
Copper binding to naturally occurring, lactam form of angiogenin differs from that to recombinant protein, affecting their activity†
D.
La Mendola
*a,
F.
Arnesano
*b,
Ö.
Hansson
c,
C.
Giacomelli
a,
V.
Calò
b,
V.
Mangini
b,
A.
Magrì
d,
F.
Bellia
d,
M. L.
Trincavelli
a,
C.
Martini
a,
G.
Natile
b and
E.
Rizzarelli
d
aDepartment of Pharmacy, University of Pisa, via Bonanno Pisano 6, 56126, Pisa, Italy. E-mail: lamendola@farm.unipi.it
bDepartment of Chemistry, University of Bari “A. Moro”, via E. Orabona 4, 70125 Bari, Italy. E-mail: Fabio.arnesano@uniba.it
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, Medicinaregatan 9C, PO Box 462, SE-40530 Göteborg, Sweden
dInstitute of Biostructure and Bioimaging, CNR, via P. Gaifami 18, 95126 Catania, Italy
First published on 12th November 2015
Abstract
Angiogenin is a member of the ribonuclease family and a normal constituent of human plasma. It is one of the most potent angiogenic factors known and is overexpressed in different types of cancers. Copper is also an essential cofactor in angiogenesis and, during this process, it is mobilized from inside to outside of the cell. To date, contrasting results have been reported about copper(II) influencing angiogenin activity. However, in these studies, the recombinant form of the protein was used. Unlike recombinant angiogenin, that contains an extra methionine with a free terminal amino group, the naturally occurring protein present in human plasma starts with a glutamine residue that spontaneously cyclizes to pyroglutamate, a lactam derivative. Herein, we report spectroscopic evidence indicating that copper(II) experiences different coordination environments in the two protein isoforms, and affects their RNase and angiogenic activity differently. These results show how relatively small differences between recombinant and wild type proteins can result in markedly different behaviours.
Introduction
Human Angiogenin (Ang) is a member of the ribonuclease protein family though its RNase catalytic activity is unusually low.1 Ang is a normal constituent of human plasma (concentration of 250–360 μg L−1) and is over-expressed in patients affected by different types of cancers.2 It is a potent stimulator of angiogenesis and its presence is also required for cell proliferation induced by other angiogenic agents such as the vascular endothelial growth factor (VEGF).3 However, the precise molecular mechanism by which Ang affects angiogenic processes is not yet clear. The RNase activity of this protein is essential for angiogenesis stimulation and the catalytic site encompasses specifically His-13, Lys-40, and His-114 residues.4The biological role of Ang is not limited to induction of angiogenesis, as suggested by its widespread expression in all human organs and tissues,5,6 but recent findings demonstrate that Ang is down-regulated both in the mouse model of Parkinson's7 and patients affected by Alzheimer's diseases.8 Moreover, Ang has emerged as one of the key agents in amyotrophic lateral sclerosis (ALS) where it acts as a motoneuron protective factor.9,10
Also copper is known to play a key role in neurodegenerative diseases and has been shown to be an essential angiogenesis cofactor in vivo.11 Serum copper levels are raised in a wide variety of human cancers and correlate with tumor malignancy.12 So far, the targets of Ang activity and the specific role of the metal remain unclear. It is known that during angiogenesis, there is an extracellular translocation of copper,13 thus metal binding to extracellular proteins involved in angiogenesis, such as Ang, is a possible pathway through which copper takes part in the signaling process. Different relationships between Ang and copper have been reported. In one case Cu2+ binding to the protein has been found to increase its interaction with endothelial cells;14,15 the binding details of this interaction have been addressed in a study concerning the formation of copper(II) complexes with a linear peptide encompassing the putative cellular binding site (residues 60–68) of angiogenin.16 In order to probe the copper(II) binding features of the recombinant protein, some peptide fragments of its N-terminal region have been reacted with copper(II) and the results showed similarities as well as differences.17 It has also been suggested that copper and Ang are both angiogenic factors, but through different and independent biological pathways.18 It should be noted, however, that most data reported so far have been obtained using the recombinant form of Ang (r-Ang), containing an extra methionine as the first residue.14,15 In contrast, the wild-type protein (wt-Ang) starts with a glutamine residue which spontaneously cyclizes to pyroglutamate, the γ-lactam form, so that wt-Ang normally present in human plasma has no free amino terminal group.19 Recently, some of us have shown that Cu2+ increases the expression of wt-Ang and modulates its intracellular localization in HUVEC.20 In the present work, Cu2+ binding to wt-Ang has been investigated by means of several techniques, including UV-vis, CD, ESI-MS, EPR, and NMR, and compared to that of r-Ang. The wild-type protein was obtained through the specific enzymatic cleavage of the first Met residue present in the recombinant form.21 Moreover, copper perturbation of the biological activities of the two proteins, such as RNase activity and angiogenesis induction, has been determined.
Materials and methods
Human umbilical vein endothelial cells (HUVECs) and cell culture medium (EGM®-2 Bullet Kit®) were purchased from Lonza srl (Milan, Italy). All other reagents were obtained from standard commercial sources and were of the highest commercially available grade.Protein expression
A codon-optimized gene for Ang inserted into the pET22b expression vector was purchased from Sloning BioTechnology and successfully transformed into the E. coli expression host BL21. For 13C,15N-enriched Ang the bacteria were grown on minimal medium M9 with 13C-glucose and 15NH4Cl. The purification of recombinant Ang was performed using an automated chromatographic workstation (Akta prime, GE Healthcare). Cation-exchange purification was carried out by using a 15 × 1.6 cm column packed with SP Sepharose Fast Flow (GE Healthcare). After a washing step with 25 mM Tris-HCl and 0.2 M NaCl (pH 8.0), the recombinant protein was eluted with 25 mM Tris-HCl and 0.8 M NaCl (pH 8.0). The purity of the protein was evaluated by means of SDS-PAGE (Criterion XT 10% bis-Tris) and the ribonucleolytic activity was determined as described in the literature.22 The concentration of Ang was determined using ε280 = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1.
500 M−1 cm−1.
The wt-Ang was obtained incubating the recombinant protein (7–10 μM) with 1 nM Aeromonas aminopeptidase in 200 mM potassium phosphate buffer, pH 7.2, for 24 h at 37 °C under gentle shaking. The buffer was replaced with Tris-HCl 25 mM, EDTA 1 mM, and NaCl 0.1 M (pH 7.4) by dialysis (Spectra/por MWCO 6–8000) and the reaction mixture subjected to a cation-exchange purification step. The cyclization of the N-terminal glutamine residue was assessed measuring the molecular weight by means of Electrospray Mass spectrometry. The ribonucleolytic activity was determined as described in the literature to confirm the correct folding of the protein.22
CD measurements
CD spectra were obtained at 25 °C under a constant flow of nitrogen on a Jasco model 810 spectropolarimeter at a scan rate of 50 nm min−1 and a resolution of 0.1 nm. The path lengths were 0.1 (190–260 nm range) and 1 cm (290–750 nm range). The spectra were recorded as an average of 10 or 20 scans. Calibration of the instrument was performed using a 0.06% solution of ammonium camphorsulfonate in water. All the solutions were freshly prepared using double-distilled water.EPR measurements
A Bruker Elexsys E500 CW-EPR (CW = continuous wave) spectrometer driven by a PC running XEpr program under Linux and equipped with a Super-X microwave bridge, operating at 9.3–9.5 GHz, and a SHQE cavity was used. EPR spectra of frozen solutions of copper(II) complexes were recorded at 150 K by means of a ER4131VT variable temperature apparatus. EPR magnetic parameters were obtained directly from the experimental EPR spectra, calculating them from the 2nd and the 3rd line to get rid of second order effects. Instrumental settings of EPR spectra recordings were as follows: number of scans 5; microwave frequency 9.344–9.376 GHz; modulation frequency 100 kHz; modulation amplitude 0.2–0.6 mT; time constant 164–327 ms; sweep time 2.8 min; microwave power 20–40 mW; receiver gain 1 × 104–2 × 105. Ang protein concentration was 0.5 mM in 25 mM MOPS, 150 mM NaCl (pH 7.4) and in the presence of 1 mol equivalent of Cu2+. Copper(II) complexes were prepared by the addition of an appropriate amount of isotopically pure copper(II), taken from a 63Cu(NO3)2 0.05 M solution, to the protein solution.UV-visible measurements
UV-visible (UV-vis) spectra were recorded at 25 °C by using an Agilent 8453 or a Varian Cary 500 spectrophotometer. Measurements were performed using a quartz cuvette with a 1 cm path length. Spectra were collected at [Ang] = [Cu2+] = 0.5 mM, and 10 mM MOPS (pH 7.4).Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance (NMR) experiments were performed at 25 °C on 13C,15N-enriched wt- and r-Ang samples (1.0 mM concentration) in buffered (50 mM phosphate, pH 7.4) saline (100 mM NaCl) water solution (90% H2O and 10% D2O).Resonance assignment of the apoprotein was carried out with the aid of 2D TOCSY and NOESY along with 3D CBCANH and CBCA(CO)NH experiments, using the available 1H and 15N chemical shift data.23 The titration of the 1.0 mM samples of wt- and r-Ang with CuCl2 (0.1, 0.25, 0.5, 0.75, and 1.0 mol equivalents) at two different pH values (7.4 and 5.5, obtained by addition of HCl to the starting buffer solution) was followed by 1H,15N and 1H,13C heteronuclear single quantum coherence (HSQC) experiments. All spectra were collected on a Bruker Avance 600 with an Ultra Shield Plus magnet using a triple resonance probe equipped with z axis self-shielded gradient coils, and processed using the standard Bruker software (TOPSPIN) and analyzed using the program CARA (The Computer Aided Resonance Assignment Tutorial, R. Keller, 2004, CANTINA Verlag), developed at ETH-Zürich. Cross-peaks affected during Cu2+ titration were identified by comparing their intensities (I) with those of the same cross-peaks (I0) in the dataset of samples lacking Cu2+. The I/I0 ratios as a function of metal-to-protein ratio were fitted to a single-exponential decay function and the obtained decay constants k were plotted as a function of protein sequence.
Ribonucleolytic activity
The ribonucleolytic activity toward tRNA was determined by measuring the formation of perchloric acid-soluble fragments. Briefly, wt- or r-Ang (0.5 μM) and tRNA (2 mg mL−1) were incubated at pH 7.4 (33 mM MOPS, 33 mM NaCl) in the presence or absence of CuSO4 (0–15 μM). After 2 h at 37 °C, the solution (300 μL) was diluted with ice-cold 3.4% HClO4 (700 μL) and kept on ice for 10 min. Finally, the sample was centrifuged (10000 × g) for 10 min and the absorbance of perchloric acid soluble fragments was measured at 260 nm.
Tube formation assay
Matrigel (without copper(II) ions) (BD Biosciences) was thawed at 4 °C overnight and spread evenly over each well (50 μL) of a 96-well plate. The plates were incubated for 30 min at 37 °C to allow the Matrigel to gel before the cell seeding. HUVEC cells (1 × 104) from passage 3 or earlier were seeded in 100 μL of EGM-2 per well with r-Ang (100 nM) and wt-Ang (100 nM) alone or in the presence of different concentrations of CuSO4 (100–500 nM). The Endothelial Basal Medium (EBM-2) contains 5 nM CuSO4, while the final concentration of FBS in the supplemented medium will only be 2% and the non-bound copper will be negligible. When Ang was used in combination with copper, they were mixed 2 hours prior to the cell treatment. After 16 h of incubation at 37 °C, the tube structures were observed with an inverted microscope equipped with a digital camera (Nikon, Sesto Fiorentino, Italy). Two fields (magnification 4×) were captured for each sample, performed in triplicate. For each image, the total length of the tube network, the number of intact loops, and the number of branching points were quantified with the image analysis software ImageJ using the plug-in AngioJ for the angiogenesis assay.Results and discussion
Metal loading is different for r-Ang and wt-Ang, without affecting significantly their secondary structures
The far-UV CD spectra of wild-type and recombinant proteins, carried out at pH 7.4, are very similar to each other suggesting that the two proteins have the same secondary structure (Fig. S1, ESI†) which is rich in β-strands.23,24 The addition of Cu2+ ions induces a slight decrease of the CD signal in both isoforms (Fig. 1).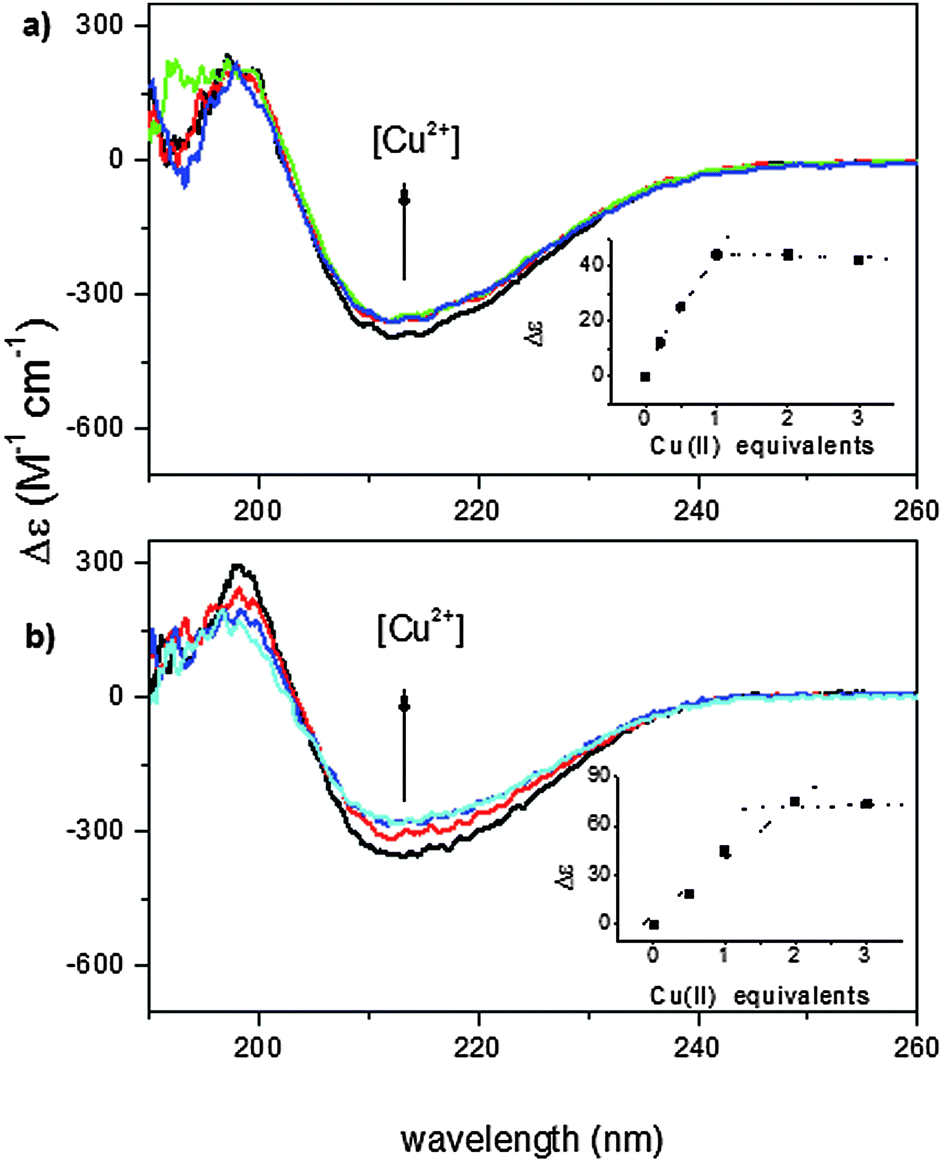 | ||
| Fig. 1 Far-UV CD spectra at pH 7.4 (MOPS buffer) for titration with Cu2+ of (a) wt-Ang (the black curve is the free protein spectrum; red, green and blue are spectra of protein after the addition of 1, 2 and 3 Cu2+ equivalents, respectively); and (b) r-Ang (the black curve is the free protein spectrum; red, cyan and blue are protein spectra after the addition of 1, 2 and 3 Cu2+ equivalents, respectively). Inset: Spectral changes as a function of copper addition monitored at λ = 212 nm. [Ang] = 50 μM. | ||
The small effect on the secondary structure suggests that Cu2+ binding may involve little structural rearrangement in the protein. The Cu2+ titration curve of wt-Ang, shown in the inset of Fig. 1a, indicates that the intensity of the CD band decreases with Cu2+ addition up to one mole equivalent; no further change occurs up to the addition of 2–3 mol equivalents of Cu2+. These data are in accord with a 1:1 metal to ligand stoichiometry. Unlike wt-Ang, the analogous binding curve for r-Ang (inset of Fig. 1b) shows a decrease of the CD band intensity up to the addition of two mol equivalents of Cu2+, suggesting that r-Ang can bind up to two Cu2+ ions.
Electrospray Ionization Mass Spectrometry (ESI-MS) measurements, carried out at physiological pH, confirm the stoichiometry of the metal–protein complexes deduced above. After the addition of only one equivalent of metal ion, both wt-Ang and r-Ang show the signals corresponding to a 1:1 Cu2+–protein adduct. By increasing the amount of copper ion up to ten equivalents, no significant changes are observed for wt-Ang whereas a species with two copper ions per protein molecule is detected for r-Ang (Fig. S2 and S3, ESI†).
The main metal binding site is different for wt-Ang and r-Ang
Previous data on the addition of one equivalent of Cu2+ to the recombinant protein showed the formation of a Cu2+ complex with a strong ligand field (band at 560 nm in the UV-vis) conceivably involving four nitrogen donors in a planar arrangement.15 In addition to imidazole and deprotonated amide nitrogens (as indicated by the far-UV CD band profiles), also the terminal amino nitrogen is likely to be involved in metal coordination.15 Unlike r-Ang, addition of one equivalent of Cu2+ to wt-Ang at pH 7.4 induces the formation of a band at a higher wavelength (610 nm) which is indicative of a lower ligand field around the metal core (Table 1).The EPR spectrum of wt-Ang in the presence of Cu2+ (1:1 molar ratio, pH 7.4) is typical of type 2 Cu2+–protein complexes (axial g matrix, Cu hyperfine coupling constant >130 × 10−4 cm−1). The calculated EPR parameters are: g∥ = 2.252 and A∥ = 185 × 10−4 cm−1 (Table 1 and Fig. 2).
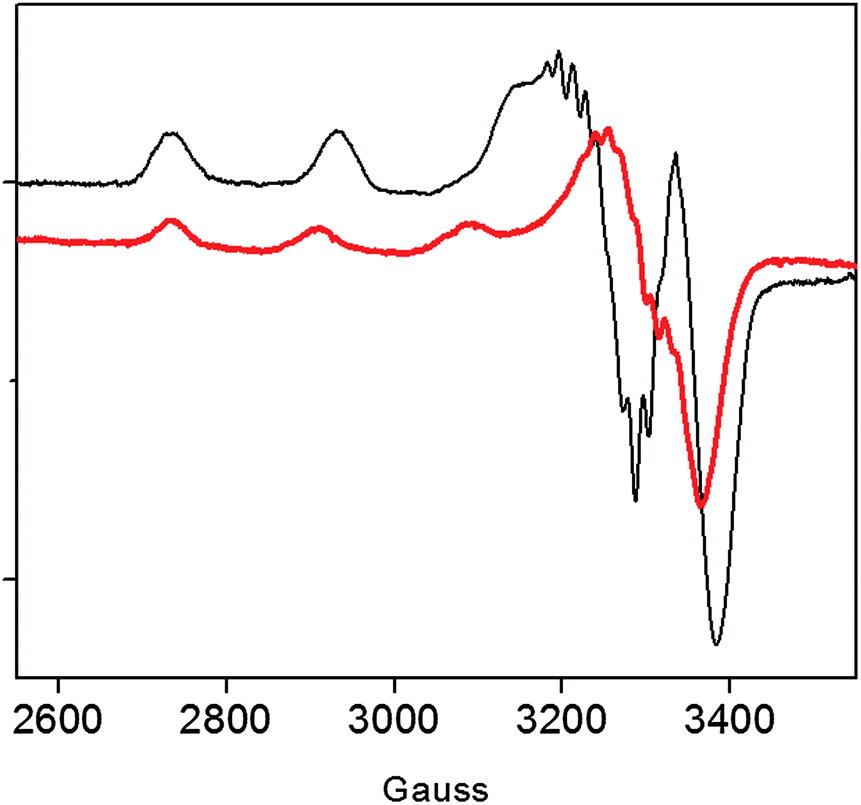 | ||
| Fig. 2 EPR spectra of Cu2+–r-Ang (black line) and Cu2+–wt-Ang (red line). [Ang] = [Cu2+] = 0.5 mM, 25 mM MOPS, pH 7.4. | ||
These Hamiltonian values are distinctly different from those previously reported for r-Ang,17 and indicate the formation of a copper complex in which the metal ion coordination shell may involve three (rather than four) nitrogen atoms with macro-chelate formation in a tetragonally distorted geometry.25,26
The UV-vis CD spectrum recorded at pH 7.4 for the Cu2+ complex formed by wt-Ang displays ligand-to-metal charge-transfer bands at 300 (Δε = −0.25), 326 (Δε = +0.19) and 364 nm (Δε = −0.05) (Fig. 3 and Table 1).
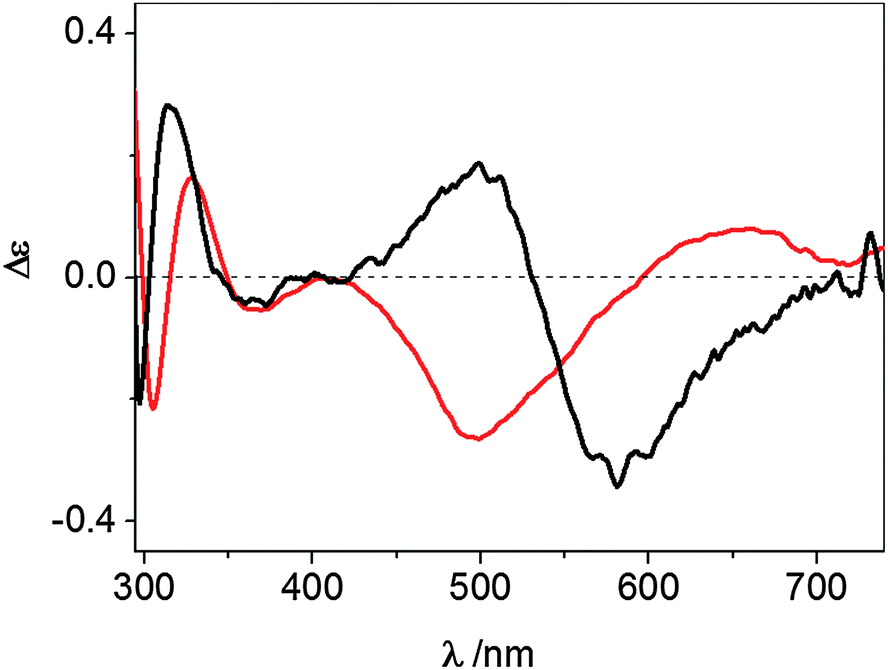 | ||
| Fig. 3 UV-vis CD spectra at pH 7.4 of Cu2+–wt-Ang (red line) and Cu2+–r-Ang (black line). [Ang] = [Cu2+] = 0.5 mM, 10 mM MOPS, pH 7.4. | ||
These spectroscopic parameters are the hallmark of the simultaneous involvement of two different protein nitrogen atoms in Cu2+ binding. The first band is diagnostic of a deprotonated amide nitrogen while the second and third bands are originated by the charge transfer from imidazole nitrogen atoms to metal ions,27 clearly suggesting the anchoring role played by the histidine residues. For both proteins the d–d bands show a cross-over signal which is originated by the metal chelate formation. The different chirality around the metal ion (Fig. 3) is attributable to the different main anchoring site, the amino N-terminus for the r-Ang protein and the histidine imidazole for the wt-Ang protein, that determine a different disposition of the binding side chains with respect to the peptide backbone.
The binding features of copper(II) complexes of two peptides encompassing the N-terminus of the two proteins further support the different coordination environments
To highlight the role of the free terminal amino group, the N-terminal fragment of Ang, encompassing residues 1–17 (QDNSRYTHFLTQHYDAK), was synthesized with and without acetylation of the amino terminus (LH and LH2, respectively) and the interaction with Cu2+ was investigated by potentiometric and spectroscopic techniques. At physiological pH, the Cu2+ complex formed by the peptide with a free terminal amino group (CuLH2) displays a UV-vis spectrum (λmax = 565 nm) similar to that given by r-Ang. In contrast, the Cu2+ complex formed by the acetylated peptide (CuLH) shows a different UV-vis spectrum (λmax = 595 nm) (Fig. S4 and S5, ESI†). On the whole, the spectroscopic data indicate an effective role of the free amino group of r-Ang in Cu2+ binding and suggest, at physiological pH, a Cu(2NIm,N−,O) chromophore for wt-Ang and a Cu(NH2,2N−,NIm) chromophore for r-Ang.The NMR parameters allow us to identify the different protein regions and amino acid residues involved in metal binding to r-Ang and to wt-Ang
In an attempt to discriminate between these two different coordination modes, NMR titration experiments (up to one equivalent addition of CuCl2) were carried out on 13C,15N-labeled wt- and r-Ang samples at physiological pH. The remarkable similarity of 1H,15N HSQC spectra indicates no significant structural changes between the two proteins (Fig. S1, ESI†).The increase of relaxation rates and NMR signal line widths of nuclei close to the paramagnetic center was exploited to obtain insights into the Cu2+ binding sites.28,29 In the case of wt-Ang, large exponential decay constants of signal intensity, as a function of Cu2+ concentration, were found for a large number of residues (Fig. S6a, Tables S1 and S2, ESI†). The most affected signals identify a region of the protein corresponding to the catalytic site, including His-13 and His-114 (Fig. 4a), while residues 60–68 and Asn-109 (the putative endothelial cell-binding site) are only marginally affected.
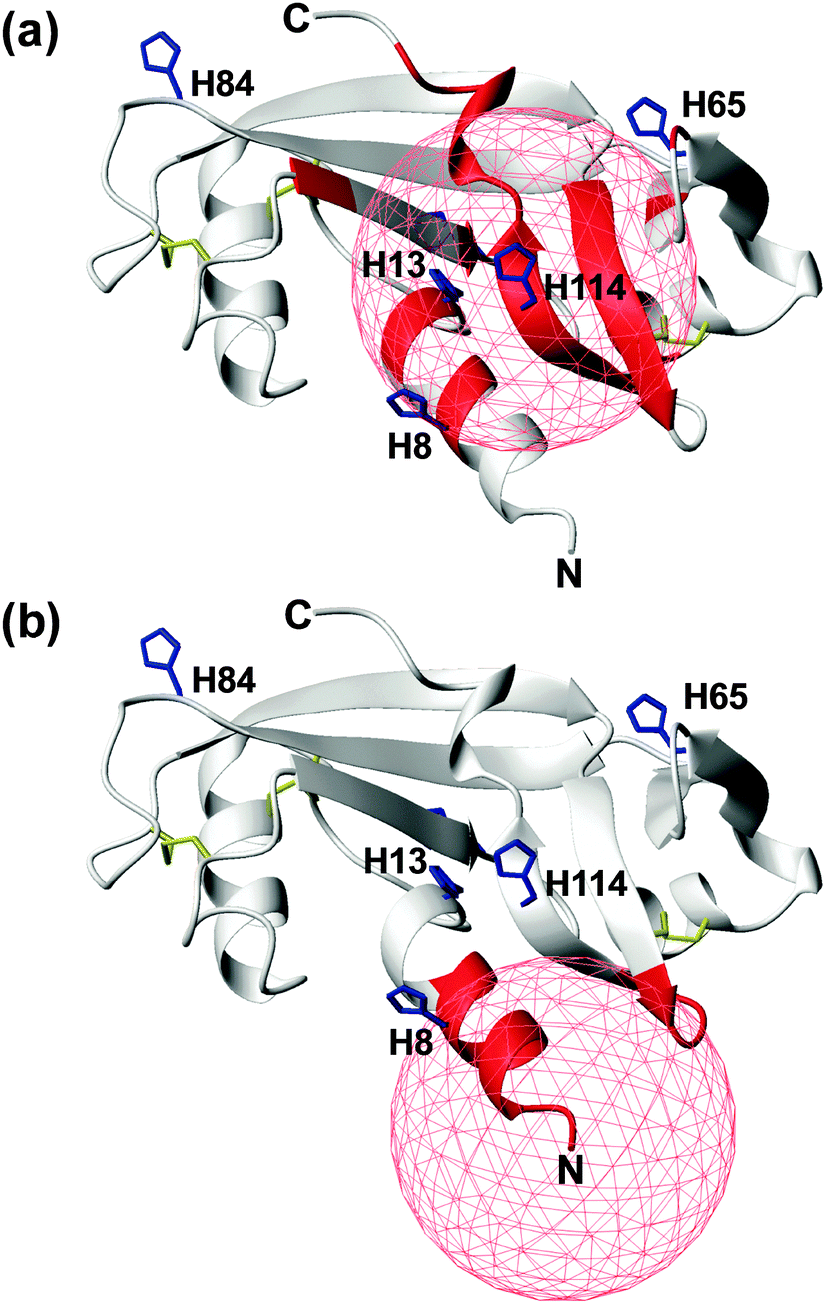 | ||
| Fig. 4 Mapping the effects of Cu2+ titration, at pH 7.4, on the NMR signals of wt- (a) and r-Ang (b) (structure: PDB ID 1H52). Residues with an exponential decay constant of signal intensity larger than average plus one standard deviation are colored in red. Histidine residues (blue sticks) and disulfide bonds (yellow sticks) are shown. A red sphere of 10 Å radius is centered on the putative Cu2+ anchoring site. | ||
These data support the presence of a Cu(2NIm,N−,O) chromophore in the wild-type protein, in which His-114 is the anchoring residue while the neighbouring side chain of His-13 may provide the additional imidazole nitrogen. The involvement of His-8 and His-65 in Cu2+ coordination is less likely, because of the greater distance of their imidazole rings from the anchoring residue in the apoprotein structure and the limited structural rearrangement occurring upon copper binding, as deduced by CD spectra.
In contrast, the addition of CuCl2 to r-Ang mainly affects the signals of residues in the N-terminal region of the protein, including His-8 (Fig. 4b and Fig. S6b, Table S2, ESI†), thus supporting a 4N metal coordination mode (NH2,2N−,NIm). Therefore in r-Ang the Cu2+ anchoring group appears to be the free amino terminus of Met, while the deprotonated amide nitrogens of Glu-1 and Asp-2 and the imidazole nitrogen of His-8 most likely complete the coordination environment of the metal ion. The pH decrease (from 7.4 to 5.5) produces dramatic effects only on the NMR spectra of r-Ang. For this reason, new Cu2+ titration experiments were performed at lower pH (5.5).
While the NMR parameters indicated that the Cu2+-binding site of wt-Ang remained unchanged (Fig. S7a and b, ESI†), those pertinent to r-Ang clearly suggest a conversion to a Cu2+ coordination mode similar to that found for wt-Ang (Fig. S7c and d, ESI†). The terminal amino group of r-Ang is protonated at pH 5.5 and therefore unable to bind Cu2+; as a consequence the imidazole nitrogens of histidine residues present in the catalytic domain become the preferred coordination site for Cu2+, as in the case of wt-Ang. 1H,13C HSQC NMR experiments performed on wt-Ang at low pH and sub-stoichiometric molar ratios of Cu2+ confirm that His-114 serves as an anchoring residue located in the protein core (Fig. S8, ESI†).
Cu2+ affects differently the RNase activity and capillary-like tube formation of r-Ang and wt-Ang
Cu2+ binding to His-13 and His-114 can prevent the participation of these conserved residues in the catalytic process. Therefore, the ensemble of NMR data suggests that Cu2+ can differently affect the RNase activity of wt- and r-Ang at physiological pH.Therefore, the catalytic activity of wt- and r-Ang was investigated in the presence of Cu2+ (Fig. 5).
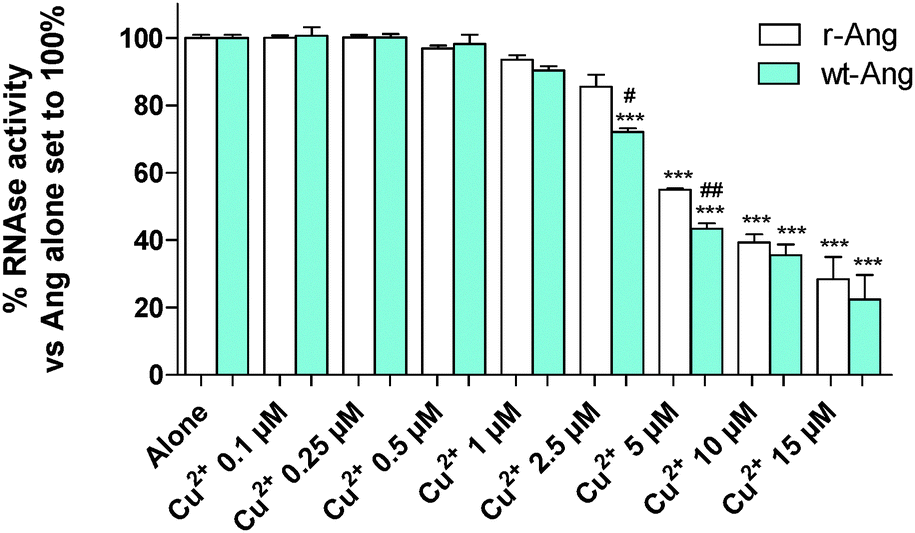 | ||
| Fig. 5 tRNA cleavage activities (2 h at 37 °C, pH 7.4, in 2 mg mL−1 tRNA) of r-Ang (white) and wt-Ang (cyan), at 0.5 μM, as a function of Cu2+ concentration. Cu2+ alone was not able to induce tRNA cleavage. Each data point is the mean of at least two experiments performed in duplicate. The significance of the differences was determined using one-way ANOVA with Bonferroni's post hoc test *P < 0.01, **P < 0.05 and ***P < 0.001 vs. the respective protein alone; #P < 0.05, ##P < 0.01 vs. wt-Ang. | ||
The enzymatic assay was carried out by a modification of the Shapiro et al. procedure.4 In the absence of Cu2+, the two proteins show similar activity, which was comparable to that reported by Holloway et al.22 at pH 7.0. The increase of Cu2+ concentration (within the range used in the literature4) induces a decrease of RNase enzymatic activity in both proteins. This effect is more pronounced for wt-Ang, where His-114 of the catalytic site is the anchoring residue for Cu2+, than for r-Ang where the anchoring group is the free amino terminus. Therefore, a larger amount of Cu2+ has to be added to r-Ang to obtain the same decrease of RNase activity observed in wt-Ang, confirming that r-Ang can bind the metal ion also through sites different from the catalytic one.
The effect of Cu2+ on the RNase activity of the protein may influence its angiogenic activity and, to this end, the capillary-like tube formation test was performed on both r-Ang and wt-Ang in the presence of Cu2+ (Fig. 6).
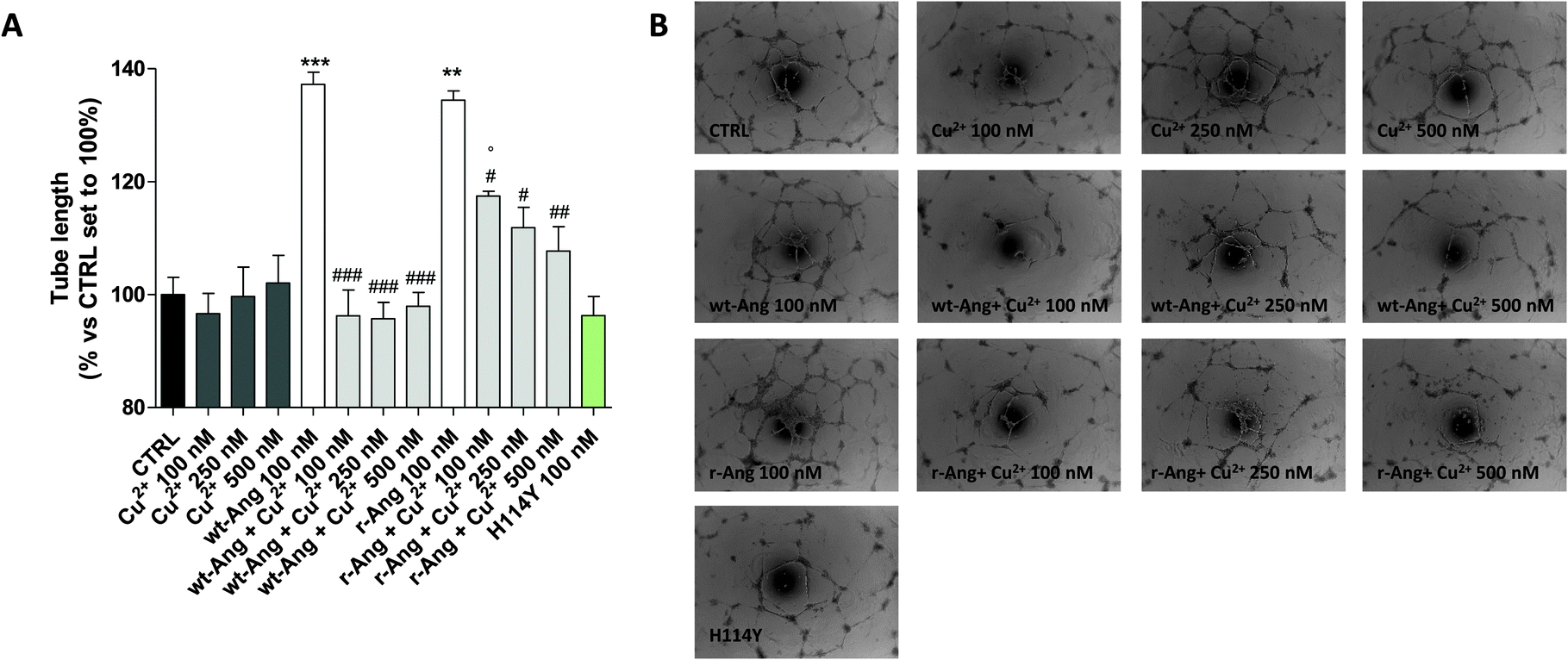 | ||
| Fig. 6 HUVEC was treated for 16 h in EGM-2 medium with r-Ang (100 nM) and wt-Ang (100 nM) alone or in the presence of different concentrations of CuSO4 (100, 250 or 500 nM). Capillary-like tube formation was observed by microscopy and quantified using the ImageJ program. (A) The tube length was quantified and expressed as a percentage versus control, set to 100%. The data are expressed as the mean ± SEM of two independent experiments performed in triplicate. The significance of the differences was determined using one-way ANOVA with Bonferroni's post hoc test **P < 0.01, ***P < 0.001 vs. the control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the respective angiogenin; °P < 0.05 vs. the respective Cu2+. (B) Representative pictures of the angiogenesis process. | ||
In the unbound form, the two protein isoforms display the same ability to induce capillary-like tube formation. Conversely, Cu2+ alone, at the used concentrations (100–500 nM), was not able to induce significant tube formation. Noteworthily, the addition of copper ions decreases the activity of both proteins. However, also in this case the effect is much sharper in wt-Ang where it is already evident at an Ang/Cu2+ ratio of 1:1. This may be indirectly correlated with the involvement of His-114 in metal ion binding. To further corroborate this hypothesis, a protein mutant, in which His-114 has been substituted with a tyrosine, has been expressed and subjected to first-methionine removal so as to have a single point mutated form of wt-Ang (H114Y). Noteworthily, the activity of wt-Ang in the presence of Cu2+ was similar to that observed in Ang H114Y (Fig. 6). This confirms that Cu2+ binding to His-114 in wt-Ang inhibits the catalytic process so that its activity becomes similar to that observed for the mutated protein H114Y.
Conclusions
Angiogenin is a main angiogenic factor, its activity being affected by copper which, in turn, has also angiogenic properties. The interaction between the metal ion and the protein might be part of the biochemical pathway which regulates both copper and angiogenin activities in vivo. The comprehensive characterization of the complex species formed between copper(II) and angiogenin is therefore a valuable support for a better understanding of their reciprocal biological influence. The protein typically used in experiments so far reported in the literature is the recombinant one. In this paper, we show that recombinant Ang binds Cu2+ through the terminal amino group of the extra methionine residue. In contrast, the wild-type protein has the amino terminus locked in a pyroglutamic ring and His-114 results to be the main Cu2+ binding site. His-114 is one of the three constituents of the RNase catalytic site that is essential to the protein for carrying out its angiogenic activity. Therefore, a small amount of copper is likely to influence directly the RNase activity and consequently the capillary-like tube formation of wt-Ang. Unlike wt-Ang, r-Ang has a free amino terminus and then an additional and stronger copper binding site at physiological pH, which is not directly involved in the protein catalytic activity. As a consequence, copper addition (up to an equimolar amount) has less influence on r-Ang activity and a higher metal concentration is required in order to observe a reduction of RNase activity comparable to that observed for wt-Ang.Our data highlight, for the first time, the relevant difference between recombinant and wild type Ang in binding copper and the influence of metal binding on Ang biological activity. The awareness of such a difference entails the need to use the wild-type form for the correct understanding of Ang–copper interaction, and, in turn, the ensuing effects on angiogenic processes.
Acknowledgements
The Italian Ministry of University and Research is gratefully acknowledged for partial support (PRIN 2010M2JARJ to D. LM, E. R., F. A.); we thank the University Consortium for Research in the Chemistry of Metal ions in Biological Systems (CIRCMSB) and COST Action CM1105. We thank Daniel Farkas for helpful advice about protein expression.References
- R. Shapiro, J. F. Riordan and B. L. Vallee, Biochemistry, 1986, 25, 3527–3532 CrossRef CAS PubMed.
- D. J. Strydom, Cell. Mol. Life Sci., 1998, 54, 811–824 CrossRef CAS PubMed.
- K. Kishimoto, S. Liu, T. Tsuji, A. K. Olson and G. F. Hu, Oncogene, 2005, 24, 445–456 CrossRef CAS PubMed.
- R. Shapiro, S. Weremowicz, J. F. Riordan and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8783–8787 CrossRef CAS.
- M. Moenner, M. Gusse, E. Hatzi and J. Badet, Eur. J. Biochem., 1994, 226, 483–490 CrossRef CAS PubMed.
- S. Li and G.-F. Hu, Int. J. Biochem. Mol. Biol., 2010, 1, 26–35 CAS.
- T. U. Steidinger, S. R. Slone, H. Ding, D. G. Standaert and T. A. Yacoubian, PLoS One, 2013, 8, e56092 CAS.
- Y. N. Kim and H. Kim, Biol. Psychiatry, 2012, 38, 116–120 CAS.
- M. J. Greenway, P. M. Andersen, C. Russ, S. Ennis, S. Cashman, C. Donaghy, V. Patterson, R. Swingler, D. Kieran, J. Prehn, K. E. Morrison, A. Green, K. R. Acharya, R. H. Brown Jr and O. Hardiman, Nat. Genet., 2006, 38, 411–413 CrossRef CAS PubMed.
- H. Kishikawa, D. Wu and G. F. Hu, Expert Opin. Ther. Targets, 2008, 12, 1229–1242 CrossRef CAS PubMed.
- J. H. Kaplan and S. Lutsenko, J. Biol. Chem., 2009, 284, 25461–25465 CrossRef CAS PubMed.
- S. A. Lowndes and A. L. Harris, J. Mammary Gland Biol. Neoplasia, 2005, 10, 299–310 CrossRef PubMed.
- L. Finney, S. Mandava, L. Ursos, W. Zhang, D. Rodi, S. Vogt, D. Legnini, J. Maser, F. Ikpatt, O. I. Olopade and D. Glesne, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2247–2252 CrossRef CAS PubMed.
- J. Badet, F. Soncin, J. D. Guitton, O. Lamare, T. Cartwright and D. Barritault, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 8427–8431 CrossRef CAS.
- F. Soncin, J. D. Guitton, T. Cartwright and J. Badet, Biochem. Biophys. Res. Commun., 1997, 236, 604–610 CrossRef CAS PubMed.
- D. La Mendola, A. Magrì, L. I. Vagliasindi, Ö. Hansson, R. P. Bonomo and E. Rizzarelli, Dalton Trans., 2010, 10678–10684 RSC.
- D. La Mendola, D. Farkas, F. Bellia, A. Magrì, A. Travaglia, Ö. Hansson and E. Rizzarelli, Inorg. Chem., 2012, 51, 128–141 CrossRef CAS PubMed.
- G. F. Hu, J. Cell. Biochem., 1998, 69, 326–335 CrossRef CAS PubMed.
- D. J. Strydom, J. W. Fett, R. R. Lobb, E. M. Alderman, J. L. Bethune, J. F. Riordan and B. L. Vallee, Biochemistry, 1985, 24, 5486–5494 CrossRef CAS PubMed.
- C. Giacomelli, M. L. Trincavelli, C. Satriano, Ö. Hansson, D. La Mendola, E. Rizzarelli and C. Martini, Int. J. Biochem. Cell Biol., 2015, 60, 185–196 CrossRef CAS PubMed.
- R. Shapiro, J. Wade Harper, E. A. Fox, H.-W. Jansen, F. Hein and E. Uhlmann, Anal. Biochem., 1998, 175, 450–461 CrossRef.
- D. E. Holloway, M. C. Hares, R. Shapiro, V. Subramanian and K. R. Acharya, Protein Expression Purif., 2001, 22, 307–317 CrossRef CAS PubMed.
- O. Lequin, H. Thuring, M. Robin and J. Y. Lallemand, Eur. J. Biochem., 1997, 250, 712–726 CAS.
- B. Crabtree, N. Thiyagarajan, S. H. Prior, P. Wilson, S. Iyer, T. Ferns, R. Shapiro, K. Brew, V. Subramanian and K. R. Acharya, Biochemistry, 2007, 46, 11810–11818 CrossRef CAS PubMed.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed.
- D. La Mendola, A. Magrì, A. M. Santoro, V. G. Nicoletti and E. Rizzarelli, J. Inorg. Biochem., 2012, 111, 59–69 CrossRef CAS PubMed.
- P. G. Daniele, E. Prenesti and G. Ostacoli, J. Chem. Soc., Dalton Trans., 1996, 3269–3275 RSC.
- D. Milardi, F. Arnesano, G. Grasso, A. Magrì, G. Tabbì, S. Scintilla, G. Natile and E. Rizzarelli, Angew. Chem., Int. Ed., 2007, 46, 7993–7995 CrossRef CAS PubMed.
- F. Arnesano, L. Banci and M. Piccioli, Q. Rev. Biophys., 2005, 8, 167–219 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis and purification; potentiometric and UV-vis characterization of peptide–copper(II) complex species; ESI-MS measurements; and NMR tables. See DOI: 10.1039/c5mt00216h |
| This journal is © The Royal Society of Chemistry 2016 |