
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide-tags for site-specific protein labelling in vitro and in vivo
Jonathan
Lotze†
a,
Ulrike
Reinhardt†
b,
Oliver
Seitz
*b and
Annette G.
Beck-Sickinger
*a
aInstitut für Biochemie, Universität Leipzig, Brüderstrasse 34, D-04103 Leipzig, Germany. E-mail: abeck-sickinger@uni-leipzig.de
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, D-12489 Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de
First published on 29th February 2016
Abstract
Peptide-tag based labelling can be achieved by (i) enzymes (ii) recognition of metal ions or small molecules and (iii) peptide–peptide interactions and enables site-specific protein visualization to investigate protein localization and trafficking.
Introduction
Visualizing the localization and trafficking of proteins in living systems is the key to understand protein functions in their native environment. Tracking and visualization by fluorescence microscopy require methods that allow the site-specific labelling of a protein of interest (POI). Established almost 20 years ago,1 the genetic fusion of a POI to an autofluorescent protein such as the green fluorescent protein (GFP) revolutionized protein imaging. The fusion of POIs with engineered, self-modifying enzymes such as SNAP-,2 CLIP-,3 TMP-,4,5 or Halo-tags6 significantly extended the scope to customized labelling by enabling the use of reporter units with unique spectroscopic signatures. Enzyme-based tags provide high specificity and enable fast labelling reactions, which is an advantage when fast biological processes ought to be analysed in time-lapse experiments. Yet, the fusion of enzymes brings along a massive increase in size (18–33 kDa). This can perturb protein trafficking and may impair protein–protein interaction networks that rely on clustering. To avoid potential interference with protein localization and protein function, major efforts are spent to decrease the tag size. The smallest tags currently accessible are provided by the incorporation of unnatural amino acids (UAA) by amber stop codon or related technologies.7,8 In addition, the ligand-directed protein labelling or traceless affinity labelling is independent of any tag.9,10 However, the establishment and incorporation of UAA in mammalian cells still is very challenging and the traceless affinity labelling requires a POI with a native binding pocket as well as the exact knowledge of the binding site and ligand.Peptide-based recognition tags offer an attractive alternative for protein labelling. The genetic fusion of peptide-tags is straightforward, the size of 0.6–6 kDa is considerably small and a variety of labelling chemistries allow the introduction of virtually arbitrary reporter groups.
Peptide-tag based labelling methods are multifaceted and can be categorised into three classes: (i) enzyme mediated labelling, (ii) metal ion- or small molecule-dependent labelling and (iii) labelling based on peptide–peptide interactions. We will discuss the characteristics of the different approaches with respect to specificity, speed, stability, tag size, toxicity and versatility. Applications as well as shortcomings will be explained. The review covers the literature until 2015 and particularly emphasizes the most recent and most frequently used developments. Additional information can be taken from other comprehensive reviews.11–14
Peptide-tag based labelling by enzymes – mechanisms and characteristics
Enzymes offer a naturally occurring sequence-specificity for site-selective modifications. Ligases such as biotin ligase, lipoic acid ligase or tubulin tyrosine ligase are capable of transferring molecular probes to a particular amino acid (aa) within a recognition sequence. Similarly, transferases can be used to transfer functional groups from a donor substrate to a specific amino acid within a peptide sequence, as demonstrated for phosphopantetheinyl transferases Sfp and AcpS, transglutaminase or the phosphocholine transferase AnkX. Another possibility is to use transpeptidase activity. Sortase A exchanges a part of the recognition sequence with a separate peptide conjugated to a labelling moiety. The specific modification of an amino acid side chain can also be achieved with formylglycine-generating enzyme, which naturally modifies a cysteine derived thiol group to an aldehyde function (see Table 1).| Enzyme | Recognition tag | Reported tag positions | Typical/reported concentrations and incubation conditions | Reported applications | One-step or two-step labelling |
|---|---|---|---|---|---|
| RT – room temperature. | |||||
| Biotin ligase (BirA) | GLNDIFEAQ![[K with combining low line]](https://www.rsc.org/images/entities/b_char_004b_0332.gif) IEWHE (AP-tag) IEWHE (AP-tag) |
N-terminal, C-terminal, internal |
BirA-coxpression enables biotinylation during protein expression; biotin detection with streptavidin reporter within 15–60 min at RT using fixed cells or at 4 °C using live cells;17,21
Ligation of 1 mM keton probe (biotin analogue) within 10–60 min at 32 °C, subsequent detection with 1 mM hydrazine probe within 10–60 min at 4 °C in live cells23 |
In vitro, in vivo (cell surface labelling with fixed or live cells), cell lysate isolated from transgenic mice | Two-step |
| Lipoic acid ligase (LplA) |
DEVLVEIETDAVLEVPGGEEE (LAP-tag 1st generation)
GFEID |
N-terminal, C-terminal, internal | Labelling with 100–350 μM lipoic acid derivative for 15–120 min; probe detection 10–20 min; reaction temperature: 21–37 °C25,28,29 | In vitro, in vivo (cell surface labelling with live cell imaging) | One-step and two-step |
| Tubulin tyrosine ligase (TTL) | VDSVEGEGEEEGEE (Tub-tag) | C-terminal | Labelling with 1 mM tyrosine derivative for 1–3 h; subsequent detection via biocompatible labelling required 4–24 h; reaction temperature: 30 or 37 °C33 | In vitro | Two-step |
| Phospho-pantetheinyl-transferase (Sfp and AcpS) |
D![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif) LEFIASKLA (ybbR-tag) LEFIASKLA (ybbR-tag)
GD GD |
N-terminal, C-terminal, internal | Labelling with 1–10 μM CoA-derivative for 20–40 min; reaction temperature: 37 °C38,39 | In vitro, in vivo (cell surface labelling with fixed or live cells) | One-step and two-step |
| Transglutaminase (TGase) |
PNP![[Q with combining low line]](https://www.rsc.org/images/entities/b_char_0051_0332.gif) LPF (Q1-tag) LPF (Q1-tag)
PKP GQ xx |
N-terminal, C-terminal, internal | Labelling with 400–500 μM cadaverine probe for 25–30 min or up to 4 h; reaction temperature: 4 or 37 °C41,46 | In vitro, in vivo (cell surface labelling with live cell imaging) | One-step and two-step |
| AnkX/Lem3 | TITSYYR |
N-terminal, C-terminal, internal | Labelling with 1 mM CDP-choline probe derivative for ≤1 h; reaction temperature: RT49 | In vitro | Two-step |
| Sortase A (SrtA) | LPxTG | C-terminal | Labelling with 1–5 mM oligoglycine probe for 1–5 h in vitro54,55/10 μM oligoglycine probe for 15–60 min in living cells; reaction temperature: 37 °C56 | In vitro, in vivo (cell surface labelling with live cell imaging) | One-step and two-step |
| Formylglycine-generating enzyme (FGE) | L![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) xPxR (aldehyde-tag) xPxR (aldehyde-tag) |
N-terminal, C-terminal, internal | Labelling with 30–500 μM hydrazine or aminooxy probes for 2 h or overnight in vitro59,60/1 mM labelling probe for 1 h in living cells; reaction temperature: RT or 37 °C61 | In vitro, in vivo (cell surface labelling with live cell imaging) | One-step and two-step |
Already in 1999 a 15 aa long acceptor peptide, termed AP-tag, was reported to serve as a substrate for E. coli derived biotin ligase (BirA). This enzyme promotes the conjugation of a biotin to a lysine side chain within the AP-tag (Fig. 1A).15 BirA-mediated biotinylation was demonstrated for many different AP-tagged proteins in cell lysates including the transcription factors GATA-1 and EKLF.16 Co-expression of AP-tagged and BirA-tagged proteins allows the so-called proximity biotinylation to study protein–protein interactions.17 This method is widely used and has enabled investigations of homo- and heterodimerization of chemokine receptors CXCR4, CCR2 and CCR5 in RPE cells18 or glycoprotein interactions during herpesvirus entry into cells.19 Of note, the method also enables investigations of proximity between two proteins expressed on different cells. For example, BirA-mediated biotinylation was used to characterize the synaptic connectivity between the presynaptic adhesion protein NRX-1 and the postsynaptic transmembrane protein NLG-1 in vivo in Caenorhabditis elegans.20 In a recent study, BirA-mediated biotinylation exposed the anterograde trafficking of calcium activated potassium channels (KCa3.1) in polarized epithelial cells (including HEK293, MDCK, LLC-PK1 and Caro-2 cells). The AP-tag was introduced into the protein and BirA was fused to an ER-retaining sequence (KDEL) to specifically label AP-tagged KCa3.1 along the anterograde pathway.21 Furthermore the endocytosis of the apical Na+/K+/2Cl− co-transporter (NKCC2) equipped with an AP-tag between the 5th and 6th transmembrane domain was measured in real time by total internal reflection fluorescence (TIRF) microscopy. After BirA-mediated biotinylation, fluorescent staining was performed with fluorescent streptavidin.22 BirA also accepts a biotin derivative containing a ketone group. A comparison of biotin ligases from different organisms revealed that Saccharomyces cerevisiae and Pyrococcus horikoshii accept alkyne and azide derivatives of biotin.23,24 The biotin modifications enable subsequent labelling of proteins such as epidermal growth factor receptor (EGFR) on live cells by formation of acyl hydrazones or Staudinger ligation.23
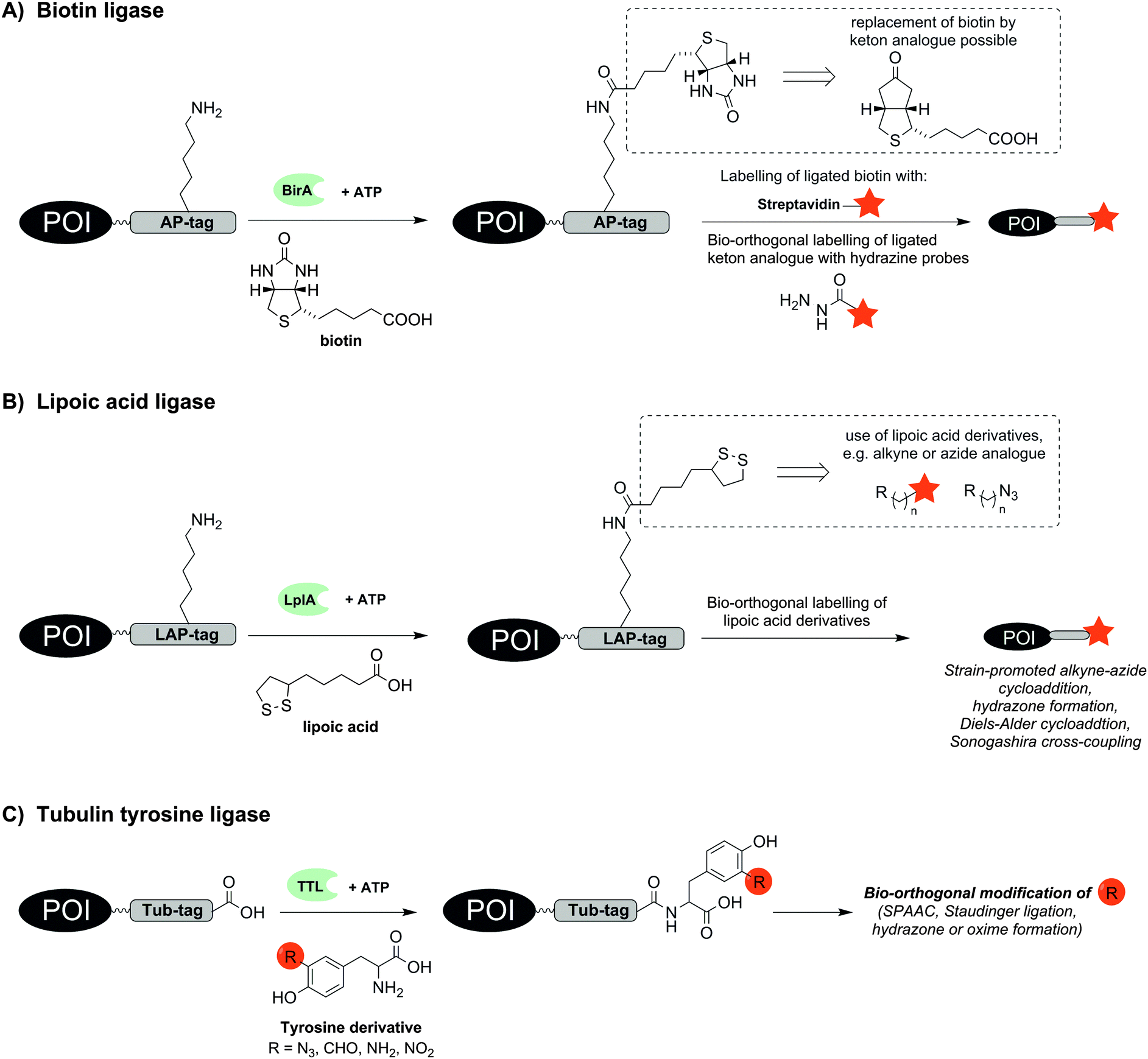 | ||
| Fig. 1 Protein labelling by means of (A) biotin ligase (BirA), (B) lipoic acid ligase (LplA) or (C) tubulin tyrosine ligase (TTL). The red star indicates the reporter group. | ||
A BirA cross-compatible technique uses E. coli derived lipoic acid ligase (LplA), which catalyses the ligation of lipoic acid to a sequence-specific lysine side chain in an ATP-dependent manner (Fig. 1B).25,26 LplA specifically recognizes the LAP-tag, which consists of 22 aa, and accepts different lipoic acid derivatives containing alkyne, azide, trans-cyclooctene, aryl aldehyde or aryl hydrazine groups as well as fluorophores.25,27–29 Strain-promoted alkyne–azide cycloaddition (SPAAC), hydrazone formation or Diels–Alder cycloaddition30 enabled the subsequent chemical modification of the lipoic acid appendage on LAP-tagged proteins such as CFP,25 BFP,28 low-density lipoprotein receptor (LDLR),25 neurexin-1β27 or actin.28 The LplA mutant LplAW37V has been reported to provide higher ligation yields than the wild-type enzyme.27 In contrast to BirA, LplA accepts fluorescently labelled conjugates and permits one-step labelling of the POI. LplA-mediated protein labelling was demonstrated on HEK293T-, HeLa and COS-7 cells. A minimized LAP-tag with only 13 aa31 was used to incorporate a p-iodophenyl derivative as new bio-orthogonal handle into proteins, which could be selectively addressed in a second step by palladium-catalysed Sonogashira cross-coupling.32 Wombacher et al. reported norbornene-conjugated lipoic acid analogues, which were introduced into LAP-tagged CFP and dihydrofolate reductase from E. coli (eDHFR) and fluorescently labelled via inverse-electron demand Diels–Alder reaction.30
The enzyme tubulin tyrosine ligase (TTL) allows the C-terminal labelling of proteins with a 14 aa hydrophilic recognition sequence called Tub-tag. TTL is a regulator of microtubule homeostasis and usually promotes the coupling of tyrosine to the α-tubulin C-terminus. Hackenberger and Leonhardt showed that TTL is also able to incorporate tyrosine derivatives containing azide, formyl, amine or nitryl groups (Fig. 1C).33 This enabled the two step labelling of GFP-specific nanobodies with biotin, fluorescent probes or polyethylene glycol (PEG) chains by means of SPAAC, Staudinger ligation, Staudinger-phosphite reactions and hydrazone or oxime forming reactions. Labelling also succeeded in E. coli lysate.
The use of transferases offers interesting alternatives to ligase-mediated labelling. Phosphopantetheinyl transferases (PPTase) such as Sfp and AcpS catalyse the transfer of a phosphopantetheinyl (Ppant) group derived from coenzyme A (CoA) to a serine residue within a specific recognition sequence (Fig. 2A).34 Owing to an impressive substrate promiscuity of Sfp and AcpS, CoA can be modified at its terminal thiol with small molecules, including biotin, sugars, peptides, porphyrin and fluorophores, the latter allowing one-step labelling similar to LplA.34–37 Furthermore, a set of three specific tags is available: ybbR-tag (11 aa), S6-tag (12 aa), preferentially recognized by Sfp and A1-tag (12 aa) preferentially recognized by AcpS.38 The ybbR-tag was successfully introduced at the N- and C-terminus, or within the protein sequence as demonstrated for eGFP.37 Orthogonal cell surface labelling with fluorescent CoA conjugates on living HeLa cells was demonstrated for S6-tagged EGFR or A1-tagged transferrin receptor 1 (TfR1).38 The S6/A1 pair of tags was also used to enable labelling of the tropomyosin receptor kinase TrkA and the p75 neurotrophin co-receptor (P75NTR) in SH-SY5Y cells.39 The A1-P75NTR construct was first labelled with biotin-CoA using AcpS and then stained with streptavidin-QDot525. Subsequently, the S6-TrkA construct was biotinylated by using Sfp followed by labelling with streptavidin-QDot655. To enable imaging of interactions between chemokines and their fluorescent protein (FP)-tagged receptors on live CHO-K1 cells, the chemokines CXCL12, CCL2 and CCL21 were expressed with a S6-tag at the C-terminus and subjected to PPTase-mediated labelling with a fluorophore–CoA conjugate.40 Co-localization of signals from the fluorophore and the FP indicated receptor–chemokine interactions.
 | ||
| Fig. 2 Protein labelling via (A) phosphopantetheinyl transferase (PPTase) Sfp or (B) phosphocholine transferase AnkX. The hydrolase Lem3 enables removal of the label. | ||
Transglutaminases (TGase) catalyse the formation of an amide bond between the carboxamide of a glutamine side chain and the ε-amino group of a lysine side chain. TGases have been used to attach small molecules to antibodies,41 modify proteins by PEGylation42 or lipidation,43 site-specifically conjugate proteins,44 as well as immobilize proteins on solid support.45 CFP and EGFR were labelled in vitro and on the surface of living cells by means of guinea pig liver TGase (gpTGase), which promotes the conjugation of labelled cadaverine (lysine analogue conjugated to biotin, AlexaFluor586 or photoaffinity moieties) with 6–7 aa long glutamine-rich Q-tags.46 Recent studies described the use of tissue TGase (TG2) or microbial TGase (mTGase).47,48 The latter provides the advantage of calcium-independent labelling. Transfer of propargylamine with subsequent CuAAC modification was demonstrated for maltose binding protein using mTGase.
Two enzymes from Legionella pneumophila were recently investigated by Hedberg et al. for the reversible covalent labelling of proteins. The first enzyme AnkX transfers a phosphocholine moiety from a cytidine diphosphate choline (CDP-choline) derivative to the second serine residue within the eight amino acid recognition sequence TITSSYYR (Fig. 2B). The second enzyme Lem3 is able to remove the label upon hydrolytic cleavage of the transferred phosphocholine from the serine side chain. The method tolerates fusion of the recognition sequence to the N- or C-terminus, as well as into internal loops as demonstrated for fluorescein labelling of model proteins such as maltose binding protein, small ubiquitin-like modifier and DrrA enzyme.49
Remarkably small tag sizes are found for sortase A and formylglycine-generating enzyme. The transpeptidase sortase A (SrtA) derived from Staphylococcus aureus specifically recognizes the short pentapeptide sequence LPxTG. SrtA cleaves the amide bond between threonine and glycine, forms a reactive thioester between the carboxyl group of threonine and an enzyme-derived cysteine and promotes the ligation of this reactive intermediate with an amino group of an oligoglycine substrate by formation of a new amide bond (Fig. 3A). First reports on SrtA as new protein engineering tool described modifications of LPETG-tagged GFP with a set of various oligoglycine substrates conjugated to peptides including unnatural amino acids, proteins or small molecules like folate.50 Subsequently, the substrate diversity was extended to PEGylation, peptide–glycosylphosphatidylinositol conjugates or cyclised peptides.51–53 SrtA-mediated labelling has been used to label proteins with fluorophores, photocrosslinkers, biotin, alkyl chains and cholesterol on intact cells such as HEK293T, CHO and HeLa cells.54–56 In a noteworthy study the bacterial toxin aerolysin was labelled with fluorophores and biotin. The ability to form homo-heptameric pores in membranes of target cells after binding to cell surface receptors was not impaired and SrtA-mediated biotinylation helped to identify aerolysin–receptor interactions, which are responsible for aerolysin uptake.57 Recently LPETG-tagged lipopeptides enabled the visualization of liposome trafficking into lung cancer.58
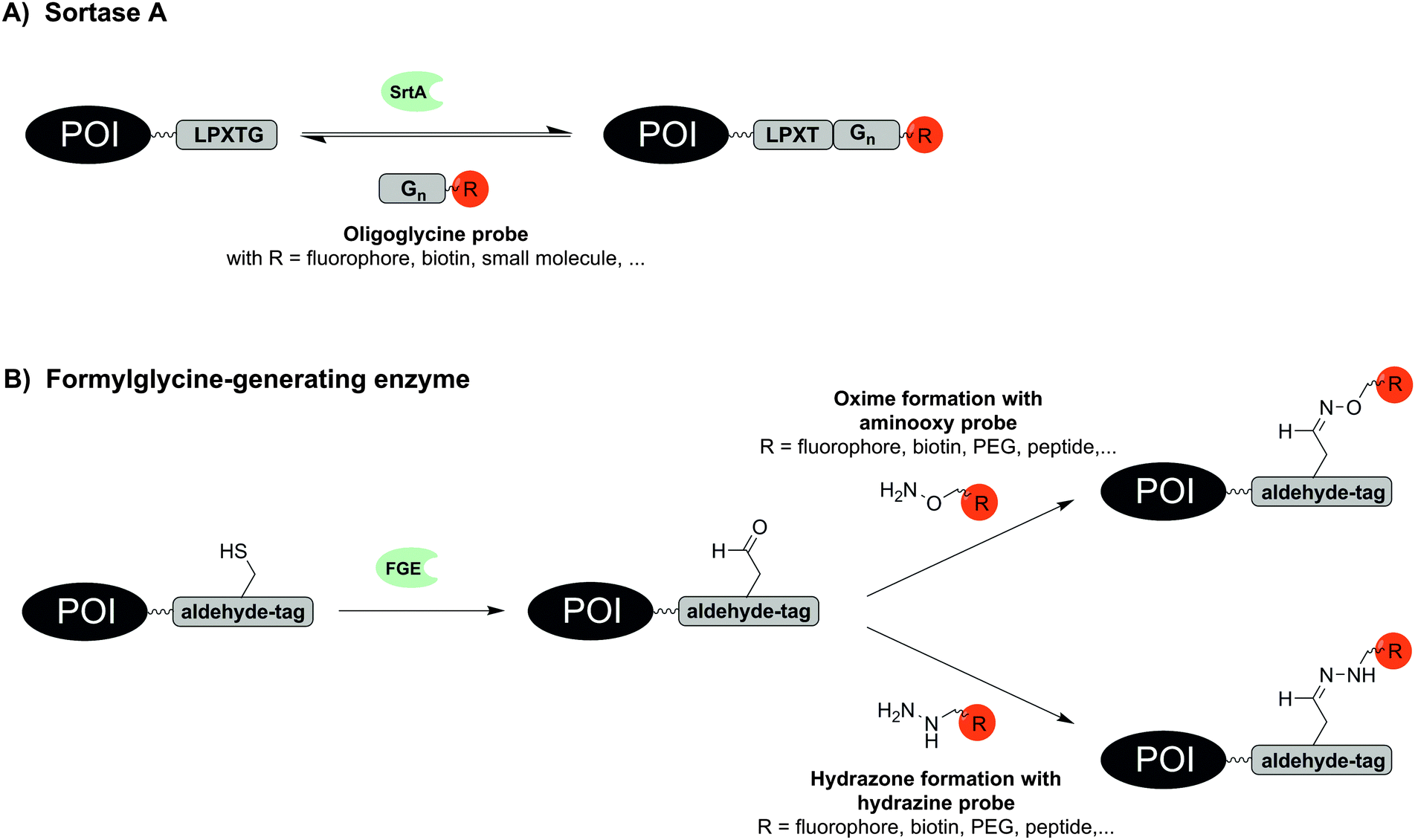 | ||
| Fig. 3 Protein labelling via (A) sortase A (SrtA) and (B) formylglycine-generating enzyme (FGE). | ||
The formylglycine-generating enzyme (FGE) naturally co- or post-translationally modifies a cysteine within the conserved recognition motif CxPxR to an aldehyde-bearing formylglycine (Fig. 3B).59 The hexapeptide sequence LCxPxR, termed “aldehyde tag”, was included at N-terminal, C-terminal or internal sites as shown for maltose binding protein, human growth hormone, bacterial sulfotransferase or antibodies. Because FGE is endogenously expressed in most prokaryotes and eukaryotes, the aldehyde moiety is formed without additional intervention. For labelling with fluorophores, biotin, peptides or PEG probes the aldehyde group was addressed in hydrazone or oxime forming reactions.59,60 The endogenous location of FGE in the endoplasmic reticulum was exploited to specifically trace aldehyde-tagged proteins such as IgG antibody, platelet-derived growth factor receptor (PDGFR) and glycoprotein CD4 along the secretory pathway.61 Recently, a natural glycosylation site within an Fc fragment was replaced by an aldehyde tag, which allowed the introduction of tailor-made glycoforms by means of oxime-forming reactions.60 Recent work with recombinantly expressed FGE suggests that CuII is required as a cofactor in order to achieve high turnover in formyl generating reactions.62
Enzymatic labelling techniques provide high specificity, relatively small tag sizes and covalently bound labels. Yet there are disadvantages. To achieve high labelling yields rather long reaction times and/or high concentrations of substrate are required. One of the fastest approaches to stain proteins in living cells was reported for BirA with a total labelling time of 20 min.23 However, the second step of this two-step labelling method had to be performed at 4 °C to reduce endocytosis of the target protein EGFR. Labelling with SrtA and fluorescent oligoglycine conjugates was reported to be achieved within 15 min on living HEK293T cells. But the concentration of the oligoglycine substrate was considerably high with 10 μM.56 At high concentrations of substrate the specificity of labelling can be at stake and excessive washing may be required to remove non-covalently bound substrate. PPTase-mediated protein labelling is even performed at 1–150 μM concentration of biotin-CoA,36,37 AlexaFluor488- or TexasRed-CoA38 probes. Most other enzyme-based labelling methods require higher concentrations of substrate. For example, 1 mM ketone-containing biotin derivative is applied in ligations by BirA.23 Some enzyme-catalysed reactions such as the transpeptidase reaction catalysed by SrtA are reversible which leads to decreases in labelling yields if the substrate is applied at low concentration.52,53 Many enzymes depend on cofactors such as ATP (e.g. BirA,23,24 LplA,25,28,31 TTL33), Mg2+ (e.g. BirA,23,24 LplA,25,28,31 TTL,33 PPTase38,39) or Ca2+ (e.g. TGase,46,47 except mTGase48) which may interfere with the biological process under investigation. With regard to the labelling time, the two-step procedures significantly prolong the process. Usual incubation times in the first step take 10–60 min (e.g. LplA,25,28 TTL,33 AnkX,49 SrtA,54–56 BirA,23 TGase46,47). The bio-orthogonal labelling reactions in the second step require up to 3 h (e.g. SPAAC, Staudinger ligation)33 or even overnight treatment (Cu-promoted alkyne–azide cycloaddition).48
However, a rapid labelling at low concentration of labelling agents is desirable in studies of fast biological processes. This feature can be obtained with peptide-tags that have high affinity for reactive metal complexes, small molecules or peptides and thereby enable reactions at high effective molarity, which provides for efficient labelling with reagents at nanomolar concentrations.
Recognition of metal ions and small molecules by peptide-tags – mechanisms and characteristics
The selectivity of labelling can be ensured by the mutual recognition between electron-deficient metal ions and electron-rich ligands. The binding of small metal ion probes, in most cases by a chelating effect, facilitates a one-step labelling of the target protein (see Table 2).| Labelling technique | Recognition tag and labelling probes | Reported tag position | Typical/reported concentrations and incubation conditions | Reported applications |
|---|---|---|---|---|
| RT – room temperature. | ||||
| Tetracysteine motif |
Tag: CCxxCC
Probes: FlAsH, ReAsH, ChoXAsH, CrAsH, Cy3As |
N-terminal, C-terminal, Internal | Labelling with 1–5 μM FlAsH probe for 30–90 min in living cells; reaction temperature: RT63,64 | Fixed and live cell staining |
| Tetraserine motif |
Tag: SSPGSS
Probe: RhoBo |
N-terminal, C-terminal, Internal | Labelling with 50 nM probe for 20 min in living cells; reaction temperature: 37 °C68 | Live cell staining |
| His-tag |
Tag: His6 or His10
Probes: fluorescent conjugates of NiII:bis/tris/tetrakis-NTA; HisZiFit or ZnII:IDA-Cy5 |
N-terminal, C-terminal | Labelling with 100–500 nM probe for 1–60 min; reaction temperature: RT72,76,77 | Fixed and live cell staining |
| Oligo-aspartate-tag |
Tags: (D4)3, CA6(D4)2, D3 (DDD or DDDXXDDD), DYKDDDDK, (DDDW)3DDD
Probes: fluorescent conjugates of ZnII:DpaTyr or NiII:DpaTyr |
N-terminal, C-terminal | Labelling with 1–2 μM for 5–7 min (non-covalent); reaction temperature: 25 °C78/20 μM probe for 12 h (covalent); reaction temperature: 4 and 20 °C79 | Live cell staining |
| Lanthanide-binding-tag |
Tags: ACADYNKDGWYEELECAA or FIDTNNDGWIEGDELLLEEG
Probe: Tb3+ |
Within target sequence | — | In vitro staining |
| Fluorette-tag |
Tag: CCGGGSKVILFERWTWEPISEGAP-GSKVILFEGGPG (ReacTR-tag)
Probes: Texas red, Rhodamine red, Oregon green 514, fluorescien |
C-terminal | Labelling with 20 μM probe for 1 h; reaction temperature: 37 °C88 | Live bacteria staining |
| dC10α-tag |
Tag: LSAAECAAREAACREAAARAGGK
Probes: maleinimide-coumarin compounds |
N-terminal, C-terminal | Labelling with 10 μM probe for 30 min; reaction temperature: 37 °C91 | Live cell staining |
One of the shortest peptide-tags is comprised of tetracysteine or tetraserine motifs. In a pioneering study, Tsien et al. showed in 1998 that organobisarsenic acid thioesters form covalent bonds with peptide-tags containing a tetracysteine core CCxxCC, most commonly CCGPCC. Upon binding to the thiol groups of the peptide-tag, the Fluorescein-Arsenical-Helix-Binders (FlAsH) increase their fluorescence approximately 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold (Fig. 4A). In order to reduce the cytotoxicity and non-specific binding of FlAsH, micromolar concentrations of 1,2-ethandiol (EDT) have to be distributed to the cells simultaneously.63,64 Derivatives such as ReAsH,64 CHoXAsH,64 CrAsH65 and Cy3As66 expand the scope of available dyes. In many cases, flanking sequences have been added to the CCxxCC motif to increase the selectivity.67 When administered in 2.5 μM concentration, the bisarsenical dyes permeate the cell membrane and enable intracellular labelling of CCxxCC tagged proteins.64 Background signals due to interactions with other thiol-containing proteins often require time-consuming washing procedures. Furthermore, the incorporation of such cysteine-rich sequences can promote incorrect disulphide-bond formation within the POI and disturb the integrity of the protein. Schepartz presented bisboronic acid dyes as alternatives of the bisarsenical dyes (Fig. 4B). A rhodamine-derived bisboronic acid (RhoBo) showed nanomolar affinity for the tetraserine motif SSPGSS and provided increases in fluorescence after binding.68 But in contrast to the tetracysteine motif, the target sequence SSPGSS is found in more than 100 human proteins, amongst them the highly abundant myosin heavy chain.
000-fold (Fig. 4A). In order to reduce the cytotoxicity and non-specific binding of FlAsH, micromolar concentrations of 1,2-ethandiol (EDT) have to be distributed to the cells simultaneously.63,64 Derivatives such as ReAsH,64 CHoXAsH,64 CrAsH65 and Cy3As66 expand the scope of available dyes. In many cases, flanking sequences have been added to the CCxxCC motif to increase the selectivity.67 When administered in 2.5 μM concentration, the bisarsenical dyes permeate the cell membrane and enable intracellular labelling of CCxxCC tagged proteins.64 Background signals due to interactions with other thiol-containing proteins often require time-consuming washing procedures. Furthermore, the incorporation of such cysteine-rich sequences can promote incorrect disulphide-bond formation within the POI and disturb the integrity of the protein. Schepartz presented bisboronic acid dyes as alternatives of the bisarsenical dyes (Fig. 4B). A rhodamine-derived bisboronic acid (RhoBo) showed nanomolar affinity for the tetraserine motif SSPGSS and provided increases in fluorescence after binding.68 But in contrast to the tetracysteine motif, the target sequence SSPGSS is found in more than 100 human proteins, amongst them the highly abundant myosin heavy chain.
 | ||
| Fig. 4 Protein labelling by reactions between (A) tetracysteine tag and a bisarsenical profluorescent probe or (B) tetraserine tag and a bisboronate. | ||
Improved specificity can be achieved by metal-chelating tags, which recognise rather unnatural repetitive sequences such as the His6-, His10-tag or the oligo-aspartate tag. The hexahistidine tag (His6-tag), originally developed as an affinity tag for protein purification, is as short as the tetracysteine or tetraserine motif and known to interact tightly with transition-metal complexes, e.g. NiII:nitrilotriacetic acid (NiII:NTA).69 Since the interactions between metal ion and ligand are noncovalent, high mutual affinities are required to achieve stable labelling. To further increase the stability of the label, Tampé, Piehler and colleagues developed multivalent complexes carrying two to four NTA moieties (called bis-, tris- and tetrakis-NTA, Fig. 5A).70 The trisNTA ligands have exceptionally high affinities for His-tags (NiII:trisNTA·His10: KD = 0.1 nM).71 The attachment of bright fluorophores allowed the single-molecule super-resolution microscopy of actin and lamin filaments in CHO-K1 cells.72 Allbritton designed a trifunctional probe comprised of a NiII:NTA guiding unit and a photo-reactive arylazide group which enabled the covalent linkage with a His6-tagged murine dihydrofolate reductase (mDHFR) in vitro. The third functionality was either a biotin or a DNA strand for further conjugation or immobilization of the POI in vitro.73 Trifunctional probes containing a reporter dye were used by Auer et al. for the covalent labelling of a His6-tagged interleukin-4 receptor on the surface of living cells.74 A covalent modification of His-tagged proteins was recently introduced by Tørring and Gothelf. The so-called DNA-templated protein conjugation (DTPC) describes the binding of a trisNTA-oligonucleotide to a His6-tagged POI. This allows site-selective recognition by a second complementary oligonucleotide carrying a reactive N-hydroxysuccinimide (NHS) moiety, which enables subsequent crosslinking to a lysine residue in close proximity within the POI.75 This was demonstrated for His6-tagged serotransferrin and native IgG1 antibodies, which contain a histidine cluster. Tsien et al. found that the hexahistidine tag not only binds NiII:NTA probes, but also a ZnII complex (HisZiFiT-ZnII) with a KD ≈ 40 nM.76 Since the diamagnetic ZnII is not a fluorescence quencher (other than the paramagnetic NiII), fluorescein was derivatized in a way to directly bind two Zn2+ ions. Incubation with 100 nM HisZiFiT in buffer with 1 μM free Zn2+ and two subsequent washing steps allowed the labelling of cell surface-exposed His6-STIM1-CFP. Hamachi et al. introduced a binuclear ZnII:iminodiacetic acid (IDA) complex to which a Cy5 fluorophore was attached via an amide bond. Two His10-tagged G protein-coupled receptors (B2R and m1AchR) could be stained on the surface of HEK293 cells when treated with 0.5 μM ZnII:IDA–Cy5 conjugate for 10 min.77
 | ||
| Fig. 5 Protein labelling via interactions between (A) a histidine tag and a fluorescent NiII:trisNTA compound or (B) an oligo-aspartate tag (e.g. CA6D4 tag) and a fluorescent ZnII:DpaTyr conjugate. | ||
In 2006 Hamachi et al. introduced the oligo-aspartate tag (D4 tag) and the corresponding multinuclear ZnII:bis((dipicolylamino)methyl)tyrosine (ZnII:DpaTyr) complexes.78 A binuclear ZnII:DpaTyr had a moderate affinity for the D4 tag (KD = 1.4 μM). But by elongation of the tag (D4x3 tag) and utilization of the dimeric ZnII:DpaTyr–FITC complex (harbouring four ZnII ions) at 2 μM concentration, the fluorescence labelling of the muscarinic acetylcholine receptor (m1AChR) was feasible on live cells. However, the interaction is non-covalent and the label will dissociate at prolonged times after labelling. To prevent undesired label losses, a nucleophilic cysteine residue was added to the D4 tag and the binuclear ZnII:DpaTyr complex was equipped with an α-chloroacetyl group (Fig. 5B). After incubation for 12 h with 20 μM of the reactive ZnII:DpaTyr–TAMRA conjugate, the probes were found to enter E. coli cells and covalently label intracellular CA6D4-tagged maltose binding protein.79 The D4/ZnII:DpaTyr system was reported to be orthogonal to the His6/NiII:NTA complex.78 Later it was described that NiII:DpaTyr complexes show remarkably high affinity for the FLAG tag (DYKDDDDK), the D3 tag (DDD) and D3x2 tag (DDDXXDDD), with binding affinities around 100-fold stronger than for the D4/ZnII:DpaTyr pair.80,81 More recently, Komatsu et al. designed a Tb3+-binding peptide (LBP) de novo with aspartic acid as Tb3+-binding sites and tryptophan as sensitizing dye. The 15-mer sequence DDDWDDDWDDDWDDD was genetically fused to the C-terminus of glutathione S-transferase (GST) and used for affinity purification on a Tb3+-loaded column, as well as for in-gel detection of the fusion protein, rendering the LBP especially useful due to its dual functionality.82
While transition metal ions such as NiII, CoII and ZnII confer high affinity for recognition tags as described in the labelling techniques above, lanthanide ions add further desirable features; long-lived luminescence in the presence of sensitizing chromophores, excellent X-ray scattering powers and some of them even magnetic properties for NMR studies (e.g. GdIII, DyIII, TbIII, TmIII).83 This prompted a search for peptide-tags with high affinity for lanthanides to proteins. Imperiali et al. optimized a sequence found in a loop region of the calmodulin protein family, by means of a split-and-pool library and identified an 18-mer lanthanide-binding tag (LBT, ACADYNKDGWYEELECAA). With a KD value of around 220 nM for the binding of Tb3+, the LBT tag could be used for concentration determination in cell lysate and for in-gel visualization of a LBT–ubiquitin fusion protein.84 Later, Imperiali also found cysteine-free LBTs with nanomolar affinities for Tb3+ (YIDTNNDGWYEGDELLA, KD = 57 nM;85 FIDTNNDGWIEGDELLLEEG, KD = 19 nM86).
The search for peptide sequences that enable fluorescence labelling of proteins without added enzymes was extended from metal binding tags to tags, which have the ability to directly bind small molecule fluorescent probes. Already in 1998, Nolan et al. discovered by phage display a panel of Texas red, Rhodamine red, Oregon green 514 and fluorescein binding peptides, named “Fluorettes” (Fig. 6).87 Amongst them, the TexasRed-binding peptides (TR) showed the highest, subnanomolar affinities. Jäschke converted the TR peptide into a reactive tag (ReacTR, sequence: CCGGGSKVILFEGPAGRWTWEPISEGAPGSKVILFEGGPG) by adding two cysteine residues, which can undergo a proximity-induced nucleophilic substitution reaction with the N-α-chloroacetamide-conjugated TexasRed. After incubation with 10 μM reactive TexasRed probe for 1 h, the tagged maltose binding protein (MBP-ReacTR) showed bright fluorescence in the cytosol of transfected E. coli cells.88
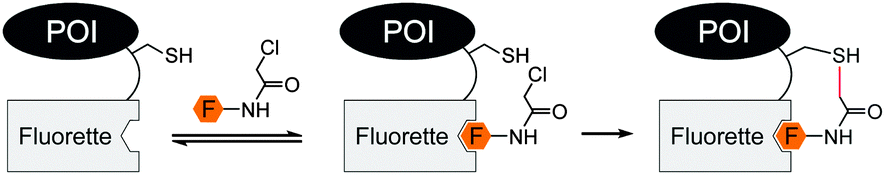 | ||
| Fig. 6 Covalent labelling with reactive Fluorette (fluorophore binding peptide) tag. | ||
Barbas et al. subjected phage-displayed peptide libraries to a reaction-based selection. A 21-mer peptide (rpf1368, sequence: CHNHQKATCRRMRSRETSVKK) was identified which formed an enaminone with 1,3-diketone derivatives. The reaction rates were rather slow with a labelling yield of 90% after 10 h.89 Also employing phage display, Weiss et al. searched for peptides that react with hydrazides. The resulting hydrazine reactive peptide (HyRe; sequence: HKTNHSCHKREQEHCRVTTT) was fused to the T4 lysozyme and underwent selective modification with 1 mM biotin hydrazide as well as with 1 mM rhodamine B hydrazide after 1 h reaction time in a crude cell lysate.90
The rational design of a maleinimide reactive α-helical peptide led to the dC10α tag (sequence: LSAAECAAREAACREAAARAGGK), in which two cysteine residues are separated by two turns of an α-helix. The minimalistic probe molecule comprises a coumarin fluorophore and a dimaleinimide moiety that quenches the fluorescence until both maleinimide groups undergo thiol addition during the labelling reaction. Owing to the small size, the probe was able to enter HEK293T cells after incubation for 30 min at 10 μM concentration and label both, C-terminally dC10α-tagged histone H2B as well as N-terminally tagged actin.91
The small molecule-binding peptide-tags provide properties that allow protein labelling in complex mixtures. However, for the achievement of a high affinity tag metal complexes seem to require less amino acid residues than small molecules (e.g. 6 aa tag for FlAsH vs. 20 aa HyRe tag for rhodamine B hydrazine). High affinity usually provides faster labelling at lower concentrations of labelling agent and, therefore, the reported labelling times for metal-chelating tags in living cells can be as short as 5–7 min in case of oligo-aspartate tagged (D4)3-m1AChR78 or 20 min for tetracysteine-tagged proteins.64 Moreover, metal-chelating tags require significant lower amounts of labelling probes with the lowest of 100 nM, as demonstrated for His10/NiII:trisNTA pair.72 These characteristics demonstrate advantages, also in comparison to enzymatic labelling techniques, however, the labelling of the POI remains non-covalent and thus reversible, hindering unambiguous analyses of low abundance proteins. The reported attempts to establish a covalent linkage between tag and probe employed moderately reactive groups to avoid background labelling. The low reactivity typically resulted in slow rates of covalent labelling, which is disadvantageous for pulse-chase-type experiments.
Peptide and protein based recognition by peptide-tags – mechanisms and characteristics
One way to improve the binding affinity of a tag sequence for the labelling agent is to increase its size. This led to the notion of peptide-binding peptide-tags. Peptides can bind other peptides with high mutual affinity (see Table 3). One example for high affinity peptide–peptide interactions is the coiled-coil motif, in which helical peptides wrap around each other to form a superhelix. In 1996, Hodges et al. designed a coiled-coil peptide de novo.92 The dimerization domain consists of five repeating heptads of the sequences EVSALEK for the negatively charged E-coil and KVSALKE for the positively charged K-coil. Electrostatic interactions drive the high-affinity heterodimer formation (KD = 1 nM). Using the E-coil as genetically encoded tag, the peptides were used for the purification, immobilization and detection of peptides expressed in E. coli. In 2008, Matsuzaki reported the artificial, heterodimeric coiled-coil peptides K3 (KIAALKE)3, K4 (KIAALKE)4, E3 (EIAALEK)3 and E4 (EIAALEK)4 – originally developed by Hodges et al.93 – as tag/probe pair for the live cell imaging of cell surface receptors. The stability of the E3/K3 coiled-coil is in the nanomolar range (KD ≈ 70 nM). With a KD ≈ 6 nM the E3/K4 coiled-coil provided even higher stability. This allowed the labelling of an E3-tagged prostaglandin E2 receptor EP3β subtype (EP3βR) upon incubation with only 20 nM TMR-conjugated K3 and K4 probe peptide within 5 min.94 The method was used to investigate the oligomerization state of two membrane receptors by staining the E3-tagged receptors with a mixture of fluorophore-modified K4 peptides.95,96 To avoid a potential loss of label during prolonged imaging periods, the coiled-coil motif was repurposed as a scaffold that instructs reactions to form a covalent bond. Ball et al. substituted amino acids at the interface of the E3/K3 coiled-coil pair to position a dirhodium catalyst on one coil so that addition of a diazo agent induces the alkylation of a tryptophan residue in the other coil.97 The E3gW-tagged maltose binding protein was selectively biotinylated in cell lysate by treatment with 5 μM K3a,eRh2 catalyst for 16 h in presence of 100 μM biotin-diazo reagent (Fig. 7A).98 Xia et al. also harnessed the concept of proximity-induced reactivity by modifying the E3-coil with a nucleophilic thiol provided by a cysteine residue within the E3-coil (CCE probe) and the K3-coil with an electrophilic α-chloroacetyl moiety (CCK probe) (Fig. 7B). Positioning of the two reactive residues at the interface of the coiled-coil promoted the ligation between probe peptide and CCE-tagged cell-surface receptors. For increased reaction rates, the labelling probe had to be used as CCK-1-dimer, still requiring more than 2 h to reach complete ligation and resulting in a 79 aa long protein label.99 In 2015, Matsuzaki et al. presented a reduction-free, amine-reactive crosslinking reaction between modified E/K coiled-coil peptides. A carboxy sulfosuccinimidyl ester linker was added to the peptide meant to serve as a labelling agent (R3CL probe) and a lysine residue was introduced to the E-peptide (ER3 tag) (Fig. 7C). Within 20 min a significant crosslinking was observed between 150 μM R3CL probes and ER3-tagged β2-adrenergic receptors on the surface of CHO cells.100| Labelling technique | Recognition tag including peptide probes | Reported tag position | Typical/reported concentrations and incubation conditions | Reported applications | Labelling modes |
|---|---|---|---|---|---|
| (* = temperature was not explicitly specified) RT – room temperature. | |||||
| E3/K3 or E3/K4 |
Tag: (EIAALEK)3
Probe: dye-(KIAALKE)3, dye-(KIAALKE)4 |
N-terminal | Labelling with 20–60 nM for 2 min; reaction temperature: 37* °C94 | Cell surface labelling, live cell imaging | Non-covalent |
| CCE/CCK |
Tag: GGGE-CAALEKE-VAALEKE-VAALEK (CCE)
Probe: GGGK-XAALKEK-VAALKEK-VAALKE (CCK) |
N-terminal | Labelling with 0.2–1 μM probe for 20 min; reaction temperature: RT99 | Cell surface labelling, live cell staining, fixed for imaging | Covalent |
| E3gW/K3a,eRh2 |
Tag: EISALEKWISALEQEISALEK (E3gW)
Catalyst: KISALKQK |
C-terminal | Labelling with 5 μM K3 catalyst and 100 μM biotin-diazo probe for 16 h; reaction temperature: 4 °C98 | In vitro | Covalent |
| ER3/R3CL |
Tag: M(EIAALER)3-GK (ER3)
Probe: dye-(RIAALRE)3-GC (R3CL) |
N-terminal | Labelling with 150 μM probe for 20 min; reaction temperature: 25 °C100 | Cell surface labelling, live cell imaging | Covalent |
| Cys-E3/K3 thioester |
Tag: C(EIAALEK)3
Probe: dye-thioester-(KIAALKE)3 |
N-terminal | Labelling with 100 nM probe for 2–5 min; reaction temperature: RT101,103 | Cell surface labelling, live cell imaging | Covalent |
| Leucine zipper |
ZIP-tag: ALEKELA-EAEKELA-ELEKELA-GGCGG-ALEKKLA-ELEKKAA-ELEKKLA
Probe: ALKKKLA-EzKKKLA-ELKKKLA |
N-terminal | Labelling with 0.1–1 μM probe for 4–15 min; reaction temperature: 25 or 37 °C104,106 | Cell surface labelling, in combination with CPP intracellular labelling, live cell imaging | Non-covalent and covalent |
| BTX-/SA-tag |
BTX-tag: WRYYESSLEPYPD
SA-tag: MDEKTTGWRGGHVVE-GLAGELEQLRARLEHHPQGQREP |
N-terminal | Labelling with 1–2 μg ml−1 BTX for 30 min; reaction temperature: 17 or 37* °C109 | Cell surface labelling, live or fixed cell imaging | Non-covalent |
| DnaE intein |
Npu: Nint aa 1–123, Cint aa 103–138
Ava: Nint aa 1–102, Cint aa 103–138 |
N-terminal, C-terminal | Npu: labelling with 2 μM Cint peptide for 1 h; reaction temperature: 37* °C114/Ava: labelling with 2.5 μM Cint for 1 h116 | Intracellular labelling via fusion to CPP | Covalent |
| AceL/Ter intein | Nint aa 1–25, Cint aa 26–129 | N-terminal | Labelling with 45 μM Nint peptide >1 h; reaction temperature: 8, 25 or 37 °C117 | In vitro | Covalent |
![[E with combining low line]](https://www.rsc.org/images/entities/b_char_0045_0332.gif)
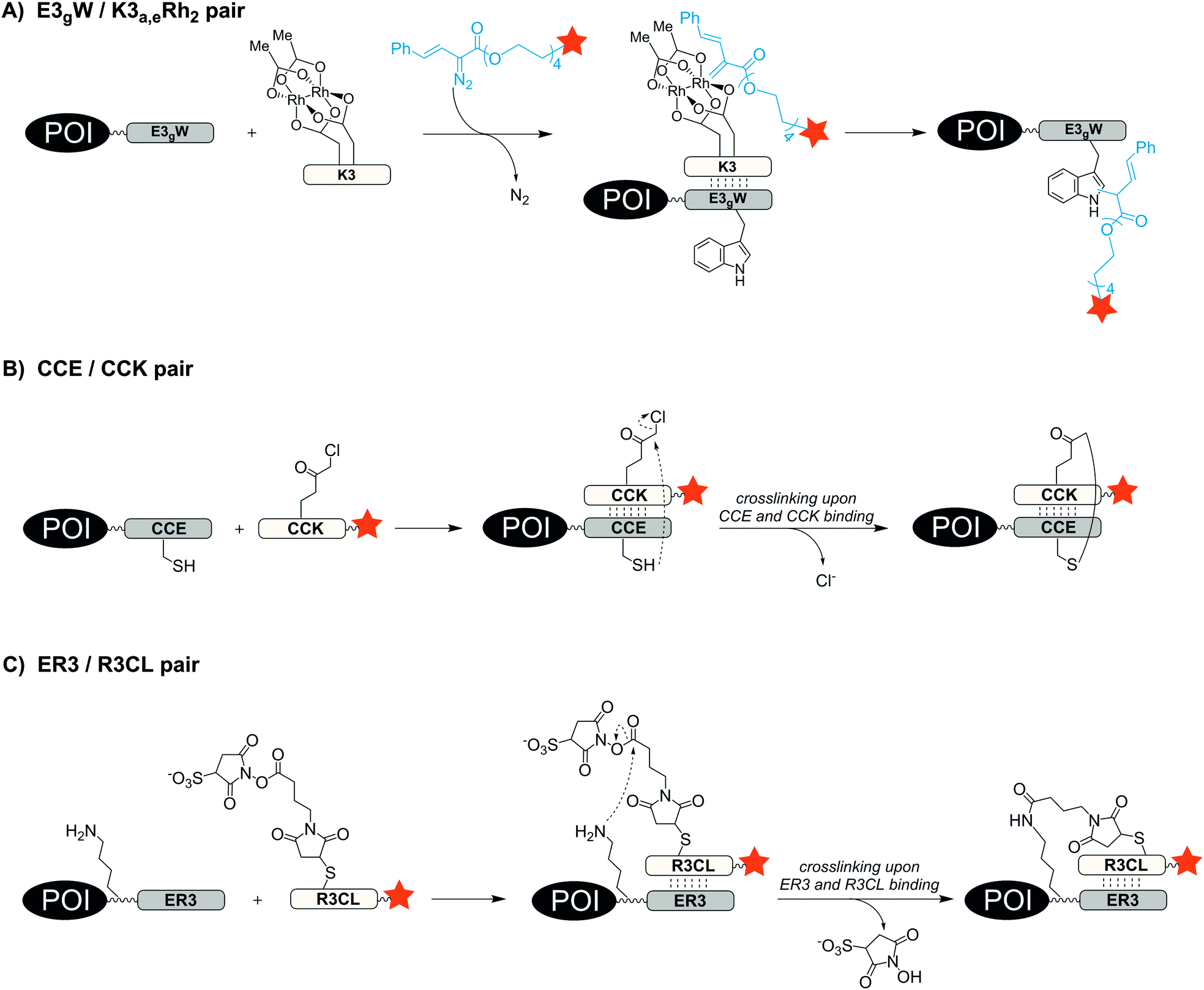 | ||
| Fig. 7 Protein labelling via formation of coiled-coils. (A) Covalent labelling via rhodium-catalyst containing K3 peptide and diazo probe, (B) proximity induced covalent cross-linking between electrophilic α-chloroacetyl group and thiol side chain and (C) proximity induced amine-reactive crosslinking. | ||
With an aim to find a very fast biocompatible reaction for covalent protein labelling that adds only little mass, Seitz et al. designed a coiled-coil-promoted acyl transfer reaction, which also bypasses the ligation between tag and probe. For this purpose, an N-terminal cysteine residue was added to the E3 tag and fused to the POI. A thioester-linked fluorophore was conjugated to the N-terminus of the K3 probe.101 The formation of the parallel coiled-coil triggered the proximity-induced transfer of the dye to the E3 tag according to mechanism known from nucleic acid templated native chemical ligation.102 Due to the high reactivity of the arylmercapto-linked thioesters and the high effective molarity induced in the end-of-helix arrangement of functional groups, the transfer of the fluorophore from the K3 probe to the Cys-E3-tag succeeded within 2 min reaction time at 100 nM labelling probe (Fig. 8). The approach was applied to live cell imaging studies, in which a variety of G protein-coupled receptors (including the human neuropeptide Y receptors 1, 2, 4, 5, human neuropeptide FF receptors 1 and 2 and human dopamine receptor 1) were labelled at the cell surface. The modular nature of the thioester-containing K3 probe facilitates the use of different fluorophores or reporter groups, as it was shown for TAMRA, ATTO488, AF350 and biotin labels.103 In this study, the authors also demonstrated that the covalent linkage between reporter dye and target protein was important to monitor receptor internalization and recycling in living HEK293 cells.
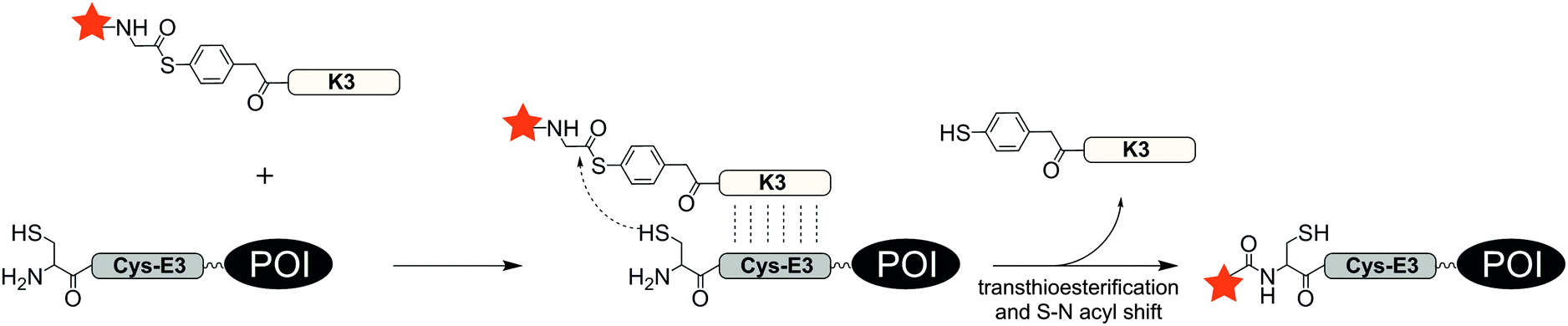 | ||
| Fig. 8 Covalent protein labelling via coiled-coil induced reporter group transfer. | ||
Another small and stable coiled-coil scaffold is the leucine zipper. Based on the crystal structure of a coiled-coil trimer of GCN4, Tamamura et al. developed in 2009 artificial leucine zipper peptides with high mutual affinity (ZIP tag/probe pair, KD = 18 nM) for the fluorescence imaging of ZIP-fused membrane proteins. The 23 aa probe helix is equipped with an environmentally sensitive dye (4-nitrobenzo-2-oxa-1,3-diazole, NBD), the other two tag helixes are connected via a loop sequence and offer a hydrophobic pocket for the dye (A2-tag, 49 aa) (Fig. 9A). The chemokine receptor CXCR4 was genetically fused to the A2-tag and expressed in CHO cells. Incubation of the cells with 1 μM NBD-probe peptide led to a fluorescence change.104 To increase the chemical and biological stability, Tamamura et al. later incorporated an α-chloroacetyl group to the probe peptide that can crosslink to a cysteine residue in the loop region of the A2-tag.105 For intracellular protein targets, an octaarginine sequence was attached to the C-terminus of the probe peptide.106
 | ||
| Fig. 9 Protein labelling (A) with the leucine zipper tag, which allows covalent labelling and turn-on of fluorescence, (B) by split-intein mediated reactions or (C) by cell-penetrating split-intein mediated reactions. | ||
Peptide-based recognition of genetically encoded protein tags beyond coiled-coil interactions was introduced by Huganir et al. in 2004. The labelling system relied on α-bungarotoxin (BTX, 74 aa); a small protein from snake venom.107 Peptide sequences that bind to BTX were initially selected by phage display and modified for high affinity binding.108 The resulting 13 aa BTX-binding site (BBS, sequence: WRYYESSLEPYPD, KD ≈ 60 nM109) was placed in the extracellular domain of the AMPA receptor for analysing the expression and membrane trafficking of the tagged receptor. At the same time, Sanes et al. not only used the BTX-binding site, but also the streptavidin-binding sequence (SBP, sequence: MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP,110KD ≈ 25 nM109) to stain the C-terminally tagged vesicle-associated protein (VAMP2) on the surface of live cells by incubation with the respective fluorophore-protein (BTX or SA) conjugates.109 Attempts to use BTX binding site or SBP for covalent labelling have not been reported but seem rather challenging given the size of BTX and SA.
The development of peptide-based tag/probe pairs for protein labelling seems to be characterised by a trade-off between the specificity of a tag and its size. Split intein-mediated protein labelling circumvents this correlation because intein excises itself after ligation of the flanking sequences (Fig. 9B). One intein fragment should be short in order to allow efficient solid-supported peptide synthesis. While such split inteins have been created artificially, they usually lack sufficient splicing performance and solubility.111 The naturally occurring Npu DnaE intein is one of the fastest inteins reported with a half-life of only 1 min.112 In 2012, Camarero et al. equipped the 36 aa C-intein (Cint) of Npu DnaE with a FRET pair in order to quench the fluorescence in the unspliced state. Using a commercial protein delivery system, the intracellular labelling of transcription factor Yin Yang 1 took place within 2 h using the FRET-quenched protein trans-splicing reaction (PTS).113 In 2015, Kwon engineered a Npu DnaE intein and synthesized a Cint with a photo-protected ester-linkage for controlled activation, a FRET pair for turn-on fluorescence detection and an octaarginine as cell penetrating peptide (Fig. 9C).114 As a proof of concept, labelling of maltose binding protein via PTS was carried out in HeLa cells. Detection of fluorescence was observed within the cells after 30 min incubation time. Beside the Npu DnaE intein other fast split inteins were described such as the Ava DnaE intein.115 In a recent study, histone H2B was fused to a 102 aa long Ava DnaE N-intein (Nint) and expressed in HEK293T cells. Live cell imaging was demonstrated after 1 h incubation with Npu DnaE Cint carrying different reporter groups. Npu DnaE Cint was used instead of Ava DnaE Cint due to higher synthesis yields.116 Mootz et al. engineered an atypical naturally occurring split intein with an Nint fragment consisting of 25 aa only and a Cint of 100 aa.117 Recently, Tampé et al. significantly improved the affinity of a Ssp DnaB M86 split intein composed of an Nint with 11 aa and a Cint with 143 aa. The Nint was C-terminal modified with trisNTA moieties and the Cint was N-terminal fused to a His6-tag which increased the affinity up to 40 fold and allowed protein trans-splicing at nanomolar concentrations.118
Intein technology is interesting, because the tag is excised upon labelling. Yet, to retain the benefits of small peptide-tags, one short, POI-connected intein fragment has to be combined with a rather large intein fragment that brings in the labelled extein. This construct must be recombinantly expressed and conjugated with a reporter group, which may limit the application.
Conclusion
The ideal protein labelling method should proceed with high tag specificity, require only small tag sizes (to avoid interference with protein function), succeed within short incubation times (to enable investigations of fast biological processes in pulse-chase-type experiments), confer stability of labelling (preferably by formation of covalent linkages to allow measurements over prolonged time scales), enable versatility as far as the choice of labelling groups is concerned and lay little toxic burden onto the cell or the cellular process under scrutiny. None of the tag-based methods discussed is without shortcomings and there is, at current, no silver bullet available for protein labelling. Enzymatic methods provide high specificity, small tag size and covalent labels, but with the enzymes currently used rather high substrate concentrations and long incubation times are required. The stability and acquisition of the enzymes itself are not always without problems and labelling of intracellular proteins remains challenging. Nevertheless, the growing number of publications in which enzymes such as BirA are used, for example to study protein–protein interactions by proximity biotinlyation demonstrate that problems are manageable and that the method allows applications that are difficult to be performed by other means. Notably short incubation times are feasible with metal ion recognition tags. Recent studies demonstrate that the placement of bright dyes such as AlexaFluor 647 in immediate vicinity of the protein via the His6–10 tags and high affinity NiII:trisNTA complexes facilitates super resolution microscopy of fixed and permeabilized cells. Ideally, the peptide-tag would be targeted by a membrane permeable probe. Though numerous studies demonstrate the feasibility of this approach, the available peptide-tags require rather long sizes to confer affinities for fluorescent dyes in a range that require staining with ≥10 μM concentration; which may lead to high background. Higher tag specificities are achievable with peptide based affinity reagents. For example, the de novo designed E3/K3 coiled-coil offers high affinity and selectivity at comparably small tag size (21 aa). This enables rapid labelling at low concentrations of probe. A recently developed peptide-templated acyl transfer technique combines the speed of complex formation via coiled-coil interactions with the advantages of covalent labelling. The method involves a highly reactive thioester-linked fluorophore K3 conjugate, which rapidly reacts with a cysteine terminated E3-coil. The covalent labelling is extremely fast (2–5 min) and only little cargo is added to the protein of interest. Yet at current, the method is restricted to N-terminal labelling of cell surface proteins.It is obvious that the peptide-tag based labelling methods available as of today still do not offer the robustness and wide applicability known from autofluorescent proteins and enzyme based tags (e.g. SNAP, CLIP, Halo). Yet, given the scientific need for less perturbing labelling methods, considering the pace of the current development of the field and taking into account that an ever increasing number of chemists joins the race, the authors are convinced that peptide based tags will become game changing enablers in a not too distant future.
References
- R. Rizzuto, M. Brini, P. Pizzo, M. Murgia and T. Pozzan, Curr. Biol., 1995, 5, 635–642 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa Jr., M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, ACS Chem. Biol., 2013, 8, 1704–1712 CrossRef CAS PubMed.
- L. W. Miller, Y. Cai, M. P. Sheetz and V. W. Cornish, Nat. Methods, 2005, 2, 255–257 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8650–8655 CrossRef CAS PubMed.
- H. Neumann, FEBS Lett., 2012, 586, 2057–2064 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed.
- T. Hayashi and I. Hamachi, Acc. Chem. Res., 2012, 45, 1460–1469 CrossRef CAS PubMed.
- M. Rashidian, J. K. Dozier and M. D. Distefano, Bioconjugate Chem., 2013, 24, 1277–1294 CrossRef CAS PubMed.
- Z. Ma, L. Du and M. Li, J. Med. Chem., 2014, 57, 8187–8203 CrossRef CAS PubMed.
- D. Jung, K. Min, J. Jung, W. Jang and Y. Kwon, Mol. BioSyst., 2013, 9, 862–872 RSC.
- Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2013, 52, 4088–4106 CrossRef CAS PubMed.
- D. Beckett, E. Kovaleva and P. J. Schatz, Protein Sci. Publ. Protein Soc., 1999, 8, 921–929 CrossRef CAS PubMed.
- E. de Boer, P. Rodriguez, E. Bonte, J. Krijgsveld, E. Katsantoni, A. Heck, F. Grosveld and J. Strouboulis, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7480–7485 CrossRef CAS PubMed.
- M. Fernández-Suárez, T. S. Chen and A. Y. Ting, J. Am. Chem. Soc., 2008, 130, 9251–9253 CrossRef PubMed.
- E. Steel, V. L. Murray and A. P. Liu, PLoS One, 2014, 9, e93646 Search PubMed.
- M. Lajko, A. F. Haddad, C. A. Robinson and S. A. Connolly, J. Virol. Methods, 2015, 221, 81–89 CrossRef CAS PubMed.
- M. Desbois, S. J. Cook, S. W. Emmons and H. E. Bülow, Genetics, 2015, 200, 697–705 CrossRef CAS PubMed.
- C. A. Bertuccio, S.-L. Lee, G. Wu, M. B. Butterworth, K. L. Hamilton and D. C. Devor, PLoS One, 2014, 9, e92013 Search PubMed.
- A. B. Jaykumar, P. S. Caceres, I. Sablaban, B. A. Tannous and P. A. Ortiz, Am. J. Physiol. Renal Physiol., 2016, 310, F183–F191 Search PubMed.
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- S. A. Slavoff, I. Chen and A. Y. Ting, J. Am. Chem. Soc., 2008, 130, 1160–1162 CrossRef CAS PubMed.
- M. Fernández-Suárez, H. Baruah, L. Martínez-Hernández, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef PubMed.
- D. E. Green, T. W. Morris, J. Green, J. E. Cronan and J. R. Guest, Biochem. J., 1995, 309(pt 3), 853–862 CrossRef CAS PubMed.
- J. D. Cohen, P. Zou and A. Y. Ting, ChemBioChem, 2012, 13, 888–894 CrossRef CAS PubMed.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 CrossRef CAS PubMed.
- J. Z. Yao, C. Uttamapinant, A. Poloukhtine, J. M. Baskin, J. A. Codelli, E. M. Sletten, C. R. Bertozzi, V. V. Popik and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 3720–3728 CrossRef CAS PubMed.
- M. Best, A. Degen, M. Baalmann, T. T. Schmidt and R. Wombacher, ChemBioChem, 2015, 16, 1158–1162 CrossRef CAS PubMed.
- S. Puthenveetil, D. S. Liu, K. A. White, S. Thompson and A. Y. Ting, J. Am. Chem. Soc., 2009, 131, 16430–16438 CrossRef CAS PubMed.
- S. Hauke, M. Best, T. T. Schmidt, M. Baalmann, A. Krause and R. Wombacher, Bioconjugate Chem., 2014, 25, 1632–1637 CrossRef CAS PubMed.
- D. Schumacher, J. Helma, F. A. Mann, G. Pichler, F. Natale, E. Krause, M. C. Cardoso, C. P. R. Hackenberger and H. Leonhardt, Angew. Chem., Int. Ed., 2015, 54, 13787–13791 CrossRef CAS PubMed.
- J. Yin, F. Liu, X. Li and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 7754–7755 CrossRef CAS PubMed.
- J. Yin, F. Liu, M. Schinke, C. Daly and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 13570–13571 CrossRef CAS PubMed.
- J. Yin, A. J. Lin, P. D. Buckett, M. Wessling-Resnick, D. E. Golan and C. T. Walsh, Chem. Biol., 2005, 12, 999–1006 CrossRef CAS PubMed.
- J. Yin, P. D. Straight, S. M. McLoughlin, Z. Zhou, A. J. Lin, D. E. Golan, N. L. Kelleher, R. Kolter and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820 CrossRef CAS PubMed.
- Z. Zhou, P. Cironi, A. J. Lin, Y. Xu, S. Hrvatin, D. E. Golan, P. A. Silver, C. T. Walsh and J. Yin, ACS Chem. Biol., 2007, 2, 337–346 CrossRef CAS PubMed.
- L. Marchetti, T. De Nadai, F. Bonsignore, M. Calvello, G. Signore, A. Viegi, F. Beltram, S. Luin and A. Cattaneo, PLoS One, 2014, 9, e113708 Search PubMed.
- T. Kawamura, B. Stephens, L. Qin, X. Yin, M. R. Dores, T. H. Smith, N. Grimsey, R. Abagyan, J. Trejo, I. Kufareva, M. M. Fuster, C. L. Salanga and T. M. Handel, PLoS One, 2014, 9, e81454 Search PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- A. Mero, B. Spolaore, F. M. Veronese and A. Fontana, Bioconjugate Chem., 2009, 20, 384–389 CrossRef CAS PubMed.
- H. Abe, M. Goto and N. Kamiya, Chem. – Eur. J., 2011, 17, 14004–14008 CrossRef CAS PubMed.
- T. Tanaka, N. Kamiya and T. Nagamune, FEBS Lett., 2005, 579, 2092–2096 CrossRef CAS PubMed.
- J. Tominaga, N. Kamiya, S. Doi, H. Ichinose, T. Maruyama and M. Goto, Biomacromolecules, 2005, 6, 2299–2304 CrossRef CAS PubMed.
- C.-W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542–4543 CrossRef CAS PubMed.
- C. Gnaccarini, W. Ben-Tahar, A. Mulani, I. Roy, W. D. Lubell, J. N. Pelletier and J. W. Keillor, Org. Biomol. Chem., 2012, 10, 5258–5265 CAS.
- S. K. Oteng-Pabi, C. Pardin, M. Stoica and J. W. Keillor, Chem. Commun., 2014, 50, 6604–6606 RSC.
- K. Heller, P. Ochtrop, M. F. Albers, F. B. Zauner, A. Itzen and C. Hedberg, Angew. Chem., Int. Ed., 2015, 54, 10327–10330 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- X. Guo, Q. Wang, B. M. Swarts and Z. Guo, J. Am. Chem. Soc., 2009, 131, 9878–9879 CrossRef CAS PubMed.
- R. Parthasarathy, S. Subramanian and E. T. Boder, Bioconjugate Chem., 2007, 18, 469–476 CrossRef CAS PubMed.
- Z. Wu, X. Guo and Z. Guo, Chem. Commun., 2011, 47, 9218–9220 RSC.
- J. M. Antos, G. M. Miller, G. M. Grotenbreg and H. L. Ploegh, J. Am. Chem. Soc., 2008, 130, 16338–16343 CrossRef CAS PubMed.
- M. W. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707–708 CrossRef CAS PubMed.
- T. Tanaka, T. Yamamoto, S. Tsukiji and T. Nagamune, ChemBioChem, 2008, 9, 802–807 CrossRef CAS PubMed.
- I. Wuethrich, J. G. C. Peeters, A. E. M. Blom, C. S. Theile, Z. Li, E. Spooner, H. L. Ploegh and C. P. Guimaraes, PLoS One, 2014, 9, e109883 Search PubMed.
- A. Tabata, Y. Ohkubo, N. Anyoji, K. Hojo, T. Tomoyasu, Y. Tatematsu, K. Ohkura and H. Nagamune, Anticancer Res., 2015, 35, 4411–4417 CAS.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- E. L. Smith, J. P. Giddens, A. T. Iavarone, K. Godula, L.-X. Wang and C. R. Bertozzi, Bioconjugate Chem., 2014, 25, 788–795 CrossRef CAS PubMed.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS PubMed.
- P. G. Holder, L. C. Jones, P. M. Drake, R. M. Barfield, S. Bañas, G. W. de Hart, J. Baker and D. Rabuka, J. Biol. Chem., 2015, 290, 15730–15745 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- H. Cao, B. Chen, T. C. Squier and M. U. Mayer, Chem. Commun., 2006, 2601–2603 RSC.
- H. Cao, Y. Xiong, T. Wang, B. Chen, T. C. Squier and M. U. Mayer, J. Am. Chem. Soc., 2007, 129, 8672–8673 CrossRef CAS PubMed.
- C. Hoffmann, G. Gaietta, A. Zürn, S. R. Adams, S. Terrillon, M. H. Ellisman, R. Y. Tsien and M. J. Lohse, Nat. Protoc., 2010, 5, 1666–1677 CrossRef CAS PubMed.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 438–439 CrossRef CAS PubMed.
- E. Hochuli, W. Bannwarth, H. Döbeli, R. Gentz and D. Stüber, Nat. Biotechnol., 1988, 6, 1321–1325 CrossRef CAS.
- A. Tinazli, J. Tang, R. Valiokas, S. Picuric, S. Lata, J. Piehler, B. Liedberg and R. Tampé, Chem. – Eur. J., 2005, 11, 5249–5259 CrossRef CAS PubMed.
- S. Lata, A. Reichel, R. Brock, R. Tampé and J. Piehler, J. Am. Chem. Soc., 2005, 127, 10205–10215 CrossRef CAS PubMed.
- R. Wieneke, A. Raulf, A. Kollmannsperger, M. Heilemann and R. Tampé, Angew. Chem., Int. Ed., 2015, 54, 10216–10219 CrossRef CAS PubMed.
- G. D. Meredith, H. Y. Wu and N. L. Allbritton, Bioconjugate Chem., 2004, 15, 969–982 CrossRef CAS PubMed.
- M. Hintersteiner, T. Weidemann, T. Kimmerlin, N. Filiz, C. Buehler and M. Auer, ChemBioChem, 2008, 9, 1391–1395 CrossRef CAS PubMed.
- C. B. Rosen, A. L. B. Kodal, J. S. Nielsen, D. H. Schaffert, C. Scavenius, A. H. Okholm, N. V. Voigt, J. J. Enghild, J. Kjems, T. Tørring and K. V. Gothelf, Nat. Chem., 2014, 6, 804–809 CrossRef CAS PubMed.
- C. T. Hauser and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3693–3697 CrossRef CAS PubMed.
- S. Fujishima, H. Nonaka, S. Uchinomiya, Y. A. Kawase, A. Ojida and I. Hamachi, Chem. Commun., 2011, 48, 594–596 RSC.
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452–10459 CrossRef CAS PubMed.
- H. Nonaka, S. Tsukiji, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 15777–15779 CrossRef CAS PubMed.
- H. Nonaka, S. Fujishima, S. Uchinomiya, A. Ojida and I. Hamachi, Bioorg. Med. Chem. Lett., 2009, 19, 6696–6699 CrossRef CAS PubMed.
- A. Ojida, S.-H. Fujishima, K. Honda, H. Nonaka, S.-H. Uchinomiya and I. Hamachi, Chem. – Asian J., 2010, 5, 877–886 CrossRef CAS PubMed.
- S. Sueda, S. Tanaka, S. Inoue and H. Komatsu, Anal. Biochem., 2012, 422, 52–54 CrossRef CAS PubMed.
- S. Aime, M. Botta, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19–29 RSC.
- K. J. Franz, M. Nitz and B. Imperiali, ChemBioChem, 2003, 4, 265–271 CrossRef CAS PubMed.
- M. Nitz, K. J. Franz, R. L. Maglathlin and B. Imperiali, ChemBioChem, 2003, 4, 272–276 CrossRef CAS PubMed.
- L. J. Martin, B. R. Sculimbrene, M. Nitz and B. Imperiali, QSAR Comb. Sci., 2005, 24, 1149–1157 CAS.
- M. N. Rozinov and G. P. Nolan, Chem. Biol., 1998, 5, 713–728 CrossRef CAS PubMed.
- M. Sunbul, L. Nacheva and A. Jäschke, Bioconjugate Chem., 2015, 26, 1466–1469 CrossRef CAS PubMed.
- F. Tanaka, R. Fuller, L. Asawapornmongkol, A. Warsinke, S. Gobuty and C. F. Barbas, Bioconjugate Chem., 2007, 18, 1318–1324 CrossRef CAS PubMed.
- G. M. Eldridge and G. A. Weiss, Bioconjugate Chem., 2011, 22, 2143–2153 CrossRef CAS PubMed.
- Y. Chen, C. M. Clouthier, K. Tsao, M. Strmiskova, H. Lachance and J. W. Keillor, Angew. Chem., Int. Ed., 2014, 53, 13785–13788 CrossRef CAS PubMed.
- B. Tripet, L. Yu, D. L. Bautista, W. Y. Wong, R. T. Irvin and R. S. Hodges, Protein Eng., 1996, 9, 1029–1042 CrossRef CAS PubMed.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS PubMed.
- Y. Yano, A. Yano, S. Oishi, Y. Sugimoto, G. Tsujimoto, N. Fujii and K. Matsuzaki, ACS Chem. Biol., 2008, 3, 341–345 CrossRef CAS PubMed.
- K. Kawano, Y. Yano, K. Omae, S. Matsuzaki and K. Matsuzaki, Anal. Chem., 2013, 85, 3454–3461 CrossRef CAS PubMed.
- H. Yamashita, Y. Yano, K. Kawano and K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1359–1366 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS PubMed.
- Z. Chen, B. V. Popp, C. L. Bovet and Z. T. Ball, ACS Chem. Biol., 2011, 6, 920–925 CrossRef CAS PubMed.
- J. Wang, Y. Yu and J. Xia, Bioconjugate Chem., 2014, 25, 178–187 CrossRef CAS PubMed.
- Y. Yano, N. Furukawa, S. Ono, Y. Takeda and K. Matsuzaki, Pept. Sci., 2015 DOI:10.1002/bip.22715.
- U. Reinhardt, J. Lotze, S. Zernia, K. Mörl, A. G. Beck-Sickinger and O. Seitz, Angew. Chem., Int. Ed., 2014, 53, 10237–10241 CrossRef CAS PubMed.
- T. N. Grossmann and O. Seitz, J. Am. Chem. Soc., 2006, 128, 15596–15597 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, K. Mörl, A. G. Beck-Sickinger and O. Seitz, Bioconjugate Chem., 2015, 26, 2106–2117 CrossRef CAS PubMed.
- H. Tsutsumi, W. Nomura, S. Abe, T. Mino, A. Masuda, N. Ohashi, T. Tanaka, K. Ohba, N. Yamamoto, K. Akiyoshi and H. Tamamura, Angew. Chem., Int. Ed., 2009, 48, 9164–9166 CrossRef CAS PubMed.
- W. Nomura, T. Mino, T. Narumi, N. Ohashi, A. Masuda, C. Hashimoto, H. Tsutsumi and H. Tamamura, Pept. Sci., 2010, 94, 843–852 CrossRef CAS PubMed.
- W. Nomura, N. Ohashi, A. Mori and H. Tamamura, Bioconjugate Chem., 2015, 26, 1080–1085 CrossRef CAS PubMed.
- Y. Sekine-Aizawa and R. L. Huganir, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17114–17119 CrossRef CAS PubMed.
- M. Harel, R. Kasher, A. Nicolas, J. M. Guss, M. Balass, M. Fridkin, A. B. Smit, K. Brejc, T. K. Sixma, E. Katchalski-Katzir, J. L. Sussman and S. Fuchs, Neuron, 2001, 32, 265–275 CrossRef CAS PubMed.
- C. M. McCann, F. M. Bareyre, J. W. Lichtman and J. R. Sanes, BioTechniques, 2005, 38, 945–952 CrossRef CAS PubMed.
- A. D. Keefe, D. S. Wilson, B. Seelig and J. W. Szostak, Protein Expression Purif., 2001, 23, 440–446 CrossRef CAS PubMed.
- H. D. Mootz, ChemBioChem, 2009, 10, 2579–2589 CrossRef CAS PubMed.
- J. Zettler, V. Schütz and H. D. Mootz, FEBS Lett., 2009, 583, 909–914 CrossRef CAS PubMed.
- R. Borra, D. Dong, A. Y. Elnagar, G. A. Woldemariam and J. A. Camarero, J. Am. Chem. Soc., 2012, 134, 6344–6353 CrossRef CAS PubMed.
- D. Jung, K. Sato, K. Min, A. Shigenaga, J. Jung, A. Otaka and Y. Kwon, Chem. Commun., 2015, 51, 9670–9673 RSC.
- N. H. Shah, G. P. Dann, M. Vila-Perelló, Z. Liu and T. W. Muir, J. Am. Chem. Soc., 2012, 134, 11338–11341 CrossRef CAS PubMed.
- Y. David, M. Vila-Perelló, S. Verma and T. W. Muir, Nat. Chem., 2015, 7, 394–402 CrossRef CAS PubMed.
- I. V. Thiel, G. Volkmann, S. Pietrokovski and H. D. Mootz, Angew. Chem., Int. Ed., 2014, 53, 1306–1310 CrossRef CAS PubMed.
- M. Braner, A. Kollmannsperger, R. Wieneke and R. Tampé, Chem. Sci., 2016 10.1039/C5SC02936H.
Footnote |
| † Ulrike Reinhardt and Jonathan Lotze contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |