. As can be seen from Fig. 3, further addition of sodium chloride leads to the disappearance of Q1 type at the 0.68 Si/M molar ratio and the disappearance of Q2 type at the 0.43 Si/M molar ratio. This probably is due to a combination effects; first the binding effect of sodium ions on these species and second the hydrolysis effect on small species Q1, Q2, Q3 and condensation effect of Q1, Q2.
Fig. 4–7 show the 29Si NMR spectrum for various Si/M molar ratios demonstrating effect of sodium on monomeric silica species (Q0 type surroundings) compare to the same dilutions with H2O. As can be seen, a much higher proportion (at least twice) of monomeric silicic acid (Q0) was present for the dilution with sodium chloride compare to the dilutions with H2O.
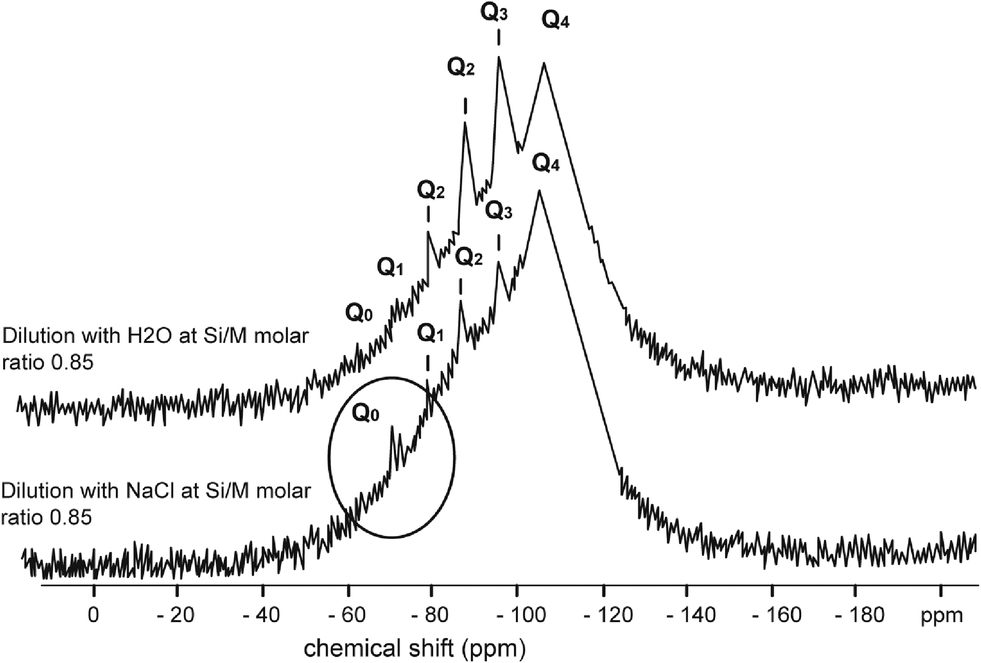 | ||
| Fig. 5 29Si NMR spectrum of sodium silicate diluted with H2O at Si/M molar ratio 0.85 (upper) and 29Si NMR spectrum of sodium silicate diluted with NaCl (1000 mg L−1) at Si/M molar ratio 0.85 (lower). | ||
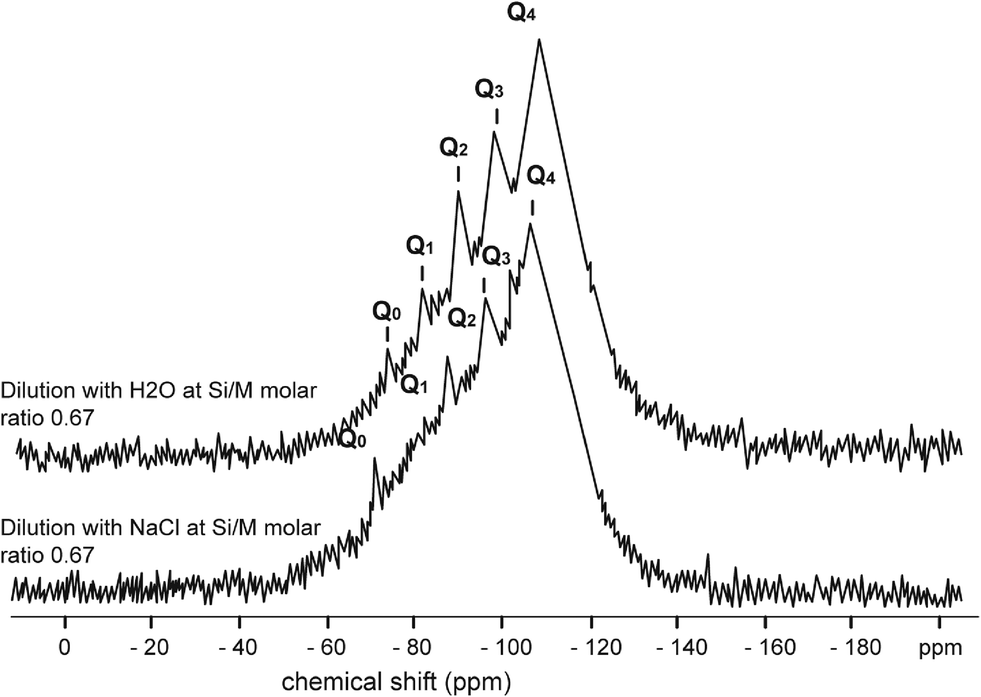 | ||
| Fig. 6 29Si NMR spectrum of sodium silicate diluted with H2O at Si/M molar ratio 0.67 (upper) and 29Si NMR spectrum of sodium silicate diluted with NaCl (1000 mg L−1) at Si/M molar ratio 0.67 (lower). | ||
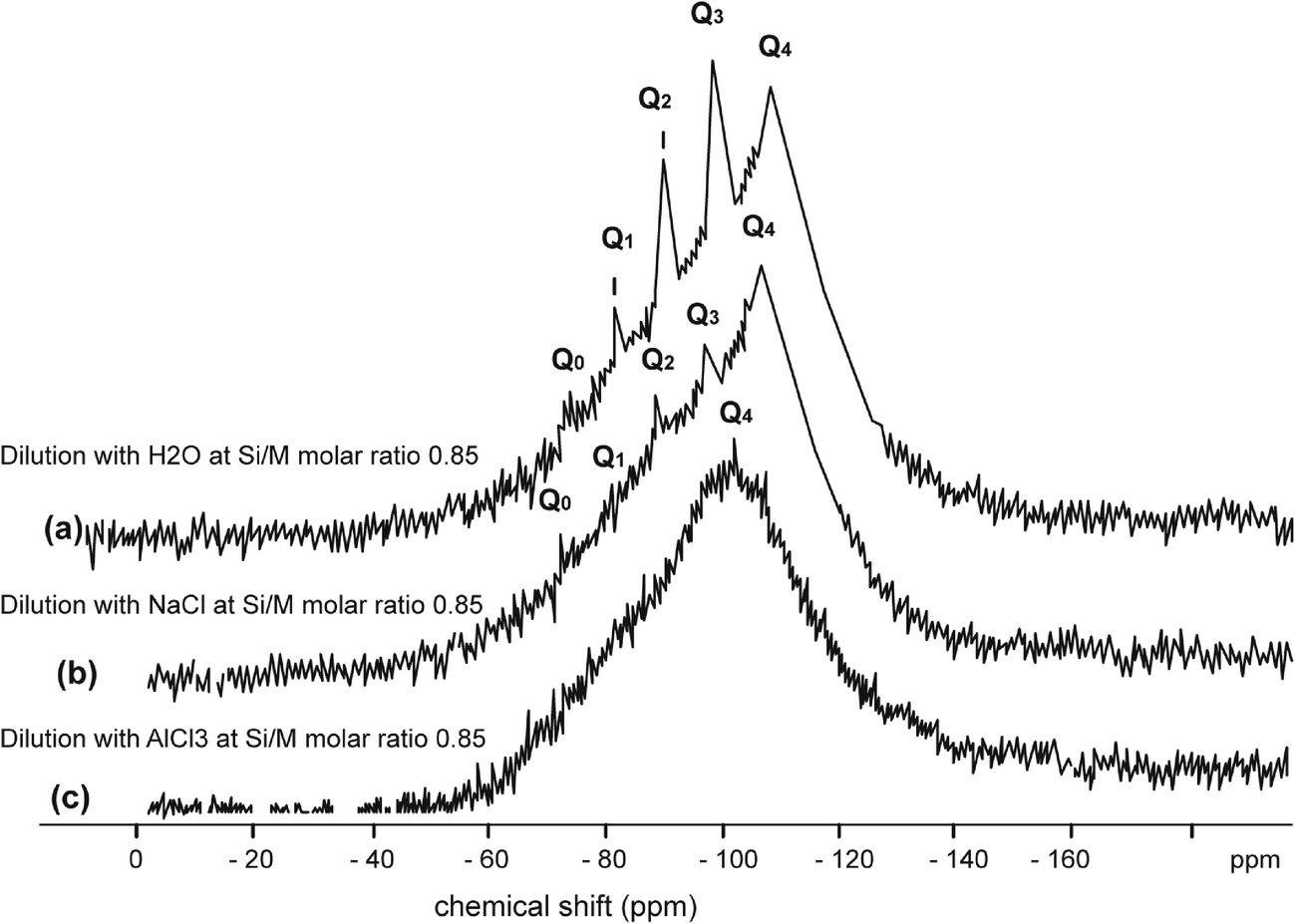 | ||
| Fig. 7 29Si NMR spectrum of sodium silicate diluted with H2O at Si/M molar ratio 0.85 (upper), (b) diluted with NaCl (1000 mg L−1) at Si/M molar ratio 0.85; (c) diluted with AlCl3 (130 mg L−1) at Si/M molar ratio 0.85 (lower). | ||
Fig. 7 illustrates silicate spectrum for dilution with H2O and dilution with sodium chloride at the Si/M molar ratio 0.85. The results clearly demonstrate a reduction of proportions of Q1, Q2 and Q3 type surroundings, but Q0 type was present in slightly higher proportion than in the baseline sample. When mother sodium silicate solution is diluted, more monomer Q0 type surrounding is released into the solution. With dilution by NaCl, liberation of monosilicic acid (Q0) from silica polymorphs, such as Q2, Q3 types, was slightly higher. This might suggest that negatively charged surface of silanol groups (Si–O–), neutralised by hydrated cations Na+, are attacked by hydroxyl anions (–OH) by which the coordination number of the silicon atom is increased to more than four. As a result, the underlying silicon bonds are weakened and monomeric silicate anions are released from the other silicate groups, including from colloidal silicate present in the solution. It is likely a similar mechanism applies to Q1 and Q2 species when these species disappeared from the spectrum.
Other potential interpretations for the loss of sensitivity of silicate species under the effect of sodium ions is that destabilisation of the –O–Si–O– bonds by Na+ occurred to the extent that some silicate species precipitated in the sample (Si NMR tube) or were transferred into Q4 type surroundings. This hypothesis is demonstrated in Fig. 8, where silica precipitation is shown within the 29Si NMR tubes numbered “1” and “2”. As can be seen, silica precipitation occurred in tubes “1” and “2” probably due to the destabilisation effect of sodium ions on dissolved silica species. No visible silica precipitation was observed in the tube “3”. The precipitation occurred after 48 hours of 29Si NMR experiment.
 | ||
| Fig. 8 The experimental solutions after 48 hours of 29Si NMR experiment: tubes “1” and “2” show the samples diluted with sodium chloride (at the 1.14 and 0.68 Si/M molar ratio respectively), tube “3” is the sample diluted with H2O at the 0.68 Si/M. | ||
Sodium silicate mother solution is already stabilised with NaOH and the majority of the silanol groups on the surface of silicate sol are covered with Na+ ions. Na+ ions are strongly adsorbed onto the silanol group, and this condition is expressed as Si–ONa. However, addition of extra sodium chloride seems to shift the equilibrium between dissolved silicate and amorphous silicate domains resulting in silicate polymerisation and precipitation as shown in Fig. 8. Na+ ions liberated as a result of dilution generate a silicate steric layer surrounding oligomer silicate such as Q2, Q3species, which leads to further binding of Na+ to the stabilizing layer.8,13,14 According to Healy (1994), the presence of increasing amounts of bound Na+ must switch off the electro steric contribution of the adsorbed polysilicate structures.8,12 This suggests that the effect of sodium ions is to attract –OH groups to Q0 type silica species and also to prevent –OH groups forming into Q2, Q3 type forcing silica to precipitate. Both effects play a major role in sodium–silicate interactions.
The results shown in Fig. 3–8 suggest that sodium ions not only shield silicate species but also sustain monomeric silica acid (possible speed up hydrolysis of weakly connected monomeric silicic acid) and depress hydrolysis and condensation processes of Q1, Q2, Q3 types by preventing access of water molecules into polymeric silica species. The trend is to either shield Si–(O–Si)4 bonds (Q4) or to hydrolyse Si–(O–Na)x bonds. This means for Q0 type surrounding, Si–OH is the preferred bond over Si–O–Na. Instead for Q1 and Q2 type surroundings, Si–O–Na is preferred over Si–OH. For Q3 type silicates with Na+ and OH− associations occurring in some combination and depending on pH, leads to ionisation of silicate species and more complex physical structures of silicate species. The mechanism is consistent with the disappearance of Q1 before Q2 before Q3 and an increase in Q0 type species. Legrand identified that the physical structure of silicate species is defined by the probability of chemical reactions that occur and can influence chemical behaviours of silicate.10,11
4.3 Effect of aluminium
The effect of residual aluminium on silicate scale formation is less known, and concerns have been expressed by industries and researchers that aluminium might have a significant impact on dissolved silicate species and silica polymerisation paths.1,2,8,9 Iler has remarked that there is a peculiar affinity between the oxides of aluminium and silicon. This affinity results from the nature of (SiO4)4− and (AlO4)5−, which is responsible for the vast range of natural aluminosilicates.2,9 Al3+ ions appear to slow down hydrolysis of silicate species.11,13 It was therefore appropriate to investigate further the effect of aluminium on dissolved silica species.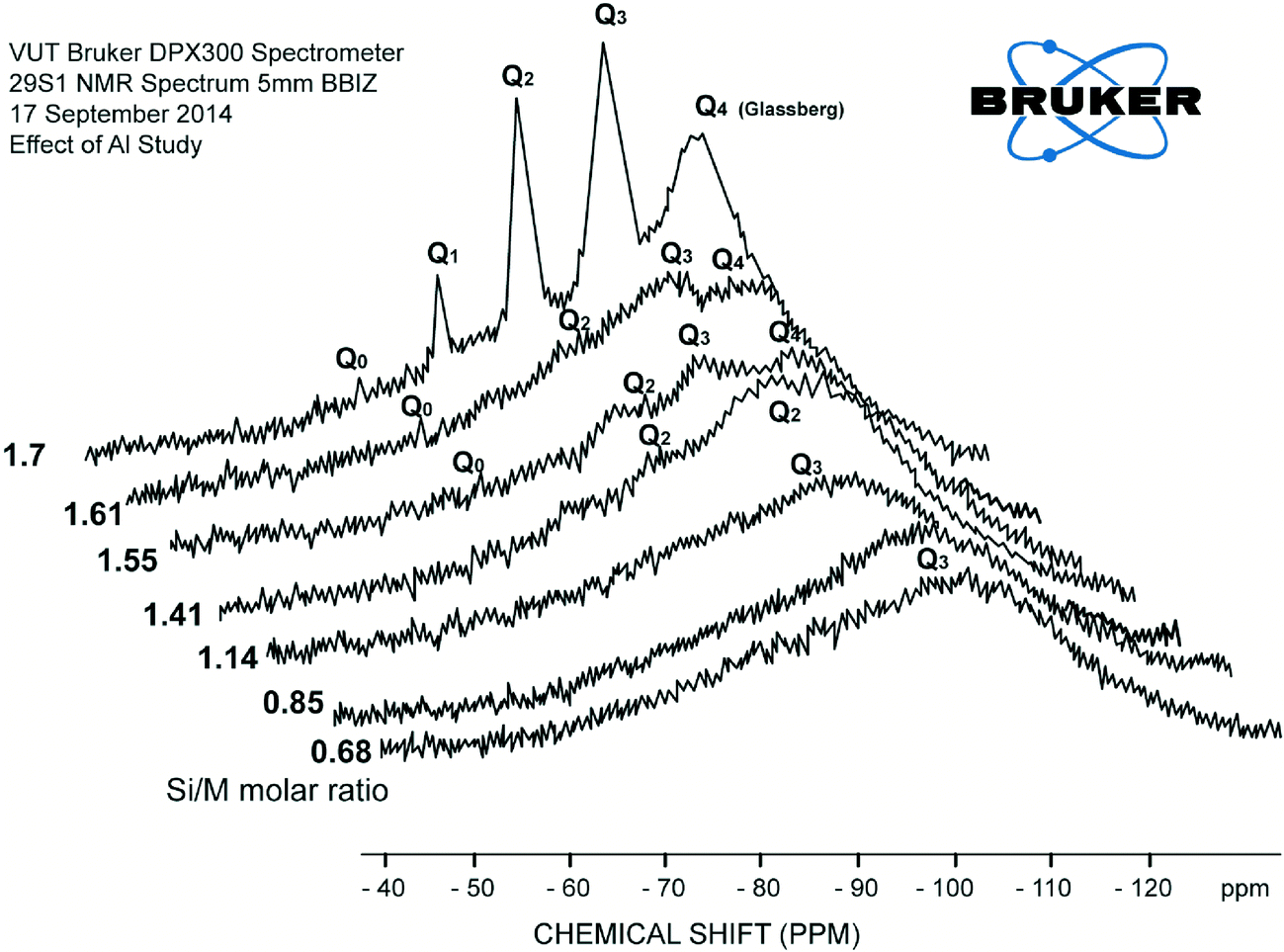 | ||
| Fig. 9 Consolidated results of 29Si NMR spectrum (Q0, Q1, Q2, Q3 type surroundings) of the samples for the range of diluted silicate solutions with AlCl3 (130 mg L−1) (at Si/M molar ratio 0.68, 0.85, 1.14, 1.41, 1.55, 1.61, 1.7). | ||
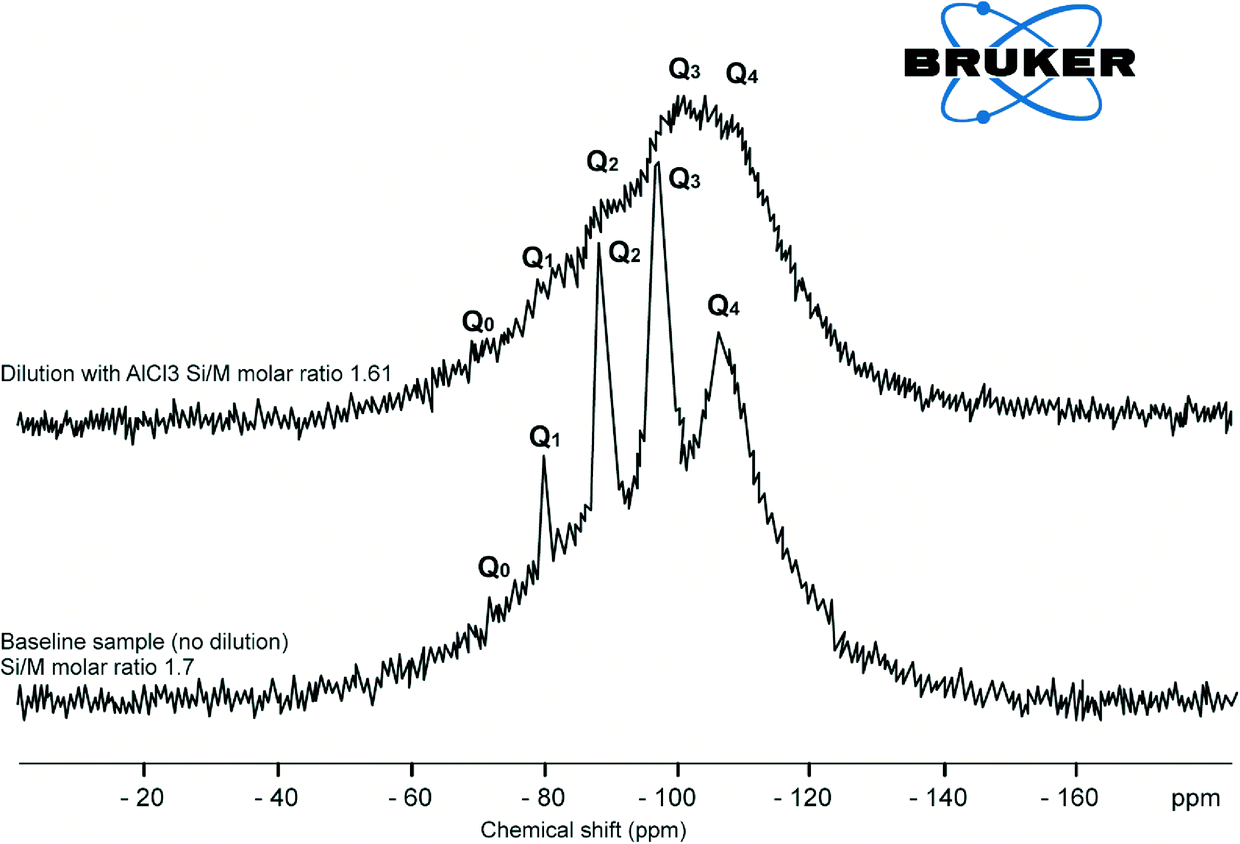 | ||
| Fig. 10 29Si NMR spectrum of baseline sample Si/M molar ratio 1.7 and the sample diluted with AlCl3 (130 mg L−1) at Si/M molar ratio 1.61. | ||
At the same time the loss of peaks for Q1, Q2, Q3 suggests that the number of possible atomic arrangements increases dramatically with the addition of aluminium, so the there are no sharp peaks, but lots of little peaks that appeared as shown in Fig. 10. Disappearance of Q0, Q1 type silica species were also an indication that Al3+ affects hydrolysis reactions and the equilibrium between species. Addition of aluminium favours condensation processes possibly because –O–Si–O–Al– bonds have higher reaction energy than –O–Si–O–Si–O– and are less mobile.11,12 Although the results do not directly confirm the formation of –O–Si–O–Al– bonds, the formation of such networks would likely shift the equilibrium between hydrolysis and condensation towards condensation. Such a shift in equilibrium is consistent with silicate and aluminium silicate precipitation on membrane surfaces when residual aluminium is present in RO feed solutions at elevated concentrations, leading to scale formation.11 This explains rapid silicate and aluminium silicate precipitation on membrane surfaces when residual aluminium is present in RO feed solutions at elevated concentrations.11
Explicit characterisation of Qn distributions by 29Si NMR is difficult in alkaline silicate solutions and even more difficult with a minor addition of aluminium. The reason for this increase in complexity is that, in contrast to dilution with deionised water or with a relatively low concentration of sodium chloride, with addition of aluminium even in very minor concentrations, it is no longer possible to match the observed Qn distribution against the known stoichiometry of the sample. This is a result of the ill-defined aluminium coordination in these samples, Fig. 9 and 10, due to multiple peaks.
It is proposed that when aluminium is added to sodium silicate solutions, aluminium hydroxide is formed and silicic acid polymerises on the aluminium hydroxide surface to form a layer of polysilicic acid.11 Once polysilicic acid is formed on the surface of aluminium hydroxide the growth of silica particles continues to the size 0.1 nm.3–5 Therefore, the Si:Al ratio in solution decreases and precipitates form as demonstrated in Fig. 11, tube “1”, where significant precipitation was observed 24 hours after the experiment. Two major observations can be drawn from these results, that the recorded chemical shifts moved into the region of Q3 type spectra while Q0, Q1 andQ2 species are no longer seen in solution, (Fig. 9). Q3 silica species are found in more amorphous type structures and is an indication that aluminium forces dissolved silicate species to precipitate quite rapidly.2–4
 | ||
| Fig. 11 The experimental solutions after 24 hours of 29Si NMR experiment: “1” is dilution with AlCl3 at the ratio 1.61 Si/M molar ratio and “2” is dilution with H2O at the 1.61 Si/M molar ratio. | ||
5. Implication for RO desalination
The results recorded in the dilution with H2O study illustrate that monomeric silicic acid may exist as isolated molecules, so called monosilicic acid Q0 type surroundings or (Si(OH)4) and Q1 type or Si2O3(OH)42−, (and as linked molecules, called polysilicic acid) and as Q2, Q3 types. Polymers consist of silicate tetrahedrons that are linked via silicon–oxygen–silicon bonds. Addition of H2O to the solution immediately initiates hydrolysis of monosilicic acid groups and as a result adjustment of the equilibrium between the rest of the silicate species.The results presented in Fig. 3, 5–7 clearly illustrate that the introduction sodium chloride into the mother sodium silicate solution reduce 29Si NMR spectrum for Q1, Q2, Q3 types. The proportion of Q3 type was significantly reduced immediately after the first addition of sodium chloride at Si/M molar ratio 1.41, presumably due to the shielding effect of sodium ions. As a result of further addition of sodium chloride, Q1 and Q2 type surroundings disappeared, and Q3 type also gradually decreased. This is consistent with the hypothesis that sodium ions tend to surround polymeric silica species discussed by Healy (1994) acting as a stabilising agent, protecting polymeric species from attack by water molecules.8,12 The results with sodium dilution showed significantly increased proportions of monomeric silicic acid (Q0) compared to similar dilutions with H2O, Fig. 4, 5 and 7. This is probably a result of sodium ions attracting water shells (discussed in detail by El-Manharawy,6,7 which initiate separation of monomeric silicic acid from polymeric silica groups. When dissolved silicate exceeds its solubility limit (supersaturation condition) the nucleation process will, in principle, be governed by interacting silanol groups that interact to form –O–Si–O–Si–O bonds.1,2,12
Under supersaturation conditions, the probability of interactions between neighbouring silanol groups to form –Si–O–Si– bonds is higher, and therefore intramolecular nucleation would be favoured.8,9 During silicate precipitation the monomer groups [SiO4]4− and [SiO6]4− randomly pack and rapid growth results in non-periodic structures, Fig. 1. In medium to high salinity waters dissolved and polymerised silicate species will be surrounded by sodium ions as shown in Fig. 12.
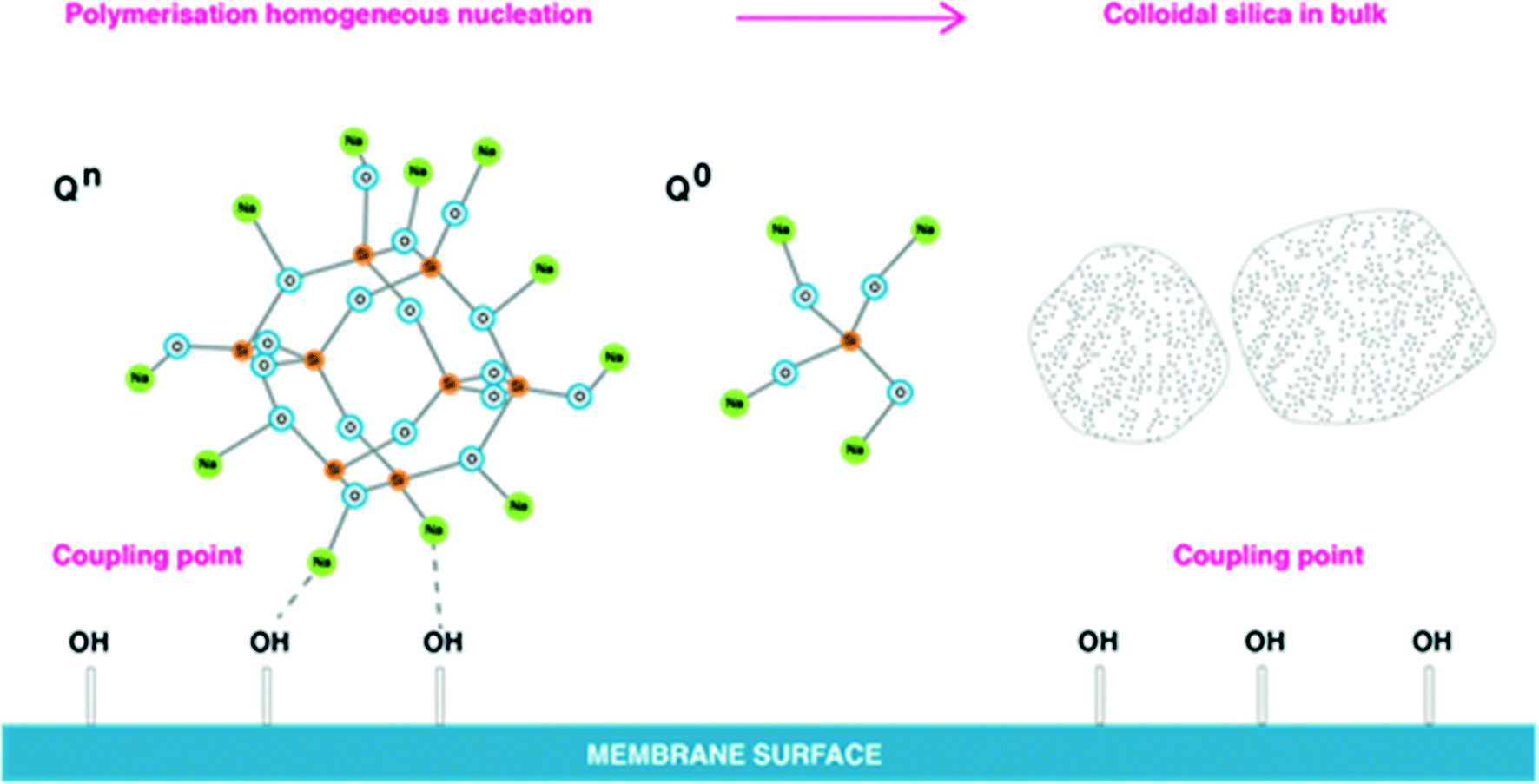 | ||
| Fig. 12 Dissolved silica polymerisation in CP layer on the membrane surface in medium and high salinity waters without presence of aluminium. | ||
Sodium ions attract to silicate, but also can interact with –OH groups present on membrane surfaces (usually in abundance), Fig. 12, that act as coupling points. Na–O–Si–O–Si–O– bonds are stronger then –O–H–O–Na bonds, as according to Healy, the more silicon atoms present in silicate polymeric structures the stronger the sodium binding layer attached to silicate. In this case, silicate polymerisation is likely to occur homogeneously in bulk solution.8,15,16 As depicted in Fig. 12, polymeric silicate species may aggregate into colloidal silica in bulk solution. Such polymerised silicate is unlikely to deposit on membrane surfaces due to sodium ions creating barriers between –OH groups and silicate, Fig. 12. Monomeric silicic acid can potentially deposit solely on the membrane surface, but the reverse of this process will be apparent as it is likely the dissolution (hydrolysis) process will dominate for monomeric silicic acid, Fig. 12. It is know that monomeric silicic acid can coat natural organic matters present on membranes, and this would increase the likelihood of silicate deposition on membrane surfaces.3,8,11
A number of parallel and competitive reactions are expected to occur in solution before dissolved silicate precipitates as a result of destabilisation of the equilibrium between silicate species. Healy suggests that sodium ions have a number of effects on silicate species.2,8 For instance, Na+ ions in small concentrations (<8 g L−1) attract water molecules and sustain further dissolution (hydrolysis) of monosilicic acid increasing the concentration of Q0 type and at the same time preventing access to polymeric silicate structures such as Q2, Q3 types. Applying this concept to medium and high salinity waters, dissolved silicate can be difficult to remove effectively due to sodium ions preventing access to silicate. Ultimately effective dissolved silicate removal from a groundwater by coagulation, for instance, might suggest that dissolved silicate was present in solely monomeric form. Experimental and analytical observations by others found high concentrations of monomeric silicic acid in high salinity groundwater in Tunisia, Egypt, Qatar, and Oman where RO desalination presents even bigger challenges due to extreme salinity of groundwater (NaCl = 100–200 g L−1).6,7
The effect of aluminium ions on silicate species, shown in Fig. 9 and 10, is reduced 29Si NMR spectra peak proportion possibly due to silicate precipitation as aluminium silicates. It is proposed that, aluminium ions can over time disassemble polymeric silicate structures due to (AlO4)−5 having similar bonds to Si with oxygen, so Al can easily access polymeric silicate species. Once aluminium silicates are formed, aluminium will bond with –OH groups on membrane surfaces, leading to an irreversible process of scale formation on the membrane surface as shown in Fig. 13. Further research is required to definitively identify the formation of Al–O–Si bonds.
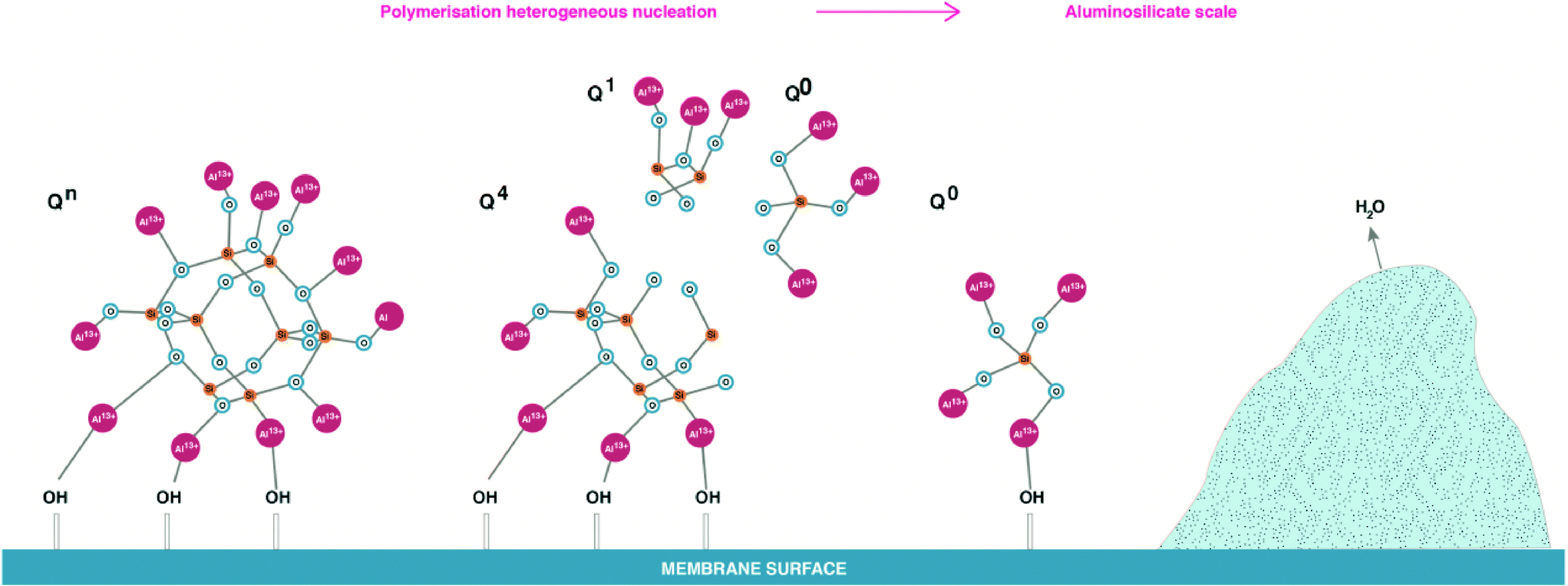 | ||
| Fig. 13 Proposed polymerised silica under effect of aluminium and final aluminium silicate deposition of RO membrane surfaces. | ||
The outcome of these hypotheses is that opposite to the effect of sodium ions on silicate species, aluminium ions force re-arrangement of silicate species leading potentially to precipitation, Fig. 13. This is supported by the results in Fig. 10, where major peaks representing Q1, Q2, Q3 types transformed into small, multiple peaks. It appears that in the presence of aluminium ions, monomeric silicic acid and other dissolved silicate species can deposit on the membrane surface without following the classical silica polymerisation path (Q0 → Q1 → Q2 → Q3 → Q4) described by Iler.9 This is likely to occur in water when dissolved aluminium present in the RO feed. Once silicate is fully polymerised and deposited on the RO membrane surface, it is likely that the formed cake will be difficult to remove due to aluminium silicate attaching to the membrane surface via OH groups which became a part of the scale structure. It is suggested that overtime aluminium will tend to substitute H2O from the cake leading to irreversible scale deposit, Fig. 13.
Most natural waters have a range of cations and anions which influence polymeric structures of silicate species, and that affect silicate precipitation patterns. The effect of aluminium on dissolved silica species recorded in this work showed that aluminium ions can interact with dissolved silicate species such as Q2, Q3 types forcing re-arrangement of species. Both sodium and aluminium are present in natural waters, particularly high salinity natural waters, and affect the speciation of silicates and precipitation patterns as can be concluded from the results recoded here.
Structural bonding of the silicate species, or how many silicon atoms are bound to other silicon atoms via oxygen, is complicated because diverse variables affect concurrent reactions in different ways. The hydrolysis of –O–Si–O–Si–O– linkages, however, is often considered as the key reaction dominating many geochemical processes. These bonds play a key role in many silicate transformations including silicate scale formation on the membrane surface.
6. Conclusion
The main outcomes can be summarised as follows:As a result of dilution with deionised water, dissolved silicate species gain or lose monomeric silicic acid (Q0 type surroundings). A gradual decrease of 29Si NMR spectrum intensity or proportions of Q0 increased and Q1, Q2 and Q3 decreased, indicating that hydrolysis or dissolution of monomeric silicic acid occurred immediately. A consistent proportional decrease of 29Si NMR spectrum for each species indicates that there is an equilibrium between species at the Si/M molar ratio 1.7, which changed with further dilutions from the Si/M molar ratio 1.55 to 0.11.
Here it is shown that addition of sodium chloride rather than water slightly increased the release of monomeric silicic acid (Q0) and decreased hydrolysis of more complex silicate species (Q2, Q3) suggesting sodium prevents water molecular to access Q2, Q3 types.
The effect of sodium ions on silicate species suggests that Q0 type surrounding Si–OH is the preferred bond over Si–O–Na, while for Q1 and Q2 type surroundings Si–O–Na is preferred over Si–OH. For Q3 type both reactions with Na+ and OH− may occur in combination.
Undoubtedly, a profound effect of aluminium ions was recorded on silicate species, as there was a loss of sensitivity in the 29Si NMR spectrum. It is proposed that the presence of aluminium on silicate species has three impacts: (a) aluminium ions connect to silicate, Al–O–Si–O, resulting in a loss of sensitivity of 29Si NMR spectrum, (b) aluminium forced re-arrangement of species perhaps into smaller groups, which also led to a loss of sensitivity of 29Si NMR spectrum, and (c) it is likely some silicate species Q1, Q2 type precipitated as aluminium silicate. There was a clear indication of a structural shift of silicate species towards condensation (precipitation reaction) processes, as precipitates were observed after prolonged times (48 hours).
A significant impact of minor concentrations of aluminium into relatively rich in silicon sodium silicate solutions is indicative that even at low Al:Si ratios aluminium can have a significant impact on dissolved silicate species.
References
- H. Bergna and W. Roberts, Colloidal Silica: Fundamentals and Applications, CRC Press, 2005, pp. 131–128 Search PubMed.
- H. Bergna, The Colloid Chemistry of Silica, American Chemical Society, Washington, DC, 1996, pp. 24–123 Search PubMed.
- M. Dietzel, Depolymerisation von hochpolymererkieselsaure in waBrigerLosung. Dissertation, Universitat Gottingen, Germany, 1993, p. 93 Search PubMed.
- M. Dietzel, Gelostepolymere und monomereKieselsauren und die WechselwirkungmitGibbsit und Fe–O–OH-Festphasen. Habilitations - Schrift, Universitat Gottingen, Germany, 1998, p. 93 Search PubMed.
- M. Dietzel, Dissolution of silicates and the stability of polysilicic acid, Geochim. Cosmochim. Acta, 1998, 64(19), 3275–3281 CrossRef.
- S. El-Manharawy and A. Hafez, Technical management of RO system, Desalination, 2000, 131, 173–188 CrossRef CAS.
- S. El-Manharawy and A. Hafez, Dehydration model for RO-membrane fouling: a preliminary approach, Desalination, 2003, 153, 95–107 CrossRef CAS.
- T. Healy, Stability of Aqueous Silica Sols, School of Chemistry, The University of Melbourne, Australia, 1994, pp. 12–16 Search PubMed.
- R. Iler, The chemistry of silica - Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry, Wiley-Interscience, New York, 1976, pp. 866–868 Search PubMed.
- A. Legrand, The surface properties of silicas, John Wiley & Sons, 1999, pp. 23–34 Search PubMed.
- L. Lunevich, Silica fouling in coal seam gas water in reverse osmosis desalination, PhD thesis, Institute for Sustainability and Innovation, Victoria University, Melbourne, Australia, 2015, pp. 34–56 Search PubMed.
- Y. Smolin, Structure of water soluble silicates with complex cations, Institute of silicate chemistry of the USSR Academia of Science, Leningrad, 1968, pp. 12–14 Search PubMed.
- S. Sjöberg, L. Ohman and N. Ingri, Equilibrium and structural studies of silicon(IV) and aluminium(III) in aqueous solution. II. Polysilicate formation in alkaline aqueous solution. A combined potentiometric and 29Si NMR study, Acta Chem. Scand., 1985, A39, 93–107 CrossRef.
- S. Sjöberg, Silica in aqueous environments, J. Non-Cryst. Solids, 1996, 196, 51–57 CrossRef.
- S. Sjöberg, A. Nordin and N. Ingri, Equilibrium and structural studies of silicon(IV) and aluminium(III) in aqueous solution. II. Formation constants for the monosilicate ions SiO(OH)3− and SiO2(OH)22−. A precision study at 25 °C in a simplified seawater medium, Mar. Geol., 1981, 10(6), 521–532 Search PubMed.
- S. Sjöberg, N. Ingri, A. Nenner and L. OlofÖhman, Equilibrium and structural studies of silicon(IV) and aluminium(III) in aqueous solution. 12. A potentiometric and 29Si-NMR study of silicon tropolonates, J. Inorg. Biochem., 1985, 24(4), 267–277 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |