
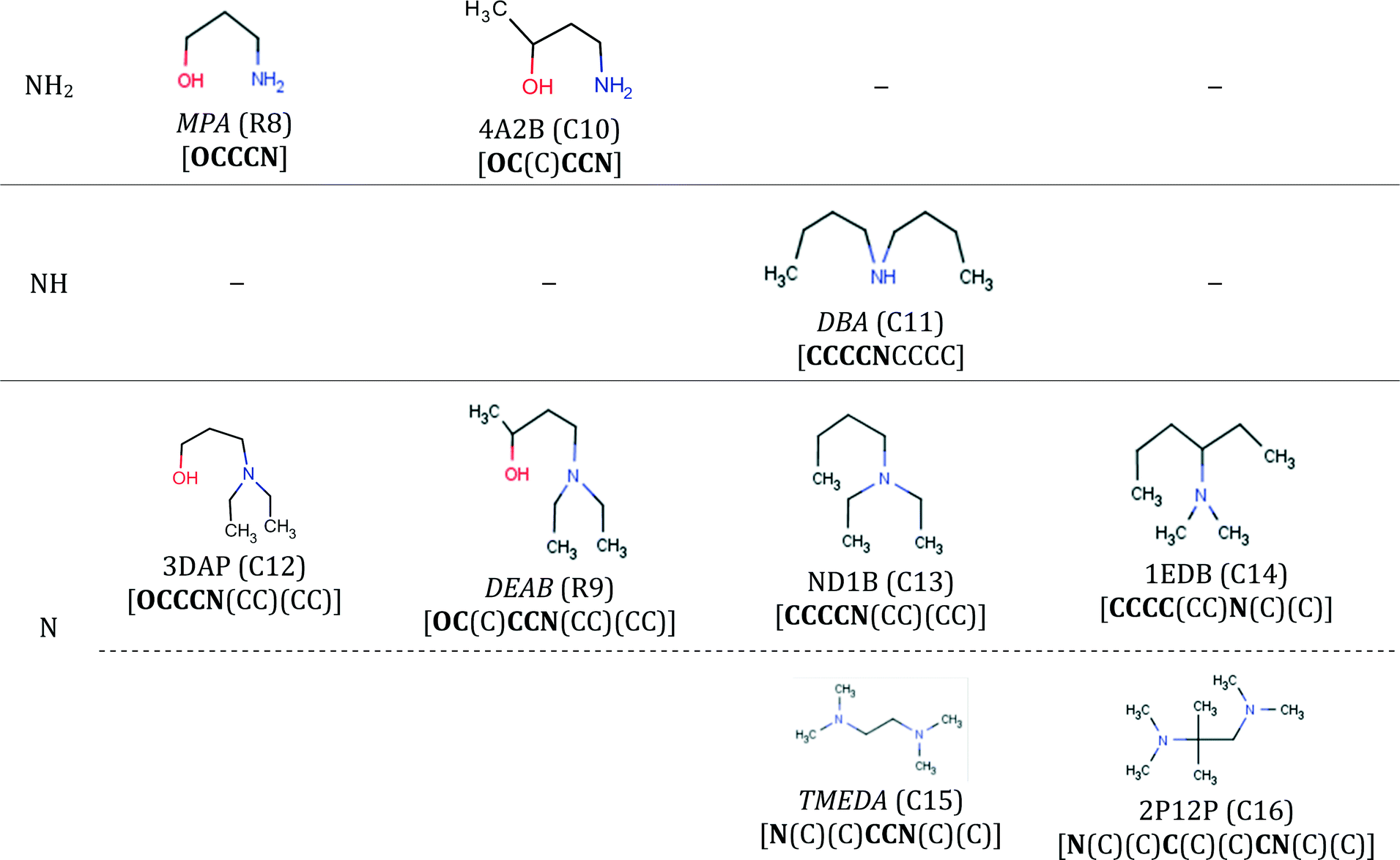
Computer-aided molecular design and selection of CO2 capture solvents based on thermodynamics, reactivity and sustainability†
Athanasios I.
Papadopoulos
a,
Sara
Badr
b,
Alexandros
Chremos‡
c,
Esther
Forte
c,
Theodoros
Zarogiannis
ad,
Panos
Seferlis
*ad,
Stavros
Papadokonstantakis
be,
Amparo
Galindo
c,
George
Jackson
 c and
Claire S.
Adjiman
c
c and
Claire S.
Adjiman
c
aChemical Process and Energy Resources Institute, Centre for Research and Technology-Hellas, 57001 Thermi, Greece
bSwiss Federal Institute of Technology, Institute for Chemical and Bioengineering, Zurich, Switzerland
cImperial College London, Department of Chemical Engineering, Centre for Process Systems Engineering, London SW7 2AZ, UK
dDepartment of Mechanical Engineering, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: seferlis@auth.gr
eDepartment of Energy and Environment, Chalmers University of Technology, Kemivägen 4, 41296 Gothenburg, Sweden
First published on 10th August 2016
Abstract
The identification of improved carbon dioxide (CO2) capture solvents remains a challenge due to the vast number of potentially-suitable molecules. We propose an optimization-based computer-aided molecular design (CAMD) method to identify and select, from hundreds of thousands of possibilities, a few solvents of optimum performance for CO2 chemisorption processes, as measured by a comprehensive set of criteria. The first stage of the approach involves a fast screening stage where solvent structures are evaluated based on the simultaneous consideration of important pure component properties reflecting thermodynamic, kinetic, and sustainability behaviour. The impact of model uncertainty is considered through a systematic method that employs multiple models for the prediction of performance indices. In the second stage, high-performance solvents are further selected and evaluated using a more detailed thermodynamic model, i.e. the group-contribution statistical associating fluid theory for square well potentials (SAFT-γ SW), to predict accurately the highly non-ideal chemical and phase equilibrium of the solvent–water–CO2 mixtures. The proposed CAMD method is applied to the design of novel molecular structures and to the screening of a data set of commercially available amines. New molecular structures and commercially-available compounds that have received little attention as CO2 capture solvents are successfully identified and assessed using the proposed approach. We recommend that these solvents should be given priority in experimental studies to identify new compounds.
Design, System, ApplicationWe propose a computer aided molecular design (CAMD) method which employs optimization to support the synthesis and selection of high performance molecules for use in process systems and to guide experimental efforts. The method can be used to address challenging applications where a) the desired molecules exhibit phase and chemical equilibria, b) numerous combinations of molecules need to be evaluated, and c) multiple criteria must be considered to capture the effects of molecular chemistry on the process system performance. The method is applied to the design of solvents in chemical absorption processes for the separation of carbon dioxide (CO2) from gas streams. The molecular design problem is first approached via a fast screening stage where molecules are evaluated based on the simultaneous consideration of multiple performance indices pertaining to thermodynamics, reactivity and sustainability. A few high-performance solvents are further evaluated using an advanced group contribution equation of state to predict reliably the highly non-ideal equilibrium behavior of solvent–water–CO2 mixtures. Several promising novel solvents for CO2 capture are proposed and can now be assessed experimentally. The proposed method can readily be applied to other chemical absorption processes to accelerate the identification of novel solvents. |
1. Introduction
In view of the pressing need to curtail carbon dioxide (CO2) emissions to the atmosphere in a world that remains highly dependent on carbon-based fuels for its energy provision, a large number of technological solutions for CO2 capture are currently being investigated.1 Chemical absorption processes2,3 represent a well-established industrial technology for CO2 capture compared to many of the alternatives that are being developed: it can be retrofitted onto existing plants and the conditions for both absorption and solvent regeneration (desorption) are relatively easy to meet. However, some major downsides must be overcome to enable the widespread adoption of this technology. These include the high energy input required for solvent regeneration and negative environmental impacts associated with the solvents, to name but a few.4Many of these issues can be linked to the solvents most commonly used in such processes, namely, amine-based organic compounds. In view of the shortcomings of these solvents, significant efforts are being devoted to identify solvents that can significantly improve the economic and sustainability performance of chemisorption processes, compared to existing options. The identification of improved solvents, however, is very challenging due to a) the highly non-ideal solvent–CO2–water chemical interactions, b) the countless combinations of potential capture solvents and blend candidates and c) the complex interactions between the solvent and the process that require the simultaneous consideration of multiple criteria5 in the selection of solvents with optimum capture features. To address this last aspect, one must avoid focusing on a narrow property target (e.g. Oexmann and Kather6 argue convincingly against searching for low heat of absorption solvents) and instead consider a comprehensive set of properties related to thermodynamics, kinetics and sustainability.
A large number of experimental studies have been undertaken to identify solvents and solvent blends that perform better than the current benchmark solvent, aqueous solutions of monoethanolamine (MEA). Based on the collective experience of a large community of researchers, several rules of thumb have been formulated to help generate molecular structures that are likely to perform well as CO2 chemisorption solvents. For example, Singh et al.7–9 and Singh and Versteeg10 performed a large set of experiments to derive useful insights into the effects of amine structure on important parameters such as absorption rate and solvent capacity. Other researchers have developed qualitative design rules which can be used for the heuristic screening of solvents, and a selection of these heuristics is listed in Table 1, with more details provided in section A in the ESI.† While qualitative relationships that link structure and properties offer a very useful guide to search the vast space of possibilities, they also reduce the diversity of the options considered, thereby decreasing the potential for novel designs. Additionally, they do not reduce the significant cost of obtaining a measure of the performance of the postulated solvents, which requires the synthesis and characterization of each new solvent. For instance, the laboratory-based investigation of DEAB has required extensive effort as reflected in publications related to compound identification11,12 and investigation.13–16 Thus, in addition to acquiring high-quality data for different solvents, there is a need to accelerate the search using computational methods.17
| Label | Evidence | Compound type(s) | Structure–property relationship |
|---|---|---|---|
| (1) | Puxty et al.18 | Hydroxylamines | 2–4 carbons between hydroxyl and amine functional groups increase the absorption capacity. |
| Yamada et al.19 | |||
| Tontiwachwuthikul et al.12 | |||
| (2) | Singh and Versteeg10 | Diamines | Longer chains between amine groups, up to 6 carbons, increase basicity and improve desorption. |
| Aronu et al.20 | |||
| (3) | Singh and Versteeg10 | Hydroxylamines | At least 2 carbon atoms between amine and hydroxyl or first amine and amine branch improve desorption. |
| Singh et al.8,9 | Diamines | ||
| (4) | Singh and Versteeg10 | Linear hydroxylamines | Up to 4 carbon atoms between hydroxyl and amines improve desorption. |
| (5) | Zhang et al.21 | Branched alkylamines | An alkyl branch at the α carbon improves the kinetics; at the β carbon, it leads to precipitation. |
| (6) | Sartori et al.22 | Sterically hindered amines | • A primary amine is hindered when the amino group is attached to a tertiary carbon; |
| • A secondary amine is hindered when the amino group is attached to at least one secondary or tertiary carbon. |
Systematic computer-aided molecular design (CAMD) methods offer a promising alternative route to the identification of solvents that should be investigated experimentally, by allowing the fast, cost-effective, and automated evaluation of a larger and more diverse set of solvent properties compared to what is possible through purely experimental studies.23 CAMD methods have been particularly successful in addressing some of the challenges in the design and selection of solvents for a range of separation systems and other applications; they have been used to explore a very wide range of solvent structures, leading to the identification of either novel molecular structures or conventional, but previously overlooked, optimum molecules. The reader is referred to a few general works24–27 and references therein to gain a historical perspective.
Because many of the predictive models used are relatively simple and yet cover a very broad set of molecular structures, uncertainty has emerged as an important issue in CAMD. Approaches to mitigate the impact of uncertainty have been previously proposed within systematic CAMD methods for the design of solvents as separation28–30 and reaction31 media, of polymers,32 and of heat exchange fluid mixtures.33 An uncertainty quantification method for property predictions through GC was also recently proposed.34,35
While research on CAMD methods has led to the development of many sophisticated and successful CAMD techniques, the deployment of a systematic approach in the context of chemisorption is not without difficulty. The models typically used in chemisorption process design, while of high accuracy, are specific to a given solvent and require extensive experimental data on each solvent to be studied; this is the case of models of the thermodynamics and kinetics of aqueous alkanolamine and CO2 mixtures (e.g. eNRTL models36–38) and of the kinetics of degradation reactions.39–41 A few predictive models that relate solvent molecular structure or solvent blend composition to properties, and hence performance, are available to support CAMD activities. As a result, recent research efforts to screen CO2 capture solvents quantitatively have mostly been based on the use of simple approximation models or statistical correlations.42,43 While such approaches led to some promising results, the narrow range of performance criteria considered and the relatively low accuracy of the predictive models have not allowed the full benefits of in silico solvent design to be realized.
Encouragingly, three CAMD-based approaches have been reported for the design of CO2 capture solvents for chemisorption processes. Eden and co-workers44,45 have proposed solvents to replace MDEA in an absorption/desorption process, using a range of physical properties to define the optimum molecular design space44 and developing a new CAMD algorithm.45 Salazar et al.46 used different physical properties as performance criteria to identify a few promising solvents. These solvents were then evaluated based on their stripping energy requirement, calculated using ASPEN Plus®47 process models and with the reaction and thermodynamic models derived in part from similarity with MEA and DEA (diethanolamine) and in part by using undisclosed NRTL48 parameters derived based on UNIFAC49 groups to model the liquid phase non-ideality.
Among these CAMD-based works, only Salazar et al.46 considered the multicomponent chemical and phase equilibria that are central to solvent performance as an explicit part of the design. However, to the best of our knowledge, the physical model used has not been validated against experimental data and its predictive capabilities are unknown. In this context, the increasing availability of reliable predictive models of relevance to chemisorption provides opportunities to enhance CAMD approaches to CO2 capture solvent design by broadening the range of performance metrics and increasing their reliability. Chemical reactivity, for instance, has been predicted using continuum solvation models, among other computational chemistry methods, for the calculation of the amine base strength as a measure of molecular reactivity.50,51 Although such models are computationally expensive, they could, in the future, be included in CAMD approaches that embed quantum mechanical models.52,53
Another interesting route is the use of a predictive equation of state, such as the statistical associating fluid theory equation for potentials of variable range, SAFT-VR,54,55 which has been applied within a CAMD methodology to the design of solvents for the separation of CO2 from a natural gas stream through physical absorption using a single family of compounds56 or within a group contribution version57 for a broader range of compounds.58 The use of the SAFT platform to design CO2 capture solvents for physical absorption was also proposed by Bardow et al.,59 Oyarzún et al.,60 Stavrou et al.61 and Lampe et al.62 using the PC-SAFT EoS, the perturbed-chain version of SAFT63 and the PCP-SAFT EoS, its extension to polar fluids.64–66 In the context of chemisorption, the SAFT thermodynamic platform has been used to predict the phase and chemical equilibria of CO2 capture fluids through a parameterization of a group contribution version of the statistical associating fluid theory for potentials of variable range equation of state, SAFT-γ SW.67,68 This approach has been applied successfully to model primary alkanolamines and their mixtures with water and CO2.69–71 With such a model, it becomes possible to treat aqueous solutions of CO2 and amines for which no data are available and to investigate the effect of decision variables such as composition and temperature, as well as the performance of mixtures containing several amine components.
Drawing on these advances, we aim in our work to identify a set of promising solvents that offer a simultaneously high performance in many criteria associated with effective chemical absorption of CO2 and solvent regeneration. For the first time in CO2 capture research, we take an approach that combines thermodynamic behaviour, reactivity and sustainability as performance criteria in solvent design or selection and that includes predictive models of the phase and chemical equilibria of the solvents identified. Furthermore, we recognise the inherent uncertainty in many of the methods we use and propose a strategy to address this issue and reduce the risk of premature elimination of solvent candidates. The proposed approach can easily be extended to include other aspects not considered here provided that appropriate structure–property models are available (e.g. the selectivity of the solvent towards CO2 can be predicted using thermodynamic models similar to those used to predict CO2 loading). Other considerations not accounted for here, such as propensity to oxidative degradation, can be investigated for the solvents in the final set obtained. The proposed computer-based solvent design framework is applied in two ways: (i) a database of commercially available molecules is screened systematically for high-performance capture options (solvent selection) and (ii) novel molecules that are found to be optimum capture solvents are identified (solvent design).
The article is organised as follows. In section 2, an overview of the proposed systematic CAMD methodology is given. The approach is generic and can be applied to different molecular design applications depending on the properties and property models selected. In section 3, the specific choices required for the application of the method to CO2 capture are discussed. The solvents identified are presented in section 4; the approach is assessed based on the predicted performance of the solvents generated relative to MEA, their novelty, and any indication from the literature that they are viable options.
2. Overview of generic design and selection framework
An overview of the proposed generic framework for solvent design and selection is presented in this section, together with a description of each step. Our aim is to enable the identification of efficient CO2 capture solvents by investigating the following two cases:• The selection of solvents from a database of pre-existing chemical compounds (selection problem).
• The design of novel solvents using an optimization-based CAMD method (design problem).
Both cases are addressed within a single framework whose major characteristics include the use of a wide range of properties as criteria for solvent selection and design, the use of simple models to develop an initial list of candidate solvents, the explicit consideration of uncertainty in decision-making and the pruning of the list of candidate solvents using a more advanced predictive model (here, we use the SAFT-γ SW EoS). Uncertainty is accounted for by generating a distribution of solvent ranks that results from the use of different prediction models for the same properties. The distribution of ranks is widely used in non-parametric statistics72 to detect patterns in data without requiring information on error distributions or other assumptions on the data under consideration. These ideas are implemented within a systematic, two-stage approach, as illustrated in Fig. 1. Each step is introduced in the remainder of this section.
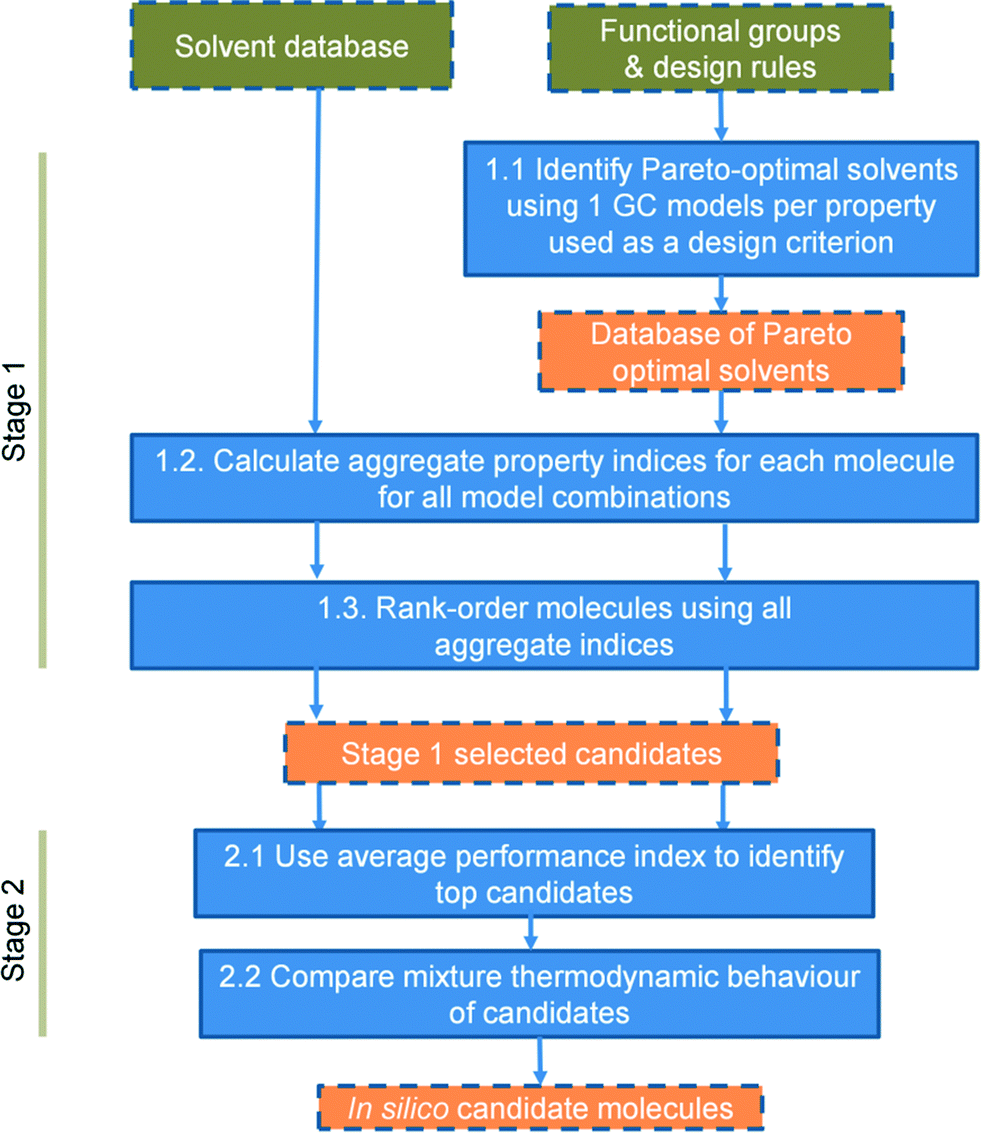 | ||
| Fig. 1 Unified computer-aided molecular design framework for selection (left) or design (right). | ||
2.1 Stage 1 of the methodology
The aim of stage 1 is to quickly identify and eliminate solvents of inferior performance, producing a “long list” of candidates. In the design problem, the algorithm used73 can rapidly evaluate around 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular structures to generate a few tens of solvents comprising the “long list”. Solvents are designed and/or evaluated using GC and non-GC-based models for the prediction of properties that are specifically selected to reflect the major driving forces for CO2 capture. The selected properties provide a set of solvent performance measures that link the chemical structure of the solvent to thermodynamic, reactivity and sustainability characteristics of importance in the capture/regeneration process (see section 3 for details).
000 molecular structures to generate a few tens of solvents comprising the “long list”. Solvents are designed and/or evaluated using GC and non-GC-based models for the prediction of properties that are specifically selected to reflect the major driving forces for CO2 capture. The selected properties provide a set of solvent performance measures that link the chemical structure of the solvent to thermodynamic, reactivity and sustainability characteristics of importance in the capture/regeneration process (see section 3 for details).
To explain how the aggregate indices are computed, let us consider a set of properties Pr = {1, …, Np} that are linked to performance. Let each property j ∈ Pr be calculated from a set of available models lj = {1, …, Nmdj} consisting of a total of Nmdj models per property. The number of all possible model combinations is given by
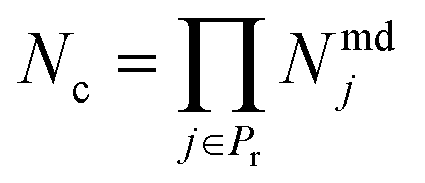 | (1) |
Furthermore consider a solvent i in the set G = {1, …, Ns} of Ns candidate molecules. One can then define an aggregate property index Ji,k for solvent i based on the kth model combination, k ∈ {1, …, Nc}, corresponding to some choice lj of model (lj ∈ {1, …, Nmdj}) for a property j:
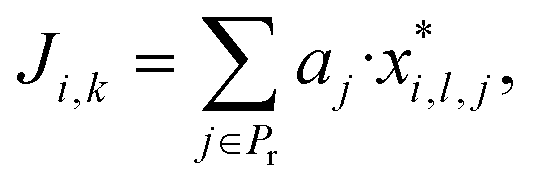 | (2) |
The set of indices Ji,k, i ∈ {1, …, Ns}, k ∈ {1, …, Nc}, may be represented as a matrix J of dimensions (Ns × Nc) with each model combination k (column of J) indicating the performance of each solvent i (row of J). As an example of matrix J, assume a set G of Ns = 4 solvents is used and a set Pr of Np = 2 properties with Nmd1 = 2 and Nmd2 = 3 models (hence, Nc = 6) is available for the calculation of properties 1 and 2, respectively. The elements of matrix J are calculated as shown in Table 2.
| Solvent | k = 1, j = {1, 2} and l1 = 1, l2 = 1 | k = 2, j = {1, 2} and l1 = 1, l2 = 2 | … | k = 6, j = {1, 2} and l1 = 2, l2 = 3 |
|---|---|---|---|---|
| 1 | J 1,1 = a1x*1,1,1 + a2x*1,2,1 | J 1,2 = a1x*1,1,1 + a2x*1,2,2 | … | J 1,6 = a1x*1,1,2 + a2x*1,2,3 |
| 2 | J 2,1 = a1x*2,1,1 + a2x*2,2,1 | J 2,2 = a1x*2,1,1 + a2x*2,2,2 | … | J 2,6 = a1x*2,1,2 + a2x*2,2,3 |
| 3 | J 3,1 = a1x*3,1,1 + a2x*3,2,1 | J 3,2 = a1x*3,1,1 + a2x*3,2,2 | … | J 3,6 = a1x*3,1,2 + a2x*3,2,3 |
| 4 | J 4,1 = a1x*4,1,1 + a2x*4,2,1 | J 4,2 = a1x*4,1,1 + a2x*4,2,2 | … | J 4,6 = a1x*4,1,2 + a2x*4,2,3 |
To this end, the elements in each column of J are sorted in ascending order independently for every column (model combination) k, yielding a matrix J′ of independently rank-ordered columns. The transformations needed to go from J to J′ can be used to construct a matrix of ranked lists, L, in which an entry Lr,k is an integer that denotes the identity of the rth ranked solvent according to model combination k. Consider an example with the same dimensionality as that in Table 2: the elements of matrix L will be of the form presented in Fig. 2a. The solvents may follow a different order for different k values (i.e. different combinations of models). For example, solvent 3 ranks first for k = 2, second for k = 1, 3, 5, 6, and third for k = 4. In addition, notice that all four solvents appear in the top 2 positions (ranks 1 and 2). For this small solvent set, the number of appearances of each solvent in the top 2 positions can be used to select solvents to be taken forward for further analysis, as shown in Fig. 2b: solvents 1 and 3 show the strongest predicted performance, closely followed by solvent 2, while solvent 4 appears to be an under-performer.
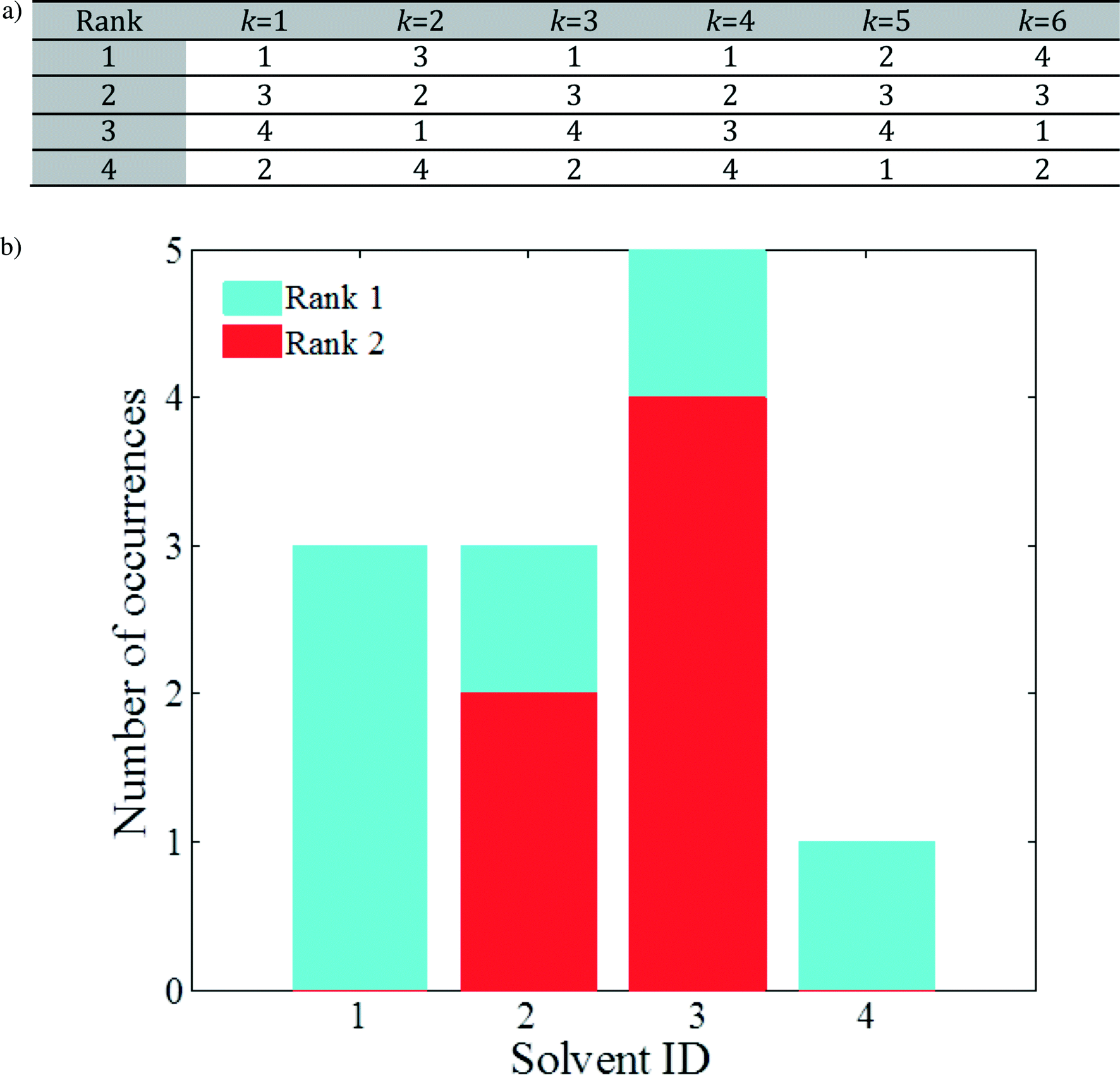 | ||
| Fig. 2 a) Example of rank-ordered solvents in matrix L where each entry represents the identity of a solvent. b) Number of occurrences of each molecule in the top 2 solvents, based on (a). | ||
The frequent appearance of specific solvents at the top implies an agreement in the predictions made by multiple different models. On the other hand, even solvents that appear with lower frequency at the top are also considered due to the use of the distribution of ranks to select candidate molecules. This allows a reduction of the original solvent list without bias due to uncertainty in the predictions. This is important because there is often little or no prior knowledge on the predictive accuracy of the models employed. As a result, agreement in the predictions obtained from different models does not necessarily imply a higher accuracy than in the case where fewer models result in similar or different predictions. With the proposed approach, assumptions related to accuracy are avoided. Further details are provided in section B.2 in the ESI.†
The multi-criterion approach adopted for stage 1, which combines a range of properties and multiple property models, provides a robust evaluation of the solvents in the initial candidate list, in spite of the use of relatively simple models. As a result, a few effective solvents emerge as the best performers: this reduced solvent list is progressed to stage 2.
2.2 Stage 2 of the methodology
In stage 2, the most promising candidates from stage 1 are further evaluated using a combination of ranking, structure–property relationships and, where feasible, more detailed predictive models to assess mixture behaviour. This allows the solvent list to be narrowed down further and priorities to be set for further research and experimental investigation.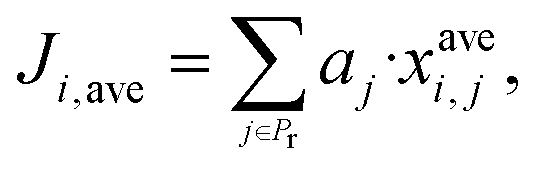 | (3) |
3. Application of proposed framework to CO2 chemisorption
In this section, we discuss the specific application of the generic framework presented in section 2 to the selection and design of CO2 capture solvents. At the heart of the framework is the identification of relevant property criteria spanning thermodynamics, reactivity and sustainability, as well as appropriate models to relate these to the solvent molecules; the models and criteria relevant to stage 1 are introduced in section 3.1, while those for stage 2 are discussed in section 3.2. In section 3.3, we define the “molecular search space”, i.e. the rules that define the new and existing molecules that are considered as potential capture solvents.3.1 Selection of relevant properties and property models for stage 1
Numerous solvent properties can be considered as screening criteria to select optimum CO2 capture solvents; Hoff et al.74 recommended a large set of relevant properties, independently of the effort required for their calculation. Several authors7–10,12,42,43 have provided significant evidence that, prior to using rigorous models or lab-scale experimentation, simpler models, indices or guidelines facilitate a quick yet reliable screening of CO2 capture solvents. Several properties are selected for stage 1 based a) on their potential to reflect important thermodynamic, reactivity and sustainability characteristics relevant to this application, b) on the availability of models that enable their fast calculation, and c) on the availability of sufficient data so that these models may be applied for a wide range of molecular structures. These properties are summarized in Table 3, together with stage 2 properties, and their connections with broader criteria for design5 are highlighted. The property models used are presented in Tables B1 and B2 in the ESI.† The rationale for the property choices made and the relevant criteria are described in the remainder of this section.| Criterion | Impact on absorption/desorption process |
|---|---|
| Thermodynamics | |
| –CO2 solubility | Ability and capacity to dissolve CO2/all design and operating parameters are affected (CAPEX and OPEX, energy consumption) |
| • Solubility parameter (δ) (stage 1) | |
| • Chemical and phase equilibria (stage 2) | |
| –Vapour pressure | Solvent losses (OPEX and environmental impact) |
| • Amine pressure (Pvp) (stage 1) | |
| • Phase equilibrium (stage 2) | |
| –Liquid heat capacity (Cp) (stage 1) | Sensible heat/reboiler duty/heat exchanger size (CAPEX and OPEX, energy consumption) |
| –Density | Equipment size-capacity (directly CAPEX) |
| • Amine density (ρ) (stage 1) | |
| • Phase equilibrium (stage 2) | |
| –Surface tension (σ) (stage 1) | Mass transfer coefficient-packing material characteristics (directly CAPEX) |
| –Viscosity (n) (stage 1) | Mass transfer coefficient-packing material characteristics (directly CAPEX) |
| –Boiling point temp. (Tbp) (stage 1) | Solvent evaporation losses/reboiler duty (OPEX, energy consumption, environmental impact) |
| –Melting point temp. (Tm) (stage 1) | Solvent solidification (directly OPEX) |
| Reactivity | |
| –Amine basicity (pKa) (stage 1) | Solvent ability to react and solvent kinetics/all design and operating parameters are affected (CAPEX and OPEX, energy consumption) |
| –Chemical equilibrium (stage 2) | |
| Sustainability | |
| –Environmental health and safety hazards (EHS) (stage 1) | Inherent hazards of solvent in the capture processes/cradle-to-gate environmental impact of solvent (per kg of solvent used)/environmental impact of the reclaimer waste (e.g. characterization as hazardous waste according toxicity and corrosion indices) if landfilled |
| –Cumulative energy demand (CED) (stage 1) | |
| –Global warming potential (GWP) (stage 1) | |
| –Eco-indicator 99 (EI-99) (stage 1) | |
The vapour pressure (Pvp) is an indicator of solvent losses and should be as low as possible. Several amine-based solutions used as solvents present a high Pvp; hence, water sprinklers are often used in the absorption column to reduce losses.
The liquid heat capacity (Cp) plays an important role in determining the heat required to regenerate the amine-based solution during desorption. This heat can be divided into three components:79 the sensible heat, the heat of vaporization (mostly that of water, which is the main component evaporated in the reboiler), and the heat of absorption necessary to desorb one mole of CO2. The sensible heat represents the heat required to raise the temperature of the solution from the absorption to the desorption temperature. This is directly proportional to the heat capacity, Cp, of this solution. Hence, a solution containing an amine with a low Cp may be assumed to contribute towards reducing the sensible heat requirements. Cp is predicted via a GC method80 developed specifically for amine-based solvents used in CO2 capture.
The density (ρ), surface tension (σ) and viscosity (n) of the liquid are three properties tightly associated with the design and operating features of the absorption column. In particular, the solvent liquid density, ρ, should be high because this leads to reduced solvent flow rate, equipment size and pumping power requirements. Furthermore, ρ, σ, and n have an effect on the mass transfer coefficient, which increases with increasing values of the density and decreasing values of the surface tension and viscosity, as shown, for instance, in Dvorak et al.81
The melting point temperature (Tm) should be lower than the lowest absorption/desorption temperature to avoid solvent solidification under processing conditions.
The boiling point temperature (Tbp) should be higher than the highest absorption/desorption temperature to avoid excessive vaporization of the pure solvent under processing conditions.
T m and Tbp are considered in the calculations as an average from Marrero and Gani82 and Hukkerikar et al.34
The cumulative energy demand (CED), measured in MJ equivalent, is a resource-oriented indicator which reflects the total primary energy demand during the production cycle and therefore also reflects to some extent the economic impacts.89
The global warming potential (GWP),90 measured in kg CO2 equivalent, is a damage-oriented indicator reflecting the effect of greenhouse gas emissions, considered here for a period of 100 years.
The Eco-indicator 99 (EI-99)91 provides an aggregated score (i.e. EI-99 points) for the end-point impact categories of human health, ecosystem quality and resource consumption.
The required cradle-to-process gate data for the environmental impact of the production of solvents are either obtained from the Ecoinvent database92 or estimated using the state-of-the-art FineChem tool93 from molecular descriptors. The FineChem tool provides estimations with an average error of 30–40% but is very useful in the case of data gaps, especially for the design of new molecules.
The EHS hazard categories represent the safety hazards regarding accidental release of energy or material, the health aspects associated with the long term effects on workers due to workplace exposure and the environmental hazards referring to the damage to flora and fauna. Koller et al.94 used a prioritized list of physical and chemical substance properties to estimate a comprehensive set of characteristic dangerous properties for each hazard category. In the safety category, the properties include mobility, fire and explosion and acute toxicity effects. In the health category, properties include irritation and chronic toxicity effects. In the environmental category, properties include air and water mediated effects as well as persistency and the potential for accumulation in the food chain.
Note that the environmental index of the solvent also refers to a part of the environmental impact of the reclaimer waste, if landfilled (e.g. sludge including the part of the solvent that is not recycled by the reclaimer). The toxicity and corrosion indices used in the EHS characterize indirectly the reclaimer waste as hazardous or not (and thus requiring special treatment). The LCA indices (i.e. CED, GWP and EI-99) refer to the environmental impact of the make-up solvent, i.e. per kg of solvent. The LCA relevant reclaimer impact is partly described by the solvent physical and hazard properties, but also from the amount of purge stream, which depends on the amount and type of degradation products not included in this study. Furthermore, energy related gate-to-gate sustainability aspects are represented by many of the indicators used for solvent design and selection (e.g. boiling point and vapour pressure of the solvent affect not only the reboiler duty but also the reclaimer energy demands).
A comprehensive sustainability assessment should also include the degradation potential of the solvent under the capture process conditions, as well as the type of degradation products. These issues are directly associated with the amount of solvent make-up (i.e. the complementary factor of the “per-kg of solvent” LCA indices) and indirectly with the environmental burden of the reclaimer (i.e. because of the purged amount treated by the reclaimer in order to avoid accumulation of the degradation products in the main capture system). Moreover, certain degradation products (e.g. nitrosamines) are associated with additional health hazards to those described by the solvent inherent health indices. However, there is severe lack of mechanistic studies relating degradation potential to the molecular structure of the solvent. Some rules have been proposed for instance, requiring the presence of secondary amines for the formation of stable nitrosamines or supporting the use of tertiary amines (typically in blends because of their slower absorption kinetics) because they have much lower nitrosation rates. However, they are too broad and qualitative to play a useful role in a CAMD approach.
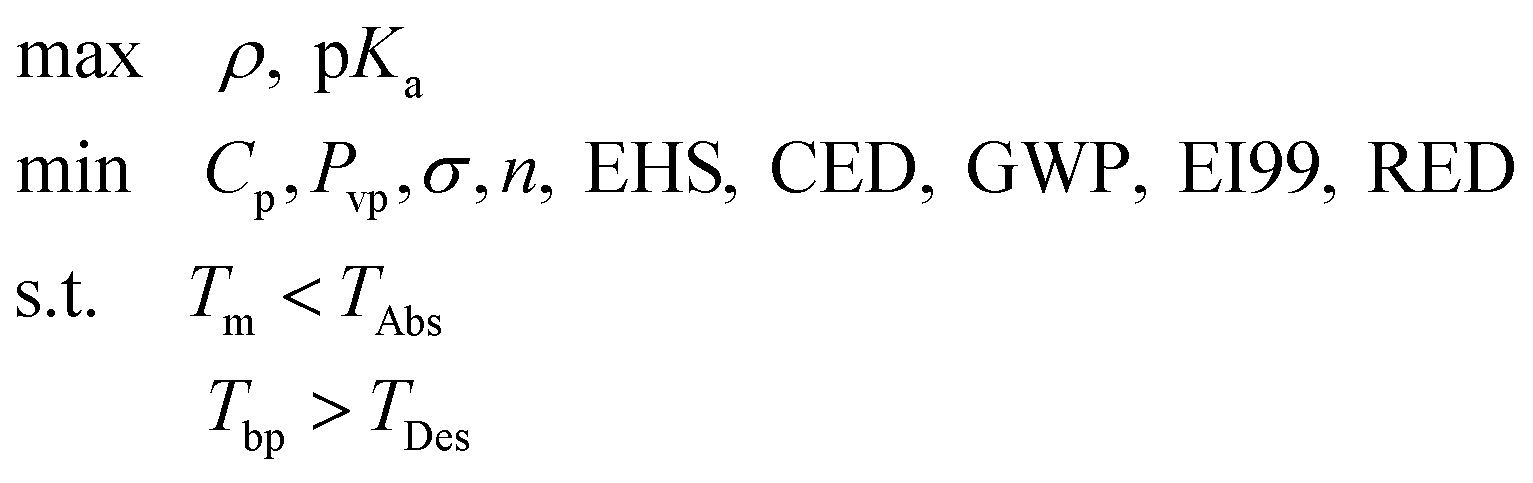 | (4) |
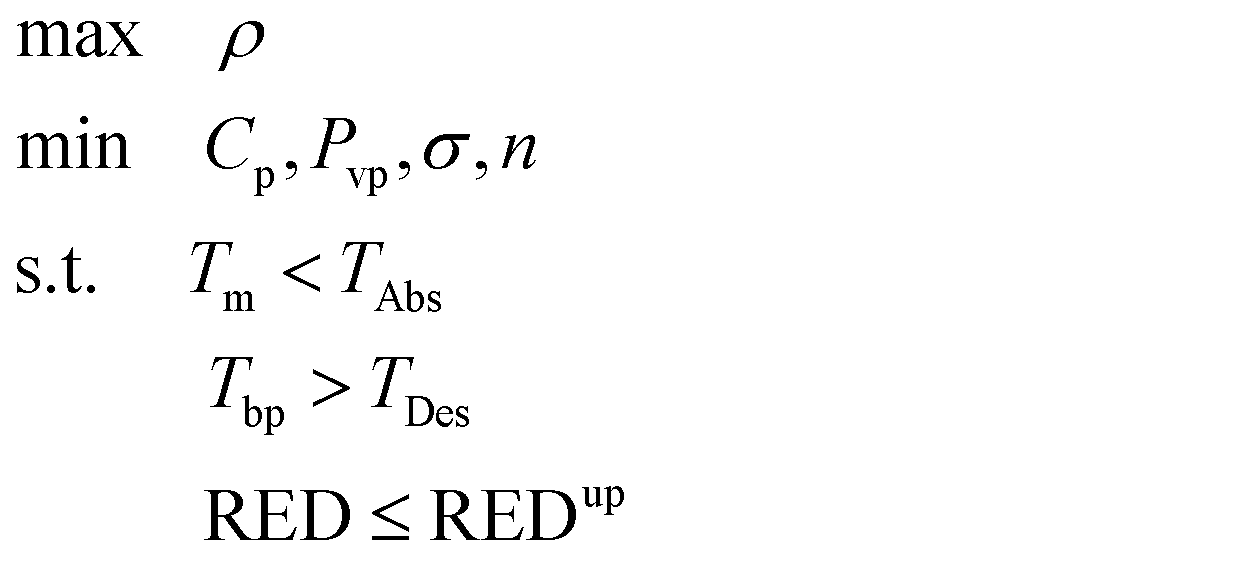 | (5) |
3.2 Selection of relevant models for stage 2
At stage 2 (step 2.2), more detailed models are required to estimate mixture behaviour and, in particular, the absorption capacity of the candidate solvents. Most models of the thermodynamic behaviour of reactive mixtures of aqueous solvents and CO2 require extensive experimental data on the specific solvent of interest. However, more predictive models have recently been derived from the SAFT95,96 equation of state. A systematic approach to the modelling of the thermodynamics of CO2 chemisorption solvents based on limited data has been developed for monoethanolamine (MEA)97 and n-alkylamines98 and further extended by Rodriguez et al.99 using the SAFT-VR EoS applied to various aqueous mixtures of alkanolamines and carbon dioxide. To enhance the predictive capabilities of this approach, models to predict the phase behaviour and thermophysical properties of aqueous alkanolamine mixtures with CO2 using the GC formalism embedded within the SAFT-γ SW framework67,68 have been proposed.69–71 This provides a much firmer basis for the design of novel solvents, enabling the design of molecular structure and mixture composition to be varied. Thus, in step 2.1, the fluid-phase behaviour of mixtures of the candidate solvents with carbon dioxide and water is evaluated using the SAFT-γ SW EoS, provided that the relevant building blocks are available in the corresponding group contribution table (cf. section D in the ESI†). Both phase and chemical equilibria are taken into account simultaneously via this model, enabling a more detailed assessment of the selected solvents. Partial pressure profiles are used to assess the relative performance of the solvents in terms of absorption potential. Saturated vapour pressures and densities can also be evaluated with the approach.3.3 The molecular search space
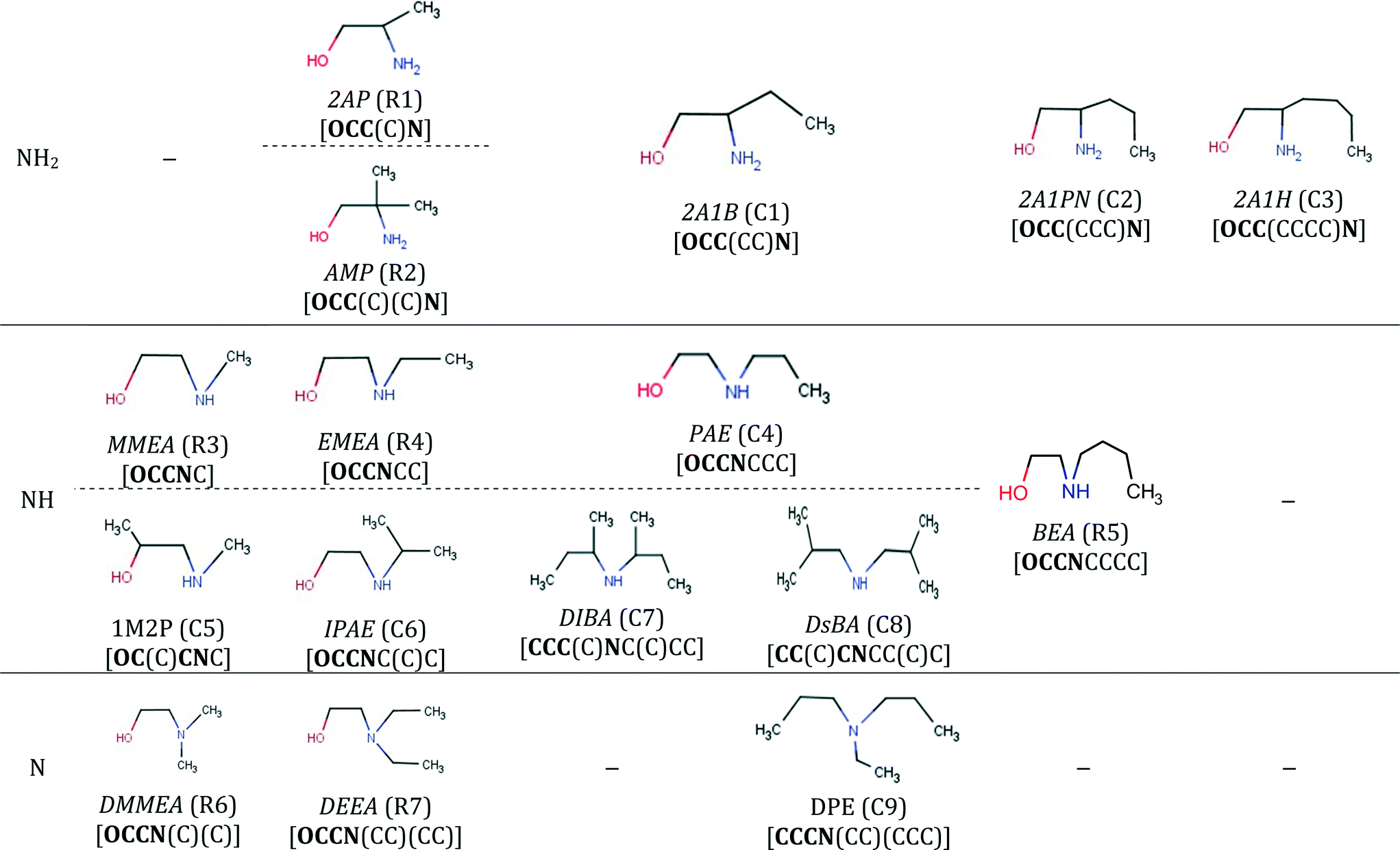
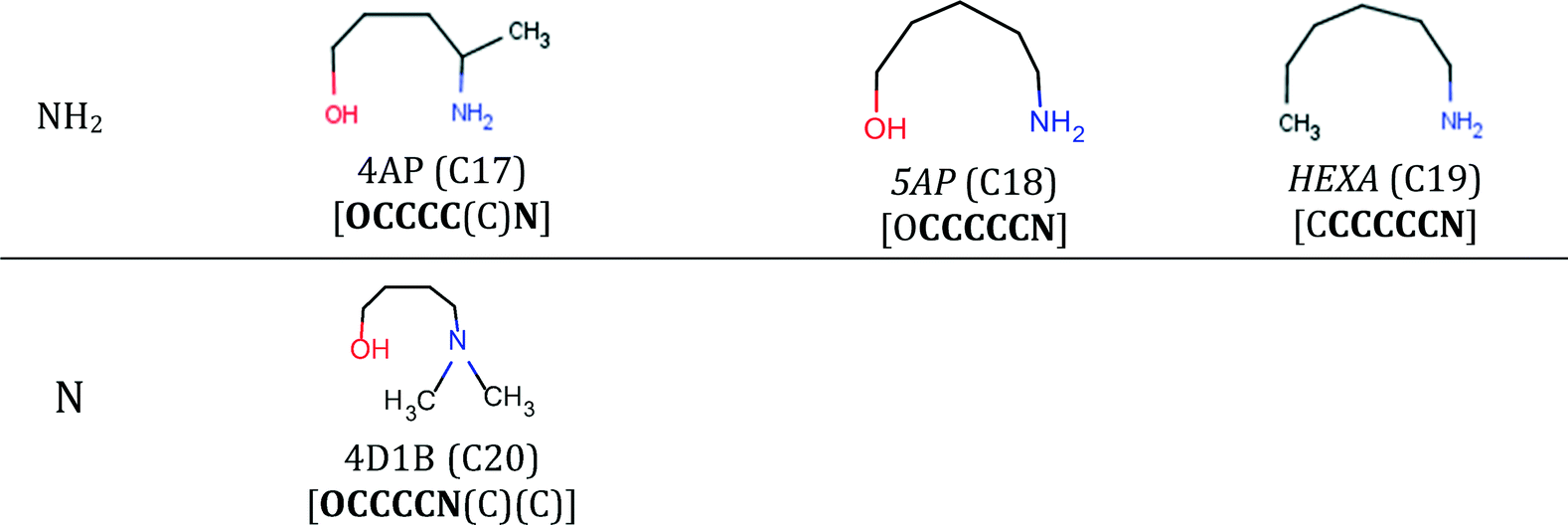
The final specification required to enable the application of our approach to CO2 capture solvents is the definition of the search space. The search space for solvent design is defined by a set of 13 functional groups, namely, CH3–, –CH2–, >CH–, >C<, –OH, –CH2–NH2, –CH2–NH–, –CH2–N<, >CH–NH2, >CH–NH–, CH3–NH–, CH3–N<, and ≥C–NH2. These groups are chosen due to the availability of the input data required for the calculation of pure-component molecular properties through GC methods and their frequent occurrence in existing CO2 capture solvents.Using these 13 groups as building blocks, a database of existing molecules is also developed by conducting a broad search for acyclic, aliphatic and hydroxylamines available in an in-house data repository at ETH Zurich, in publicly available databases100,101 and in the commercial catalogue of Sigma-Aldrich.102 The search results in a database of 126 amines and hydroxylamines, each with a unique CAS registry number.
The database is partitioned into two classes: firstly, the reference class (R), containing 25 solvents previously considered extensively in CO2 capture in the open literature (Table E1 in the ESI†), serves as a benchmark to assess the viability of the new capture solvents proposed in our work and to assess the potential performance improvements; secondly, the commercial class (C) containing 101 acyclic, aliphatic amines for which we had limited or no knowledge regarding their suitability as CO2 capture solvents, prior to this work (Tables E2, E3 and E4 in the ESI†). The C-class solvents are assumed to be commercially available because they were retrieved from public and commercial databases. The database is not exhaustive but provides a varied set of amines with different structural characteristics (e.g. primary, secondary, tertiary, branching at different positions).
4. Results and discussion
The proposed systematic approach is applied in selection mode, using classes R and C, and in design mode, using the set of 13 functional groups. In this section, we present the top solvents identified after stages 1 and 2 and analyse the results with the aim to address the following two questions:1. How successful is the proposed approach at identifying realistic solvents for CO2 capture?
2. Which molecules are most promising for further investigation as CO2 capture solvents?
4.1 Selection of top candidate solvents from the reference and commercial classes
|
|
|
| Solvent | Source | Investigated properties |
|---|---|---|
| 2AP (R1) | Da Silva103 | Carbamate stability |
| Fernandes et al.104 | Protonation constant, standard molar enthalpy and entropy | |
| AMP (R2) | Sartori et al.22 | Chemical behaviour due to steric hindrance |
| Harbou et al.105 | Process behaviour (e.g. reboiler duty, plant operation) | |
| Zheng et al.106 | Reaction kinetics | |
| MMEA (R3) | Ma'mun et al.107 | Absorption rate |
| Suda et al.108 | Absorption rate, mass transfer coefficient | |
| Ali et al.109 | Reaction kinetics | |
| EMEA (R4) | Kumar110 | Equilibrium solubility |
| Yamada et al.19 | Absorption capacity | |
| BEA (R5) | Ma'mun et al.107 | Absorption rate |
| Yamada et al.19 | Absorption capacity | |
| Ali et al.109 | Reaction kinetics | |
| DMMEA (R6) | Tong111 | Equilibrium solubility |
| Versteeg et al.3 | Reaction kinetics | |
| DEEA (R7) | Sutar et al.112 | Equilibrium solubility and reaction kinetics |
| Xu et al.113 | Equilibrium solubility and heat of absorption | |
| MPA (R8) | Henni et al.114 | Reaction kinetics |
| Dong et al.115 | Equilibrium solubility | |
| DEAB (R9) | Sema et al.11,13 | Equilibrium solubility and reaction kinetics |
| Maneeintr et al.14 | Equilibrium solubility and cyclic capacity | |
| Naami et al.15 | Mass transfer coefficient in packing material | |
| Shi et al.16 | Vapour liquid equilibrium model |
| Solvent | Main characteristics | Source | |
|---|---|---|---|
| 2AP | (R1) | The closest similar structure is MIPA which presents very similar solubility to MDEA under the same conditions but has higher pKa than MDEA. | Rebolledo-Morales et al.122 |
| MMEA | (R3) | Higher absorption rate and capacity than MEA. | Ma'mun107 |
| EMEA | (R4) | Higher CO2 loading than MEA, DEA and MMEA (at 30% w/w) over a wide range of pressures. | Kumar110 |
| MPA | (R8) | Higher reaction rate and absorption capacity than MEA. In mixtures with AMP, it exhibits considerably reduced corrosiveness, oxidative degradation and formation of nitrosamines than mixtures of MEA and AMP. | Henni et al.114 |
| Dong et al.115 | |||
| Hoff et al.123 | |||
| DEAB | (R9) | Designed specifically for high CO2 capacity, reaction constant similar to AMP and DEA but higher than MDEA and much lower regeneration energy than MEA. | Sema et al.11 |
| Maneeintr et al.14 | |||
| 2A1B | (C1) | Exhibiting high stability and low corrosiveness in mixtures with MDEA. | Rooney116 |
| 2A1PN | (C2) | Similar to 2A1B. | Rooney116 |
| 1M2P | (C5) | Not traced as a CO2 capture solvent. The structure is similar to MMEA. | — |
| IPAE | (C6) | Slightly higher absorption capacity but lower reaction rate than the similar PAE which exhibits a high absorption capacity, similar to EMEA and BEA. | Yamada et al.19,120 |
| DBA | (C11) | A biphasic solvent exhibiting phase change and regeneration at 90 °C, hence of much lower energy requirements than usual. | Zhang et al.21 |
| 4AP | (C17) | Not traced as a CO2 capture solvent. The structure is similar to 2AP. | — |
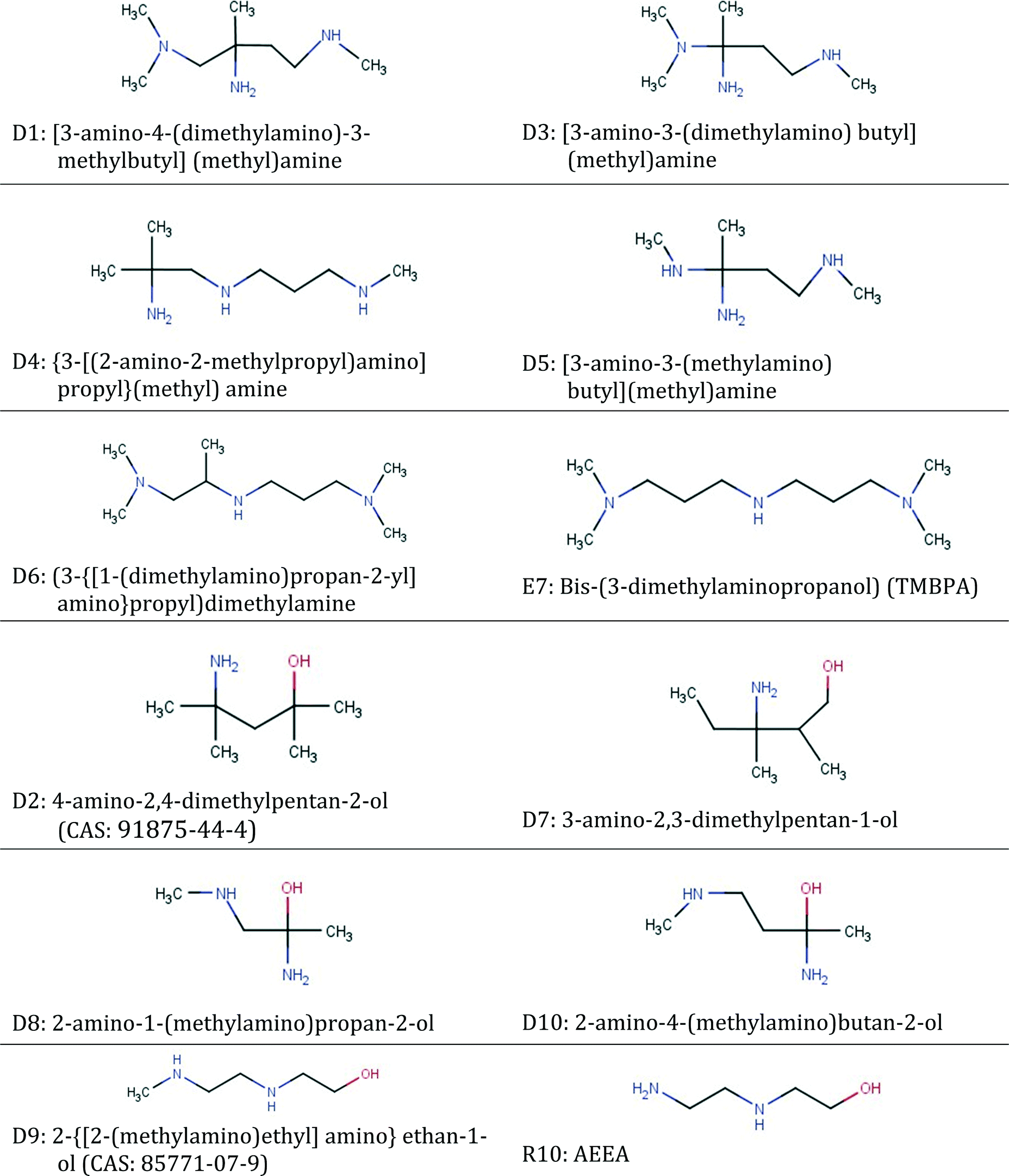
| ID | Groups in molecule | Frequency of groups |
|---|---|---|
| D1 | [–CH3, >CH2, –>C–NH2, –CH2–NH–, –CH2–N<] | [4 1 1 1 1] |
| D2 | [–CH3, >CH2, >C<, –OH, –>C–NH2] | [4 1 1 1 1] |
| D3 | [–CH3, >CH2, –>C–NH2, –CH2–NH–, CH3–N<] | [3 1 1 1 1] |
| D4 | [–CH3, >CH2, –>C–NH2, –CH2–NH–] | [3 2 1 2] |
| D5 | [–CH3, >CH2, –>C–NH2, CH3–NH–, –CH2–NH–] | [2 1 1 1 1] |
| D6 | [–CH3, >CH2, >CH–, –CH2–NH–, –CH2–N<,] | [5 1 1 1 2] |
| D7 | [–CH3, >CH2, >CH–, –OH, –>C–NH2] | [3 2 1 1 1] |
| D8 | [–CH3, –OH, –>C–NH2, –CH2–NH–] | [2 1 1 1] |
| D9 | [–CH3, >CH2, –OH, –CH2–NH–] | [1 2 1 2] |
| D10 | [–CH3, >CH2, –OH, –>C–NH2, –CH2–NH–] | [2 1 1 1 1] |
|
MEA, which was included in class R, is not found amongst the top solvents due to its low environmental health and safety performance (it exhibits a high EHS value). When sustainability indices are not considered, MEA appears in the top 15 solvents. Solvents in class R have been investigated to a wider extent in the published literature compared to C class solvents with respect to their CO2 absorption capacity and/or reaction kinetics as well as other properties (Table 7). Additional details are provided in sections E and F in the ESI.†
An examination of the solvents in class C shows that:
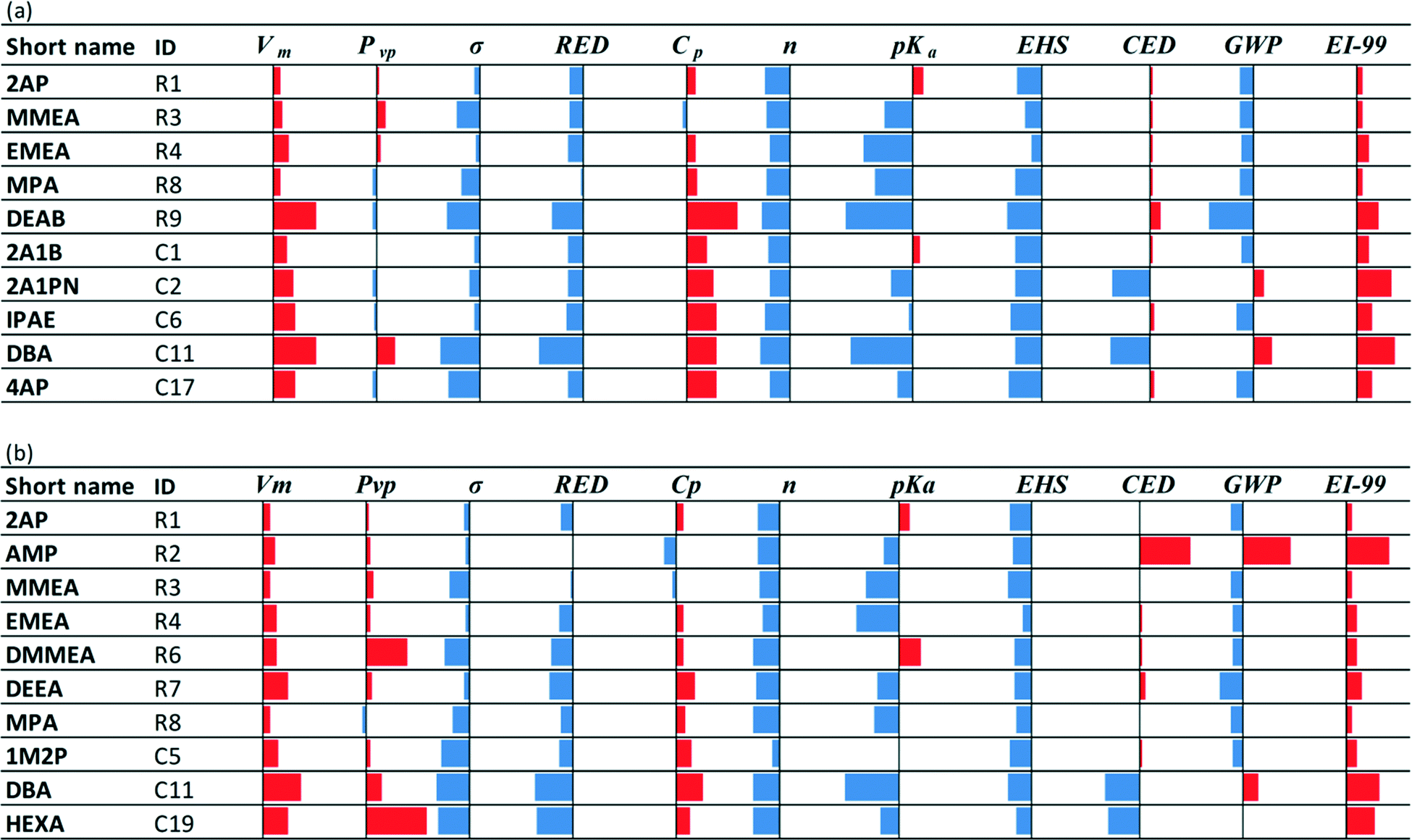
• Nine of the twenty class C solvents are potentially novel capture options: very limited or no evidence that these molecules have been considered for application to CO2 capture could be found in the open literature. The performance of these solvents for each property is shown in Fig. 3, relative to MEA.
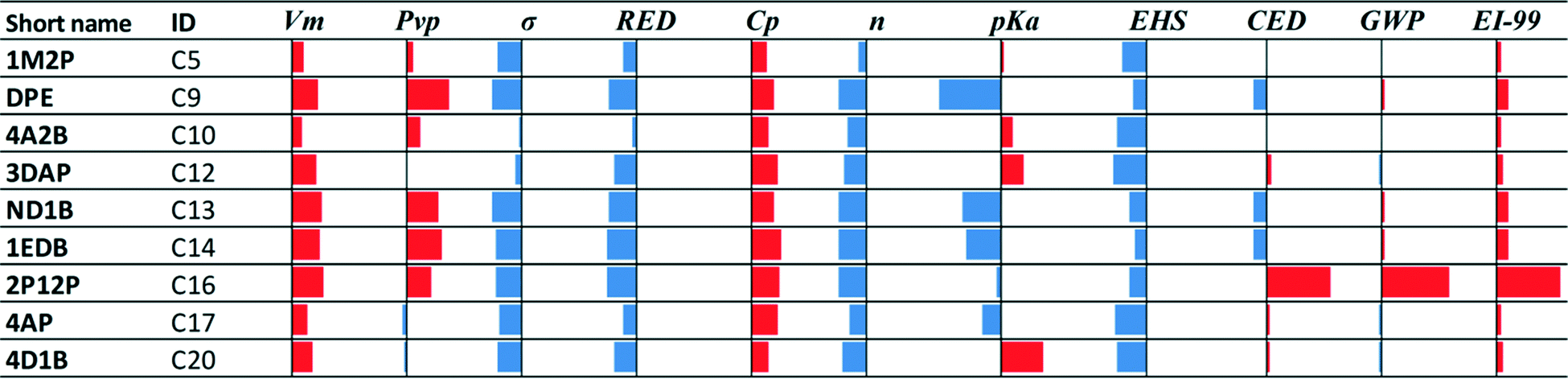 | ||
| Fig. 3 Comparative performance with respect to MEA (solid vertical lines) of commercial molecules which have not been traced (or for which data may be rare) as CO2 capture solvents. The blue bars to the left of the solid vertical lines indicate better performance than MEA. The red bars to the right of the solid vertical lines indicate worse performance than MEA. | ||
• Solvents 2A1B, 2A1PN and 2A1H have been considered in mixtures with methyldiethanolamine (MDEA) where they were tested experimentally in terms of stability and corrosiveness with respect to carbon steel.116 The tests were motivated by the discovery that an aqueous mixture comprising a tertiary alkanolamine and a primary alkanolamine with a secondary carbon atom attached to the amino group (such as 2A1B, 2A1PN and 2A1H) is not only effective in removing acid gases, but it also exhibits unexpectedly low degradation, corrosiveness and metal solubility properties. However, very few details are disclosed on these compounds; hence, they may be worthy of further investigation in mixtures with MDEA and other amines of similar structure.
• Solvents DIBA, DsBA, DBA and HEXA are phase-change solvents21,117,118 associated with significant reductions in the desorption energy requirements because they can be partly separated in a non-thermal process after absorption and before entering the desorption column. This class of solvents contains also several other options,118 which are very promising for use either in their pure aqueous form or in mixtures. A review of other types of phase-change solvents is available in Wang and Li.119
• Solvents PAE, IPAE,120 TMEDA121 and 5AP7–9 have been investigated considerably less than the other solvents and mainly with respect to their CO2 absorption capacity; hence, they may be worthy of further investigation.
Note that the solubility parameter values of solvents 4A2B, 3DAP, ND1B, 1EDB and 2P12P are within 14–17 (J cm−3)1/2 which is the range of solubility parameter values exhibited by the biphasic solvents HEXA, DsBA and DBA.7–9,21 These solvents are therefore expected to exhibit low solubility in water and may also be biphasic candidates. Finally, the EHS index shows clearer, more favourable trends than the LCA metrics (Tables C1a and C1b in the ESI†) for these solvents. In this regard, 1EDB exhibits much lower acute and chronic toxicity values than the rest of the solvents in this set, and while DPE, 4A2B and 4D1B have higher acute toxicity indices, they are still less than half the MEA index value. DPE, 4A2B 2P12P and 4D1B, however, all have flash points lower than 35 °C.
 | ||
| Fig. 4 Comparative results for the top 10 solvents selected based on (a) the set of equations in eqn (4) and (b) the set of equations in eqn (4) but without considering sustainability indices EHS, CED, GWP, and EI-99. The results are standardized with respect to MEA (shown by a solid vertical line). The blue bars to the left of the solid vertical lines indicate better performance than MEA. The red bars to the right of the solid vertical lines indicate worse performance than MEA. Solvents in the reference class (R) are reported first, followed by solvents in the commercial class (C), regardless of their ranking in the top 10. | ||
• Vm, Cp, EI-99: the performance of almost all solvents deteriorates compared to MEA. Vm reflects density; hence, all solvents exhibit lower density than MEA.
• Pvp and CED: the performance of all solvents is slightly better or slightly worse than MEA in Fig. 4a, while it deteriorates when sustainability indices are not considered (Fig. 4b).
• σ, RED, n, pKa: the performance of almost all solvents improves compared to MEA. When sustainability is not considered (Fig. 4b), σ and n of the selected solvents appear to be better than for the solvents in Fig. 4a, while pKa appears to be slightly worse.
• EHS and GWP: the performance is worse when sustainability indices are not considered, but better performance than MEA is observed in most cases.
As seen in Fig. 4, 2AP, MMEA, EMEA and MPA are repeated in both cases, indicating that they are not affected by the variation in the employed performance indices. DBA is the only solvent from the C class that is shared between the two sets in Fig. 4. The consideration of sustainability indices has a significant effect on the selection of solvents from the C class. The evaluation of the selected solvents based on mixture behaviour (step 2.2) will be considered in section 4.3, where solvents that have been found via selection or design are compared.
Focusing on sustainability performance, the EHS index generally shows clearer trends than the LCA metrics (Tables C2a and C2b in the ESI†) for the top 10 solvents. DMMEA, DEEA, DBA and HEXA are flammable with flash points lower than 50 °C. DEEA is a recognized skin sensitizer. HEXA also exhibits aquatic toxicity that is an order of magnitude higher than MEA. 2A1PN, 2AIB, IPAE, 4AP, and DEAB are predicted to have lower acute toxicity than MEA due to their relatively high oral lethal dosage (LD50oral) values. On the other hand, MEA is characterized by higher toxicity indices due to its low immediately dangerous to life and health (IDLH) threshold value generated by the National Institute for Occupational Safety and Health (NIOSH). This indicator, however, is not available for the rest of the molecules in the data set.
4.2 Design of top candidate solvents from functional groups
The resulting database of designed structures (class D) is analyzed using the same strategy as classes C and R. The matrices J′ and L are thus of size 157 × 252. The calculation of the thermodynamic properties is carried out using first-order functional groups except for the pKa that can only be calculated using a specific structure as input in the Marvin 6.0.5 (ref. 86) software. For this purpose, we use the isomers exhibiting the highest pKa value as the representative for each solvent structure. As in the case of solvent selection, top structures are identified using the criteria set out in formulation (4): five structures are obtained with the use of the sustainability indices and five without.
The set of candidates identified in this way consists of the 10 molecular structures (listed in Table 9) appearing most frequently with top ranks and representing 114 isomers. The structures are reported in the form of functional group combinations with their frequency of appearance in the molecule because this is how they are designed using the CAMD method. The results in Table 9 provide a first indication of the group combinations which may favour CO2 capture.
Structures D1–D5 are obtained using eqn (4) without considering the sustainability indices. Structures D2 and D7–D10 are obtained using the entire set of equations in eqn (4). Some of the structures obtained are multifunctional amines containing up to 3 amine groups, while some contain only a single amine group. The fact that only D2 is common to both sets of structures shows that the consideration of sustainability plays an important role in assessing the performance of the designed solvents. Structure D6 is not in the top 5 structures of the case examined in Fig. 5a, due to undesirably high Cp. It is reported here because one of its isomers is a novel molecule which closely resembles bis-(3-dimethylaminopropanol) (TMBPA).20 This is a solvent containing 3 amine groups and that has been found experimentally to exhibit very favourable CO2 capture features in mixtures with piperazine, including 70% higher cyclic capacity and 41% higher CO2 removal than a 5 M MEA solution.20
 | ||
| Fig. 5 Comparative performance of novel solvents designed using the proposed CAMD approach with respect to MEA: a) solvents D1–D5 were obtained using the set of equations in eqn (4) without considering the sustainability indices (although their values are reported here) and b) solvents D2 and D7–D10 were obtained using the entire set of equations in eqn (4). TMBPA (E7) is a commercial solvent previously tested as a CO2 capture option.20 D6 was not in the top choices, but it is reported here to compare its performance to that of E7 since the structure of a D6 isomer is very similar to E7. Fig. 5a compares solvents without considering sustainability; hence, these properties are not reported for E7 and D6. | ||
The performance of structures D1–D10 relative to MEA is presented in Fig. 5. Except for EHS, all the sustainability indices point to lower performance than MEA. This is a reasonable outcome because the sustainability indices were not considered during CAMD in step 1.1 but as a subsequent measure to identify the solvents with lower impacts among those obtained from CAMD. In this case (Fig. 5b), EHS and EI-99 improved, but CED and GWP slightly deteriorated, possibly due to the effect of the remaining property indices.
Furthermore, the apparent trade-off between the EHS and LCA indices could be attributed to the fact that molecules which are structurally larger and more complex than MEA would have longer production chains with higher environmental impacts, while they could be more stable with higher boiling points and thus lower mobility index values. The mobility index is also used as a correction factor for the toxicity and flammability indices to account for the relative ease of vapour formation promoting the risk of inhalation or catching fire. All the molecules in Table 9 show improved toxicity indices and slightly better flammability indices due to the improved mobility indices. However, they suffer from worse environmental indices such as aquatic toxicity and persistency in the environment. This could be a result of the more complex structure that is less naturally occurring and thus less prone to degradation in the environment. Tables C3a and C3b in the ESI† present, respectively, the LCA metrics and the various hazard indices and the parameters used to calculate them for the molecular structures listed in Table 9.
Isomers D2, D7, D8, D9 and D10 all contain a hydroxyl. D2 is a commercially available structure with a CAS registry number of 91875-44-4 and is the solvent shared between the two cases investigated in Fig. 5. It can be said to exhibit steric hindrance based on structure–property relationship (6) in Table 1. These molecules all contain one or two primary or secondary amine groups. They are combined with a quaternary carbon atom; hence, they also resemble AMP, except for D9 which is a simpler hydroxyl-diamine. In isomers D2, D7, D8 and D10, structure–property relationship (1) appears as [NCCO] or [NCCCO], while structure–property relationship (2) also appears in D10. D9 is a commercially available amine with a CAS number of 85771-07-9. The structure is very similar to AEEA (shown next to D9 in Table 10) which exhibits absorption capacity, CO2 reactivity, and energy efficiency higher than those of MEA, while it has also low vapour pressure.126
4.3 SAFT-based evaluation of selected solvents (step 2.2)
The sets of selected and designed solvents are now considered jointly for further analysis. The SAFT-γ SW EoS is used to predict the phase behaviour of all solvents in these sets, which can be modelled with the approach using the current set of group interactions available (section D in the ESI†). These are MMEA, EMEA, MPA, 1M2P, DBA and D9. Comparisons with experimental data, where available, and with the performance of MEA are provided.Several authors have measured the solubility of CO2 in aqueous mixtures of MMEA127–129 and EMEA110 under conditions of chemical and vapour–liquid equilibria. In Fig. 6a and b, we compare SAFT-γ SW predictions for MMEA mixtures and EMEA mixtures, respectively, with experimental data. The agreement with experiments is good considering these calculations are purely predictive and that the group parameters have been characterised based on data for other alkanolamines at lower loadings.71 In particular, the relative solubility of CO2 in the different solvents is found to be reliable. Furthermore, both models and experiments indicate that CO2 loading (cf. Figure D1 in the ESI†) decreases with solvent concentration at moderate to high pressures but increases with solvent concentration at low pressures. The qualitative agreement achieved provides a useful basis for solvent selection. We note that the accuracy of the model could be improved by developing group interaction parameters that account for the proximity effects between the groups of relevance to MMEA and EMEA, as has been done successfully for other groups.70,71
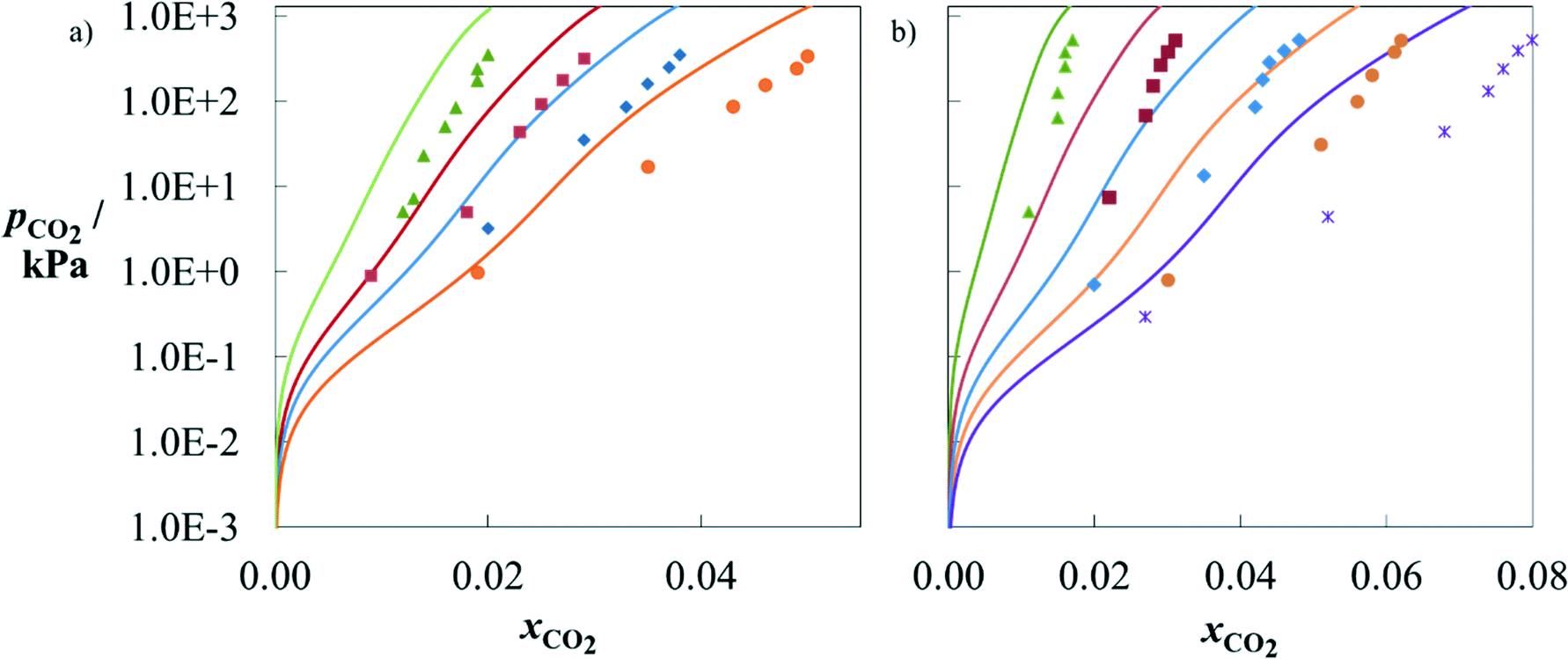 | ||
| Fig. 6 Partial pressure of CO2 as a function of mole fraction of CO2 in aqueous solutions of alkanolamine + H2O + CO2. a) MMEA at 303.1 K. The symbols correspond to experimental data110,127 for aqueous solutions of MMEA of various weight percentages in mass: 6.8 wt% (green), 11 wt% (red), 14 wt% (blue) and 19 wt% (orange). b) EMEA at 303.1 K. The symbols correspond to experimental data117 for aqueous solutions of EMEA of various weight percentages in mass: 6 wt% (green), 12 wt% (red), 18 wt% (blue), 24 wt% (orange) and 30 wt% (purple). The solid curves correspond to SAFT-γ SW calculations for the same water:solvent ratios. | ||
A further comparison with experimental data can be seen in Fig. 7 for the solubility of CO2 in aqueous solutions of MPA. The data for this mixture at two temperatures (T = 313.15 K and 393.15 K) were used in the estimation of a number of group parameters;70 and therefore, only the calculations at 343.15 K and 373.15 K can be seen as predictions. SAFT-γ is found to provide a very good description of the effect of temperature on the solubility of CO2 in this case.
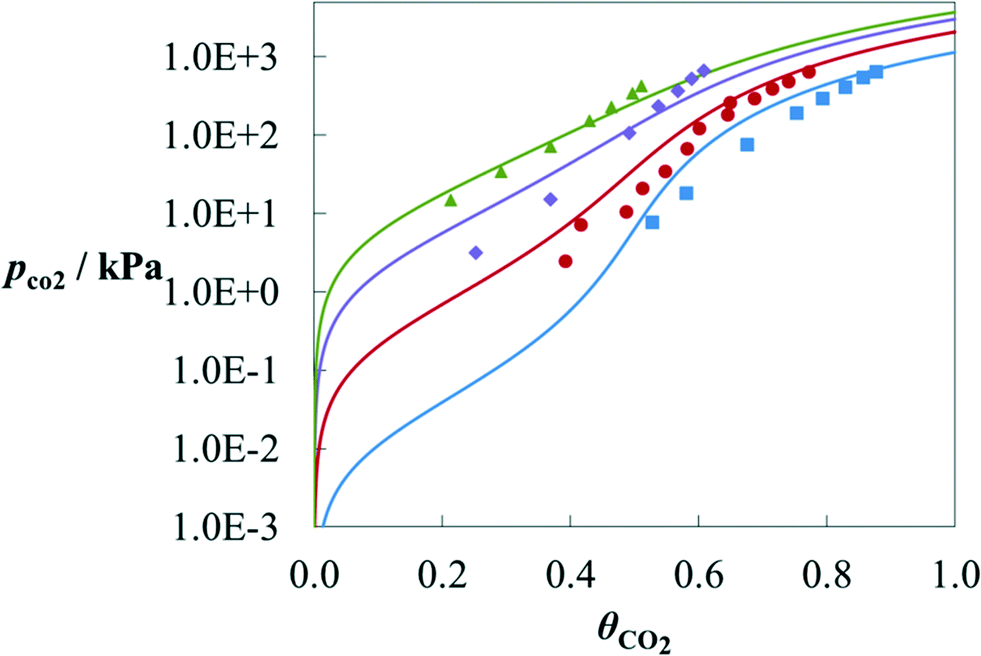 | ||
| Fig. 7 Solubility of CO2 in a 4 M (∼30 wt% in mass) aqueous solution of MPA at T = 313.15 K (blue), 343.15 K (red), 373.15 K (purple) and 393.15 K (green) as a function of the partial pressure of CO2 along the vapour–liquid equilibrium of the ternary mixture MPA + H2O + CO2. The solubility is represented as CO2 loading θCO2, defined as the moles of CO2 absorbed in the liquid phase per mole of amine in the liquid. The symbols correspond to experimental data115 for the corresponding temperatures. The solid curves correspond to SAFT-γ SW calculations. | ||
A comparison of the predicted performance of SAFT-γ for a number of high-performing solvents is presented in Fig. 8. These solvents are compared here based on the solubility of CO2 in aqueous solutions of a given mass percentage. At a low temperature (T = 313.15 K) typical of an absorption process, there is a range of pressures at which all solvents are predicted to reach higher solubility loadings than MEA for a given partial pressure of carbon dioxide. The range of pressures at which this occurs is wider for DBA and D9. Solvents such as MMEA, EMEA and 1M2P present very similar solubility curves; this can be attributed to the similarities in their molecular structures (see Table 4). At a high temperature (T = 393.15 K) typical of a desorption process, and at solubility loadings lower than 0.5, most of the solvents appear to outperform the regeneration capacity of MEA, as they lead to partial pressures of CO2 higher than those obtained with MEA. At high loadings, MEA leads to only slightly higher partial pressures than MMEA, EMEA, 1M2P and MPA.
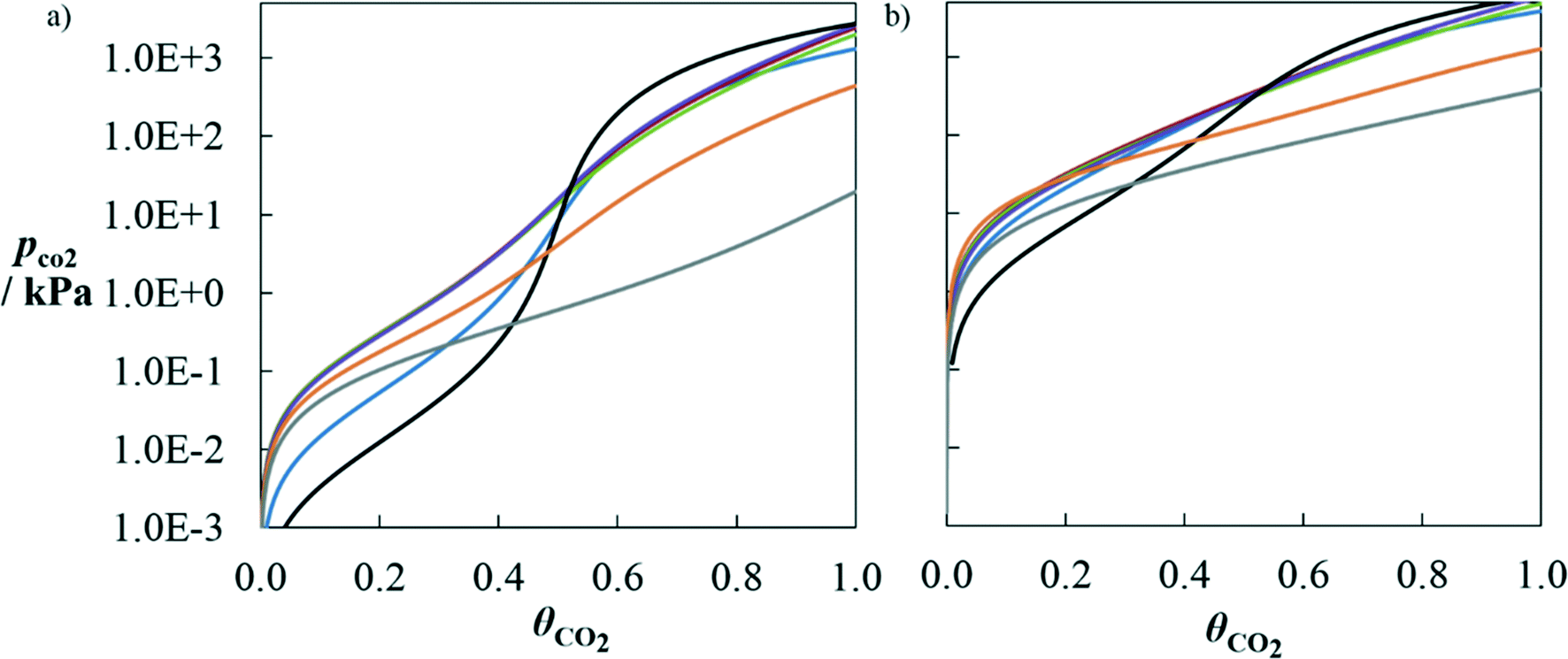 | ||
| Fig. 8 Solubility of CO2 in 30 wt% (in mass) aqueous solutions of MPA (blue), MMEA (red), EMEA (green), 1M2P (purple), MEA (black), DBA (orange) and D9 (grey) at a) T = 313.15 K and b) 393.15 K as a function of the partial pressure of CO2 along the vapour–liquid equilibrium of each ternary mixture alkanolamine + H2O + CO2. The solubility is represented as CO2 loading θCO2, defined as the moles of CO2 absorbed in the liquid phase per mole of amine in the liquid. The solid curves correspond to SAFT-γ SW calculations. | ||
The performance predictions obtained for the designed or selected solvents with SAFT-γ SW therefore confirm that these solvents should be investigated further for their CO2 capture potential.
5. Conclusions
A systematic two-stage approach to molecular design has been proposed and applied to the discovery of CO2 capture solvents. The approach is based on multiple criteria, is applicable to the identification of molecules from databases or functional groups, combines simple group contribution models with the molecular-based SAFT framework, and takes model uncertainty into account. It has been used to generate a prioritised list of candidate molecules from hundreds of thousands of possible compounds.The proposed methodology has clearly led to the identification of important candidate solvents for CO2 capture, including novel compounds. Several of these have been tested experimentally in the published literature and recommended as highly performing alternatives to MEA, indicating that the proposed approach leads to the rapid identification of useful compounds. Others have received little attention or have yet to be considered as alternatives for CO2 capture despite their commercial availability indicated by their CAS registry number, demonstrating that the proposed approach can help generate novel leads. For example in the cases of 2A1B, 2A1PN and 2A1H only a patent that mentions very good stability and reduced corrosiveness compared to MEA when they are used in mixtures with MDEA exists; however, no equilibrium or reaction kinetic data are reported. In several cases, the structural and property resemblance of these candidates with state-of-the-art solvents recently considered as CO2 capture options provides significant motivation to undertake further experimental testing. Without prior intention or use of any specific constraints, the selection procedure also resulted in the identification of several thermomorphic biphasic solvents. Such solvents enable desorption at temperatures much lower than the 120 °C employed in the MEA case, hence requiring a significantly lower regeneration load.
The novel solvents designed using the proposed CAMD approach combine structural characteristics that are known empirically to promote both high CO2 absorption capacity and increased reaction rates. D6 is a novel structure proposed by CAMD which is very similar to TMBPA, a multi-functional amine identified in the published literature as a solvent of high CO2 absorption capacity and reaction rate in mixtures with piperazine. The isomers corresponding to D2 and D9 are commercially available solvents. This is clear evidence that the systematic CAMD approach employed in this work points towards promising CO2 capture options, despite the initial use of simple property prediction models. This is further confirmed by the phase and chemical equilibrium predictions obtained with the SAFT-γ SW equation of state. For example, D9 exhibits very high performance as it actually outperforms all the other amines in the commercial set for a wide range of pressures. The fact that such an assessment of absorption and desorption potential can be made without recourse to any experimental data on the candidate solvents, and indeed without ever synthesizing the proposed molecules, is a significant benefit of the use of a predictive model such as SAFT-γ SW, relative to approaches based on more qualitative rules for solvent selection.
Due to the use of sustainability metrics in different stages of the methodology, a clear opportunity has been identified to develop less hazardous solvents than MEA. However, the LCA metrics do not always point in the same direction as safety metrics, and the improvement potential with respect to MEA is limited to specific solvents and metrics. Of course, it has to be noted that these LCA metrics refer only to the life cycle impact of solvent production, and therefore, the extent of solvent fugitive losses and degradation has been neglected in this analysis, despite its known importance in the case of MEA. Additionally, other more significant life cycle impacts (e.g. the solvent regeneration life cycle impact) are indirectly covered by the thermodynamic properties considered in CAMD.
In our current work, we have shown that considering multiple and diverse criteria in the design of solvents using a CAMD method can lead to novel molecules. Building on this approach, the space of molecules to be explored and the range of criteria for the in silico assessment of candidate compounds could both be extended. For instance, the consideration of higher order groups in the group contribution models employed could increase the accuracy of the property predictions and allow the more reliable identification of the most suitable isomers as CO2 capture options. Predictive group contribution property models that provide accurate caloric information56 could also be used to achieve a more direct assessment of energy consumption and operating cost. Furthermore, the proposed uncertainty quantification method could be expanded to account for additional properties, especially those associated with sustainability. Sustainability metrics in the form of group contribution models combined with uncertainty quantification could also be considered during CAMD to enable a more focused search as well as to increase the robustness of the obtained results. Since the results obtained from CAMD also provide insights into the structural combinations that favour CO2 capture, this could be used to prioritize the efforts to develop new group contribution parameters for the SAFT-γ SW equation, in the form of new functional groups, or of combinations of existing groups to form second-order groups and increase prediction accuracy. Such developments can provide further guidance for experimental studies and help to accelerate the search for better solvents.
Disclaimer
Certain commercial equipment, instruments, or materials (or suppliers, or software) are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.Acknowledgements
The authors are grateful to the Commission of the European Union (project FP7-ENERGY-2011-282789) and the Engineering and Physical Sciences Research Council (EPSRC) of the UK (grants EP/E016340, EP/J014958/1 and EP/J003840/1) for financial support.References
- N. Mac Dowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3(11), 1645–1669 CAS.
- F. Bougie and M. C. Iliuta, J. Chem. Eng. Data, 2012, 57, 635–669 CrossRef CAS.
- G. F. Versteeg, L. A. J. Van Dijck and W. P. M. Van Swaaij, Chem. Eng. Commun., 1996, 144(1), 113–158 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and R. J. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- I. Kim and H. F. Svendsen, 6th Korea CCS Conference, Jan 27–29, Jeju-do, S. Korea, 2016, http://www.koreaccs.or.kr/esub02_2/data/down/year/2016/gubun/MOUTH/page/3/id/1093/nnu/2 (accessed May 2016) Search PubMed.
- J. Oexmann and A. Kather, Int. J. Greenhouse Gas Control, 2010, 4(1), 36–43 CrossRef CAS.
- P. Singh, J. P. M. Niederer and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 5–10 CrossRef CAS.
- P. Singh, D. W. F. Brilman and M. J. Groeneveld, Energy Procedia, 2009, 1(1), 1257–1264 CrossRef CAS.
- P. Singh, J. P. M. Niederer and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2009, 87, 135–144 CAS.
- P. Singh and G. F. Versteeg, Process Saf. Environ. Prot., 2008, 86, 347–359 CrossRef CAS.
- T. Sema, A. Naami, Z. Liang, R. Idem, P. Tontiwachwuthikul, H. Shi, P. Wattanaphan and A. Henni, Chem. Eng. Sci., 2008, 81, 251–259 CrossRef.
- P. Tontiwachwuthikul, A. G. H. Wee, R. Idem, K. Maneeintr, G.-J. Fan, A. Veawab, A. Henni, A. Aroonwilas and A. Chakma, US Pat., 7910078, 2011 Search PubMed.
- T. Sema, A. Naami, Z. Liang, R. Idem and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2011, 50(24), 14008–14015 CrossRef CAS.
- K. Maneeintr, R. Idem, P. Tontiwachwuthikul and A. G. H. Wee, Energy Procedia, 2009, 1, 1327–1334 CrossRef CAS.
- A. Naami, M. Edali, T. Sema, R. Idem and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2012, 51(18), 6470–6479 CrossRef CAS.
- H. Shi, T. Sema, A. Naami, Z. Liang, R. Idem and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2012, 51(25), 8608–8615 CrossRef CAS.
- L. Espinal, D. L. Poster, W. Wong-Ng, A. J. Allen and M. L. Green, Environ. Sci. Technol., 2013, 47(21), 11960–11975 CrossRef CAS PubMed.
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427–6433 CrossRef CAS PubMed.
- H. Yamada, F. A. Chowdhury, Y. Matsuzaki, K. Goto, T. Higashii and S. Kazama, Energy Procedia, 2013, 37, 499–504 CrossRef CAS.
- U. E. Aronu, H. F. Svendsen and K. A. Hoff, in Proceedings of Distillation Absorption 2010, ed. A. B. de Haan, H. Kooijman and A. Górak, 2010, pp. 151–156 Search PubMed.
- J. Zhang, Y. Qiao and D. W. Agar, Energy Procedia, 2012, 23, 92–101 CrossRef CAS.
- G. Sartori, W. S. Ho, D. W. Savage, G. R. Chludzinski and S. Wlechert, Sep. Purif. Rev., 1987, 16(2), 171–200 CrossRef CAS.
- L. E. K. Achenie, R. Gani and V. Venkatasubramanian, Computer Aided Chemical Engineering, Computer aided molecular design: Theory and practice, Elsevier, 2002, vol. 12 Search PubMed.
- R. Gani, in Chemical Product Design: Toward a Perspective through Case Studies, ed. K. M. Ng, R. Gani and K. Dam-Johansen, Elsevier, 2007, vol. 14, pp. 435–458 Search PubMed.
- L. Y. Ng, F. K. Chong and N. G. Chemmangattuvalappil, Comput. Chem. Eng., 2015, 81, 115–129 CrossRef CAS.
- C. S. Adjiman, A. Galindo, E. N. Pistikopoulos, M. C. Georgiadis and V. Dua, Process Systems Engineering, Molecular Systems Engineering, Wiley-VCH, Germany, 2010, vol. 6 Search PubMed.
- R. Gani, Comput. Chem. Eng., 2004, 28, 2441–2457 CrossRef CAS.
- K. J. Kim and U. M. Diwekar, Ind. Eng. Chem. Res., 2002, 41(18), 4479–4488 CrossRef CAS.
- K. J. Kim and U. M. Diwekar, Ind. Eng. Chem. Res., 2002, 41(18), 1285–1296 CrossRef CAS.
- W. Xu and U. M. Diwekar, Ind. Eng. Chem. Res., 2005, 44(18), 7138–7146 CrossRef CAS.
- M. Folić, C. S. Adjiman and E. N. Pistikopoulos, AIChE J., 2007, 53, 1240–1256 CrossRef.
- C. D. Maranas, AIChE J., 1997, 43(5), 1250–1264 CrossRef CAS.
- A. I. Papadopoulos, M. Stijepovic, P. Linke, P. Seferlis and S. Voutetakis, Ind. Eng. Chem. Res., 2013, 52, 12116–12133 CrossRef CAS.
- A. S. Hukkerikar, B. Sarup, A. Ten Kate, J. Abildskov, G. Sin and R. Gani, Fluid Phase Equilib., 2012, 321, 25–43 CrossRef CAS.
- A. S. Hukkerikar, S. Kalakul, B. Sarup, D. M. Young, G. Sin and R. Gani, J. Chem. Inf. Model., 2012, 52(11), 2823–2839 CrossRef CAS PubMed.
- C. C. Chen, J. F. Boston, H. I. Britt and L. B. Evans, AIChE J., 1982, 23, 588–596 CrossRef.
- Y. Song and C. C. Chen, Ind. Eng. Chem. Res., 2009, 48, 7788–7797 CrossRef CAS.
- Y. Zhang, H. Que and C. C. Chen, Fluid Phase Equilib., 2011, 311, 67–75 CrossRef CAS.
- H. Lepaumier, D. Picq and P. L. Carrette, Ind. Eng. Chem. Res., 2009, 48, 9061–9067 CrossRef CAS.
- H. Lepaumier, D. Picq and P. L. Carrette, Ind. Eng. Chem. Res., 2009, 48, 9068–9075 CrossRef CAS.
- H. Lepaumier, S. Martin, D. Picq, D. Delfort and P. L. Carrette, Ind. Eng. Chem. Res., 2010, 49, 4553–4560 CrossRef CAS.
- F. J. G. Retief, MSc. Thesis, Stellenbosch University, South Africa, 2012 Search PubMed.
- F. Porcheron, A. Gibert, P. Mougin and A. Wender, Environ. Sci. Technol., 2011, 45(6), 2486–2492 CrossRef CAS PubMed.
- S. Bommareddy, N. G. Chemmangattuvalappil, C. C. Solvason and M. R. Eden, Comput. Chem. Eng., 2010, 34(9), 1481–1486 CrossRef CAS.
- N. G. Chemmangattuvalappil and M. R. Eden, Ind. Eng. Chem. Res., 2013, 52(22), 7090–7103 CrossRef CAS.
- J. Salazar, U. Diwekar, K. Joback, A. H. Berger and A. S. Bhow, Energy Procedia, 2013, 37, 257–264 CrossRef.
- ASPEN Plus®, 2015, http://www.aspentech.com/products/engineering/aspen-plus/(accessed December 2015).
- H. Renon and J. M. Prausnitz, AIChE J., 1968, 14, 135–144 CrossRef CAS.
- A. Fredenslund, R. L. Jones and J. M. Prausnitz, AIChE J., 1975, 21, 1086–1099 CrossRef CAS.
- K. Z. Sumon, A. Henni and A. L. L. East, Ind. Eng. Chem. Res., 2012, 51, 11924–11930 CrossRef CAS.
- E. F. Da Silva and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2007, 1(2), 151–157 CrossRef CAS.
- I. Stanescu and L. E. K. Achenie, Chem. Eng. Sci., 2006, 61, 6199–6212 CrossRef CAS.
- H. Struebing, Z. Ganase, P. G. Karamertzanis, E. Siougkrou, P. Haycock, P. M. Piccione, A. Armstrong, A. Galindo and C. S. Adjiman, Nat. Chem., 2013, 5, 952–957 CrossRef CAS PubMed.
- A. Gil-Villegas, A. Galindo, P. J. Whitehead, S. J. Mills, G. Jackson and A. N. Burgess, J. Chem. Phys., 1997, 106(10), 4168–4186 CrossRef CAS.
- A. Galindo, L. A. Davies, A. Gil-Villegas and G. Jackson, Mol. Phys., 1998, 93(2), 241–252 CrossRef CAS.
- F. E. Pereira, E. Keskes, A. Galindo, G. Jackson and C. S. Adjiman, Comput. Chem. Eng., 2011, 35(3), 474–491 CrossRef CAS.
- V. Papaioannou, T. Lafitte, C. Avendaño, C. S. Adjiman, G. Jackson, E. A. Müller and A. Galindo, J. Chem. Phys., 2014, 140, 054107 CrossRef PubMed.
- J. Burger, V. Papaioannou, S. Gopinath, G. Jackson, A. Galindo and C. S. Adjiman, AIChE J., 2015, 61(10), 3249–3269 CrossRef CAS.
- A. Bardow, K. Steur and J. Gross, Ind. Eng. Chem. Res., 2010, 49, 2834–2840 CrossRef CAS.
- B. Oyarzún, A. Bardow and J. Gross, Energy Procedia, 2011, 4, 282–290 CrossRef.
- M. Stavrou, M. Lampe, A. Bardow and J. Gross, Ind. Eng. Chem. Res., 2014, 53(46), 18029–18041 CrossRef CAS.
- M. Lampe, M. Stavrou, J. Schilling, E. Sauer, J. Gross and A. Bardow, Comput. Chem. Eng., 2015, 81, 278–287 CrossRef CAS.
- J. Gross and G. Sadowski, Ind. Eng. Chem. Res., 2001, 40(4), 1244–1260 CrossRef CAS.
- J. Gross, AIChE J., 2005, 51(9), 2556–2568 CrossRef CAS.
- J. Gross and J. Vrabec, AIChE J., 2006, 52(3), 1194–1204 CrossRef CAS.
- M. Kleiner and J. Gross, AIChE J., 2006, 52(5), 1951–1961 CrossRef CAS.
- A. Lymperiadis, C. S. Adjiman, G. Jackson and A. Galindo, Fluid Phase Equilib., 2008, 274, 85–104 CrossRef CAS.
- A. Lymperiadis, C. S. Adjiman, A. Galindo and G. Jackson, J. Chem. Phys., 2007, 127(23), 234903 CrossRef PubMed.
- A. Chremos, E. Forte, V. Papaioannou, A. Galindo, G. Jackson and C. S. Adjiman, Chem. Eng. Trans., 2013, 35, 427–432 Search PubMed.
- A. Chremos, E. Forte, V. Papaioannou, A. Galindo, G. Jackson and C. S. Adjiman, Fluid Phase Equilib., 2016, 407, 280–297 CrossRef CAS.
- A. Chremos, A. Galindo, G. Jackson and C. S. Adjiman, Predictive SAFT-γ SW models for application in the development in absorption processes for carbon capture, Unpublished work, 2016 Search PubMed.
- W. Mendenhall, Statistics for the engineering and computer sciences, Dellen Pub. Co., San Francisco, 2nd edn, 1988 Search PubMed.
- A. I. Papadopoulos and P. Linke, AIChE J., 2006, 52(3), 1057–1069 CrossRef CAS.
- K. A. Hoff, E. F. Da Silva, I. Kim, A. Grimstvedt and S. Ma'mun, Energy Procedia, 2013, 37, 292–299 CrossRef CAS.
- J. H. Hildebrand, J. Am. Chem. Soc., 1929, 51, 66–80 CrossRef CAS.
- C. M. Hansen, J. Paint Technol., 1967, 39, 104–117 CAS.
- E. Stefanis and C. Panayiotou, Int. J. Thermophys., 2008, 29(2), 568–585 CrossRef CAS.
- C. M. Hansen, Prog. Org. Coat., 2004, 51(1), 77–84 CrossRef CAS.
- J. Oexmann, PhD Thesis, Technischen Universität Hamburg-Harburg, Germany, 2011 Search PubMed.
- A. V. Rayer, A. Henni and P. Tontiwachwuthikul, Can. J. Chem. Eng., 2012, 90, 367–376 CrossRef CAS.
- B. I. Dvorak, D. F. Lawler, J. R. Fair and N. E. Handler, Environ. Sci. Technol., 1996, 30(3), 945–953 CrossRef CAS.
- J. Marrero and R. Gani, Fluid Phase Equilib., 2001, 183, 183–208 CrossRef.
- Y. Mergler, R. Rumley-van Gurp, P. Brasser, M. de Koning and E. Goetheer, Energy Procedia, 2011, 4, 259–266 CrossRef CAS.
- U. E. Aronu, H. F. Svendsen, K. A. Hoff and O. Juliussen, Energy Procedia, 2009, 1, 1051–1057 CrossRef CAS.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, Netherlands, 1981 Search PubMed.
- Marvin 6.0.5, http://www.chemaxon.com, (accessed April 2015).
- J. Szegezdi and F. Csizmadia, presented at 27th ACS National Meeting, Anaheim, California, USA, 2004 Search PubMed.
- F. Csizmadia, A. Tsantili-Kakoulidou, I. Panderi and F. Darvas, J. Pharm. Sci., 1997, 86(7), 865–871 CrossRef CAS PubMed.
- Verein-Deutcher-Ingenieure, Vdi-richtlinie 4600: Cumulative Energy Demand, Terms, Definitions, Methods of Calculation, Beuth Verlag GmbH, Berlin, 1997 Search PubMed.
- Intergovernmental Panel on Climate Change (IPCC), Climate change 2007 synthesis report, 2007, https://www.ipcc.ch/pdf/assessment-report/ar4/syr/ar4_syr.pdf (accessed February 2016) Search PubMed.
- M. Goedkoop and R. Spriensma, The eco-indicator 99: a damage orientated method for life-cycle impact assessment, Methodology report 2000a, Pre-Consultants, https://www.pre-sustainability.com/download/EI99_annexe_v3.pdf (accessed February 2016) Search PubMed.
- Ecoinvent database, http://www.ecoinvent.org/database/database.html (accessed February 2016) Search PubMed.
- G. Wernet, S. Papadokonstantakis, S. Hellweg and K. Hungerbühler, Green Chem., 2009, 11, 1826–1831 RSC.
- G. Koller, U. Fischer and K. Hungerbuehler, Ind. Eng. Chem. Res., 2000, 39(4), 960–972 CrossRef CAS.
- W. G. Chapman, K. E. Gubbins, G. Jackson and M. Radosz, Fluid Phase Equilib., 1989, 52, 31–38 CrossRef CAS.
- W. G. Chapman, K. E. Gubbins, G. Jackson and M. Radosz, Ind. Eng. Chem. Res., 1990, 29(8), 1709–1721 CrossRef CAS.
- N. Mac Dowell, F. Llovell, C. S. Adjiman, G. Jackson and A. Galindo, Ind. Eng. Chem. Res., 2010, 49, 1883–1899 CrossRef CAS.
- N. Mac Dowell, F. E. Pereira, F. Llovell, F. J. Blas, C. S. Adjiman, G. Jackson and A. Galindo, J. Phys. Chem. B, 2011, 115(25), 8155–8168 CrossRef CAS PubMed.
- J. Rodriguez, N. Mac Dowell, F. Llovell, C. S. Adjiman, G. Jackson and A. Galindo, Mol. Phys., 2012, 110, 1325–1348 CrossRef CAS.
- NIST (National Institute of Standards) Chemistry Webbook http://webbook.nist.gov/chemistry (accessed February 2016).
- EPA (Environmental Protection Agency), http://www.epa.gov (accessed February 2016).
- Sigma-Aldrich (Commercial catalogue), http://www.sigmaaldrich.com/catalog (accessed February 2016).
- E. F. Da Silva, Energy Procedia, 2011, 4, 164–170 CrossRef CAS.
- D. Fernandes, W. Conway, X. Wang, R. Burns, G. Lawrance, M. Maeder and G. Puxty, J. Chem. Thermodyn., 2012, 51, 97–102 CrossRef CAS.
- I. Harbou, H. P. Mangalapally and H. Hasse, Int. J. Greenhouse Gas Control, 2013, 18, 305–314 CrossRef.
- C. Zheng, B. Zhao, K. Wang and G. Luo, AIChE J., 2015, 61, 4358–4366 CrossRef CAS.
- S. Ma'mun, PhD Thesis, NTNU, Norway, 2005 Search PubMed.
- T. Suda, M. Iijima, H. Tanaka, S. Mitsuoka and T. Iwaki, Environ. Prog., 1997, 16, 200–207 CrossRef CAS.
- S. H. Ali, S. Q. Merchant and M. A. Fahim, Sep. Purif. Technol., 2002, 27(2), 121–136 CrossRef CAS.
- G. Kumar, PhD Thesis, National Institute of Technology, Rourkela, Odisha, India, 2013 Search PubMed.
- D. Tong, PhD Thesis, Imperial College London, UK, 2012 Search PubMed.
- P. N. Sutar, P. D. Vaidya and E. Y. Kenig, Chem. Eng. Sci., 2013, 100, 234–241 CrossRef CAS.
- Z. Xu, S. Wang, G. Qi, A. A. Trollebø, H. F. Svendsen and C. Chen, Int. J. Greenhouse Gas Control, 2014, 29, 92–103 CrossRef CAS.
- A. Henni, J. Li and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2008, 47, 2213–2220 CrossRef CAS.
- L. Dong, J. Chen and G. Gao, J. Chem. Eng. Data, 2010, 55, 1030–1034 CrossRef CAS.
- P. C. Rooney, US Pat., 6165432A, 2000 Search PubMed.
- J. Zhang, Y. Qiao and D. W. Agar, Chem. Eng. Res. Des., 2012, 90, 743–749 CrossRef CAS.
- J. Zhang, PhD Thesis, Technical University of Dortmund, Germany, 2013 Search PubMed.
- X. Wang and B. Li, in Novel Materials for Carbon Dioxide Mitigation Technology, ed. F. Shi and B. Morreale, Elsevier, Amsterdam, 2015, pp. 3–22 Search PubMed.
- H. Yamada, F. A. Chowdhury, K. Goto and T. Higashii, Int. J. Greenhouse Gas Control, 2013, 17, 99–105 CrossRef CAS.
- Z. Bouzina, A. Negadi, I. Mokbel, J. Jose and L. Negadi, presented at AICHE Annual Meeting, Pittsburgh, USA, 2012 Search PubMed.
- M. G. Rebolledo-Morales, M. E. Rebolledo-Libreros and A. Trejo, J. Chem. Thermodyn., 2011, 43(5), 690–695 CrossRef CAS.
- A. K. Hoff, T. Mejdell, I. Kim, A. Grimstvedt and E. F. Silva, WIPO Pat., WO 2014/086988A1, 2014 Search PubMed.
- S. Asad Rahman, M. Bashton, G. L. Holliday, R. Schrader and J. M. Thornton, J. Cheminf., 2009, 1(12), 1–13 Search PubMed.
- D. J. Cook and L. B. Holder, Mining Graph Data, Wiley & Sons Inc., New York, 2006 Search PubMed.
- M. Stec, A. Tatarczuk, D. Spiewak and A. Wilk, J. Solution Chem., 2014, 43, 959–971 CrossRef CAS PubMed.
- G. Kumar and M. Kundu, Can. J. Chem. Eng., 2012, 90(3), 627–630 CrossRef CAS.
- R. Pacheco, A. Sanchez, M. D. La Rubia, A. B. Lopez, S. Sanchez and F. Camacho, Ind. Eng. Chem. Res., 2012, 51(13), 4809–4818 CrossRef CAS.
- H. A. M. Haider, R. Yusoff and M. K. Aroura, Fluid Phase Equilib., 2011, 303(2), 162–167 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details are provided as supporting information regarding all the molecular structures considered together with a discussion of the compounds in terms of their structural characteristics and likely CO2 capture potential based on literature sources. See DOI: 10.1039/c6me00049e |
| ‡ Current address: Materials Science and Engineering Division, National Institute of Standards and Technology, Gaithersburg, MD, 20899, USA. |
| This journal is © The Royal Society of Chemistry 2016 |