
Contemplating a role for titanium in organisms
Mark R.
Zierden
and
Ann M.
Valentine
*
Department of Chemistry, Temple University, Philadelphia, PA 19122, USA. E-mail: ann.valentine@temple.edu; Fax: +1 215 204 7836; Tel: +1 215 204 1532
First published on 12th November 2015
Abstract
Titanium is the ninth most abundant element in the Earth's crust and some organisms sequester it avidly, though no essential biological role has yet been recognized. This Minireview addresses how the properties of titanium, especially in an oxic aqueous environment, might make a biological role difficult to recognize. It further considers how new -omic technologies might overcome the limitations of the past and help to reveal a specific role for this metal. While studies with well established model organisms have their rightful place, organisms that are known avid binders or sequesterers of titanium should be promising places to investigate a biological role.
 Mark Zierden | Mark Zierden was raised in northern New Jersey, near the New York State border. He earned his BS in chemistry from Stockton University in 2010 where he studied thermochromic nickel complexes with Rogers Barlatt. He also performed research on the glass transitions of alkali-borate glasses under the guidance of Steve Feller at Coe College. He is currently a fifth-year graduate student at Temple University. He has been an NSF GK-12 Fellow and currently volunteers at a local High School teaching lessons and performing demonstrations. He is also the co-host of Stereotopical Chemistry, a chemistry-based podcast, aimed at educating non-scientists about current chemical research. His current research is focused on the binding interactions of several Rhodococcus species with titanium dioxide. |
 Ann Valentine | Ann Valentine earned a BS in chemistry in 1993 at the University of Virginia and a PhD in 1998 with Stephen J. Lippard at MIT. She was a postdoctoral fellow with Stephen J. Benkovic at Penn State University from 1998–2001. At Temple University, her research group focuses on how nature manages hydrolysis-prone elements, including titanium. She is the recipient of the 2009 Paul Saltman Award for Metals in Biology and a 2014 American Institute of Chemists Chemical Pioneer Award. She was the 2015 Honors Professor of the Year at Temple University. |
1. Introduction
There is no known essential role for titanium in the biology of any organism. However, there are studies across many fields that suggest Ti is at least biologically active.1 Most titanium on Earth is locked up in insoluble minerals, such that one might wonder how titanium could participate in biorelevant chemistry. Titanium is the ninth most abundant element in the crust at 0.6%,2 so even a small percentage of solubilized material presents a large amount of metal.3 Most of the titanium present in the environment exists as rutile, a crystal form of TiO2, or as ilmenite, FeTiO3.3,4 Geologically, titanium is often considered immobile although recent evidence suggests it may be mobile in rocks under weathering conditions5,6 and in soils.7 Soluble titanium ranges from 4 pM in surface ocean waters,8–10 where it is far under-saturated, to 100 μM in hot spring waters.11 In both fresh water and seawater, thermodynamic models assume the predominance of TiO(OH)2.12 Complexation by organic ligands is important in at least some natural environments.1 TiO2 nanoparticles in both fresh water and marine systems agglomerate, forming micrometer size particles, or are incorporated into marine snow where particle feeders may ingest them.13 Taken together, these various environments present the opportunity for organisms to use titanium in the form of soluble hydroxide species, as organically complexed species, or in mineral form.Proteins have evolved to use the metals that are, or at least were at the time of those proteins' evolution, the most accessible.14–16 Some active centers of metalloenzymes resemble the structures of minerals presumed to be present in precipitates from hydrothermal solutions in the ocean billions of years ago.17,18 Considering this use of available material and the vast abundance of titanium on the planet, it is conceivable that nature might have made use of this element, for example during evolution of the earliest prokaryotes. It is clear that some extant organisms do use and contain titanium in certain structures.
Titanium is capable of biologically interesting chemistry but has inherent characteristics that make it difficult to identify in biological systems. Modern experimental methods offer the possibility of identifying new roles for metals like titanium in bioinorganic chemistry. The metal and its ions bring a set of complications for common separation and -omic techniques. Choice of experimental system will be crucial, whether a well-studied and familiar model system or a lesser-known organism known to be associated with high titanium concentrations.
2. Historical identification of essential elements
Many bioessential elements were identified because humans experienced symptoms from an accidental dietary deficiency, as for iron or cobalt, or exhibited diseases of metal mismanagement, like Wilson's disease for copper. Some metals, like chromium, were rigorously excluded from the diet to look for signs that the excluded metal was essential. Some metal ions in biology have conspicuous properties that make them easy to identify, like the color of blue copper proteins or the multiline EPR spectra of some manganese proteins.Even in recent years, the list of metals having a native biological role continues to grow. In the oceans where zinc is scarce, a diatom species has a carbonic anhydrase that utilizes cadmium in its active site instead of zinc.19 Some methanotrophs natively feature the usually toxic lanthanides lanthanum and cerium instead of calcium in the active site of their methanol dehydrogenase.20 Each of these discoveries was foreshadowed by the novel metals' binding in the active sites of the class of enzymes in question, cadmium to zinc enzymes and lanthanides to calcium enzymes. But a form with a unique requirement for cadmium and for lanthanide, respectively, was a significant development.
3. Characteristics of titanium relevant to biology
The most stable oxidation state of titanium in an aqueous oxic environment is Ti(IV), which shares characteristics such as ionic radius with Al(III) and Fe(III).1 These metals also share a thermodynamic preference for similar binding sites, though Ti(IV) is more strongly Lewis acidic (Fig. 1).21 Ti(IV) can associate with some Fe(III) proteins. In vitro Ti(IV) binds more tightly than Fe(III) to the iron transport protein human serum transferrin.22 The iron storage protein ferritin can also biomineralize titanium.23,24 Like those other ions, Ti(IV) is prone to hydrolysis and hydrolytic precipitation, though its insolubility is not as extreme as is often assumed.1 Binding to small or large biomolecules increases the solubility of Ti(IV).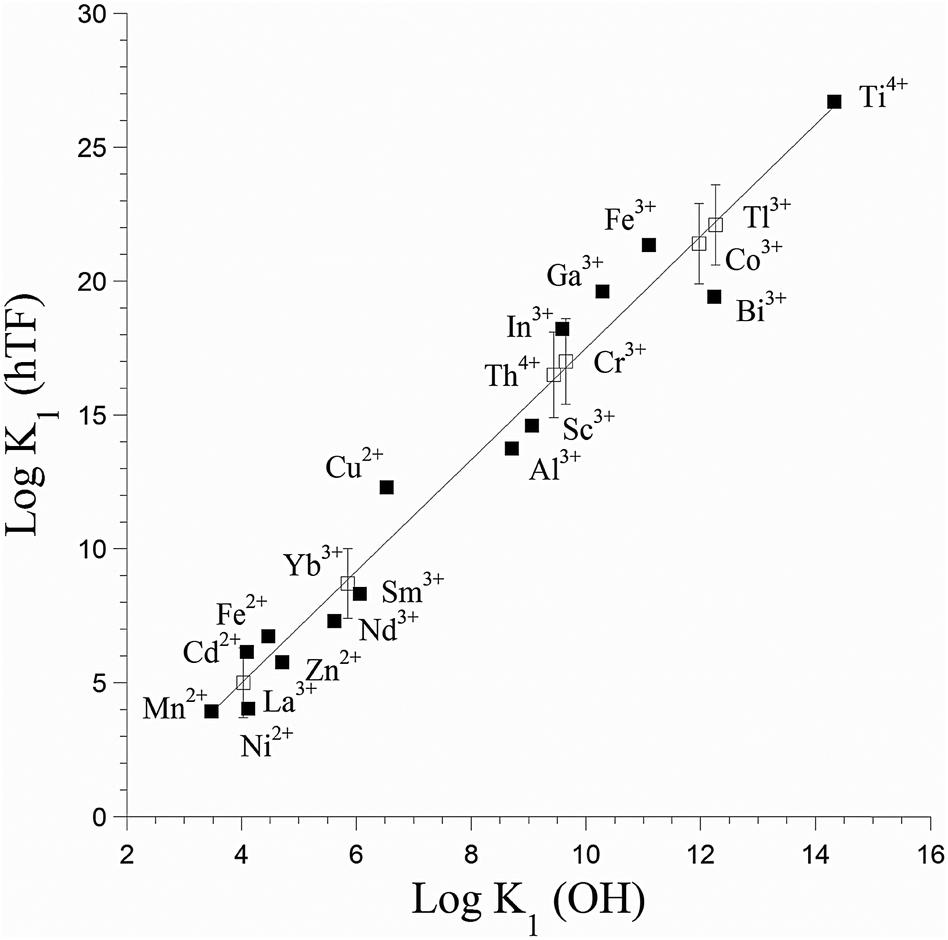 | ||
| Fig. 1 Correlation of binding constants for metal binding to human serum transferrin (hTF) with metal Lewis acidity, as quantified by K1 (OH). Filled squares represent experimental values, while open squares represent estimated ones. Adapted from data in ref. 21 and 22. | ||
Turning to kinetics, Ti(IV) complexes exhibit a wide range of ligand exchange rates. Hydroxyl and water ligands are very labile and exchange with rate constants on the order of thousands per second.25 But essentially no exchange is observed when the Ti ligand is the hexadentate siderophore enterobactin.26 Falling somewhere in the middle of these extremes, over minutes to hours, are the rates for exchange with small bioligands like ascorbate or citrate, or with proteins like transferrin.22,27
Just considering the thermodynamic stability and kinetic lability of metal–ligand complexes ignores the powerful compartmentalization control that living cells exhibit over different metal species in vivo.28,29 Some cells use physical compartmentalization to overcome the inherent thermodynamic preferences of a protein binding site. Once a complex is removed from the compartmentalized environment, though, it is important to appreciate the fundamental thermodynamics and kinetics of the metal ion binding to its ligand(s).
4. The possible roles of titanium
Against the backdrop of this fundamental chemistry, Nature would sequester Ti if it were useful to the organism. Some biochemical processes for which titanium might be suitable (or even uniquely excellent) are suggested by the many ways humans and human chemists use this element.The powerful Lewis acidity of Ti(IV) could be deployed to deprotonate difficult-to-deprotonate substrates in a metalloenzyme active site. As a benchmark to illustrate its power, water molecules coordinated to Ti(IV) are deprotonated below pH 0.1,30 In chemical catalysis outside biology, titanium plays this role often and well, for example as a catalyst for aldol reactions.31
Depending on the ligand environment, a Ti4+/Ti3+ couple would be suitable for low-potential redox, for example in an electron transfer chain or redox metalloenzyme. The potential of the Ti4+/Ti3+ couple in acid is close to 0 V vs. NHE,32 but ligands that tightly bind the oxidized form and stabilize it with respect to hydrolytic precipitation can tune the potential over a range down to at least −1 V.1,33,34
Titanium might assume a role in the construction or repair of soft biomaterials, for example in the acsidians that are avid accumulators of the element. Ascidians are also called tunicates because of their protective covering, the tunic.35 The tunic varies between species but is composed of fibrous cellulose components linked with proteins.36 Metals and polyphenolic secondary metabolites called tunichromes may be involved in tunic formation or wound repair.37 Possible metal ligands in the tunic include the tunichromes, proteins or modified proteins (including those containing DOPA side chains) and carbohydrates. The tunic also contains free wandering blood cells from the ascidian's open circulatory system. Some of these cells have high metal concentrations. Some are high in polyphenol content, and probably contain high concentrations of tunichromes, either with or without associated metals. During wound healing, these cells coagulate and lyse, and electron dense fibers form.38 The metal may have a regulating effect by acting as a stabilizer of the component monomers for polymer formation, as a catalyst for polymerization, or as a regulator of the process.
Titanium minerals are widespread and stable. Titanium dioxide is a widely used pigment, and the oxide forms as a passivating coating when titanium metal and its alloys are used in medical implants.1 Mineralized or biomineralized titanium could provide a protective coating or defense for organisms. One place titanium dioxide has found promising use is in dye-sensitized solar cells.39 It is exciting to think that Nature has had the raw materials to take advantage of this chemistry for billions of years, though no natural TiO2 solar cell is yet known.
Titanium might serve an antimicrobial function for an organism,1 either in the presence or absence of light. UV irradiation of some Ti compounds, especially in aqueous environments, generates singlet oxygen and superoxide anion. Both species are damaging cellular oxidants.
Finally, titanium may afford ultraviolet protection for organisms, in effect forming a sunscreen. TiO2 is used as a commercial sunscreen because of its light-scattering properties. Depending on the identity of the ligand or the size of the particle, light absorption or scattering by a Ti compound or mineral may provide a similar protective effect. The tunic of the ascidian Lissoclinum patella absorbs UV-B (290–320 nm) but not visible light, providing a protective environment for a symbiotic photosynthetic prokaryote.40,41 The symbiont provides oxygen to the ascidian, and cannot survive the UV exposure outside of the tunic. This particular ascidian species has not been associated with elevated Ti levels, though other ascidians are avid accumulators.
5. Detecting titanium
Detecting Ti(IV) can be difficult; it is d0, diamagnetic, and exhibits no EPR signal. Most titanium complexes are colorless or have indistinct ligand-to-metal charge transfer bands in the ultraviolet region. There are no practically useful NMR or radioisotopes,1 and use of X-ray absorption spectroscopy (XAS) has been limited. Older elemental analysis methods such as flame atomic absorption (AA) were relatively insensitive to titanium. Importantly, in such single-element techniques, researchers had to be specifically looking for titanium to find it.Techniques like Inductively Coupled Plasma – Mass Spectrometry (ICP-MS) are more sensitive than AA and are multi-element. But other kinds of considerations sometimes interfere with the detection of Ti. The excellent international GEOTRACES project42 strives to characterize biogeochemical cycles of many trace elements. A few of the associated studies have focused on titanium,10,43–45 but much of the collection apparatus was constructed from titanium, presumably because it is quite inert and because there was relatively less interest in focusing on Ti as an experimental element.
Finally and in general, the inherent difficulty in characterizing metalloproteomes is well known.46–51 In one important study,46 although no Ti was detected in the growth medium, a relatively high Ti concentration (1.6 μM) was detected in the cytoplasmic extract of Pyrococcus furiosus. This cytoplasmic Ti concentration was fourth among trace metals after Fe, Zn, and W, and was higher than Ni, Co, or Mn. But Ti was no longer detected after a relatively mild anion exchange separation; the fate of the Ti is not clear. Possibly it was not ever bound to protein, or it was very labile. This result led us to consider how the bioinorganic behaviour and properties of titanium might manifest during the application of emerging -omic methods to identify novel metalloproteins.
In a plausible experimental plan, an experimental organism would be chosen, driven by the desire for a well-studied model system or for a system already known to be associated with Ti. The biomolecules would be subjected to a separation method like liquid chromatography or gel electrophoresis. A downstream elemental analysis such as ICP-MS or a metal-specific sensor would identify which biomolecule(s) were associated with titanium. Minimal disruption of binding sites would be necessary. Finally, the biomolecules would be identified by some means. Each of these steps deserves special consideration in light of the properties and aqueous behavior of titanium.
6. Model organisms
There are two options when deciding on where to search for putative titanium metalloproteins: well-studied generic model organisms or established sequesterers of titanium such as diatoms and ascidians. Each of these routes offers distinct advantages.6.1. Well-studied model organisms
The large body of work on organisms like E. coli and yeast allows for a better chance at identifying proteins using a shotgun style of proteomics.46 Studying the metalloproteomes of P. furiosus, E. coli and S. solfataricus allowed for high throughput and quick identification of isolated proteins. There is some work on the effect of titanium compounds on the growth of yeast.52 But as the cost of genome sequencing continues to decrease, the availability of the organism's full genome ceases to be an impediment to the use of a more unusual system.6.2. Avid marine sequesterers of titanium
 | ||
| Fig. 2 Titania formation by recombinant silafins, rSil1L and rSilC. SEM images of precipitates formed by (a) 53 μM rSil1L (b–d) 32 μM rSilC in sodium phosphate/citrate buffer solution (50 mM, pH 7). In (a) the particles fuse together forming an interconnected network. (b) rSilC-titania contains two different structures; (c) smaller hollow particles and (d) larger particles with frequent cracks. Scale bars: 1 μM (a and c) 20 μM (b) and 5 μM (d). Adapted from ref. 61. | ||
6.3. Organisms associated with titanium minerals
Many organisms exploit inorganic minerals for structural support, or as sensors or instruments.74,75 Although titanium minerals are abundant, examples of organisms associating with them are rare. In one classic text on biomineralization, titanium appears on a list of biomineralized elements with a reference to unpublished work by the author on ilmenite found in a prokaryote.74 The identity of the prokaryote is unknown (S. Mann, personal communication).Organisms produce biominerals with varying degrees of control or collect them as detrital materials from the environment. Mineralization of titanium would require a mechanism for sequestration from the environment and then active direction of mineralization. Collection from the environment would require a specific interaction between molecules on the cell-surface of the organism with the titanium mineral surface. There are a few possibilities about how the latter molecular recognition process might work. Peptides might have evolved to bind specifically to TiO2 mineral surfaces.76,77 Glycopeptides, phosphopeptides and phosphoproteins demonstrate a particularly high affinity for TiO2,78,79 and nucleosides for TiO2/ZrO2.80 The specifics of these biomolecule–surface interactions are being explored.81–83
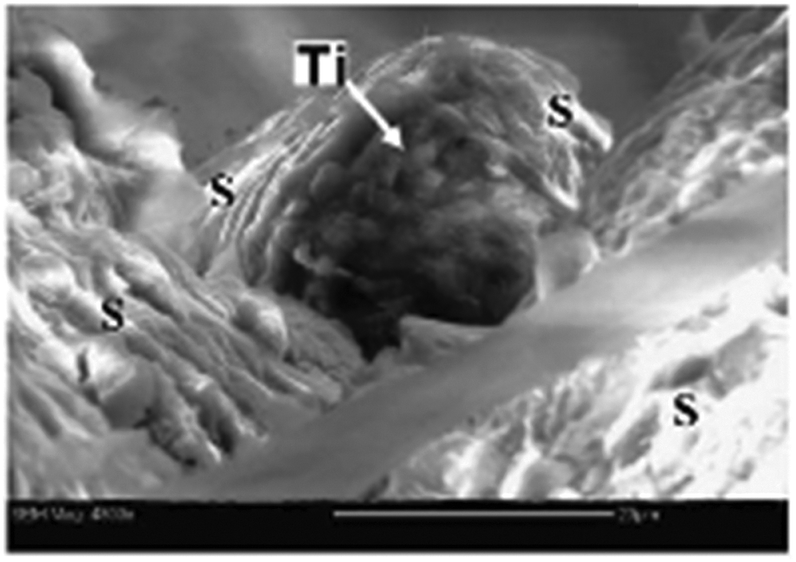 | ||
| Fig. 3 ESEM image of a titanium particle (arrow) from the roof of a hornet's comb cell. “S” marks the saliva fibers. Scale bar = 20 μM. Adapted from ref. 86. | ||
Bacteria from the Mediterranean Sea were isolated by exploiting their ability to adhere to TiO2 very strongly.92 A Gram positive species was isolated, Rhodococcus ruber GIN-1, which adhered to TiO2 selectively over other metal oxides in under 1 min, at pH values between 1 and 9, and at temperatures between 4 and 80 °C.92 Cell adhesion to the mineral was not disrupted by dilute acids, alcohols, and cationic or nonionic detergents. A cell surface homolog of the normally cytosolic protein dihydrolipoamide dehydrogenase was among the proteins implicated in adhesion.93
7. Separations and detection
Biomolecules are often subjected to a separation step and characterized by one of several techniques. Putative titanium biomolecules might fare better or worse than other metallobiomolecules in some of these procedures.7.1. Separations effect on ion occupancy
During any of these separation techniques, it would be important to monitor the titanium inventory. If the metal did dissociate from an associated biomolecule, it might bind very tightly to a column or, alternatively, not bind at all, depending on speciation.
7.2. Methods of identification
A variety of techniques can identify new metalloproteins. Simple searches of genomic or proteomic data require prior knowledge of a predicted metal binding sequence, and thus are not suitable for the identification of new motifs.101 Accurate prediction of metalloproteins is limited to closely homologous well-characterized proteins. A recent metallomic study demonstrated that some metal binding motifs have not yet been recognized, even for common biologically relevant metals like iron.46 Phage display identifies sequences with high affinity,102 and phage display has identified TiO2 binding sequences,76,103 but those specific sequences are not apparent in the proteomic databases. There are affinity proteomic methods that allow for detection of proteins in their natural environment,104 but an appropriate affinity material must be chosen. Identifying a metal binding site deep within a folded protein remains a challenge. There are imaging techniques that can be useful in identifying metalloproteins.105,106 Recently, promising metal-specific sensors capable of in vivo detection were developed.107 While each of these methods has its advantages, some of the most versatile methods for identifying and characterizing metalloproteins involve mass spectrometry after protein separations, which we will focus on. The immense growth of available genome sequences has led to a rapid rise in the power of bioinformatics. The combination of this vast knowledge with chromatography and mass spectrometry is a powerful tool in determination of metalloproteins.8. Conclusion
Much data suggests that titanium is biologically relevant but it has not been demonstrated to be essential for any organism. Evolution has directed proteins to use accessible metals and the abundance of titanium mineral and dissolved titanium argues for the existence of a titanium metalloprotein. The high concentrations of titanium in organisms and the chemical processes it is known to participate in support this idea. The identification of a true titanium metalloprotein is not an easy task. Titanium is normally a difficult metal to handle in an aqueous oxic environment. Attempting to manipulate a putative titanium metalloprotein without disturbing its protein environment complicates matters. The considerations outlined here for titanium apply more generally to many metal ions, especially those that are Lewis acidic and hydrolysis prone. This group includes established biometals like iron, which despite decades of study still occurs in coordination environments not yet recognized.46 By understanding both how titanium is known to interact with organisms and how it interacts with local protein environments, it may be possible to address the essentiality of this quite abundant element.Acknowledgements
This work was supported by the U.S. National Science Foundation (CHE-1412373) and by the U.S. National Science Foundation award #0841377 for a graduate traineeship for MRZ.References
- K. M. Buettner and A. M. Valentine, Chem. Rev., 2012, 112, 1863–1881 CrossRef CAS PubMed.
- J. Emsley, The Elements, Clarendon Press, Oxford, UK, 3rd edn, 1998 Search PubMed.
- A. KabataPendias and A. B. Mukherjee, Trace Elements from Soil to Human, 2007 Search PubMed.
- C. W. Correns, in Handbook of Geochemistry, ed. K. H. Wedepohl, Springer, Berlin, 1969–1978, vol. 2 Search PubMed.
- X. Du, A. W. Rate and M. A. M. Gee, Chem. Geol., 2012, 330, 101–115 CrossRef.
- T. Taboada, A. M. Cortizas, C. Garcia and E. Garcia-Rodeja, Geoderma, 2006, 131, 218–236 CrossRef CAS.
- S. Cornu, Y. Lucas, E. Lebon, J. P. Ambrosi, F. Luizao, J. Rouiller, M. Bonnay and C. Neal, Geoderma, 1999, 91, 281–295 CrossRef CAS.
- K. J. Orians, E. A. Boyle and K. W. Bruland, Nature, 1990, 348, 322–325 CrossRef CAS.
- K. W. Bruland and M. C. Lohan, in The Oceans and Marine Geochemistry, ed. H. Elderfield, Elsevier-Pergamon, Oxford, 2003, pp. 23–47 Search PubMed.
- A. Dammshaeuser, T. Wagener, D. Garbe-Schoenberg and P. Croot, Deep Sea Res., Part I, 2013, 73, 127–139 CrossRef CAS.
- M. R. Van Baalen, Chem. Geol., 1993, 110, 233–249 CrossRef CAS.
- D. R. Turner, M. Whitfield and A. G. Dickson, Geochim. Cosmochim. Acta, 1981, 45, 855–881 CrossRef CAS.
- J. J. Doyle, V. Palumbo, B. D. Huey and J. E. Ward, Water, Air, Soil Pollut., 2014, 225, 2106 CrossRef.
- C. L. Dupont, S. Yang, B. Palenik and P. E. Bourne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17822–17827 CrossRef CAS PubMed.
- R. E. M. Rickaby, Philos. Trans. R. Soc. London, Ser. A, 2015, 373, 20140188 CrossRef PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- B. Herschy, A. Whicher, E. Camprubi, C. Watson, L. Dartnell, J. Ward, J. R. G. Evans and N. Lane, J. Mol. Evol., 2014, 79, 213–227 CrossRef CAS PubMed.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Nat. Rev. Microbiol., 2008, 6, 805–814 CAS.
- T. W. Lane, M. A. Saito, G. N. George, I. J. Pickering, R. C. Prince and F. M. M. Morel, Nature, 2005, 435, 42 CrossRef CAS PubMed.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. Op den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef CAS PubMed.
- H. Y. Li, P. J. Sadler and H. Z. Sun, Eur. J. Biochem., 1996, 242, 387–393 CAS.
- A. D. Tinoco and A. M. Valentine, J. Am. Chem. Soc., 2005, 127, 11218–11219 CrossRef CAS PubMed.
- M. T. Klem, J. Mosolf, M. Young and T. Douglas, Inorg. Chem., 2008, 47, 2237–2239 CrossRef CAS PubMed.
- F. F. Amos, K. E. Cole, R. L. Meserole, J. P. Gaffney and A. M. Valentine, J. Biol. Inorg. Chem., 2013, 18, 145–152 CrossRef CAS PubMed.
- P. Comba and A. Merbach, Inorg. Chem., 1987, 26, 1315–1323 CrossRef CAS.
- T. Baramov, K. Keijzer, E. Irran, E. Moesker, M.-H. Baik and R. Suessmuth, Chem. – Eur. J., 2013, 19, 10536–10542 CrossRef CAS PubMed.
- K. M. Buettner, J. M. Collins and A. M. Valentine, Inorg. Chem., 2012, 51, 11030–11039 CrossRef CAS PubMed.
- S. Tottey, K. J. Waldron, S. J. Firbank, B. Reale, C. Bessant, K. Sato, T. R. Cheek, J. Gray, M. J. Banfield, C. Dennison and N. J. Robinson, Nature, 2008, 455, 1138–U1117 CrossRef CAS PubMed.
- R. Kudva, K. Denks, P. Kuhn, A. Vogt, M. Mueller and H.-G. Koch, Res. Microbiol., 2013, 164, 505–534 CrossRef CAS PubMed.
- L. Ciavatta, D. Ferri and G. Riccio, Polyhedron, 1985, 4, 15–22 CrossRef CAS.
- R. Mahrwald, Aldol Reactions, Springer, Netherlands, Dordrecht, 2009, p. 73 Search PubMed.
- J. Schmets, J. Van Muylder and M. Pourbaix, in Atlas of Electrochemical Equilibria in Aqueous Solutions, ed. M. Pourbaix, Pergamon Press, Oxford, 1966, p. 213 Search PubMed.
- R. Uppal, C. D. Incarvito, K. V. Lakshmi and A. M. Valentine, Inorg. Chem., 2006, 45, 1795–1804 CrossRef CAS PubMed.
- B. A. Borgias, S. R. Cooper, Y. B. Koh and K. N. Raymond, Inorg. Chem., 1984, 23, 1009–1016 CrossRef CAS.
- I. Goodbody, Adv. Mar. Biol., Academic Press, New York, 1974, vol. 12, p. 2 Search PubMed.
- U. Welsch, in Biology of the Integument 1: Invertebrates, ed. J. Bereiter-Hahn, K. Matolsky and S. Richards, Springer-Verlag, New York, 1984, p. 800 Search PubMed.
- S. W. Taylor, B. Kammerer and E. Bayer, Chem. Rev., 1997, 97, 333–346 CrossRef CAS PubMed.
- E. Hirose, Y. Taneda and T. Ishii, Dev. Comp. Immunol., 1997, 21, 25–34 CrossRef CAS PubMed.
- B. Oregan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. L. DionisioSese, M. Ishikura, T. Maruyama and S. Miyachi, Mar. Biol., 1997, 128, 455–461 CrossRef CAS.
- M. L. Dionisio-Sese, T. Maruyama and S. Miyachi, Mar. Biotechnol., 2001, 3, 74–79 CrossRef CAS PubMed.
- GEOTRACES, http://www.geotraces.org, 2015.
- A. Dammshaeuser, T. Wagener and P. L. Croot, Geophys. Res. Lett., 2011, 38, L24601 Search PubMed.
- P. L. Croot, Anal. Chem., 2011, 83, 6395–6400 CrossRef CAS PubMed.
- S. Poehle, K. Schmidt and A. Koschinsky, Deep Sea Res., Part I, 2015, 98, 83–93 CrossRef CAS.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole, II, F. E. Jenney, Jr., W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–U718 CrossRef CAS PubMed.
- J. Estellon, S. O. de Choudens, M. Smadja, M. Fontecave and Y. Vandenbrouck, Metallomics, 2014, 6, 1913–1930 RSC.
- D. Fu and L. Finney, Expert Rev. Proteomics, 2014, 11, 13–19 CrossRef CAS PubMed.
- A. Lothian, D. J. Hare, R. Grimm, T. M. Ryan, C. L. Masters and B. R. Roberts, Front. Aging Neurosci., 2013, 5, UNSP 35 Search PubMed.
- E. A. Roberts and B. Sarkar, Curr. Opin. Clin. Nutr. Metab. Care, 2014, 17, 425–430 CrossRef CAS PubMed.
- W. Shi, M. Punta, J. Bohon, J. M. Sauder, R. D'Mello, M. Sullivan, J. Toomey, D. Abel, M. Lippi, A. Passerini, P. Frasconi, S. K. Burley, B. Rost and M. R. Chance, Genome Res., 2011, 21, 898–907 CrossRef CAS PubMed.
- J. Hegoczki, B. Janzso and A. Suhajda, Acta Aliment., 1995, 24, 181–190 Search PubMed.
- J. P. Riley and I. Roth, J. Mar. Biol. Assoc. U. K., 1971, 51, 63–72 CrossRef CAS.
- C. Jeffryes, T. Gutu, J. Jiao and G. L. Rorrer, ACS Nano, 2008, 2, 2103–2112 CrossRef CAS PubMed.
- J. H. Martin and G. A. Knauer, Geochim. Cosmochim. Acta, 1973, 37, 1639–1653 CrossRef CAS.
- M. S. Chauton, L. M. B. Skolem, L. M. Olsen, P. E. Vullum, J. Walmsley and O. Vadstein, J. Appl. Phycol., 2015, 27, 777–786 CrossRef PubMed.
- S. A. Skrabal, W. J. Ullman and G. W. Luther, Mar. Chem., 1992, 37, 83–103 CrossRef CAS.
- A. Scheffel, N. Poulsen, S. Shian and N. Kroeger, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3175–3180 CrossRef CAS PubMed.
- Y. Yan, D. Wang and P. Schaaf, Dalton Trans., 2014, 43, 8480–8485 RSC.
- G. J. Bedwell, Z. Zhou, M. Uchida, T. Douglas, A. Gupta and P. E. Prevelige, Biomacromolecules, 2015, 16, 214–218 CrossRef CAS PubMed.
- N. Kroger, M. B. Dickerson, G. Ahmad, Y. Cai, M. S. Haluska, K. H. Sandhage, N. Poulsen and V. C. Sheppard, Angew. Chem., Int. Ed., 2006, 45, 7239–7243 CrossRef CAS PubMed.
- M. Cargnello, T. R. Gordon and C. B. Murray, Chem. Rev., 2014, 114, 9319–9345 CrossRef CAS PubMed.
- Y. Lang, F. del Monte, B. J. Rodriguez, P. Dockery, D. P. Finn and A. Pandit, Sci. Rep., 2013, 3, 3205 Search PubMed.
- T. Ueki and H. Michibata, Coord. Chem. Rev., 2011, 255, 2249–2257 CrossRef CAS.
- J. P. Gaffney and A. M. Valentine, Dalton Trans., 2011, 40, 5827–5835 RSC.
- J. H. Swinehar, W. R. Biggs, D. J. Halko and N. C. Schroede, Biol. Bull., 1974, 146, 302–312 CrossRef.
- E. P. Levine, Science, 1961, 133, 1352–1353 CAS.
- D. A. Roman, J. Molina and L. Rivera, Biol. Bull., 1988, 175, 154–166 CrossRef CAS.
- M. Sugumaran and W. E. Robinson, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2012, 163, 1–25 CrossRef CAS PubMed.
- L. C. Dorsett, C. J. Hawkins, J. A. Grice, M. F. Lavin, P. M. Merefield, D. L. Parry and I. L. Ross, Biochemistry, 1987, 26, 8078–8082 CrossRef CAS.
- J. D. Popham and J. M. Dauria, Mar. Pollut. Bull., 1982, 13, 25–27 CrossRef CAS.
- A. Arias, A. Giangrande, M. C. Gambi and N. Anadon, Mediterr. Mar. Sci., 2013, 14, 162–171 Search PubMed.
- D. Fattorini, A. Notti, M. Nigro and F. Regoli, Environ. Sci. Pollut. Res., 2010, 17, 220–228 CrossRef CAS PubMed.
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 41, 21–40 CrossRef CAS.
- K. I. Sano and K. Shiba, J. Am. Chem. Soc., 2003, 125, 14234–14235 CrossRef CAS PubMed.
- L. Agosta, G. Zollo, C. Arcangeli, F. Buonocore, F. Gala and M. Celino, Phys. Chem. Chem. Phys., 2015, 17, 1556–1561 RSC.
- Q. Sheng, X. Li, W. Yin, L. Yu, Y. Ke and X. Liang, Anal. Methods, 2013, 5, 7072–7080 RSC.
- X. Zhao, Q. Wang, S. Wang, X. Zou, M. An, X. Zhang and J. Ji, J. Proteome Res., 2013, 12, 2467–2476 CrossRef CAS PubMed.
- Q. Wu, D. Wu and Y. Guan, Anal. Chem., 2014, 86, 10122–10130 CrossRef CAS PubMed.
- S. Steckbeck, J. Schneider, L. Wittig, K. Rischka, I. Grunwald and L. C. Ciacchi, Anal. Methods, 2014, 6, 1501–1509 RSC.
- Y. Razvag, V. Gutkin and M. Reches, Langmuir, 2013, 29, 10102–10109 CrossRef CAS PubMed.
- A. I. K. Eriksson, K. Edwards and V. A. Hernandez, Analyst, 2015, 140, 303–312 RSC.
- I. Stokroos, L. Litinetsky, J. J. L. van der Want and J. S. Ishay, Nature, 2001, 411, 654 CrossRef CAS PubMed.
- J. S. Ishay, K. Riabinin, M. Kozhevnikov, H. van der Want and I. Stokiroos, Biomacromolecules, 2003, 4, 649–656 CrossRef CAS PubMed.
- J. S. Ishay, Z. Barkay, N. Eliaz, M. Plotkin, S. Volynchik and D. J. Bergman, Naturwissenschaften, 2008, 95, 333–342 CrossRef CAS PubMed.
- A. M. Horst, A. C. Neal, R. E. Mielke, P. R. Sislian, W. H. Suh, L. Maedler, G. D. Stucky and P. A. Holden, Appl. Environ. Microbiol., 2010, 76, 7292–7298 CrossRef CAS PubMed.
- M. A. Kiser, H. Ryu, H. Jang, K. Hristovski and P. Westerhoff, Water Res., 2010, 44, 4105–4114 CrossRef CAS PubMed.
- L. Petrone, Adv. Colloid Interface Sci., 2013, 195, 1–18 CrossRef PubMed.
- M. J. McWhirter, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2003, 19, 3575–3577 CrossRef CAS.
- M. J. McWhirter, A. J. McQuillan and P. J. Bremer, Colloids Surf., B, 2002, 26, 365–372 CrossRef CAS.
- Y. Shabtai and G. Fleminger, Appl. Environ. Microbiol., 1994, 60, 3079–3088 CAS.
- G. Gertler, I. Brudo, R. Kenig and G. Fleminger, Materialwiss. Werkstofftech., 2003, 34, 1138–1144 CrossRef CAS.
- M. S. Jimenez, L. Rodriguez, J. R. Bertolin, M. T. Gomez and J. R. Castillo, Anal. Bioanal. Chem., 2013, 405, 359–368 CrossRef CAS PubMed.
- A. Raab, B. Ploselli, C. Munro, J. Thomas-Oates and J. Feldmann, Electrophoresis, 2009, 30, 303–314 CrossRef CAS PubMed.
- A. B. Nowakowski, W. J. Wobig and D. H. Petering, Metallomics, 2014, 6, 1068–1078 RSC.
- C. M. H. Ferreira, I. S. S. Pinto, E. V. Soares and H. M. V. M. Soares, RSC Adv., 2015, 5, 30989–31003 RSC.
- E. Z. Jahromi, W. White, Q. Wu, R. Yamdagni and J. Gailer, Metallomics, 2010, 2, 460–468 RSC.
- A. Sanz-Medel, A. B. S. Cabezuelo, R. Milacic and T. B. Polak, Coord. Chem. Rev., 2002, 228, 373–383 CrossRef CAS.
- A. Sarmiento-Gonzalez, J. Ruiz Encinar, A. M. Cantarero-Roldan, J. M. Marchante-Gayon and A. Sanz-Medel, Anal. Chem., 2008, 80, 8702–8711 CrossRef CAS PubMed.
- Y. Valasatava, A. Rosato, G. Cavallaro and C. Andreini, J. Biol. Inorg. Chem., 2014, 19, 937–945 CrossRef CAS PubMed.
- J. Pande, M. M. Szewczyk and A. K. Grover, Biotechnol. Adv., 2010, 28, 849–858 CrossRef CAS PubMed.
- S. R. Meyers, P. T. Hamilton, E. B. Walsh, D. J. Kenan and M. W. Grinstaff, Adv. Mater., 2007, 19, 2492–2498 CrossRef CAS.
- O. Stoevesandt and M. J. Taussig, Expert Rev. Proteomics, 2012, 9, 401–414 CrossRef CAS PubMed.
- D. Raimunda, T. Khare, C. Giometti, S. Vogt, J. M. Argueello and L. Finney, Metallomics, 2012, 4, 921–927 RSC.
- K. M. Dean, Y. Qin and A. E. Palmer, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 1406–1415 CrossRef CAS PubMed.
- E. L. Que, R. Bleher, F. E. Duncan, B. Y. Kong, S. C. Gleber, S. Vogt, S. Chen, S. A. Garwin, A. R. Bayer, V. P. Dravid, T. K. Woodruff and T. V. O'Halloran, Nat. Chem., 2015, 7, 130–139 CrossRef CAS PubMed.
- J. Zhou, Y. Hu, Y. Lin, H. Liu and P. Xie, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2957–2962 CrossRef CAS PubMed.
- J. V. Olsen, B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen and M. Mann, Cell, 2006, 127, 635–648 CrossRef CAS PubMed.
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1–35 CrossRef CAS PubMed.
- J. C. Tran, L. Zamdborg, D. R. Ahlf, J. E. Lee, A. D. Catherman, K. R. Durbin, J. D. Tipton, A. Vellaichamy, J. F. Kellie, M. Li, C. Wu, S. M. M. Sweet, B. P. Early, N. Siuti, R. D. LeDuc, P. D. Compton, P. M. Thomas and N. L. Kelleher, Nature, 2011, 480, 254–258 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |