
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3-Ketosphinganine provokes the accumulation of dihydroshingolipids and induces autophagy in cancer cells†
Yadira F.
Ordóñez‡
a,
Jèssica
González‡
a,
Carmen
Bedia§
 a,
Josefina
Casas
a,
José Luis
Abad
a,
Antonio
Delgado
ab and
Gemma
Fabrias
*a
a,
Josefina
Casas
a,
José Luis
Abad
a,
Antonio
Delgado
ab and
Gemma
Fabrias
*a
aConsejo Superior de Investigaciones Científicas (CSIC), Institut de Química Avançada de Catalunya (IQAC–CSIC), Research Unit on Bioactive Molecules (RUBAM), Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: gemma.fabrias@iqac.csic.es
bUniversity of Barcelona (UB), Faculty of Pharmacy, Pharmaceutical Chemistry Unit (CSIC Associated Unit), Avda. Joan XXIII s/n, 08028 Barcelona, Spain
First published on 24th February 2016
Abstract
Although several reports describe the metabolic fate of sphingoid bases and their analogs, as well as their action and that of their phosphates as regulators of sphingolipid metabolizing-enzymes, similar studies for 3-ketosphinganine (KSa), the product of the first committed step in de novo sphingolipid biosynthesis, have not been reported. In this article we show that 3-ketosphinganine (KSa) and its dideuterated analog at C4 (d2KSa) are metabolized to produce high levels of dihydrosphingolipids in HGC27, T98G and U87MG cancer cells. In contrast, either direct C1 O-phosphorylation or N-acylation of d2KSa to produce dideuterated ketodihydrosphingolipids does not occur. We also show that cells respond to d2KSa treatment with induction of autophagy. Time-course experiments agree with sphinganine, sphinganine 1-phosphate and dihydroceramides being the mediators of autophagy stimulated by d2KSa. Enzyme inhibition studies support that inhibition of Des1 by 3-ketobases is caused by their dihydroceramide metabolites. However, this effect contributes to increasing dihydrosphingolipid levels only at short incubation times, since cells respond to long time exposure to 3-ketobases with Des1 overexpression. The translation of these overall effects into cell fate is discussed.
Introduction
Sphingolipids (SLs) are a major class of constituents of eukaryotic cell membranes. In addition to a structural role, some sphingolipids are bioactive and control vital biological functions by regulating signal transduction pathways involved in several processes (i.e. apoptosis, adhesion, autophagy, cell proliferation, differentiation, migration, senescence, etc.).1–12The regulation of sphingolipid metabolism is critical for normal cell function and its dysregulation has been implicated in pathophysiological conditions. Ceramide (Cer), the central molecule in SL metabolism, can be generated by several mechanisms including both catabolic and anabolic pathways.1 One catabolic pathway involves sphingomyelinase-mediated hydrolysis of sphingomyelins. In another catabolic route, complex glycosphingolipids are degraded by specific hydrolases to monohexosylceramides (MHCer) (glucosylceramide and galactosylceramide), which are then hydrolyzed by specific β-glucosidases and galactosidases, respectively, to produce Cer. Another mechanism is the anabolic de novo pathway, which begins with the condensation of serine and palmitoyl-CoA to form 3-ketosphinganine (KSa), a reaction catalyzed by serine palmitoyltransferase (SPT). Then, 3-ketosphinganine reductase (KSR) mediates the reduction of KSa to sphinganine (Sa), which is transformed into dihydroceramide (dhCer) through acylation by ceramide synthases (CerS). In the last step of the de novo pathway, Cer is formed through the introduction of an (E)-4 double bond into dhCer by dihydroceramide desaturase (Des1). Once formed, Cer can be degraded through the catabolic route, which involves N-deacylation to sphingosine (So) by ceramidases, further phosphorylation to sphingosine 1-phosphate (S1P) and final irreversible cleavage by S1P lyase.
The metabolic fate of several free bases has been studied over the years. Dragusin et al.13 investigated the metabolism of D-erythro-sphinganine (Sa) in several cell lines and compared it to that of its stereoisomer safingol (D-threo-Sa). Venkataraman and Futerman14 compared the metabolism of L-erythro- and L-threo-Sa in cultured cells and in subcellular fractions. Finally, the metabolic fates of 1-deoxysphinganine and 1-deoxysphingosine have also been examined,15,16 as well as those of ω-azidosphingosine17 and ω-alkynylsphingosine.18 Although enzymatic conversions of KSa into Sa,19 ketosphingosine20 and sphingosine (So)21 were reported in cell free systems in the early 70s, only the reduction step has been later extensively confirmed in cell culture. Surprisingly, putative N-acylation of KSa and further formation of 3-ketodihydrosphingolipids, as well as KSa C1 O-phosphorylation, have not been analyzed.
On the other hand, the regulation of SL metabolizing-enzymes by SL metabolites has been reported. For instance, cytosolic S1P inhibits de novo Cer synthesis,22 which has been recently reported to occur through negative regulation of SPT by sphingosine kinase 123 and it inhibits acid sphingomyelinase.24 In addition, D-erythro-So, but not L-threo-So, decreases SPT activity in primary cultured cerebellar cells.25
In this article, using both KSa and [4,4-2H2]-KSa (d2KSa), a dideuterated analog of KSa, we show that the 3-ketobase is used to produce SLs de novo and directed to the catabolic pathway, both prior to reduction to Sa (Fig. 1), and that this results in an overall accumulation of dihydrosphingolipids. Metabolic transformations by direct N-acylation and C1 O-phosphorylation do not seem to occur (Fig. 1). Furthermore, we report that Des1 activity is reduced by short cell treatment with KSa and d2KSa by their dihydroceramide and/or ceramide metabolites. Finally, in agreement with previous reports supporting the role of dhCer as a mediator of autophagy, we show that autophagy is induced by d2KSa in three different cell lines.
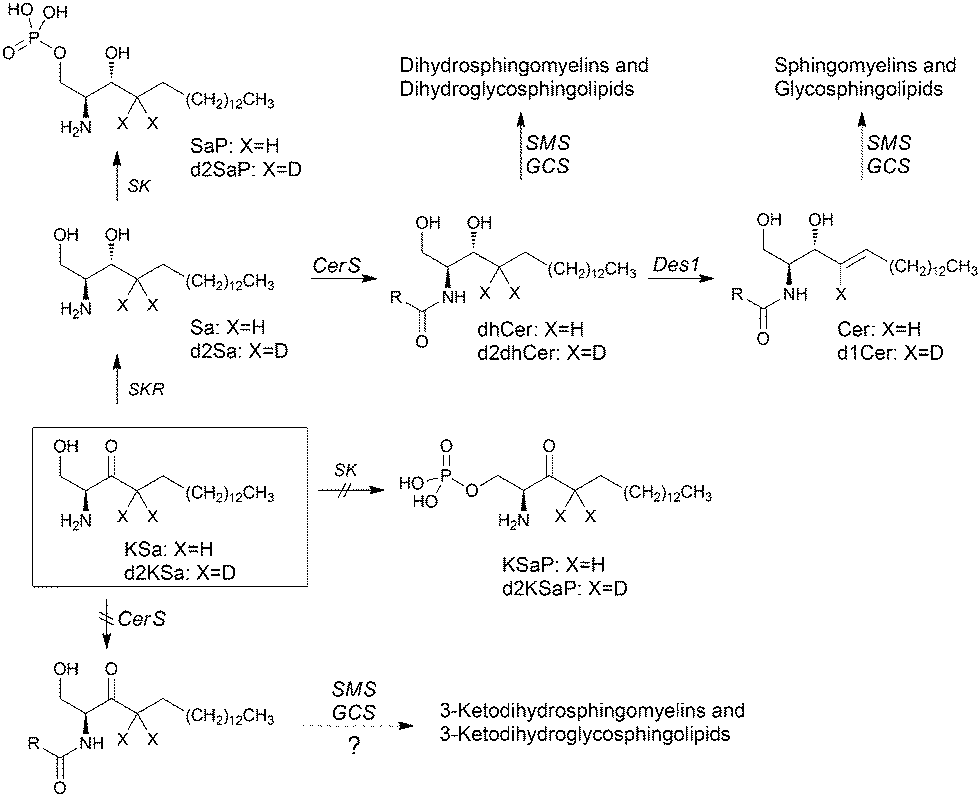 | ||
| Fig. 1 Possible metabolism of exogenous 3-ketosphinganine. The interrogation sign indicates that the reaction has not been demonstrated. Enzymes are abbreviated as follows: CerS, ceramide synthases; Des1, dihydroceramide desaturase; GCS, glucosylceramide synthase; SK, sphingosine kinases; SKR, 3-ketosphinganine reductase; and SMS, sphingomyelin synthase. | ||
Results
3-Ketosphingoid bases induce changes in the sphingolipidome in HGC27 cells
Cell viability experiments showed that KSa was cytotoxic to HGC27 cells with CC50 and CC25 values (24 h) of 19.7 ± 3.3 μM and 12.6 ± 6.4 μM, respectively (mean ± SD of three experiments in triplicate). In these cells, KSa at 5 μM modified sphingolipid composition and this modification was already significant at the earliest time point examined (3 h) (Fig. 2). Exogenous KSa was metabolized to afford high levels of Sa, which decreased over incubation time (Fig. 2A). Sa was then incorporated into both the catabolic and the de novo Cer synthesis pathways (see Fig. 1). Thus, high amounts of Sa 1-phosphate (SaP) were produced at 3 and 6 h of treatment, to decrease at longer incubation times (Fig. 2B). On the other hand, Sa was acylated to dhCer, whose levels reached a maximum at 6 h post-treatment (Fig. 2C). A similar pattern was observed for dihydrosphingomyelin (dhSM) (Fig. 2D) and lactosyldihydroceramide (LdhCer) (Fig. 2E),¶ although in both cases the difference between 3 and 6 h was higher than that for dhCer. Desaturation of dhCer to Cer was also found to occur with a maximum at 6 h post-treatment (Fig. 2F). Although MHCer (ESI,† Fig. S1A), LCer (ESI,† Fig. S1B) and So (ESI,† Fig. S1C) followed a similar tendency, the differences were not always statistically significant. Finally, SM did not change significantly over the time of treatment with KSa (ESI,† Fig. S1D). These overall data indicate that catabolism of Sa occurs more rapidly than its incorporation into the de novo pathway for SL synthesis. In order to obtain a clearer picture of the metabolism of exogenous KSa, the metabolic transformation of the labeled KSa analog d2KSa was investigated. The use of this probe allows discriminating between the metabolites produced from the exogenous precursor and those already present in cells at the time of treatment. Labeled sphingolipids produced from d2KSa followed similar patterns to those found with unlabeled KSa (Fig. 2G). Thus, prior to reduction to [4,4-2H2] Sa (d2Sa), the probe was incorporated into the catabolic route to afford high levels of [4,4-2H2] SaP (d2SaP), which reached a maximum at 3 h. In contrast, biosynthesis of SLs de novo occurred at later time points, with [4,4-2H2] dhCer (d2dhCer) exhibiting maximum levels at 3 and 6 h and [4,4-2H2] dihydrosphingomyelin (d2dhSM) and [4,4-2H2] lactosyldihydroceramide (d2LdhCer) peaking at 16 h. Regarding monodeuterated sphingolipids, [4-2H] Cer (d1Cer) was detected at lower levels than those of natural Cer formed from KSa (Fig. 2G and F, respectively). This result is in agreement with the reported primary isotope effect in the desaturation of a 4,4-dideuterated dhCer.26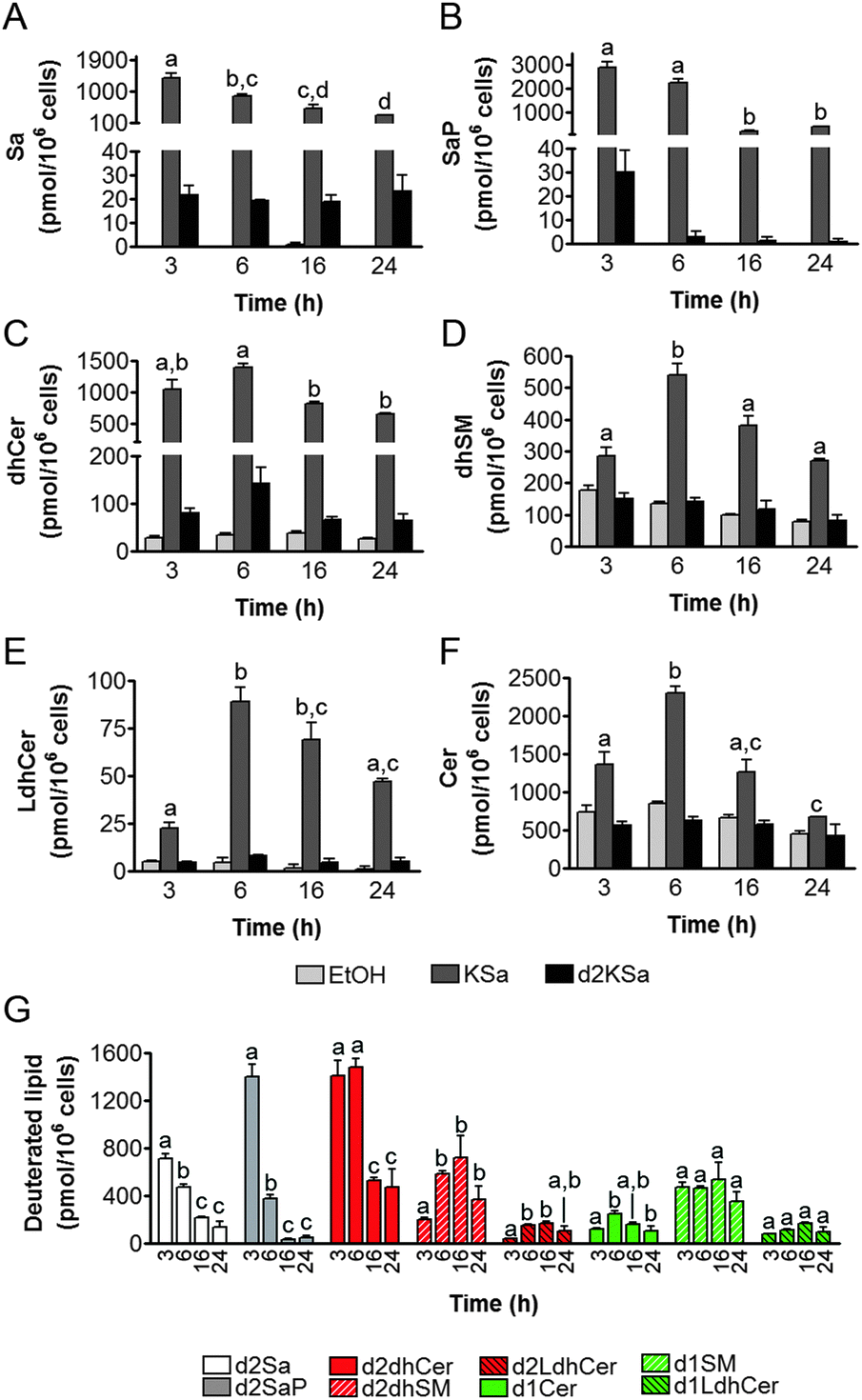 | ||
| Fig. 2 Effect of KSa and d2KSa on sphingolipid levels in HGC27 cells. Cells were treated with KSa (5 μM), d2KSa (5 μM) or vehicle for specified time periods and then cells were collected and processed for LC/MS lipid analysis. All species were analyzed under ESI-positive mode. (A—F) Natural sphingolipids. (G) Deuterated sphingolipids. Data correspond to the mean ± SD of three independent experiments in triplicate. Lipid production over the time of treatment was analyzed using the one-way ANOVA test (A and F, P < 0.0001; B, P < 0.0002; E, P < 0.0003; D, P < 0.0004; C, P < 0.0017; G/d2Sa and G/d2SaP, P < 0.0001; G/d2dhCer, P < 0.0002; G/d2dhSM, P < 0.038; G/d2LdhCer, P < 0.0073; G/d1Cer, P < 0.0136) followed by Bonferroni's multiple comparison test. Different letters atop the bars denote statistically significant differences between means at different times at P < 0.05 as found by this test. | ||
Putative N-acylation and C1 O-phosphorylation of d2KSa were also investigated. Selection of ions corresponding to the deuterated 3-ketosphingolipid analogs (ESI,† Table) did not provide any evidence for direct N-acylation of d2KSa and further formation of deuterated ketodihydrosphingomyelins and glucosylketodihydroceramides. Likewise, dideuterated 3-ketosphiganine 1-phosphate was not detected in the chromatograms. An authentic synthetic sample of N-hexadecanoyl-3-ketosphinganine was used as standard to predict the expected retention times of the putative 3-ketodihydrosphingolipids. The absence of N-hexadecanoyl-3-ketosphinganine and dideuterated N-hexadecanoyl-3-ketosphinganine in extracts from cells treated with KSa and d2KSa, respectively, is shown as an example in ESI,† Fig. S2.
Inhibition of Des1 by the 3-ketobases
Although the above results support that the observed increase in dihydrosphingolipids provoked by exogenous KSa and d2KSa was due to their metabolism, the possibility that Des1 was impaired by the 3-ketobases could not be ruled out. To assess this issue, Des1 activity was measured in both intact cells and cell lysates in the presence of KSa, d2Ksa or some downstream metabolites.As shown in Fig. 3A, live cells co-incubated for 4 h with KSa and the Des1 substrate, namely N-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]sphinganine (dhCerC6NBD), produced significantly less N-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]sphingosine (CerC6NBD)27 than controls (EtOH). Des1 inhibition was also provoked by d2KSa and XM462 (positive control). However, when cells were treated with the probes for 24 h and then with the substrate for 4 additional hours, significantly higher desaturation occurred in cells treated with d2KSa than with KSa, which was not significantly different from controls (Fig. 3B). XM462 showed Des1 inhibitory activity, proving that the enzyme is active and responds to inhibition under the experimental conditions of the assay. These results suggested that long time treatment with d2KSa might induce overexpression of Des1. To judge this possibility, Des1 mRNA from cells exposed to d2KSa, KSa and the vehicle for 24 h was determined by qPCR. As shown in Fig. 3C, significantly higher Des1 mRNA levels were present in cells treated with d2KSa than with the vehicle. Although the Des1 transcript also increased after treatment with KSa, this increase was only marginally significantly different from controls (P = 0.058). These results support that although Des1 inhibition may contribute to the accumulation of dihydrosphingolipids brought about by the 3-ketobases at short time points, impaired Des1 activity does not contribute to increase dihydrosphingolipid levels at long incubation times.
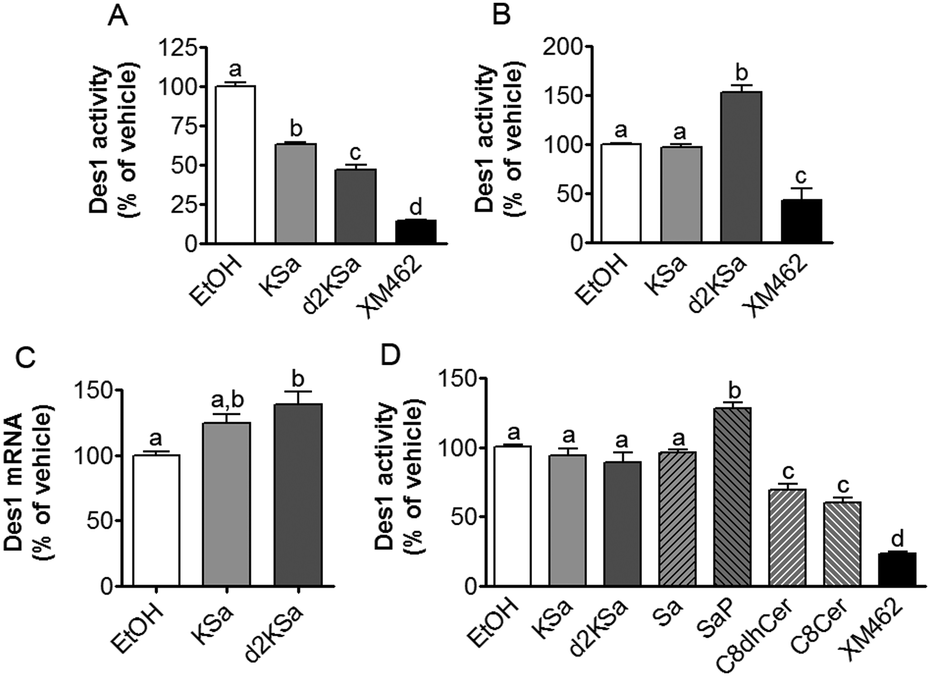 | ||
| Fig. 3 Effect of KSa and d2KSa on Des1 activity and expression. Inhibition was determined in both intact cells (A and B) and cell lysates (D) by measuring the desaturation of dhCerC6NBD (10 μM) to CerC6NBD.27 (A) Intact cells were incubated for 4 h with the substrate together with KSa (5 μM), d2KSa (5 μM), XM462 (10 μM) (positive control of inhibition) or ethanol (control). (B) Intact cells were incubated for 24 h with KSa (5 μM), d2KSa (5 μM), XM462 (10 μM) (positive control of inhibition) or ethanol (control), and then the substrate was added and further incubated for 4 h. (D) The assay in cell lysates was carried out by incubation of protein (equivalent to 106 cells per replicate) and the substrate in the absence (EtOH, vehicle) or presence of 10 μM of test compounds and 120 μM of NADH as reported.27 In all cases, the formation of CerC6NBD was determined by HPLC coupled to a fluorescence detector. Data correspond to the mean ± SD of three (A and B) or six (D) independent experiments in triplicate. Data were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison post-test. In all cases, ANOVA P < 0.0001. Different letters denote statistically significant differences between means at P < 0.05 as found by this test. (C) Effect of KSa (5 μM/24 h) and d2KSa (5 μM/24 h) on the mRNA levels (as determined by quantitative real-time PCR) of Des1. Data correspond to the mean ± SD of three independent experiments in duplicate. Data were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison post-test. ANOVA P < 0.01. Different letters denote statistically significant differences between means at P < 0.05 as found by this test. | ||
The effect of KSa and d2KSa on Des1 in a cell-free system was also analyzed. Since lipid analysis showed the formation of diverse metabolites in cells, the Des1 inhibitory activity of downstream sphingolipid metabolites was also determined. As shown in Fig. 3D, neither KSa nor d2KSa reduced Des1 activity. Likewise, Des1 was not inhibited by Sa and SaP. However, both N-octanoylsphinganine (C8dhCer) and N-octanoylsphingosine (C8Cer) reduced Des1 activity to 70 and 60% of control, respectively, at equimolar concentrations with the substrate. In these experiments, XM462 elicited Des1 inhibitory activity, proving that the enzyme is active and responds to inhibition under the experimental conditions of the assay. These results support that dhCer and/or Cer are the actual Des1 inhibitory species in intact cells subjected to short time treatments with the 3-ketobases (Fig. 3A).
Physiological effect of 3-ketosphingoid bases
We previously reported that 4,4-dideuterated dhCer induced autophagy in HGC-27 cells and that this induction was preceded by an increase in d2dhCer of different chain lengths.28 This result supported a connection between dhCer increase and autophagy. Since both KSa and d2KSa induce an increase in dhCer, we investigated whether autophagy was also activated by the 3-ketosphingoid bases. For an unambiguous interpretation of results, these experiments were carried out with d2KSa because this compound induces an increase in d2dhCer (Fig. 2G) with no increase in natural Cer levels (Fig. 2F) and a poor increase in monodeuterated Cer levels (Fig. 2G), while both dhCer (Fig. 2C) and Cer (Fig. 2F) augment in treatments with KSa. As shown in Fig. 4A and B, HGC27 cells treated with 12 μM d2KSa for 6 h exhibited a significant rise in the levels of LC3-II over controls and this increase was enhanced by co-incubation with protease inhibitors, which block the degradation of proteins included in the autophagolysosome. These results support that d2KSa stimulates the autophagic flux. However, LC3-II did not augment in cells treated with 12 μM d2KSa for 24 h (data not shown), but cells exhibited signs of apoptotic cell death (Fig. 4C and D). However, other experiments using different techniques must be carried out to confirm the apoptosis outcome. In order to determine the scope of the proautophagic action of the 3-ketobases, the d2KSa activity at increasing LC3-II levels was also determined in two glioblastoma cell lines. Western blot analysis of proteins from T98G cells treated with d2KSa (12 μM)|| for 6 h showed increased levels of LC3-II as compared to controls treated with the vehicle. Moreover, this increase was enhanced in the presence of protease inhibitors (Fig. 5A and C). Similar results were found in U87MG cells upon exposure to d2KSa (20 μM/6 h)|| (Fig. 5E and G). With these cells, immunofluorescence studies with an antibody that recognizes preferentially the lipidated form of endogenous LC3 revealed an increased accumulation of immunostained LC3-II puncta in cells treated with the compounds (Fig. 5B and F), indicating that d2KSa induces autophagosome formation. Importantly, under conditions found to induce autophagy, d2KSa modified the sphingolipidome in both cell lines. As found with HGC27 cells (see above), d2KSa provoked an increase in d2dhCer, d2dhSM and d2LdhCer levels. Also similarly to HGC27 cells, the levels of d1Cer (Fig. 5D and H) were lower than the levels of Cer (ESI,† Fig. S3), likely due to a primary kinetic isotope effect at C4 in the Des1-catalyzed reaction.26 In contrast to HGC27 and U87MG cells, T98G cells produced very low amounts of d2SaP (Fig. 5D), indicating either that phosphorylation is very rapid and no longer evident at 6 h after treatment or that sphingosine kinases are poorly active in these cells under the experimental conditions of the assay. These overall results indicate that d2KSa stimulates the production of dihydrosphingolipids and the autophagic flux also in T98G and U87MG cell lines, supporting that these effects are not cell specific. In contrast to HGC27 cells, increasing d2KSa incubation time did not result in apoptosis in T98G and U87MG cells (ESI,† Fig. S4), but in an increased number of necrotic cells (positive to propidium iodide, but not to Annexin V, staining) as compared to vehicle treated controls. Early and late apoptosis were induced by N-octanoylsphingosine (C8Cer), which was used as positive control of the apoptotic response in these cells. | ||
| Fig. 4 Effect of d2KSa in HGC27 cells. Cells were treated with 12 μM d2KSa for either 6 h (A and B) or 24 h (C and D). In (A) and (B), treatments were carried out in the absence (−PI) or presence (+PI) of protease inhibitors. Cells were lysed and proteins were analyzed by the Western blot technique using an anti-LC3 antibody. (A) Western blot image corresponding to one representative experiment. (B) Histograms showing the relative fold-changes of LC3-II/Actin over control (EtOH). In (C) and (D), cells were processed and analyzed by flow cytometry after propidium iodide/FITC-Annexin V staining. (C) Representative cell distribution plots. (D) Histogram showing the percentage of alive, apoptotic and necrotic cells. Data correspond to three experiments in duplicate (A and B) or triplicate (C and D). In (B), data were analyzed by the one-way ANOVA test (P < 0.0001) followed by Bonferroni's multiple comparison test. Different letters denote statistically significant differences between means at P < 0.05 as found by this test. In (D), asterisks indicate statistical significance vs. vehicle at P < 0.05 (unpaired two-tailed t test). | ||
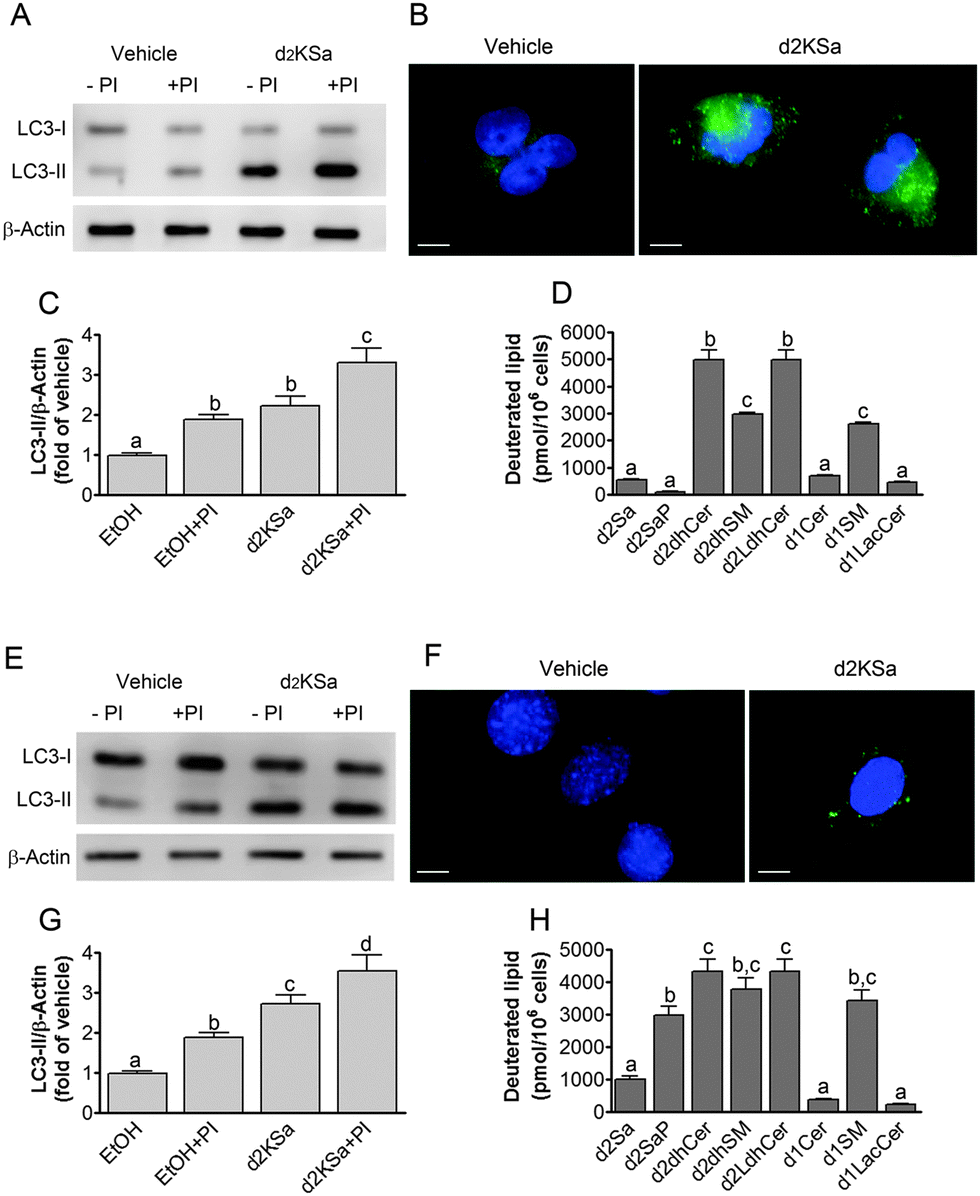 | ||
| Fig. 5 Effect of d2KSa on autophagy and deuterated lipid levels in glioblastoma cells. Autophagy was determined by Western blot (A, C, E, G) and immunofluorescence (B and F) analysis of LC3-II in T98G (A–C) and U87MG (E–G) cells treated with d2KSa (A, 12 μM/6 h; B, 20 μM/6 h) or vehicle in the presence or absence of protease inhibitors (PI: E64D and Pepstatin A) as detailed in the Experimental section. (A and E) Western blot images corresponding to representative experiments. (B and F) Representative fluorescence microscopy images showing the formation of LC3-II puncta in T98 (B) and U87 (F) cells after treatment with d2KSa (B, 12 μM/6 h; F, 20 μM/6 h) or vehicle. Scale bar: 10 μm. (C and G) Histograms showing the relative fold-changes of LC3-II/Actin over control (EtOH) in WB analyses. (D and H) Deuterated sphingolipids present in T98G (D) and U87MG (H) cells treated with d2KSa (A, 12 μM/6 h; B, 20 μM/6 h). Data correspond to three experiments in duplicate (A–C and E–G) or triplicate (D and H). In (C), (D), (G) and (H) data were analyzed by the one-way ANOVA test (P < 0.0001 in all cases) followed by Bonferroni's multiple comparison test. Different letters denote statistically significant differences between means at P < 0.05 as found by this test. | ||
Discussion
Although several reports describe the metabolic fate of sphingoid bases and their analogs,13,14,16,17 as well as their action and that of their phosphates as regulators of sphingolipid metabolizing enzymes,22–25 similar studies for KSa, the product of the first committed step in sphingolipid biosynthesis, have not been reported. Using both natural KSa and mass labeled d2KSa, in this article we show that exogenous KSa follows the expected metabolic pathways being directed to the catabolic route and used to produce SLs de novo, both prior to reduction to Sa. Furthermore, previously unreported direct N-acylation of d2KSa and C1 O-phosphorylation have not been found to occur. These results may indicate that CerS and sphingosine kinases do not accept KSa as substrate. However, this does not agree with the reported ability of CerS to accept a wide range of sphingoid bases as substrates, including fumonisins,29,30 deoxysphingoid bases,16,31 azidosphingoid bases17 and jaspine B (Cingolani et al., manuscript in preparation). On the other hand, sphingosine kinases have also been found to phosphorylate several unnatural sphingoid bases, such as, for instance, the immunomodulatory sphingolipid analog FTY720.32,33 A more plausible explanation is that KSa is readily reduced to Sa as soon as it is available, either from exogenous sources or endogenously generated by SPT action, so that sufficient amounts of the ketobase substrate are never accessible for CerS- and SK-mediated transformation. The fact that free KSa was not detected in lipid extracts, while very high amounts of Sa, its reduction product, were found at early time points supports the fast reduction hypothesis.Lipid analysis and Des1 activity studies support that Des1 inhibition by substrates (dhCer or d2dhCer) may contribute to the accumulation of dihydrosphingolipids brought about by the 3-ketobases in the first hours of treatment, but not in long incubation times. This conclusion is based on the findings that only C8dhCer and C8Cer, but not KSa, Sa and SaP, decrease Des1 activity in cell lysates. Furthermore, although C8Cer inhibits Des1 in cell lysates and ceramide levels increase in treatments with KSa, d2KSa is a more potent Des1 inhibitor in cell lysates but deuterated ceramide levels increase poorly in treatments with d2KSa. The latter finding is against Cer being the Des1 inhibitory species in treatments with the 3-ketobases. Finally, the 3-ketobases lowered Des1 activity in intact cells only in co-incubation with the Des1 substrate for a short time period, but not after long pre-incubation times. Interestingly, the latter conditions resulted in increased Des1 expression, mainly in d2KSa treatments, in which augmented Des1 activity was also evidenced. Increased Des1 expression likely occurs as a response of cells to the accumulation of dhCer at long incubation times with the 3-ketobases. The higher overexpression provoked by d2KSa as compared to natural KSa can be explained considering that d2KSa is metabolically converted into d2dhCer, which is desaturated at a 7-fold lower rate than natural dhCer.26 Higher overexpression of Des1 would be induced by d2dhCer rather than by dhCer (formed from exogenous KSa) to cope with the enzyme clogging provoked by the slowly processed deuterated substrate.
Both Cer and dhCer have been reported to exhibit important biological functions. While the roles of Cer in mediating antimitogenic actions have been recognized for a number of years,1–4,34–37 the biological actions of dhCer have been disclosed more recently.5 They include induction of cell cycle arrest,38 delay28 and autophagy39–44 and inhibition of Cer-induced channel formation in mitochondria with final mitigation of the apoptotic effect of Cer.45 In addition to both amides, phosphorylated sphingolipids such as long chain base 1-phosphates8–11,46–47 and Cer 1-phosphate12,48,49 are also biologically active. In the first case, experimental evidence supports the idea that the location of S1P production dictates its functions. Thus, while S1P generated in the cytosol by the action of sphingosine kinase 1 stimulates cell proliferation and inhibits de novo Cer synthesis, S1P produced in the ER by sphingosine kinase 2 promotes Cer synthesis through the salvage pathway and induces apoptosis.22 In this article we report that d2KSa induces autophagy at short incubation times, when a large increase in Sa, SaP and dhCer occurs, but not at long incubation times, when those dihydrosphingolipids undergo a significant decrease. In contrast, dhSM and dhLCer experienced the highest increase at long incubation times, when autophagy was no longer induced by d2KSa, arguing against the role of both lipids in autophagy induction by d2KSa. This correlation supports the argument that Sa, SaP and/or dhCer are involved in the pro-autophagic activity of d2KSa. Autophagy activation by dhCer5–7,41 and Sa42,50 has been reported and our results add further support to the role of both sphingolipids as mediators of autophagy. Cer and S1P have also been reported to stimulate autophagy.51 However, Cer and S1P (data not shown) do not increase upon d2KSa treatment over controls, which does not support the role of both species as mediators of autophagy induction by d2KSa treatment. Regarding SaP, although differential effects of SaP versus S1P have been described,9–11 SaP has never been reported to induce autophagy. Although d2SaP also increases in parallel with autophagy induction by d2KSa in HGC27 and U87MG cell lines, such an increase is not observed in T98G cells, while autophagy is also stimulated in these cells. These results are against SaP being involved in autophagy induction.
Although autophagy is activated as a cell survival response to harmful stimuli, cells can eventually die if the pro-autophagic input is severe or long lasting. Resveratrol has been shown to induce autophagy without any sign of cell death in HGC27 cells with the preferential production of C16dhCer.28 On the other hand, sphingolipidomic analyses of four T-cell acute lymphoblastic leukemia cell lines revealed strong positive correlations between cytotoxicity associated with increased autophagy and levels of C22 and C24dhCer.43 Moreover, Sa has been reported to induce autophagic cell death42,50 and also to mediate the cytotoxicity of fumonisin B152 and fenretinide.53 We speculate that in our cell models, d2KSa exhibits cytotoxicity because the balance between protective (C16d2dhCer) and cytotoxic (d2Sa, C22d2dhCer and C24d2dhCer) species is tilted toward the latter. Further studies are ongoing to study the role of different sphingolipid species in autophagy induction and outcome.
Materials and methods
Materials
Minimum essential medium (MEM), Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), BSA, Trypsin–EDTA, non-essential amino acids, NADH, and pepstatin A were purchased from Sigma. E64D was from Enzo and the polyvinylidene difluoride (PVDF) membrane was from Roche. ECL Prime Western Blotting Detection Reagent was purchased from GE Healthcare. The Annexin V-FITC Early Apoptosis Detection Kit was purchased from Cell Signaling. Internal standards for lipidomics were from Avanti Polar Lipids. Laemmli buffer and acrylamide were from BioRad, and SDS was from Fluka. Antibodies: anti-LC3 was purchased from Abcam (reference Ab48394), β-actin was from Sigma (reference A2228) and HRP-secondary antibodies were from GE Healthcare (anti-mouse, reference RPN4201V; anti-rabbit, reference NA934V). Compounds KSa, d2KSa, dhCerC6NBD27 and XM46227 were synthesized in our laboratories.Synthesis of KSa and d2KSa
KSa was prepared following a reported protocol.54Cell culture
The human gastric cancer cell line HGC 27 was cultured at 37 °C in 5% CO2 in minimum essential medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids, and penicillin (100 U ml−1) and streptomycin (100 μg ml−1). Cells were routinely grown to a maximum confluence of 60%. Human glioblastoma cell lines T98G and U87MG were cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin (100 U ml−1) and streptomycin (100 μg ml−1).Cell viability
Cell viability was measured in triplicate samples by the MTT assay. Cells were seeded in 96 well plates at a density of 1 × 105 cells per ml and then subjected to various treatments for 24 h. At the end of the treatments MTT was added to each well and incubated for 3 h. The supernatant was aspirated, and the formazan crystals were dissolved in DMSO. Absorbance was measured at 570 nm.LC3-II analysis by Western blot
For LC3-II protein analysis 1 × 105 cells were plated in 6 well plates and were allowed to adhere for 24 h. Cells were pre-incubated with protease inhibitors (E64D 10 μg ml−1 and Pepstatin A 5 μg ml−1). After 2 h, cells were treated with d2KSa at the specified concentrations or EtOH as control for 6 h. After treatments, cells were directly lysed in Laemmli sample buffer and heated at 95 °C for 5 min. Samples were loaded onto a 12% polyacrylamide gel, separated by electrophoresis at 140 V/1 h and transferred onto a PVDF membrane (100 V/1 h). Unspecific binding sites were then blocked with 5% milk in TBST (LC3) or 3% BSA in TBST (Actin). Anti-LC3 antibody was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 in 5% milk in TBST. Anti-Actin antibody was diluted 1:2000 in 3% BSA in TBST. Membranes were incubated overnight at 4 °C under gentle agitation. After washing with TBST, membranes were probed with the corresponding secondary antibody for 1 h at room temperature (LC3: anti-rabbit diluted 1:1000 in 3% BSA in TBST; Actin:anti-mouse diluted 1:10000 in 5% milk in TBST). Antibody excess was eliminated by washing with TBST, and protein detection was carried out using ECL and membrane scanning with a LI-COR C-DiGit® Blot Scanner. Band intensities were quantified using LI-COR Image Studio Lite Software.
:1000 in 5% milk in TBST. Anti-Actin antibody was diluted 1:2000 in 3% BSA in TBST. Membranes were incubated overnight at 4 °C under gentle agitation. After washing with TBST, membranes were probed with the corresponding secondary antibody for 1 h at room temperature (LC3: anti-rabbit diluted 1:1000 in 3% BSA in TBST; Actin:anti-mouse diluted 1:10000 in 5% milk in TBST). Antibody excess was eliminated by washing with TBST, and protein detection was carried out using ECL and membrane scanning with a LI-COR C-DiGit® Blot Scanner. Band intensities were quantified using LI-COR Image Studio Lite Software.
Immunofluorescence
Cells were grown on coverslips in 6 well plates at a density of 1 × 105 cells per ml and allowed to attach. The medium was replaced with fresh medium containing d2KSa (T98 cells, 12 μM/6 h; U87MG cells, 20 μM/6 h) or vehicle (EtOH). After treatments cells were washed with PBS and fixed in 4% paraformaldehyde (25 °C/20 min). After washing the cells with PBS, aldehydes were quenched with 50 mM NH4Cl in PBS for 10 min. Cells were washed again with PBS, and permeabilized and blocked with 3% BSA/0.1% Triton X-100 in PBS (25 °C/30 min). After washing with PBS, cells were incubated with 40 μg ml−1 of anti-LC3 antibody in PBS overnight at 4 °C. Following incubation, cells were washed and treated with FITC conjugated secondary antibody diluted 1:100 in PBS (25 °C/1.5 h). The punctate pattern of LC3 was visualized under a Nikon Eclipse 90i fluorescence microscope at 60× objective magnification.
Apoptosis
Cells were seeded in 6 well plates (1 × 105 cells per ml), and after 24 h, they were treated for 24 h with either d2KSa at the specified concentrations or EtOH (negative control). After treatments, cells were collected by trypsinization, washed twice with PBS and centrifuged (200 rpm/3 min). The cell pellet was resuspended in 96 μL of Annexin V binding buffer, 1 μL of FITC-Annexin V and 12.5 μL of propidium iodide. After 10 min on ice in the dark, apoptosis was determined using a Guava easyCyteTM flow cytometer (Merck Millipore).Lipid analyses
Cells were seeded at 1 × 105 cells per ml in 6 well plates (1 ml per well) and were allowed to adhere for 24 h. The medium was replaced with fresh medium containing the test compounds at the specified concentrations or EtOH as control. After specified time periods, the medium was removed; cells were washed with 400 μl PBS and harvested with 400 μl Trypsin–EDTA and 600 μl of medium. Sphingolipid extracts, fortified with internal standards (N-dodecanoylsphingosine, N-dodecanoylglucosylsphingosine, N-dodecanoylsphingosylphosphorylcholine, C17-sphinganine and C17-sphinganine 1-phosphate, 0.2 nmol each), were prepared and analysed as reported by UPLC-TOF MS.40Des1 activity assay
The effect of compounds on Des1 activity was studied in both HGC27 cell lysates and intact cells as reported.40Quantitative RT-PCR
Total RNA was isolated from cells using the RNeasy® Mini Kit from the QIAGEN protocol. RNA concentration was measured by spectrophotometric absorption at 260 nm using a NanoDrop ND-8000 spectrophotometer. RNA was treated with DNAse I to remove genomic DNA contamination. Quantities from 1 μg to 100 ng of DNAse I-treated RNA were retrotranscribed to cDNA using Superscript® II Reverse Transcriptase from Invitrogen and stored at −20 °C. cDNA preparations were used to quantify specific transcripts in a LightCycler® 480 Real Time PCR System, using SYBR®Green Mix (Roche, Germany) and the following pairs of primers: Des1 (F, 5′-CTATGCGTTTGGCAGTTGCA-3′, and R, 5′-CAGTTGCCAAAGGCAGCATT-3′) and GAPDH (F, 5′-ACCATCTTCCAGGAGCGAGA-3′, and R, 5′-GATGGCATGGACTGTGGTCA-3′). For all genes, the initial PCR steps were: 10 min at 95 °C, followed by 45 cycles of 10 s melting at 95 °C and 30 s annealing/extension at 60 °C. The final step was 1 min incubation at 60 °C. All reactions were performed in triplicate. Relative mRNA abundances of the different genes were calculated from the second derivative maximum of their respective amplification curves (Cp, calculated in duplicate). Cp values for target genes (Tg) were compared to the corresponding values for the GAPDH reference gene to obtain the ΔCp values (ΔCp = CpGAPDH − CpTg).Conclusions
In conclusion, we have reported that KSa and its deuterated analog d2KSa induce the production of high levels of dihydrosphingolipids resulting from both the probe metabolism and, under certain conditions, inhibition of Des1 activity. Time-course experiments show that, among the several dihydrosphingolipids, Sa/d2Sa, SaP/d2SaP and dhCer/d2dhCer reach maximum levels at the earliest time points examined (3 and 6 h), while dhSM/d2dhSM and LdhCer/d2LdhCer increase at longer incubation times (16 and 24 h). Autophagy induction by d2KSa, which occurs at short (6 h) but not long (24 h) times of d2KSa treatment, correlates with Sa and dhCer being the mediators of autophagy stimulated by d2KSa. Finally, we could not find any evidence for the direct N-acylation or C1 O-phosphorylation of KSa.Acknowledgements
Partial financial support from the “Ministerio de Ciencia e Innovación”, Spain (Grants SAF2011-22444), “Ministerio de Economía y Competitividad” (CTQ2014-54743-R), CSIC (Grant PIE 2008801034) and Fundació La Marató TV3 (Grant 112130 and 112132) is acknowledged. A PhD fellowship from SENESCYT-Ecuador to Y. F. O. is also acknowledged. We thank Pedro Rayo for his excellent technical assistance.Notes and references
- S. F. Morad and M. C. Cabot, Nat. Rev. Cancer, 2013, 13, 51–65 CrossRef CAS PubMed.
- J. A. Chavez and S. A. Summers, Cell Metab., 2012, 15, 585–594 CrossRef CAS PubMed.
- I. Petrache, D. N. Petrusca, R. P. Bowler and K. Kamocki, Proc. Am. Thorac. Soc., 2011, 8, 492–496 CrossRef CAS PubMed.
- X. Li, K. A. Becker and Y. Zhang, Cell. Physiol. Biochem., 2010, 26, 41–48 CrossRef CAS PubMed.
- G. Fabrias, J. Muñoz-Olaya, F. Cingolani, P. Signorelli, J. Casas, V. Gagliostro and R. Ghidoni, Prog. Lipid Res., 2012, 51, 82–94 CrossRef CAS PubMed.
- M. Casasampere, Y. F. Ordoñez, A. Pou and J. Casas, Chem. Phys. Lipids, 2016 Search PubMed , in press.
- M. M. Siddique, Y. Li, B. Chaurasia, V. A. Kaddai and S. Summers, J. Biol. Chem., 2015, 290, 15371–15379 CrossRef CAS PubMed.
- R. L. Proia and T. Hla, J. Clin. Invest., 2015, 125, 1379–1387 CrossRef PubMed.
- S. Bu, M. Yamanaka, H. Pei, A. Bielawska, J. Bielawski, Y. A. Hannun, L. Obeid and M. Trojanowska, FASEB J., 2006, 20, 184–186 CAS.
- S. Bu, Y. Asano, A. Bujor, K. Highland, F. Hant and M. Trojanowska, Arthritis Rheum., 2010, 62, 2117–2126 CAS.
- S. Bu, B. Kapanadze, T. Hsu and M. Trojanowska, J. Biol. Chem., 2008, 283, 19593–19602 CrossRef CAS PubMed.
- A. Gomez-Muñoz, P. Gangoiti, L. Arana, A. Ouro, I.-G. Rivera, M. Ordoñez and M. Trueba, Biochim. Biophys. Acta, 2013, 1831, 1060–1066 CrossRef PubMed.
- M. Dragusin, C. Gurgui, G. Schwarzmann, J. Hoernschemeyer and G. van Echten-Deckert, J. Lipid Res., 2003, 44, 1772–1779 CrossRef CAS PubMed.
- K. Venkataraman and A. H. Futerman, Biochim. Biophys. Acta, 2001, 1530, 219–226 CrossRef CAS.
- H.-U. Humpf, E.-M. Schmelz, F. I. Meredith, H. Vesper, T. R. Vales, E. Wang, D. S. Menaldino, D. C. Liotta and A. H. Merrill, J. Biol. Chem., 1998, 273, 19060–19064 CrossRef CAS PubMed.
- J. L. Abad, I. Nieves, P. Rayo, J. Casas, G. Fabriàs and A. Delgado, J. Org. Chem., 2013, 78, 5858–5866 CrossRef CAS PubMed.
- M. Garrido, J. L. Abad, G. Fabriàs, J. Casas and A. Delgado, ChemBioChem, 2015, 16, 641–650 CrossRef CAS PubMed.
- P. Haberkant, F. Stein, D. Höglinger, M. J. Gerl, B. Brügger, P. P. Van Veldhoven, J. Krijgsveld, A. C. Gavin and C. Schultz, ACS Chem. Biol., 2015, 11, 222–230 CrossRef PubMed.
- W. Stoffel, D. Lekim and G. Sticht, Hoppe-Seyler's Zeitschrift für Physiol. Chemie, 1968, 349, 1637–1644 CrossRef CAS.
- Y. Fujino and M. Nakano, Biochim. Biophys. Acta, 1971, 239, 273–279 CAS.
- M. Nakano and Y. Fujino, Biochim. Biophys. Acta, 1973, 296, 457–460 CAS.
- M. Maceyka, H. Sankala, N. C. Hait, L. Stunff, H. Liu, R. Toman, C. Collier, M. Zhang, L. S. Satin, A. H. Merrill, S. Milstien and S. Spiegel, J. Biol. Chem., 2005, 280, 37118–37129 CrossRef CAS PubMed.
- D. Siow, M. Sunkara, A. Morris and B. Wattenberg, Adv. Biol. Regul., 2015, 57, 42–54 CrossRef CAS PubMed.
- A. Gómez-Muñoz, J. Kong, B. Salh and U. P. Steinbrecher, FEBS Lett., 2003, 539, 56–60 CrossRef.
- E. C. Mandon, G. van Echten, R. Birk, R. R. Schmidt and K. Sandhoff, Eur. J. Biochem., 1991, 198, 667–674 CrossRef CAS PubMed.
- C. K. Savile, G. Fabriàs and P. H. Buist, J. Am. Chem. Soc., 2001, 123, 4382–4385 CrossRef CAS PubMed.
- J. M. Munoz-Olaya, X. Matabosch, C. Bedia, M. Egido-Gabás, J. Casas, A. Llebaria, A. Delgado and G. Fabriàs, ChemMedChem, 2008, 3, 946–953 CrossRef CAS PubMed.
- V. Gagliostro, J. Casas, A. Caretti, J. L. Abad, L. Tagliavacca, R. Ghidoni, G. Fabrias and P. Signorelli, Int. J. Biochem. Cell Biol., 2012, 44, 2135–2143 CrossRef CAS PubMed.
- H. Harrer, E. L. Laviad, H. U. Humpf and A. H. Futerman, Mol. Nutr. Food Res., 2013, 57, 516–522 CAS.
- M. Seiferlein, H.-U. Humpf, K. A. Voss, M. C. Sullards, J. C. Allegood, E. Wang and A. H. Merrill, Mol. Nutr. Food Res., 2007, 51, 1120–1130 CAS.
- H. U. Humpf, E. M. Schmelz, F. I. Meredith, H. Vesper, T. R. Vales, E. Wang, D. S. Menaldino, D. C. Liotta and A. H. Merrill, J. Biol. Chem., 1998, 273, 19060–19064 CrossRef CAS PubMed.
- S. W. Paugh, S. G. Payne, S. E. Barbour, S. Milstien and S. Spiegel, FEBS Lett., 2003, 554, 189–193 CrossRef CAS PubMed.
- A. Billich, F. Bornancin, P. Dévay, D. Mechtcheriakova, N. Urtz and T. Baumruker, J. Biol. Chem., 2003, 278, 47408–47415 CrossRef CAS PubMed.
- A. Morales and J. C. Fernandez-Checa, Mini-Rev. Med. Chem., 2007, 7, 371–382 CrossRef CAS PubMed.
- S. A. Saddoughi and B. Ogretmen, Adv. Cancer Res., 2013, 117, 37–58 CrossRef CAS PubMed.
- T. D. Mullen and L. M. Obeid, Anti Canc. Agents Med. Chem., 2012, 12, 340–363 CrossRef CAS.
- M. N. Nikolova-Karakashian and K. A. Rozenova, Adv. Exp. Med. Biol., 2010, 688, 86–108 CrossRef CAS PubMed.
- J. M. Kraveka, L. Li, Z. M. Szulc, J. Bielawski, B. Ogretmen, Y. A. Hannun, L. M. Obeid and A. Bielawska, J. Biol. Chem., 2007, 282, 16718–16728 CrossRef CAS PubMed.
- P. Signorelli, J. M. Munoz-Olaya, V. Gagliostro, J. Casas, R. Ghidoni and G. Fabriàs, Cancer Lett., 2009, 282, 238–243 CrossRef CAS PubMed.
- F. Cingolani, M. Casasampere, P. Sanllehi, J. Casas, J. Bujons and G. Fabrias, J. Lipid Res., 2014, 55, 1711–1720 CrossRef CAS PubMed.
- M. M. Siddique, Y. Li, L. Wang, J. Ching, M. Mal, O. Ilkayeva, Y. J. Wu, B. H. Bay and S. A. Summers, Mol. Cell. Biol., 2013, 33, 2353–2369 CrossRef CAS PubMed.
- Q. Jiang, X. Rao, C. Y. Kim, H. Freiser, Q. Zhang, Z. Jiang and G. Li, Int. J. Cancer, 2012, 130, 685–693 CrossRef CAS PubMed.
- M. W. Holliday, S. B. Cox, M. H. Kang and B. J. Maurer, PLoS One, 2013, 8, e74768 CAS.
- S. Rodriguez-Cuenca, N. Barbarroja and A. Vidal-Puig, Biochim. Biophys. Acta, 2015, 1851, 40–50 CrossRef CAS PubMed.
- J. Stiban, D. Fistere and M. Colombini, Apoptosis, 2006, 11, 773–780 CrossRef CAS PubMed.
- V. A. Blaho and T. Hla, J. Lipid Res., 2014, 55, 1596–1608 CrossRef CAS PubMed.
- K. Mendelson, T. Evans and T. Hla, Development, 2013, 141, 5–9 CrossRef PubMed.
- L. A. Hoeferlin, D. S. Wijesinghe and C. E. Chalfant, Handb. Exp. Pharmacol., 2013, 153–166 CAS.
- I.-G. Rivera, M. Ordoñez, N. Presa, A. Gomez-Larrauri, J. Simón, M. Trueba and A. Gomez-Muñoz, Toxins, 2015, 7, 1457–1466 CrossRef CAS PubMed.
- J. Noack, J. Choi, K. Richter, A. Kopp-Schneider and A. Régnier-Vigouroux, Cell Death Dis., 2014, 5, e1425 CrossRef CAS PubMed.
- C. Bedia, T. Levade and P. Codogno, Anti Canc. Agents Med. Chem., 2011, 11, 844–853 CrossRef CAS.
- J. M. Soriano, L. González and A. I. Catalá, Prog. Lipid Res., 2005, 44, 345–356 CrossRef CAS PubMed.
- Z. Mao, W. Sun, R. Xu, S. Novgorodov, Z. M. Szulc, J. Bielawski, L. M. Obeid and C. Mao, J. Biol. Chem., 2010, 285, 29078–29090 CrossRef CAS PubMed.
- A. Bielawska, Z. Szulc and Y. A. Hannun, Methods Enzymol., 2000, 311, 518–535 CAS.
- L. A. Cowart, Z. Szulc, A. Bielawska and Y. A. Hannun, J. Lipid Res., 2002, 43, 2042–2048 CrossRef CAS.
- S. Grijalvo, C. Bedia, G. Triola, J. Casas, A. Llebaria, J. Teixidó, O. Rabal, T. Levade, A. Delgado and G. Fabriàs, Chem. Phys. Lipids, 2006, 144, 69–84 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary table with the exact masses corresponding to 3-ketodihydrosphingolipids and supplementary figures showing the effect of KSa and d2KSa on natural sphingolipid levels in HGC27 cells (Fig. S1), lack of N-acylation of ketosphingoid bases (Fig. S2), the effect of d2KSa on natural sphingolipid levels in T98G and U87MG cells (Fig. S3) and flow cytometry analysis of T98G and U87MG cells exposed to d2KSa (Fig. S4). See DOI: 10.1039/c5mb00852b |
| ‡ These authors contributed equally to this work. |
| § Current address: Institute of Environmental Assessment and Water Research (IDAEA-CSIC), Spain. |
| ¶ Monohexosyldihydroceramides, both natural and deuterated, and monodeuterated monohexosylceramides cannot be analysed accurately because of the interference of highly abundant sphingomyelins. |
| || Concentrations used according to cell viability data. d2KSa was cytotoxic to T98G and U87MG cells with CC50 values (mean ± SD, 3 experiments with triplicates) at 24 h of 26.9 μM (±4.2) and 29.9 μM (±9.1), respectively, and CC25 values (24 h) of 16.9 μM (±7.6) (T98G) and 15.0 μM (±4.9) (U87MG). |
| This journal is © The Royal Society of Chemistry 2016 |