
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biopolymer hybrid electrodes for scalable electricity storage
S.
Admassie
ab,
F. N.
Ajjan
a,
A.
Elfwing
a and
O.
Inganäs
*a
aBiomolecular and Organic Electronics, IFM, Linköping University, S-581 83, Linköping, Sweden. E-mail: fataj@ifm.liu.se.com; oling@ifm.liu.se
bDepartment of Chemistry, Addis Ababa University, PO Box 1176, Addis Ababa, Ethiopia
First published on 9th February 2016
Abstract
Powering the future, while maintaining a cleaner environment and a strong socioeconomic growth, is going to be one of the biggest challenges faced by mankind in the 21st century. The first step in overcoming the challenge for a sustainable future is to use energy more efficiently so that the demand for fossil fuels can be reduced drastically. The second step is a transition from the use of fossil fuels to renewable energy sources. In this sense, organic electrode materials are becoming increasingly attractive compared to inorganic electrode materials which have reached a plateau regarding performance and have severe drawbacks in terms of cost, safety and environmental friendliness. Using organic composites based on conducting polymers, such as polypyrrole, and abundant, cheap and naturally occurring biopolymers rich in quinones, such as lignin, has recently emerged as an interesting alternative. These materials, which exhibit electronic and ionic conductivity, provide challenging opportunities in the development of new charge storage materials. This review presents an overview of recent developments in organic biopolymer composite electrodes as renewable electroactive materials towards sustainable, cheap and scalable energy storage devices.
 S. Admassie | Dr Shimelis Admassie is currently an Associate Professor of Physical Chemistry at Addis Ababa University, Addis Ababa, ETHIOPIA. His research interest includes renewable energy conversion, electrochemical energy storages and sensors. |
 F. N. Ajjan | Fatima Nadia Ajjan was born in Madrid, Spain in 1986. She received her Bachelor of Chemistry degree from the Autonoma University of Madrid in 2010 and her MSc degree in Chemical science and nanotechnology from the Complutense University of Madrid in 2012, Spain. She is currently a PhD student under the supervision of Prof. Olle Inganäs at Linköping University, Sweden. Her research interests mainly focus on the field of energy conversion and storage and their applications in various energy storage devices including supercapacitor devices. |
 A. Elfwing | Anders Elfwing was born in Stockholm, Sweden in 1975. He received his M. Eng. degree in 2002 from Linköping University, Sweden. After positions at Uppsala University and Novozymes Biopharma in Lund he returned to academia in 2009. Currently, he is working on his Applied Physics PhD program under the supervision of Prof. Olle Inganäs at Linköping University, Sweden. His research interest focuses on process development for energy generation and storage. |
 O. Inganäs | Olle Inganäs is a professor of biomolecular and organic electronics, IFM, Linköpings Universitet, Sweden. He received his MSc degree in engineering physics from Chalmers University of Technology (1977), BSc degree in philosophy and economics from Göteborg University (1978), and PhD degree in applied physics at Linköping University in 1984. He was appointed as a professor in 1999. Inganäs received the Göran Gustafsson prize in physics in 1997, and was a Wallenberg Scholar in 2010. He was elected as a member of the Royal Swedish Academy of Sciences, class of physics, in 2006, and is the chairman of the Nobel committee for the prize in physics. Inganäs has focused on studies of the class of conjugated polymers throughout the areas of polymer physics, electrochemistry, electronics and optics. He has contributed to a number of start-up companies in the field of electronic polymers. His current interest includes energy conversion and energy storage with organic photovoltaic devices and organic supercabatteries, as well as the use of biopolymers as organisers of electronic polymers. |
Introduction
Biopolymers are the most abundant polymers on Earth, in the sequence of cellulose, lignin, proteins and DNA. They form the skeleton and have active functions in living systems and in green plants. Biopolymers contribute to the conversion of solar energy to biochemical energy. The chemical energy found in organic solids or liquid fuels for use in combustion is a plant product of recent photosynthesis as well as photosynthesis over millions of years that has been converted to fossil fuel deposits. The flow of carbon compounds in this greater scheme of things hinges on the combined electron and proton transfer affected by quinone molecules. Derivatives of quinones are found in the biomembranes of bacteria, plants and animals and play a decisive role in photosynthetic energy conversion and in respiration. The quinones couple the electronic charges from the active photosynthetic reaction center in photosynthetic bacteria and plants to drive proton diffusion over the biomembrane via the reduction of a diffusing quinone. The flow of electrons eventually creates a proton gradient, which is the basic element of bioenergetics. The pH difference between the two sides of the membrane is used to form ATP from ADP, the intermediate energy form in bioenergetics. Therefore, quinones are found all over the surface of the Earth in all biological systems. The simplest redox couple based on a quinone/hydroquinone couple will exchange two electrons and two protons. For a quinone, the charge density of 2 moles of electrons per 110 g or 487 mA h g−1 is obtained. A solid electrode composed of quinone, and capable of both electronic and ionic conduction, would thus have an impressive charge density. Since quinones are already fully integrated in the biological material flow they do not cause concerns for environmental accumulation and disruption. Hence, they are very attractive compared to inorganic electrode materials.Present day electrochemical energy storage systems are not fully scalable and are expensive.1 Hence, breakthroughs in the storage of electricity would enable an accelerated use of renewable electricity in electrical power grids, vehicles and in mobile electronics when far away from the grid system. It can also have a major impact in places that are far away from the electrical grids by dramatically changing the lifestyles of a large part of the global population who still lack access to electricity.2 Hence, cheap and scalable materials are desired materials for electrical energy storage that can be scaled-up to match the need coming from the growing use of intermittent electricity from wind turbines and solar photovoltaics. In this review, we will summarize some of our recent results using quinone containing biopolymer hybrids obtained from lignins and related studies that lay the foundation for developing cheap, scalable and environmentally friendly energy storage materials.
Lignin from plants and its uses
Plants build cellulose, hemicelluloses and lignins in their growth processes, and ≈1011 tons of cellulose are produced annually. Lignin forms some 20–30% of the solid weight of plants, whereas the remaining 70–80% is dominantly cellulose and hemicellulose. Softwood plants carry more lignin (closer to 30%) than hardwood (closer to 20%). Lignin is a complex three-dimensional polymer based on the combination of three monolignols, phenylpropane compounds, in different proportions, and with different compositions between plant species and plant elements3 (Fig. 1(a) and (b)). The role of lignin in these plants is that of a structural polymer, which together with cellulose and hemicellulose forms the cell walls of the plant cell (Fig. 1(c)).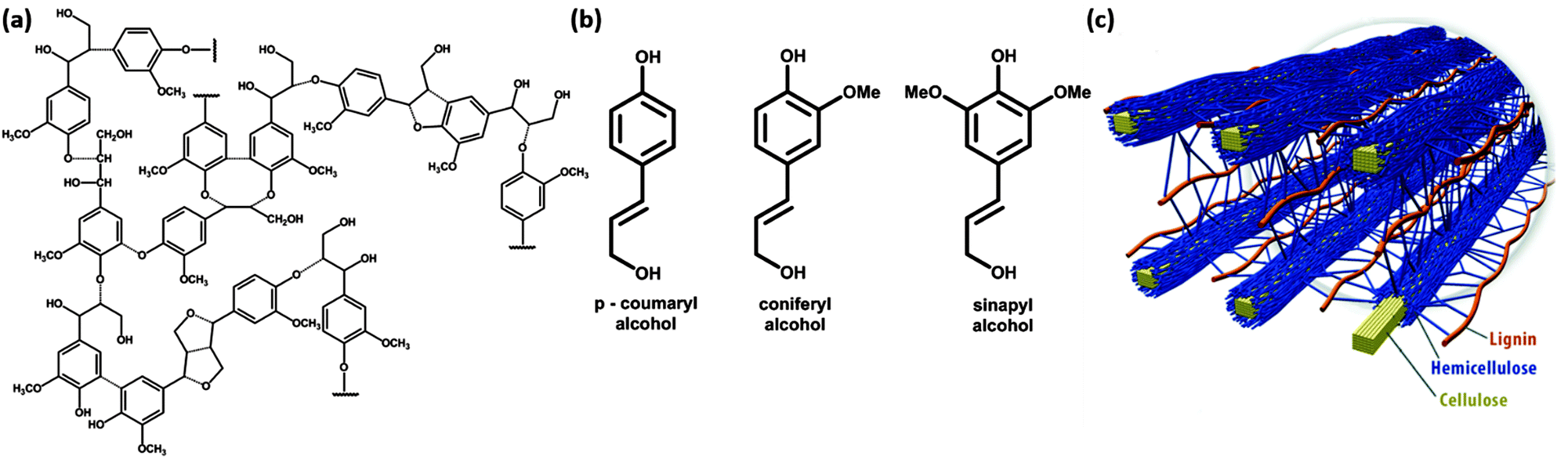 | ||
| Fig. 1 (a) Lignin polymeric structure, (b) lignin monomeric building blocks, and (c) spatial arrangement of cellulose, hemicellulose and lignin in the cell walls of lignocellulosic biomass. | ||
Aromatic lignin is the cause for the yellow color of newspaper grade paper and is due to the photochemical reaction driven by absorption of UV light in the lignin polymer. If the yellow color is not required, lignin is removed to make white paper that does not bleach over time. The aromatic phenylpropane monomers lead to a more hydrophobic polymer than cellulose. Therefore, lignins play a role in the transport of water in plants and blocks evaporation of water. The source of phenylpropane is the amino acid phenylalanine, which is glycosylated to form monolignols. These are water soluble, and can be transported across the plant membrane to form the lignin polymer, outside this membrane. Polymerisation mechanisms are debated, but radical generation on monolignols is initiated by oxidative enzymes such as peroxidases and laccases. The coupling of these radicals leads to very heterogeneous and complex three-dimensional lignin polymers.3
Cellulose is a product of the pulp and paper industry and the side product of black, brown or red liquor that contains lignin derivatives. During the sulphite extraction process, sulphonation of lignin occurs, giving a water soluble form of lignin, lignosulphonate (LS). The Kraft process leads to alkaline lignins. After separation of cellulose, the black liquor is concentrated by evaporation to allow burning of the degraded lignin and reuse of the inorganic chemicals used in the process. Thus, the main economic value of the waste product of degraded lignin is as a fuel to deliver heat for the pulp plant by burning the material. These lignin derivatives are also used as dispersant agents in concrete or oil, as stabilizers for colloidal dispersions and for synthesis of vanillin. It is also of some interest to note that the syringol and guaiacol monolignols, which are released during pyrolysis of wood, are a source of the taste of smoked foods.
Alternative routes for processing of wood are being developed in the context of the forest biorefinery, a green technology based on renewable resources for supplying organic chemicals, for further use as fuels and in organic and polymer synthesis. In a recent example of a catalytic lignocellulose biorefinery process, thermal processing of wood chips in the presence of catalysts, a refined form of pyrolysis, leads to separated chemical forms of cellulose and a lignin oil containing monomers and oligomers of the monolignols.4 This is a route that might become relevant for scaling biopolymer electrode materials.
In the next sections we will see how the use of lignins for electrical energy storage may be a critical element to free cooking from the necessity of burning wood, a present day default procedure in billions of households around the Earth, of which most do not have access to electricity. The transition to electrical cooking may thus once more require wood for electrical storage.
Electrochemistry of lignin
The insulating properties of lignin (and cellulose) are well known and introducing electronic conductivity into such materials is difficult. Hence, there are rather limited studies on the electrochemical properties of this material. The studies by Milczarek, however, showed that LS adsorbed as a monolayer on graphite electrodes could be investigated by electrochemical methods.5 One of the observations was that repeated cycling of the LS monolayer in an acidic electrolyte converted the layer to a new chemical species, a quinone. The conversion of the monolignols to their oxidized forms was suggested as the source of the quinone structures. It is possible to oxidize the syringyl (S) and guaiacyl (G) monolignols to quinone structures, as they carry methoxy structures, but not for the third monolignol phenylhydroxypropyl (H). Where these groups are formed, they may also participate in charge transfer reactions between the electrode surface and soluble species, as a modified electrode. The use of this modified electrode for electrocatalytic applications has been demonstrated.6–8Electrochemistry in a monolayer is suitable for electroanalysis. But, to make use of the quinones for charge storage, thick films must be formed on electrodes which requires considerable electronic and ionic conductivity. It is not quite plausible to make LS into a solid state material with these properties. The best approach is to make a hybrid material, where one element brings electronic conductivity and another brings the quinone redox activity. This would certainly reduce the charge density of the electrode material, but may allow the formation of thicker and more useful electrodes.
Electronic polymers
Efforts to use organic materials as electrode materials for energy storages have accelerated in the last decade.1,9–12 Among the different classes of redox active organic compounds applied for these purposes we find conjugated polymers,13 organic radical compounds, hybrid organic-sulphur materials14 and carbonyls.15 At one time a battery incorporating the conjugated polymer polyaniline was commercially available, and a number of polymer materials have seen extensive academic study.13 There is presently renewed interest in these materials for energy storage, as evidenced in the rapidly growing literature.16,17 Among these polymers, polypyrrole (pPy) appeared as a relevant candidate 30 years back, and some efforts to develop polymer electrodes for batteries and supercapacitors based on pPys were made.18,19–21 The pyrrole monomer has a lower molar mass than the thiophene or phenylene candidates for conjugated polymers also studied at that time. An analysis of the potential energy densities of these polymer batteries however indicated that they were not competitive with inorganic materials being developed at this time.22 This analysis was verified in the resounding success of the lithium ion batteries presently powering mobile electronics, which are based on inorganic insertion electrodes combined with carbon counter electrodes and organic electrolytes. Though the development of organic polymer batteries has been repeatedly attempted during these 30 years, it could not lead to industrial use.14,23–29 The present day renaissance of organic and polymer based batteries and supercapacitors is encouraging,17,30–39 but has not yet moved much beyond the performance that was prevalent in earlier years. Novel methods of materials and device assembly are however to be found, and the processability aspect is strongly emphasized.40 We demonstrated electroactive hydrogels as electrodes for supercapacitors some time since,41–46 and current developments are pursuing this avenue for higher performance and stability.47,48Some of the organic materials studied are also partly derived from biopolymers as structural materials within the electrode and electrolyte.17,31,32,39 The use of redox processes within a biomolecule is also presently studied in materials and devices. A recent demonstration of a redox flow battery using quinone in one compartment49 has attracted attention and demonstrated the versatility of quinones in making larger electrochemical devices.50 It is desirable to obtain quinone molecules and materials by renewable routes and finding sources of quinones in the realm of (cheap) biopolymers would thus appear to be an interesting route. Even more, creating conditions for electronic conduction and ionic conduction in quinone based materials would be desirable.
Conjugated polymer/lignin hybrid materials
Conjugated polymers deliver the requisite combination of electronic and ionic conductivity. Hence, we have pursued the use of lignin derivatives to create quinone functionality from a scalable biopolymer source and formed a hybrid material with conjugated polymers.Polypyrrole/lignin hybrid materials
Polypyrrole has also long been studied for the redox activity of quinones.51–53 We therefore choose pPy as a suitable host for LS, and found that polymerization of pyrrole by electrochemical methods is possible in solutions of LS and related lignin materials.54Synthesis
The electrosynthesis has to be done under conditions that will enable the electronic access to the chemical group(s) of LS that will eventually be converted to a quinone. In aqueous media the optimal synthesis conditions typically require low pH for ease of electropolymerisation of pyrrole and to form an electronically conducting host for the LS. On the other hand, some lignin derivatives will precipitate under acidic conditions, and the set of lignin derivative candidates that can be synthesized at low pH is limited. For LS, it was possible to form the composite pPy/LS materials by electropolymerisation in 0.1 M HClO4 acid. However, many of the alkaline lignin derivatives from Kraft processes precipitated under such conditions. We therefore developed alternative polymerization routes for pPy in the presence of alkaline lignins. One of these was the use of mixed solutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of aqueous HClO4 and acetonitrile as solvent for the electrolyte and lignins.55 Another approach is to use ethylene glycol as the solvent for lignins and salts during electropolymerisation.56 Both of these two routes give access to hybrids of pPy and alkaline lignins. An alternative pathway to hybrid materials with alkaline lignins was also recently reported by Leguizamon et al.57
:1) of aqueous HClO4 and acetonitrile as solvent for the electrolyte and lignins.55 Another approach is to use ethylene glycol as the solvent for lignins and salts during electropolymerisation.56 Both of these two routes give access to hybrids of pPy and alkaline lignins. An alternative pathway to hybrid materials with alkaline lignins was also recently reported by Leguizamon et al.57
Electrochemical and spectroelectrochemical properties
The galvanostatic polymerization technique was mainly used for the electrosynthesis of the hybrid materials. A polymeric product is deposited on the electrode at ≈0.6 V vs. Ag/AgCl. It is a conducting material, with an electronic conductivity of ≈1 S cm−1. The electrochemistry of this material reveals the characteristic redox wave of pPy. Repeated redox cycling in 0.1 M HClO4 leads to the growth of a redox wave at 0.5 V vs. Ag/AgCl, which will eventually become larger in amplitude than the pPy redox wave (Fig. 3).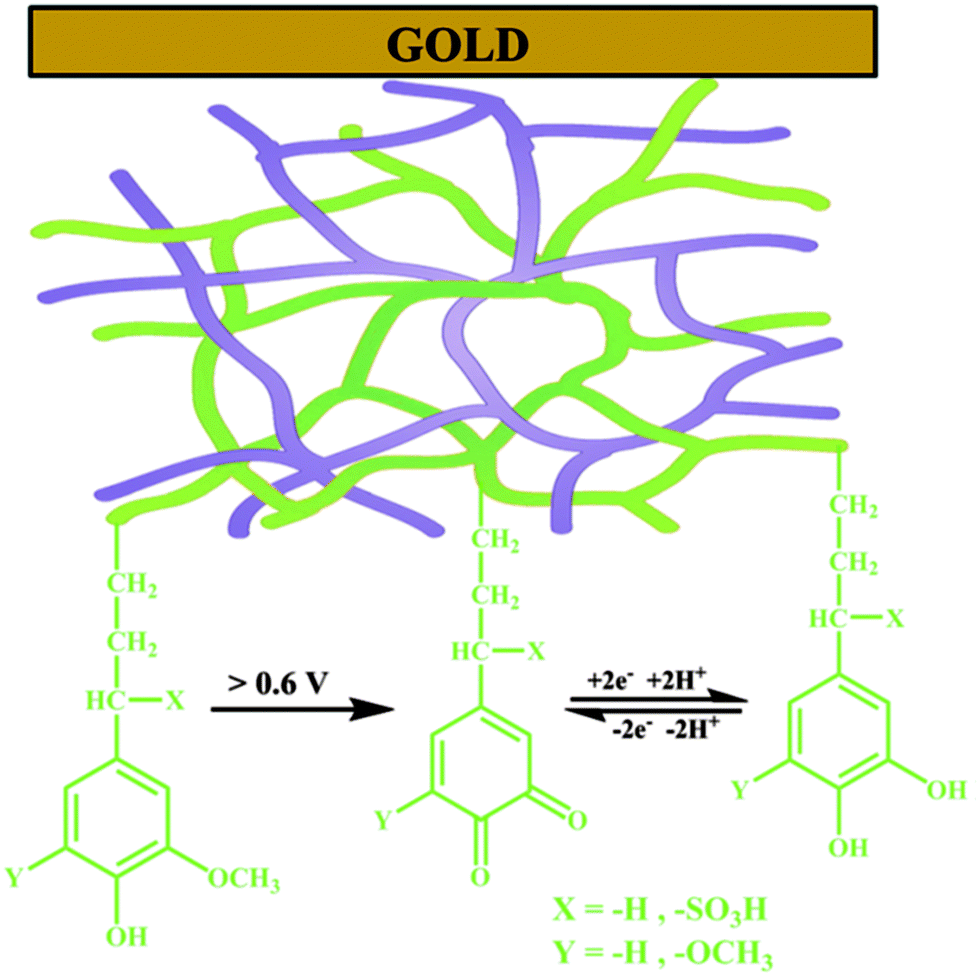 | ||
| Fig. 2 Schematic representation of an interpenetrating network of the polypyrrole (pPy)/LS biocomposite formed on a gold substrate by electrochemical polymerization of pyrrole in the presence of LS. Green lines represent lignin with the redox e−/H+ exchange reaction and the purple lines represent pPy. Reprinted with permission from ref. 58. Copyright 2015, Royal Society of Chemistry. | ||
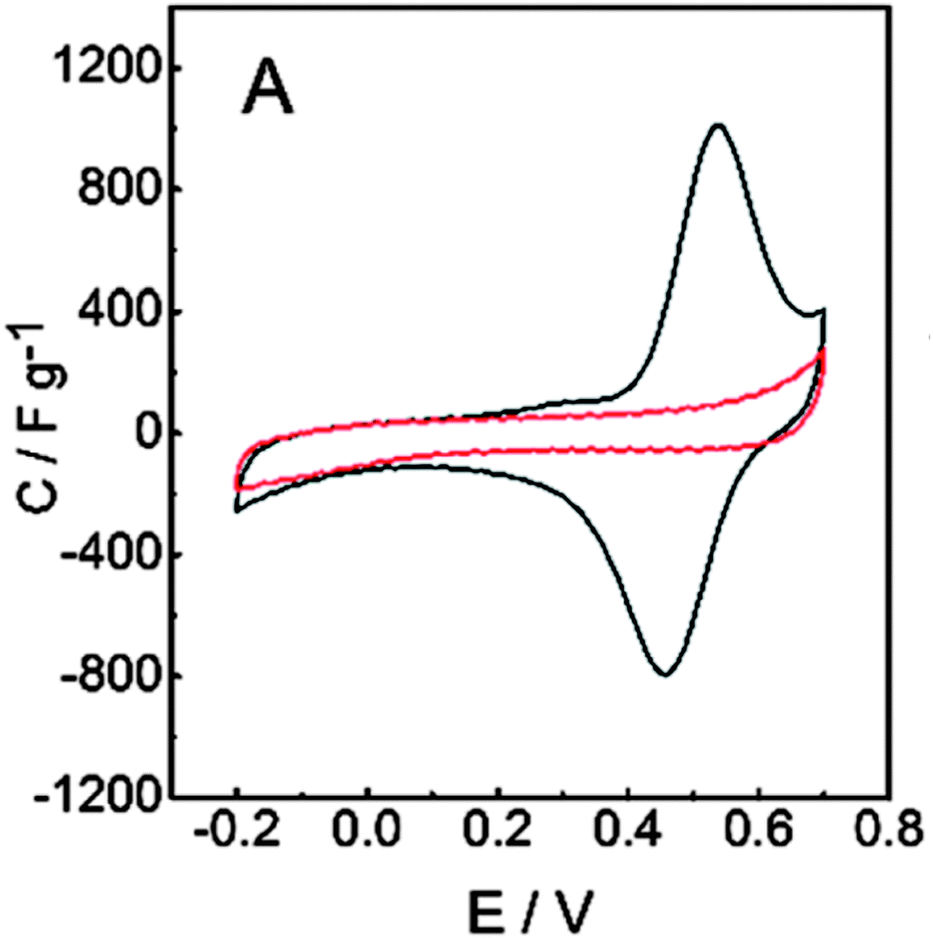 | ||
| Fig. 3 Results for the PPy/LS composite: cyclic voltammograms of (A) pPy (red line) and pPy/LS (black line). Reprinted with permission from ref. 58. Copyright 2015, Royal Society of Chemistry. | ||
The quinone group is formed through the repeated oxidation and reduction of different monolignol species incorporated in LS. The electrochemical response in different pH buffers, which shows the pH dependence expected for a one-electron, one-proton transfer reaction54 shows that this is a quinone group (Fig. 4).
 | ||
| Fig. 4 Cyclic voltammograms of the pPy/LS composite film recorded in a series of Britton–Robinson buffers of various pH values (indicated by numbers). The inset shows the dependence of the mid-peak potential on pH. All voltammograms recorded at 10 mV s−1. Reprinted with permission from ref. 54. Copyright 2012, The American Association for the Advancement of Science. | ||
The formation and conversion of the quinone group has also been investigated by FTIR spectroelectrochemistry58 where the intensity of the vibrations due to the carbonyl bonds are observed to vary with the redox state of pPy/LS (Fig. 5).
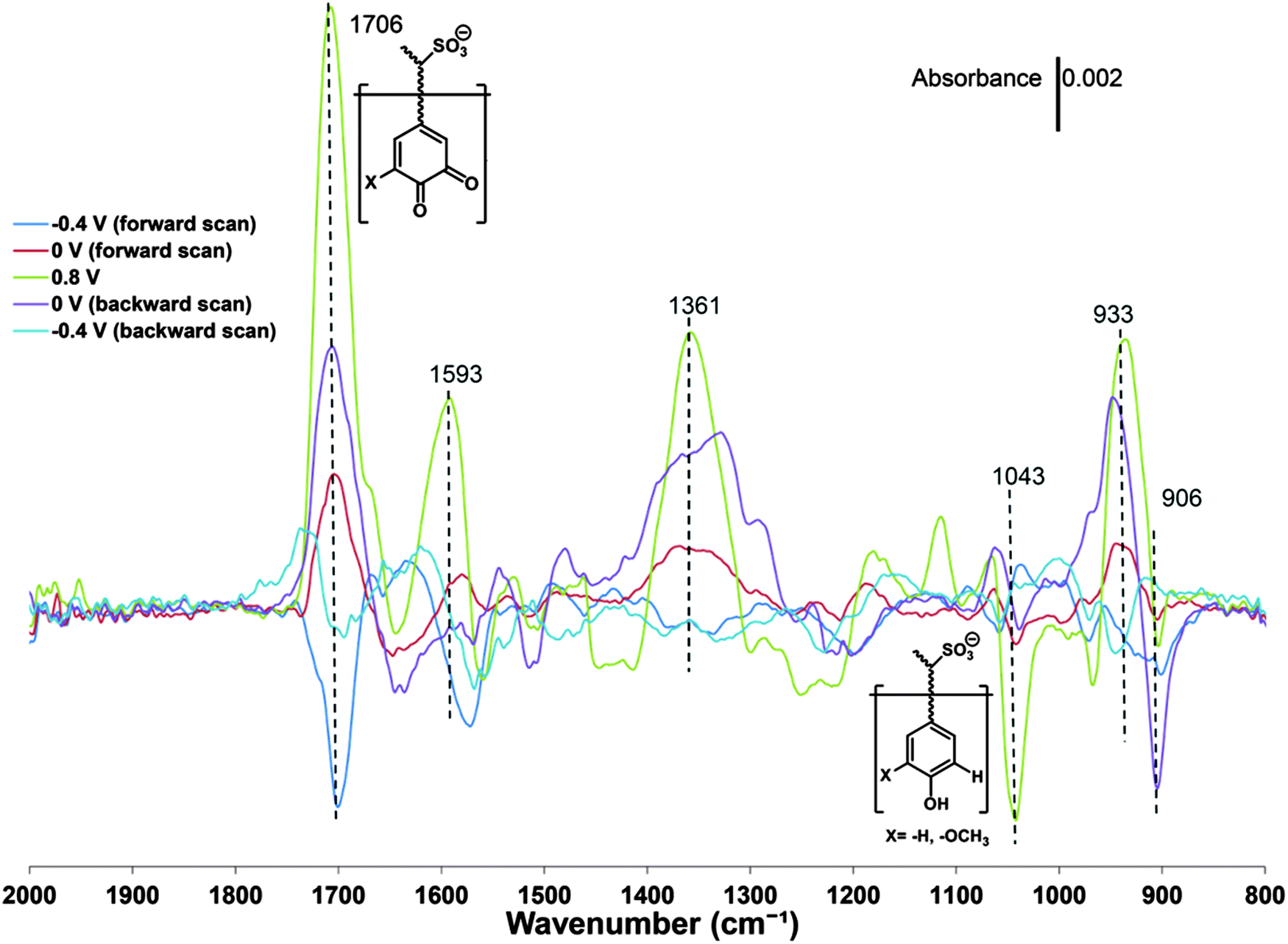 | ||
| Fig. 5 Potential-dependent ATR-FTIR difference spectra of a pPy/LS composite. Each spectrum from the in situ experiment was subtracted by the spectrum of the pristine sample. The average of the spectrum from 2000 to 1800 cm−1 was used to define the baseline for each curve. Reprinted with permission from ref. 58. Copyright 2015, Royal Society of Chemistry. | ||
The modulation of the carbonyl vibration amplitude is a proxy for the variation of quinone like groups, but also other carbonyls than those associated with a quinone can be recorded.
The conversion of monolignols to quinones as suggested in Fig. 2, where the oxidation of syringyl (S) and guaiacyl (G) monolignols leads to a quinone species, while there is no similar route for the formation of quinones from hydroxypropyl (H), leads to the conclusion that lignin derivatives from plants with high content of S and G are interesting candidates for improving the density of quinones in pPy/LS materials. Observations of this correlation has been done, starting with a set of softwood and hardwood lignin derivatives from subtropical and tropical plants (eucalyptus, acacia and African redwood (koso, or Hagenia abyssinica)).55 To determine the chemical structure of these starting materials, phosphitylation of the biopolymer and subsequent P-NMR measurements were carried out (Fig. 6). Through this method, it is possible to assign the composition of the derivatized lignin in soluble form to different chemical groups prior to integration in a biopolymer electrode and to predict the potential for formation of quinone structures. We note that the electrochemical oxidation of pPy in the presence of LS in solution may possibly cause changes to the LS chemical structure. But, since the oxidation potential of the pyrrole monomer is lower than that of LS it is not expected that oxidation will transform LS. For the first time, we can now correlate these results from NMR spectroscopy with the outcome of a biopolymer electrode incorporating this sample. The presence of S monolignol groups is correlated with a higher charge density in the composite materials.
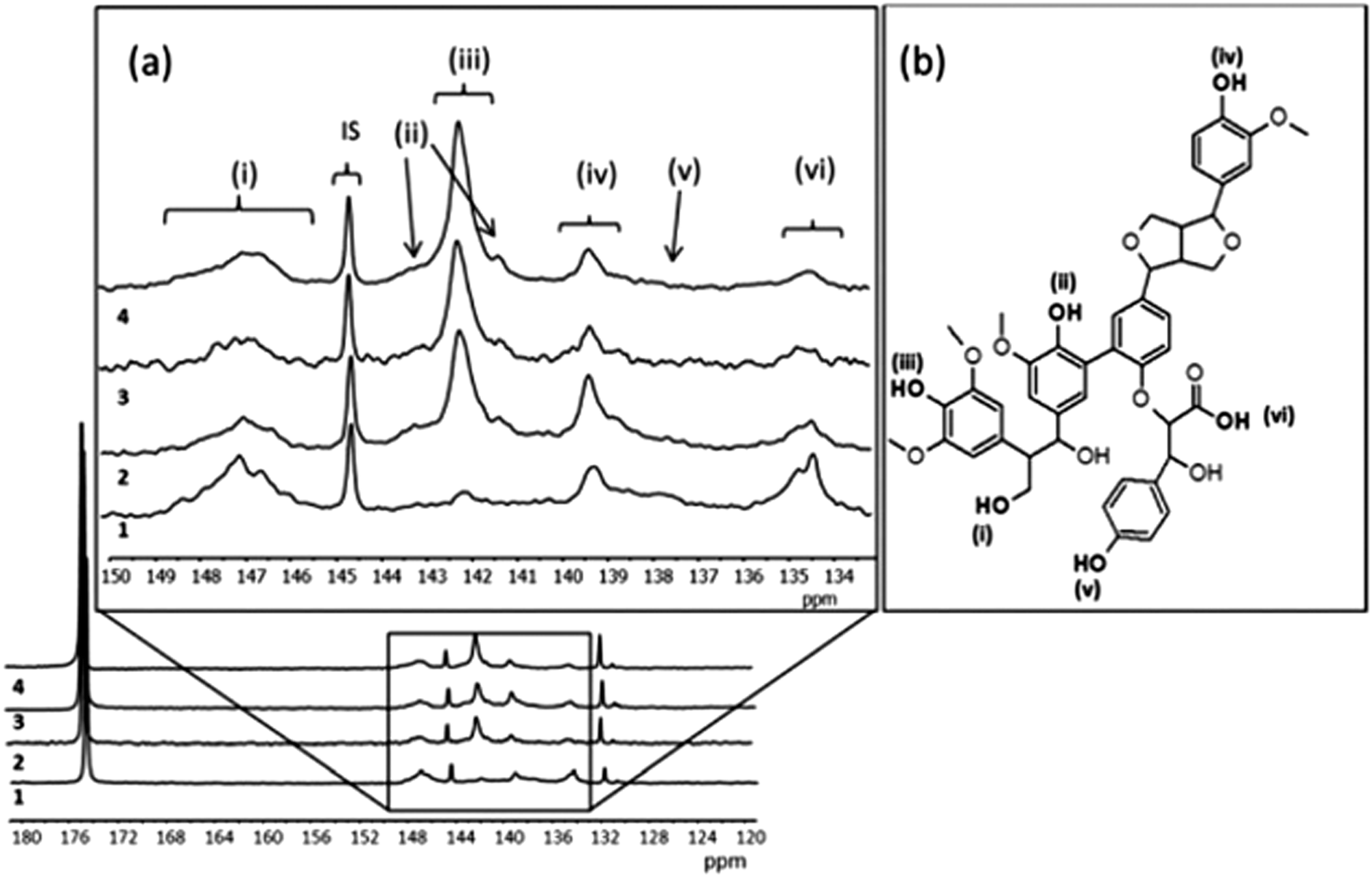 | ||
| Fig. 6 (a) Quantitative 31P NMR spectra and signal assignment of different lignins 1 (LG1), 2 (LG2), 3 (LG3), 4 (LG4). Internal standard 144.5 ppm, phosphitylating regent 174.7 ppm, water adduct 131.8 ppm. (b) The scheme of the Kraft lignin chemical structure. (i) Aliphatic hydroxyl, 145.4–150 ppm (ii) condensed phenolic hydroxyl, 141.0–141.5 and 142.5–144.0 ppm, (iii) syringyl 141.5–142.5 ppm, (iv) guaiacyl 138.5–140.2 ppm, (v) p-hydroxyphenyl at 138.0 ppm, and (vi) carboxylic acid hydroxyl 134.4–135.5 ppm. Reprinted with permission from ref. 55. Copyright 2014, Royal Society of Chemistry. | ||
The mass changes during electrochemical synthesis and operation have been followed using quartz crystal microbalances with dissipation monitoring, and used to determine the mass as well as changes in mechanical properties. The mass changes during the redox process can also be used to measure the mass of the chemical species exchanged between the electrode and electrolyte. We have shown that for a pPy/LS/polyoxometalate hybrid material,59 protons are the dominant ions exchanged during the redox process. Studying the volume change of this hybrid material by the use of a laser scanning micrometer showed that there is no significant volume change.59 Hence, the hybrid materials improved the cyclic stability by minimizing the volume expansion and compression of pPy upon redox cycling.
Hybrid material composition and nanostructure
The elemental analysis of the pPy/LS material reveals an almost 1:1 hybrid of pPy and LS, with a small contribution from the perchlorate ion. The presence of small perchlorate anions among larger LS anions during electropolymerisation leads to inclusion of both anions in the final material.
As both pPy and LS contain carbon, hydrogen, nitrogen, the contribution from sulphur is unique to LS. This result came from analysis of a single feed ratio, of pyrrole to LS in the electrochemical cell. In later experiments where the feed ratio was varied58 a minor variation of the charge capacity was obtained, but no evaluation of the contribution of the biopolymer to total mass was possible. It is therefore likely that the feed ratio may influence the hybrid material composition, and there might be options for optimization.
The nanostructure of the hybrid material has been imaged using electron microscopy methods (SEM, TEM and electron tomography). It is not possible to discern separate phases of pPy and LS in the hybrid material using these methods, though the nanostructure is that of a porous network. Presumably, the distance from the electronic polymer to LS is not large, as otherwise the transport limitations would become severe. SEM has also been used for characterizing other variants of biopolymer electrodes, and typically reports a microporous network.
Charge storage capacity
The capacitance of the biopolymer electrodes were evaluated in three-electrode systems using both cyclic voltammetry and galvanostatic charge–discharge methods. The overall integral capacitances were calculated from the cyclic voltammograms using the equation:where C is the specific capacitance (F g−1), ν is the scan rate (V s−1), Vf and Vi are the final and initial potentials (V), respectively, for the forward sweep, I is the current response and m (g) is the mass of the active materials determined from the EQCM. For the galvanostatic method, the specific capacitance (F g−1) was determined from the change in potential and discharge time using the equation:
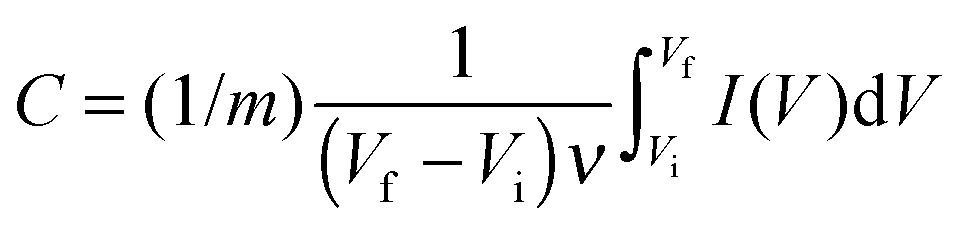
where I is the charge–discharge current, Δt is the time for discharge, m is the mass of the active material and ΔE is the potential change during discharge.
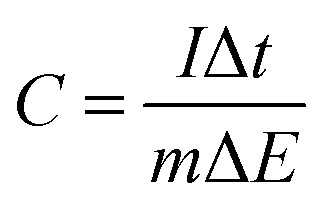
As there are two different redox species due to pPy and quinone(s), we can distinguish between capacitance in different potential ranges. The overall change in potential may also be interpreted as an averaged capacitance. Note that all these measurements have been taken for thin films of ∼0.1 μm. Thicker electrodes will reduce the numbers. The capacitance per mass obtained under these conditions is listed in Table 1.
| Sample code | Condensed unit (mmol g−1) 143.5–141.4 ppm | G unit (mmol g−1) 139.6–139.0 ppm | HQ unit (mmol g−1) 139.0–136.0 ppm | Redox functional groups (mmol g−1) | Peak molecular weighta (kDa) | Specific capacity of SLig/pPy film (pPy) |
|---|---|---|---|---|---|---|
| a Peak molecular weight from RI-SEC at 22.4–23.4 min. Reprinted with permission from ref. 67. Copyright 2015 pending to be accepted. | ||||||
| PPy | — | — | — | — | — | 28 |
| PolyS | 15.7 | — | — | 15.7 | 14.9–19.0 | 49 |
| PolyG | 6.2 | 11.7 | — | 17.9 | 14.6–18.6 | 62 |
| PolySG | 11.9 | 4.7 | — | 16.6 | 14.7–19.0 | 57 |
| PolySGHQ | 8.0 | 3.4 | 5.0 | 16.4 | 14.7–25.3 | 94 |
With the increasing discharge rate, the accessible capacity decreases. Also, the thicker the electrodes, the slower the kinetics observed, as evidenced from CVs. With thin electrodes, we observe a linear correlation between peak currents and the sweep rate; for thicker electrodes this is no longer the case, and we approach a linear dependence on the square root of the sweep rate, indicating a diffusion limited reaction. The nature of this diffusion limited species is open to discussion, and it may be electronic or ionic in nature.
From charge–discharge measurements it is possible to determine the charge flowing during oxidation and reduction of the electrode. Depending on the biopolymer electrode, the voltage range over which this charge–discharge experiment is performed may be different. For pPy/LS in 0.1 M HClO4, the voltage range was 0.53–0.1 V vs. Ag/AgCl. It was observed that extending the oxidation to higher potentials would cause a major and irreversible loss of capacity in the electrode, with a complete suppression of the quinone redox wave. The repeated cycling up to 1000 cycles between 0.7 and 0.0 V was observed to lead to loss of quinone electroactivity, This degradation reflects the same chemical changes as induced by overoxidation of pPy, a well known and documented phenomenon.60 There is a release of a degradation product from pPy into the electrolyte and this has been observed to be the same maleimide that was previously reported to be released during pPy degradation,61 or in parallel with chemical polymerisation.62 We thus conclude that loss of electronic conductivity occurs during this degradation along with loss of quinone groups, or loss of access to quinone groups.
The specific charge capacity of 70 mA h g−1 obtained for pPy/LS was ≈ twice that of a standard pPy electrode. This doubling was due to the quinone electrochemistry observed at potentials higher than the redox wave of the pPy chain. With the help of elemental analysis, we estimated the weight fraction of pPy in this material to be 50%. When considering the volume fraction, we note that percolation of the electronic conductor pPy within this composite material should make it possible to access the redox functions of quinones at lower pPy fractions. As previously discussed, we have attempted to synthesize the pPy/LS hybrid under conditions where the fraction of pPy could be smaller58 by varying the ratio between pyrrole and LS in the feed. However, we only obtained a small variation of the charge density of the composite material. Moreover, varying the solutions conditions (concentration, pH, buffer type) as well as electrosynthetic methods (potentiostatic, galvanostatic and pulse methods) did not generate major changes in charge density in the resulting materials. The difficulties when using elemental analysis to determine the material composition are considerable and precision is not high enough to allow conclusions as to variations in the stoichiometry of the resulting hybrid materials.
Adding more redox groups into pPy/LS
We have pursued inclusion of different quinone groups into these biopolymer electrodes. Prior work showed that various quinones can be incorporated inside pPy electrodes.63,64 In our case, this has been done by adding a small quinone molecule with a negative charge to the electrolyte containing pyrrole, LS and the supporting electrolyte before electropolymerization. The inclusion of the small quinone group is evident in the voltammograms of these materials.65 Different quinones can be chosen, and thus we can introduce quinones with different redox potentials side by side with the quinone derived from the LS. This has been done for anthraquinone sulfonate (AQS),65 hydroquinone (HQ), and for hydroquinone sulfonate (HQS) (Fig. 7). In all of these cases, we find evidence for inclusion of the small quinone, side by side with the quinone derived from LS. We note a recent paper on the combination of pPy with 1,4-benzoquinone, where both the quinone and pPy are redox active.66 The potential range for evaluating charge density must now be chosen to incorporate several quinones, and will be different for the different composite materials. This means that we can design an electrode to show redox activity in different potential ranges, and this way convert a voltammogram with two distinct redox processes to three and more, and onwards to an almost continuous range of redox processes all over the potential window. This may help when designing two electrode combinations of these materials, necessary for real wooden batteries, supercabatteries or supercapacitors.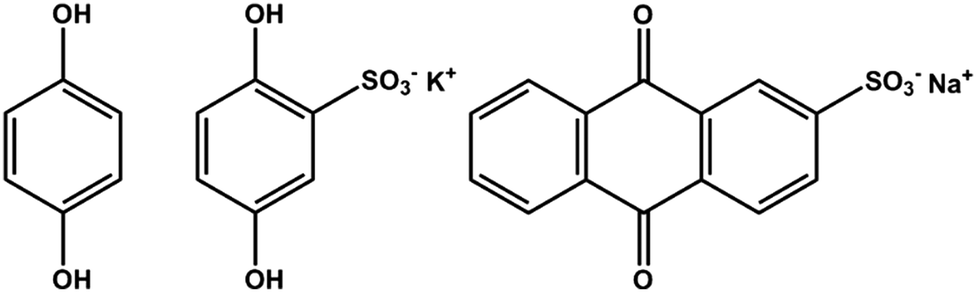 | ||
| Fig. 7 Chemical structure of hydroquinone (HQ), hydroquinone sulfonate potassium salt (HQS) and anthraquinone sulfonate sodium salt (AQS). | ||
These smaller quinones are however not trapped in the electrode and will diffuse out of the electrode during storage and operation. This is an added source of self-discharge as well as loss of capacity in these hybrid biopolymers. Crosslinking the small quinone species to something larger and immobile in the electrode appears to be a solution. We will return to this strategy in conjunction with the use of modified lignins (see Section 5.1.7).
It is observed that with the AQS incorporated, the charge density is considerably improved, to 186 mA h g−1 at 1 A g−1. Clearly, there is ample space for introducing quinone species into the biopolymer electrode. Thus, there is room to considerably improve the charge density, if we can form more quinone groups on the biopolymer (Fig. 8).
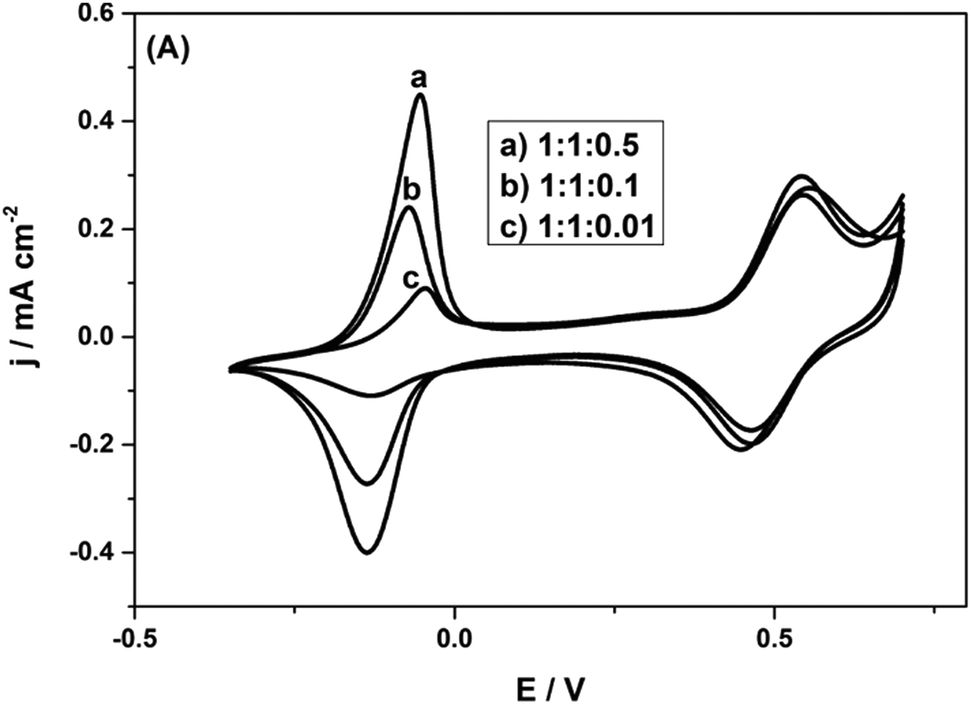 | ||
| Fig. 8 CVs of Py(Lig-AQS)(LC30) films synthesized from different concentrations of AQS with equal weight ratios of pyrrole and lignosulfonate in 0.1 M HClO4 at a scan rate of 5 mV s−1. Reprinted with permission from ref. 65. Copyright 2014, Wiley. | ||
Modifying the lignin polymer
The choices of biological sources and process chemistries for extracting the lignin derivatives are very broad. Even when limiting the choice to those process chemistries which are scaled to the industrial level, considerable variation of the outcome is expected. We have investigated lignin derivatives from many sources, from different plants and habitats, and with suppliers from commercial suppliers to industrial actors to research institutes. Ranking the lignin derivatives according to performance is easy, and some conclusions on the desired structures have been possible to obtain, for instance that the S monolignol appears to give a more redox accessible quinone. The implications from this is that adding more S monolignols by covalent attachment to a lignin scaffold may be appropriate; even more blunt is to put the quinone onto the lignin scaffold with covalent chemistry. We have had some moderate success in doing so, as documented in our recent report.56 The chemistry used for this coupling is, however, critical. Some degradation of quinone groups was also found during the covalent modification, so addition of some (potential) quinone groups also leads to subtraction of other (potential) quinones. This leads at best to a small improvement of the charge capacity of the hybrid electrode.Making lignin model polymers
The limits to performance of lignin as a precursor to the quinone redox group in hybrid electrode materials is a topic of high importance for further development. We have therefore made model polymers for lignins, exclusively using monolignols as the repeating monomer in the polymer. These synthetic lignin derivatives are formed using syringyl (S), guaiacyl (G) and hydroquinone (HQ) monomers and condensation reaction with formaldehyde.67 Homopolymers of the S, G and H kind, as well as copolymers were prepared and characterized by NMR and size exclusion chromatography (SEC). These synthetic lignins (SLig) were then integrated into polymer electrodes in the form of pPy/SLig and characterized.67 With a SLig incorporating S, G and HQ, the highest capacitance of 94 mA h g−1 was obtained in the hybrid electrodes. With the combination of carbon nanotubes (CNT) as the electronic conductor, mixed with SLig from solution, we obtained the highest performance of 72 mA h g−1 at 20% fraction of CNT. Thus percolation with the rodlike electronic conductor CNT does give access to a sizable fraction of quinone sites in SLig.Extending the electrochemistry in pPy/Lig electrodes
The interest in biopolymer electrodes lies in the low cost and scalability of the biopolymer derivative that is used. Given the electronic and ionic conductivity of these materials, and their open nanostructure, they may also be used to incorporate other redox species which are included at the molecular dispersion level. We have demonstrated this possibility with the poly(oxometalates) (POM) based on phosphomolybdenum incorporated within pPy/LS.59 The POM is included during electrochemical polymerization of pyrrole in a solution of POM and LS. Here the electrochemical processes seen in CV include three redox waves due to the transitions from Mo2+ to Mo6+, as located inside the polyoxometalate complex (Fig. 9).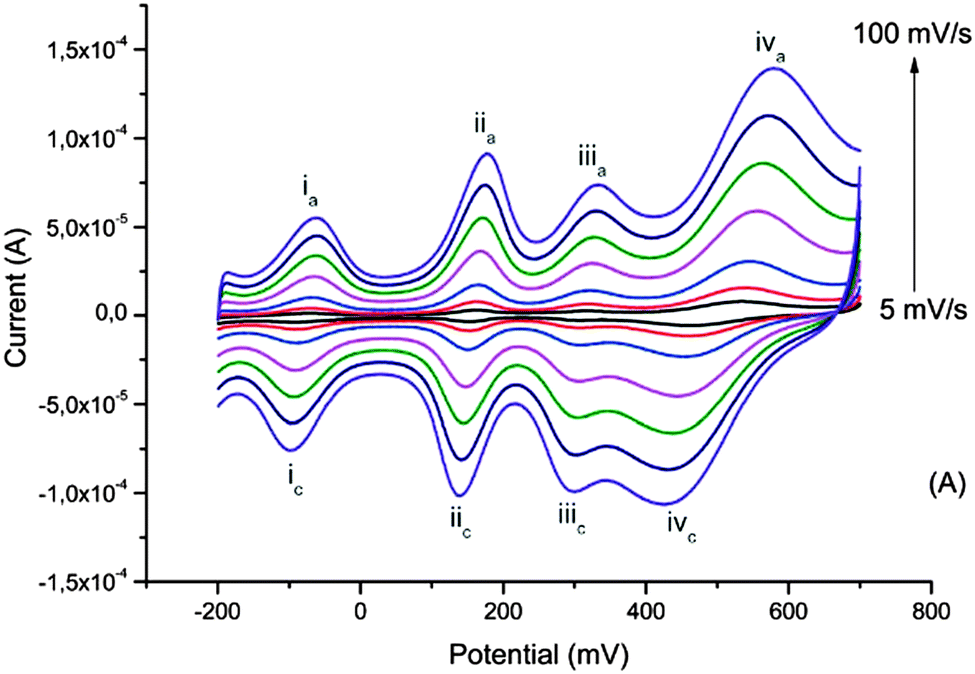 | ||
| Fig. 9 Cyclic voltammograms of the ternary composite system at different scan rates recorded in monomer- and dopant-free 0.1 M HNO3. Reprinted with permission from ref. 59. Copyright 2014, Royal Society of Chemistry. | ||
Improved charge density and capacitance are observed. In this material we have also been able to measure the masses of ions exchanged between the electrode and electrolyte, using QCM-D. The mass change is very small, and compatible with protons carried on water molecules as H3O+. The very small change in volume in this material is in agreement with this proton exchange reaction. This demonstrates that we can form a proton exchange electrode, forming a perfect electron to proton converter of relevance for bioelectronics.
The insertion of multivalent metal ions in caged structures is a pathway to extend both the electrochemical potential window as well as the charge density of the electrode material.
Problems of biopolymer electrodes
The main limitations of the biopolymer electrodes so far studied are stability/lifetime and self-discharge. Loss of activity in pPy/LS has been found to be very similar to the loss of activity of standard forms of pPy, and is associated with degradation of the pPy chain, leading to the release of low molecular weight maleimide, as first reported in the nineties.61,62 This can occur also during overoxidation, which may be part of the reason for the decrease in the redox activity of quinones which are found at the edge of the stability window for pPy. The decay of electroactivity with increasing number of cycles is associated with the formation of degraded forms of pPy. Thus, the loss of electroactivity is primarily a loss of the electronic contact to the quinone redox sites, which is also demonstrated as a diminishing quinone redox peak. This is in agreement with the release of low molecular weight species, probably maleimide, into the aqueous electrolyte. The degradation of electroactivity in the biopolymer electrode is thus inherent to the pPy system, and is dominated by this degradation.Self-discharge is the other facet of the instability of biopolymer electrodes. Polypyrrole is inherently prone to rapid self-discharge, as demonstrated in the first supercapacitor systems18 of the late 1980s and in solid state batteries with pPy electrodes.19 The self-discharge has been characterized in greater detail in subsequent work,34,38,68 and is a consequence of the degradation of the pPy chain in the oxidized state.
Given the problems of self-discharge and of loss of electroactivity upon repeated cycling, alternative polymers substituting for pPy are of interest. We are currently evaluating several polymers and finding examples where self-discharge and cycling stability are no longer limiting problems. These are materials currently under development and also promise to deliver enhanced charge capacity and capacitance. Note however that the pPy/POM/LS system was exposed to more than 2000 cycles without major loss of electroactivity.
Another problem with these hybrid electrodes is the charge and power density. While our first generation of materials have 50% of the electroactive polymer, this is not a desirable property, and it would be preferable to reduce this to much lower numbers. This is because the charge density in pPy is low, and often lower than what can be obtained from the biopolymer. It is also because of cost and scalability, as the pyrrole monomer is much more expensive and rare than the lignin derivatives. It should be feasible to generate materials with much lower fractions of pPy. Indeed, that is what we attempted to do by varying the feed ratio in the synthesis. There is not observed evidence of better charge density when the feed ratio of pyrrole/LS is obtained. A more substantial demonstration of this potential is seen with the model lignins reported. Here we have combined the synthetic lignins (SLig) with pPy, and varying the feed ratio. We have likewise prepared hybrids with the model lignins and carbon nanotubes. In this latter case, the stoichiometry of the hybrid is determined before hand. The optimal charge density was obtained with 20% of carbon nanotubes (CNTs).67 This is possible to state, because the amount of materials mixed is possible to know. That is more difficult for materials prepared by electrochemically induced synthesis, where the feed ratio does not necessary get replicated in the mass fraction of the hybrid materials. We note that with long slender CNTs the transport percolation is sufficient at much lower fraction; the maximum access to quinones found at 20% is presumably related to the access to the quinone moiety from the CNTs.
Alternative uses of hybrid biopolymer electrodes: ion extraction from water with wooden electrodes
With charge storage of renewable electricity as the most important application for wooden batteries, creation of off grid electricity systems may become feasible. These might be combined with other functions, often desired where these systems are most relevant. The water quality is deteriorating in many nations, due to pollution of water from many sources as well as due to expansion of water supply using lower water quality sources. We have demonstrated69 how biopolymer electrodes can be adapted to work in neutral aqueous media, and to extract metal ions from water in a reduction step, which are then released in the oxidation step. The mass of lead ions adsorbed and released was measured using QCM-D methods, and we could inject 70 mg lead per g of electrode material. With the appropriate design of flow batteries, this might lead to a system where ions are extracted from polluted water and released into concentrated solutions, in parallel with the charging and discharging of the wooden battery. Our study focused on lead ions (Pb2+), one of the most serious pollutants in water, but we expect that removal of other ions such as As2+, a frequent problem in ground waters in India and Bangladesh, would be equally possible.Conclusions
The use of renewable materials for charge storage is expanding, and we have not here covered all the topics, but remain limited to the combination of electronic polymers and lignin derivatives. If the limitations of renewable materials are overcome, we can achieve the development of electronic polymers with a covalently bonded quinone group, which comes very close to the situation which we try to obtain using green chemistry approaches. The derivatisation of electronic polymers with pendant redox active groups can be seen as the development and integration of the separate classes of redox polymers70 and conjugated polymers. That combination was reported some decades ago,71,72 but has once more been attempted.73–76 We also note the continued interest in biopolymer electrodes,77,78 sometimes using alternative materials for electronic communication.79 The combination of graphene with lignin80 gave a very high capacitance of 432 F g−1, far above that of graphene itself. Indeed, even without a current collector, lignin and lignin derivatives have been combined into primary batteries of very low power and current.81,82 The small aromatic biomolecule juglone(5-hydroxy-1,4-naphthalenedione), a substance produced by plants to combat other plants, has been used in sodium batteries.78 On a more technological side, thermal conversion of lignins into nanostructured carbons has been demonstrated to give high capacitance electrodes;83 note that here the quinone is thermally converted to a graphitic structure. Recent reviews have summarized the development of organic based electrodes for energy storage.15,84We have performed our studies of the hybrid pPy/lignin materials in three electrode systems, and have not focused on the evaluation of two electrode systems where one or both electrodes are based on hybrid biopolymer materials. This strategy avoided the expense of futile attempts to stabilize the supercabatteries, which indeed as we have seen in our three electrode studies, would not be sufficiently stable. Our present choices of polymers include PEDOT, a polymer of considerably better stability, but with the downside of lower charge capacity. The outcome of these materials in forming more stable supercabatteries will determine the verdict when attempting to use lignins in wooden supercabatteries. As their use in stationary storage is the goal, it may be of importance to note that the low cost of lignin will be a major asset for this most important amount of the cost of electrical storage.
Acknowledgements
We thank our colleagues working with biopolymer electrodes for all contributions and results over the last years, including Grzegorz Milczarek, Tomasz Rebis, Ting Yang Nilsson, and Doddahalli H. Nagaraju. This work has been supported by the Knut and Alice Wallenberg Foundation through projects Power Papers and a Wallenberg Scholar grant to Olle Inganäs.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- O. Inganas and S. Admassie, Adv. Mater., 2014, 26, 830–847 CrossRef PubMed.
- C. Heitner, D. R. Dimmel and J. A. Schmidt, Lignin and lignans: advances in chemistry, CRC Press, Boca Raton, FL, 2010 Search PubMed.
- S. Van den Bosch, W. S. Schutyser, R. Vanholme, T. R. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- G. Milczarek, Electroanalysis, 2007, 19, 1411–1414 CrossRef CAS.
- G. Milczarek, Electroanalysis, 2008, 20, 211–214 CrossRef CAS.
- G. Milczarek, Langmuir, 2009, 25, 10345–10353 CrossRef CAS PubMed.
- G. Milczarek, Electrochim. Acta, 2009, 54, 3199–3205 CrossRef CAS.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J. M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribiere, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- H. Chen, M. Armand, M. Courty, M. Jiang, C. P. Grey, F. Dolhem, J. M. Tarascon and P. Poizot, J. Am. Chem. Soc., 2009, 131, 8984–8988 CrossRef CAS PubMed.
- W. Walker, S. Grugeon, H. Vezin, S. Laruelle, M. Armand, J. M. Tarascon and F. Wudl, Electrochem. Commun., 2010, 12, 1348–1351 CrossRef CAS.
- P. Novak, K. Muller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–281 CrossRef CAS PubMed.
- K. Naoi, K. Kawase, M. Mori and M. Komiyama, J. Electrochem. Soc., 1997, 144, L173–L175 CrossRef CAS.
- B. Haeupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 Search PubMed.
- L. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L.-F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- G. Nystrom, A. Razaq, M. Stromme, L. Nyholm and A. Mihranyan, Nano Lett., 2009, 9, 3635–3639 CrossRef PubMed.
- A. Mohammadi, O. Inganas and I. Lundstrom, J. Electrochem. Soc., 1986, 133, 947–949 CrossRef CAS.
- P. Novak and O. Inganas, J. Electrochem. Soc., 1988, 135, 2485–2490 CrossRef CAS.
- P. Novak, O. Inganas and R. Bjorklund, J. Electrochem. Soc., 1987, 134, 1341–1345 CrossRef CAS.
- P. Novak, O. Inganas and R. Bjorklund, J. Power Sources, 1987, 21, 17–24 CrossRef CAS.
- P. Passiniemi and O. Inganas, Solid State Ionics, 1989, 34, 225–230 CrossRef CAS.
- C. Arbizzani, M. Mastragostino and L. Meneghello, Electrochim. Acta, 1996, 41, 21–26 CrossRef CAS.
- J. P. Ferraris, M. M. Eissa, I. D. Brotherston and D. C. Loveday, Chem. Mater., 1998, 10, 3528–3535 CrossRef CAS.
- Y. Gofer, J. G. Killian, H. Sarker, T. O. Poehler and P. C. Searson, J. Electroanal. Chem., 1998, 443, 103–115 CrossRef CAS.
- Y. Gofer, H. Sarker, J. G. Killian, T. O. Poehler and P. C. Searson, Appl. Phys. Lett., 1997, 71, 1582–1584 CrossRef CAS.
- D. Haringer, P. Novak, O. Haas, B. Piro and M. C. Pham, J. Electrochem. Soc., 1999, 146, 2393–2396 CrossRef CAS.
- K. Naoi, S. Suematsu and A. Manago, J. Electrochem. Soc., 2000, 147, 420–426 CrossRef CAS.
- A. Rudge, I. Raistrick, S. Gottesfeld and J. P. Ferraris, Electrochim. Acta, 1994, 39, 273–287 CrossRef CAS.
- A. Mihranyan, L. Nyholm, A. E. G. Bennett and M. Stromme, J. Phys. Chem. B, 2008, 112, 12249–12255 CrossRef CAS PubMed.
- L. Nyholm, G. Nystrom, A. Mihranyan and M. Stromme, Adv. Mater., 2011, 23, 3751–3769 CAS.
- G. Nystrom, A. Mihranyan, A. Razaq, T. Lindstrom, L. Nyholm and M. Stromme, J. Phys. Chem. B, 2010, 114, 4178–4182 CrossRef PubMed.
- G. Nystrom, M. Stromme, M. Sjodin and L. Nyholm, Electrochim. Acta, 2012, 70, 91–97 CrossRef.
- H. Olsson, E. J. Berg, M. Stromme and M. Sjodin, Electrochem. Commun., 2015, 50, 43–46 CrossRef CAS.
- H. Olsson, D. O. Carlsson, G. Nystrom, M. Sjodin, L. Nyholm and M. Stromme, J. Mater. Sci., 2012, 47, 5317–5325 CrossRef CAS.
- H. Olsson, G. Nystrom, M. Stromme, M. Sjodin and L. Nyholm, Electrochem. Commun., 2011, 13, 869–871 CrossRef CAS.
- H. Olsson, Z. Qiu, M. Stromme and M. Sjodin, Phys. Chem. Chem. Phys., 2015, 17, 11014–11019 RSC.
- H. Olsson, M. Stromme, L. Nyholm and M. Sjodin, J. Phys. Chem. C, 2014, 118, 29643–29649 CAS.
- A. Razaq, L. Nyholm, M. Sjodin, M. Stromme and A. Mihranyan, Adv. Energy Mater., 2012, 2, 445–454 CrossRef CAS.
- G. Nystrom, A. Marais, E. Karabulut, L. Wagberg, Y. Cui and M. M. Hamedi, Nat. Commun., 2015, 6, 7259–7267 CrossRef PubMed.
- S. Ghosh and O. Inganas, Adv. Mater., 1999, 11, 1214–1218 CrossRef CAS.
- S. Ghosh and O. Inganas, Synth. Met., 1999, 101, 413–416 CrossRef CAS.
- S. Ghosh and O. Inganas, J. Electrochem. Soc., 2000, 147, 1872–1877 CrossRef CAS.
- S. Ghosh and O. Inganas, Electrochem. Solid-State Lett., 2000, 3, 213–215 CrossRef CAS.
- S. Ghosh and O. Inganas, Synth. Met., 2001, 121, 1321–1322 CrossRef CAS.
- S. Ghosh, J. Rasmusson and O. Inganas, Adv. Mater., 1998, 10, 1097–1099 CrossRef CAS.
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C. K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287–9292 CrossRef CAS PubMed.
- Y. Zhao, B. Liu, L. Pan and G. Yu, Energy Environ. Sci., 2013, 6, 2856–2870 CAS.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- B. Yang, L. Hoober-Burkhardt, F. Wang, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2014, 161, A1371–A1380 CrossRef CAS.
- R. C. M. Jakobs, L. J. J. Janssen and E. Barendrecht, Electrochim. Acta, 1985, 30, 1313–1321 CrossRef CAS.
- A. Haimerl and A. Merz, J. Electroanal. Chem., 1987, 220, 55–65 CrossRef CAS.
- M. Vijayan and V. Krishnan, Electroanalysis, 1995, 7, 197–198 CrossRef CAS.
- G. Milczarek and O. Inganas, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- S. Admassie, T. Y. Nilsson and O. Inganas, Phys. Chem. Chem. Phys., 2014, 16, 24681–24684 RSC.
- T. Y. Nilsson, M. Wagner and O. Inganas, ChemSusChem, 2015, 8, 4081–4085 CrossRef CAS PubMed.
- S. Leguizamon, K. P. Diaz-Orellana, J. Velez, M. C. Thies and M. E. Roberts, J. Mater. Chem. A, 2015, 3, 11330–11339 CAS.
- F. N. Ajjan, M. J. Jafari, T. Rebis, T. Ederth and O. Inganas, J. Mater. Chem. A, 2015, 3, 12927–12937 CAS.
- S. Admassie, A. Elfwing, E. W. H. Jager, Q. Y. Bao and O. Inganas, J. Mater. Chem. A, 2014, 2, 1974–1979 CAS.
- S. Ghosh, G. A. Bowmaker, R. P. Cooney and J. M. Seakins, Synth. Met., 1998, 95, 63–67 CrossRef CAS.
- D. S. Park, Y. B. Shim and S. M. Park, J. Electrochem. Soc., 1993, 140, 609–614 CrossRef CAS.
- J. C. Thieblemont, J. L. Gabelle and M. F. Planche, Synth. Met., 1994, 66, 243–247 CrossRef CAS.
- B. Zinger, Synth. Met., 1989, 30, 209–225 CrossRef CAS.
- H. Yoneyama, Y. Ii and S. Kuwabata, J. Electrochem. Soc., 1992, 139, 28–32 CrossRef CAS.
- D. H. Nagaraju, T. Rebisz, R. Gabrielsson, A. Elfwing, G. Milczarek and O. Inganas, Adv. Energy Mater., 2014, 4, 1300443 Search PubMed.
- M. R. Arcila-Velez and M. E. Roberts, Chem. Mater., 2014, 26, 1601–1607 CrossRef CAS.
- T. Rebisz, T. Y. Nilsson and O. Inganas, J. Mater. Chem. A, 2016, 4, 1931–1940 Search PubMed.
- F. Beck and R. Michaelis, Werkst. Korros. Mater. Corros., 1991, 42, 341–347 CAS.
- S. Admassie, A. Elfwing, A. Skallberg and O. Inganas, Environ. Sci.: Water Res. Technol., 2015, 1, 326–331 CAS.
- K. Shigehara, N. Oyama and F. C. Anson, J. Am. Chem. Soc., 1981, 103, 2552–2558 CrossRef CAS.
- A. B. Kon, J. S. Foos and T. L. Rose, Chem. Mater., 1992, 4, 416–424 CrossRef CAS.
- M. V. Rosenthal, T. Skotheim and J. Warren, J. Chem. Soc., Chem. Commun., 1985, 342–343 RSC.
- C. Karlsson, H. Huang, M. Stromme, A. Gogoll and M. Sjodin, J. Phys. Chem. C, 2013, 117, 23558–23567 CAS.
- C. Karlsson, H. Huang, M. Stromme, A. Gogoll and M. Sjodin, J. Electroanal. Chem., 2014, 735, 95–98 CrossRef CAS.
- C. Karlsson, H. Huang, M. Stromme, A. Gogoll and M. Sjodin, J. Phys. Chem. C, 2014, 118, 23499–23508 CAS.
- C. Karlsson, H. Huang, M. Stromme, A. Gogoll and M. Sjodin, RSC Adv., 2015, 5, 11309–11316 RSC.
- J. Hong, M. Lee, B. Lee, D. H. Seo, C. B. Park and K. Kang, Nat. Commun., 2014, 5, 5335–5344 CrossRef CAS PubMed.
- H. Wang, P. Hu, J. Yang, G. Gong, L. Guo and X. Chen, Adv. Mater., 2015, 27, 2348–2354 CrossRef CAS PubMed.
- G. Milczarek and M. Nowicki, Mater. Res. Bull., 2013, 48, 4032–4038 CrossRef CAS.
- S. K. Kim, Y. K. Kim, H. Lee, S. B. Lee and H. S. Park, ChemSusChem, 2014, 7, 1094–1101 CrossRef CAS PubMed.
- S. V. Gnedenkov, D. P. Opra, S. L. Sinebryukhov, A. K. Tsvetnikov, A. Y. Ustinov and V. I. Sergienko, J. Ind. Eng. Chem., 2014, 20, 903–910 CrossRef CAS.
- S. V. Gnedenkov, D. P. Opra, L. A. Zemnukhova, S. L. Sinebryukhov, I. A. Kedrinskii, O. V. Patrusheva and V. I. Sergienko, J. Energy Chem., 2015, 24, 346–352 CrossRef.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Highly Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS.
- L. Zhang, Z. Liu, G. Cui and L. Chen, Prog. Polym. Sci., 2015, 43, 136–164 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |