
Protein kinase CK2 regulates metal toxicity in neuronal cells
Mohammad S.
Zaman
a,
Adam J.
Johnson
a,
Gabriele
Bobek
b,
Sindy
Kueh
b,
Cindy
Kersaitis
a,
Trevor D.
Bailey
a,
Yossi
Buskila
c and
Ming J.
Wu
*a
aSchool of Science and Health, Western Sydney University, Locked Bag 1797, Penrith, NSW 2751, Australia. E-mail: m.wu@uws.edu.au; Fax: +61 2 4620 3025; Tel: +61 2 4620 3089
bSchool of Medicine, Western Sydney University, Locked Bag 1797, Penrith, NSW 2751, Australia
cBioelectronics and Neuroscience Group, The MARCS Institute, Western Sydney University, Locked Bag 1797, Penrith, NSW 2751, Australia
First published on 25th November 2015
Abstract
Protein kinase CK2 is a pleiotropic tetrameric enzyme, regulating numerous biological processes from cell proliferation to stress response. This study demonstrates for the first time that CK2 is involved in the regulation of metal uptake and toxicity in neuronal cells. After the determination of inhibitory concentrations (IC50) for a range of metal salts (ZnSO4, Al(mal)3, CoCl2, CrO3, NaAsO2 and CaCl2) in Neuro-2a mouse neuroblastoma cells, the effect of CK2 on metal toxicity was investigated by three lines of experiments using CK2 inhibitors, metal ion specific fluorophores and siRNA-mediated knockdown of CK2 expression. The results showed that both CK2 inhibitors, 4,5,6,7-tetrabromobenzotriazole (TBB) and quinalizarin, markedly reduced the toxicity of Zn(II), Al(III), Co(II), Cr(VI) and As(III). Confocal microscopy imaging revealed that Zn(II) uptake was accompanied by the increase of intracellular Ca(II) in Neuro-2a cells treated with IC50 of ZnSO4 (240 μM), and such concurrent elevation of intracellular Zn(II) and Ca(II) was blocked by TBB and quinalizarin. The role of CK2 in metal uptake was further characterised using specific siRNA against each of the three subunits (CK2α, α′ and β) and the data demonstrate that CK2α′ is the prominent subunit regulating the metal toxicity. Finally, the role of CK2 in metal toxicity was found to be conserved in the distant species-Saccharomyces cerevisiae by employing the complete deletion mutants of CK2 (cka1Δ, cka2Δ, ckb1Δ and ckb2Δ). Taken together, these findings shed light on a new facet of CK2 functionality and provide a basis for further research on the regulation of Zn(II) and Ca(II) homeostasis by CK2.
Introduction
Protein kinase CK2 (CK2), a ubiquitous and pleiotropic serine/threonine kinase, is the first protein kinase discovered by Burnett and Kennedy in 1954.1 Many characteristics of the kinase have been discovered since then, including its biochemical structure and function, biological roles and involvement in diseases. Structurally, mammalian CK2 is usually a heterotetrameric holoenzyme comprised of dual catalytic subunits (CK2α and CK2α′) bound to a central homodimer of non-catalytic regulatory β subunits.2 However, each of the catalytic subunits can also function alone.3 A unique and enigmatic feature of CK2 is its constitutive activity, which signifies the essentiality of the kinase for basic cell growth.4,5 On top of such basal activity, CK2 can be further activated by extracellular challenges or signals, and then act upon hundreds of potential protein substrates.6 As a result, the cells can proliferate, differentiate or go into apoptosis. Any deregulation of CK2 in terms of expression level and cellular localisation can lead to cancers.7–9 Despite the accumulation of current knowledge on CK2, the complex interplay of CK2 as a holoenzyme or discrete functional monomers in the context of various stimuli remains to be explored.In this work, we focused on the role of CK2 in metal toxicity of neuronal cells. Thus far there is scant knowledge on this front. The link between CK2 and metal homeostasis has not explicitly been established until the recent findings in the model organism Saccharomyces cerevisiae and the breast cancer cell line MCF-7. Both Tun et al.10 and Jin et al.11 revealed, via genome-wide deletion mutants screening and transcriptomics in S. cerevisiae, that CK2 is related to metal toxicity. Taylor et al.12 demonstrated that the intracellular zinc concentration in the breast cancer cell line MCF-7 was increased due to the phosphorylation of endoplasmic reticulum zinc channel ZIP7 by CK2. This phosphorylation results in the gated release of Zn2+ from intracellular stores. There is no report of involvement of CK2 in regulating metal ion homeostasis of neurons.
CK2 is intrinsically involved in the biology of neurons. It is critical in mammalian developmental processes such as embryonic neural tube formation.13 CK2 is more abundant in brain tissue than in any other part of the body and its activity in adult rat tissues has been shown to be highest in brain and testis.14 Many CK2 substrates exist in synaptic and nuclear compartments. CK2 is involved in long-term potentiation in the hippocampus and neurotrophins stimulate activity of CK2 in the hippocampus.14,15 It is suggested that CK2 may play a role in neuronal adaptations to a changing environment through modulating synaptic connections, regulate specific neuronal activities and participate in survival and death decision making events.14 CK2 has also been linked to various neurodegenerative diseases, particularly AD.16 Experimental evidence demonstrates that metal ions are involved in amyloid plaque formation.17–19 Therefore, there is a likelihood that CK2 up-regulation could lead to metal ion accumulation and hence trigger amyloid aggregation.
Metal ions such as zinc [Zn(II)] and calcium [Ca(II)] are an essential part of neurobiology. Both are key second messengers in signal transduction pathways.20–22 However, a tight regulation of their homeostasis is critical to the cell's viability as their overload is detrimental. Apart from Zn(II) and Ca(II), this study also touches on hexavalent chromium [Cr(VI)], aluminium [Al(III)], arsenic [As(III)] and cobalt [Co(II)]. Chromium and cobalt toxicity has been highlighted recently by the severe health problems suffered by hip implant patients. Stainless steel, which contains chromium and cobalt, has been used in metal-on-metal hip prostheses.23 Thousands of patients in Australia and overseas have experienced pain, discomfort and memory loss after receiving the defective hip implants, due to toxic poisoning of chromium and cobalt released by metal-on-metal erosion.24 We anticipate from the outset that this work could provide useful information towards understanding the metal toxicity in neurons. Firstly we determined the inhibitory concentrations (IC50) for a range of metal salts (ZnSO4, Al(mal)3, CoCl2, CrO3, NaAsO2 and CaCl2) in Neuro-2a cells, and then investigated the effect of CK2 on metal toxicity by three lines of experiments using CK2 inhibitors (TBB and quinalizarin), metal ion specific fluorophores (FluoZin-3 AM and Fluo-4 AM) and siRNA-mediated knockdown of CK2 subunit expression. The findings demonstrate that CK2 is involved in regulating metal toxicity.
Experimental
Cell culture conditions
Mouse Neuro-2a neuroblastoma cells were cultured in high glucose DMEM (Life Technologies), supplemented with 10% foetal bovine serum (FBS), 1% antibiotics (penicillin/streptomycin) and L-glutamine. Neuro-2a cells were incubated at 37 °C with 5% CO2 and observed daily until 80% confluent. Cells were then washed with phosphate-buffered saline (PBS), trypsinised, counted using a hemocytometer and prepared for plating.Determination of the inhibitory concentration (IC50) of the metal ions
Neuro-2a cells were seeded into 96-well plates with 200 μl of complete DMEM containing 7000 cells per well. Plates were incubated in 5% CO2 at 37 °C for 32 h prior to metal treatments. Varying concentrations of metals were used, including CaCl2 (0, 0.1, 0.2, 0.4, 0.8, 1, 2 and 5 mM), ZnSO4 (0, 125, 150, 160, 180, 200, 220, 240 and 260 μM), CoCl2 (0, 150, 200, 220, 240, 260, 280, 300, 320 and 340 μM), Al(mal)3 (0, 25, 50, 75, 100, 125, 125, 150, 160, 170, 180, 190, 200 μM), NaAsO2 (0, 5, 10, 15, 20, 25, 30, 35 and 40 μM) and CrO3 (0, 5, 10, 15, 20 and 40 μM), in order to establish individual IC50. Al(mal)3 was prepared in the laboratory from aluminium chloride hexahydrate and maltol (3-hydroxy-2-methyl-4H-pyran-4-one). Briefly, for 10–15 g of Al(mal)3 complex, 15.5 g (122.8 mM) of maltol and 9.9 g (40.9 mM) of AlCl3·6H2O were dissolved in 160 ml of deionised water with mild heating applied. When all of the particulate material was dissolved, the pH was adjusted to 8.3. Subsequently, the mixture was stirred and heated to 65 °C and a fine precipitate was produced. After cooling, the off-white crystals obtained were filtered, washed several times with acetone, and dried overnight in a vacuum desiccator. The synthesised complex was confirmed with 1H Nuclear Magnetic Resonance spectroscopy. Stock solutions of metals were prepared and 10 μl of each metal concentration was applied to each well, while 10 μl of sterile H2O was used for control. Plates were then incubated for 6 h and an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) (Sigma-Aldrich) colorimetric assay was conducted to quantify Neuro-2a cell viability. Wells were treated with 50 μl of MTT (5 mg ml−1 in PBS) per well and incubated for 2 h, and then the formazan crystals were solubilized in 100 μl of dimethyl sulfoxide (DMSO) (Sigma-Aldrich) per well. The plates were shaken gently before optical absorbance at 595 nm (A595) was measured using a spectrophotometer (Multiskan EX, Thermo Electron).Inhibition of CK2 activity and metal treatment
Multiple 96-well plates were prepared containing 7000 cells per well in 200 μl of complete DMEM and incubated for 32 h. Prior to the addition of metal ions, the cells were treated with either TBB (4,5,6,7-tetrabromo-1H-benzotriazole) (Calbiochem, Millipore) or quinalizarin (Sigma-Aldrich) at a final concentration of 20 μM for 3 h. At this dose both inhibitors were not toxic according to our titration and the previous studies used the similar dose.25,26 It was then followed by 4 h incubation of metals at IC25 and IC50 concentrations. At the end of the incubation, the cell viability was measured by MTT assay as described earlier.Knockdown of CK2 gene expression by siRNA
SMARTpool siRNA targeting mouse CK2 (CK2α, CK2α′ and CK2β) were purchased from Dharmacon, USA. The targeting sequences for CK2α are UGAAUUAGAUCCACGUUUC, GCAAUUGUACCAGACGUUA, CAGAAAGCUUCGGCUAAUA, AGACUAUGACAUUCGAUUU. The targeting sequences for CK2α′ are GCAAGUAUAGUGAAGUAUU, CAACAGAGAUUGACCGCCA, GGUGGAACAAAUAUCAUUA, GAACUAUAUGGUUAUCUGA. The targeting sequences for CK2β are CAGCUGAGAUGCUUUAUGG, CCAAGUGCAUGGACGUGUA, GGUAAUGAAUUCUUCUGUG, GCAGGGAGACUUUGGCUAC. Each siRNA stock (10 μM) was prepared in siRNA buffer (pH 7.3) containing 300 mM potassium chloride, 31 mM HEPES and 2.1 mM magnesium chloride. For each siRNA transfection, two Eppendorf tubes were firstly prepared, with tube 1 containing 20 μl of 10 μM siRNA and 480 μl OPTI-MEM (Life Technologies), and tube 2 containing 20 μl lipofectamine 2000 (Invitrogen) and 480 μl OPTI-MEM. After 5 min incubation at room temperature, the content of tube 1 was pipetted into tube 2 for each siRNA and left at room temperature for 20 min. A mock tube with no siRNA containing 980 μl OPTI-MEM with 20 μl lipofectamine was also prepared as a control.Neuro-2a cells were cultured in a tissue culture flask for 32 h until 80% confluent. Cells were washed with PBS then trypsinised and 5 ml fresh complete DMEM was added. The cells were counted and 2 × 106 cells in 5 ml were prepared into each tube. The cells were centrifuged at 900g for 5 min. The supernatant was removed and cells were then washed with 5 ml PBS. Each cell pellet was re-suspended in 1 ml of each siRNA mixture which had remained at room temperature for 20 min as described earlier. Gentle mixing was performed to allow complete re-suspension of the cells. After 20 min, 9 ml of OPTI-MEM was added and the cells were thoroughly mixed and aliquoted into a 96-well plate at 100 μl per well. The plates were incubated at 37 °C with 5% CO2 for 76 h. IC25 and IC50 of the metals in 5 μl volume per well were used to treat the transfected cells in the plates and incubated for 5 h. An MTT assay was then conducted to verify cell viability.
Imaging of intracellular metal ions by confocal microscopy
Confocal imaging glass bottom culture dishes (MatTek) were firstly coated with 20 μl of gelatin on the glass surface and allowed to dry for 2 h. Neuro-2a cells in DMEM complete medium with 25 mM HEPES buffer were seeded at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per ml into each imaging dish at a final volume of 1 ml. Cells were incubated at 37 °C with 5% CO2 for 32 h. CK2 inhibitor TBB or quinalizarin at 20 μM was pipetted into all but the control dishes and allowed to incubate for 3 h. The metal treatments were followed using both IC25 (120 μM) and IC50 (240 μM) of ZnSO4 for 1 h before medium was removed, then 1 ml fresh live cell imaging medium (Life Technologies) containing 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 20 mM HEPES at pH 7.4 was added. The Fluo-4 AM calcium probe (Life Technologies) at 50 μg was dissolved in 100 μl of imaging medium, 8 μl DMSO and 2 μl of 50% pluronic acid F127 detergent. For the FluoZin-3 AM zinc probe (Life Technologies) 100 μg was dissolved in 200 μl of DMSO and 2 μl of 50% pluronic acid F127. The probe stocks were sonicated for 5 min before adding 15 μl of Fluo-4 AM or FluoZin-3 AM into imaging dishes. The confocal imaging dishes were further incubated for 1 h, the medium was then removed and a further 1 ml fresh imaging medium was added and allowed a further 30 min incubation before imaging. Images were acquired using a LSM-5 Confocal Microscope system (Carl Zeiss Pty Ltd, North Ryde, Australia), with excitation at 488 nm and emission detected at 510–530 nm. Gain settings for florescence imaging were maintained constant for each imaging probe to allow for quantification of intracellular florescence intensity through Image-J imaging software.
000 cells per ml into each imaging dish at a final volume of 1 ml. Cells were incubated at 37 °C with 5% CO2 for 32 h. CK2 inhibitor TBB or quinalizarin at 20 μM was pipetted into all but the control dishes and allowed to incubate for 3 h. The metal treatments were followed using both IC25 (120 μM) and IC50 (240 μM) of ZnSO4 for 1 h before medium was removed, then 1 ml fresh live cell imaging medium (Life Technologies) containing 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 20 mM HEPES at pH 7.4 was added. The Fluo-4 AM calcium probe (Life Technologies) at 50 μg was dissolved in 100 μl of imaging medium, 8 μl DMSO and 2 μl of 50% pluronic acid F127 detergent. For the FluoZin-3 AM zinc probe (Life Technologies) 100 μg was dissolved in 200 μl of DMSO and 2 μl of 50% pluronic acid F127. The probe stocks were sonicated for 5 min before adding 15 μl of Fluo-4 AM or FluoZin-3 AM into imaging dishes. The confocal imaging dishes were further incubated for 1 h, the medium was then removed and a further 1 ml fresh imaging medium was added and allowed a further 30 min incubation before imaging. Images were acquired using a LSM-5 Confocal Microscope system (Carl Zeiss Pty Ltd, North Ryde, Australia), with excitation at 488 nm and emission detected at 510–530 nm. Gain settings for florescence imaging were maintained constant for each imaging probe to allow for quantification of intracellular florescence intensity through Image-J imaging software.
Confocal imaging was also conducted for CK2 subunit knockdown using CK2α, CK2α′ and CK2β siRNA. siRNA transfected Neuro-2a cells (1.20 × 105 cells per ml) prepared as previously described were seeded into each imaging dish. Cells were incubated at 37 °C with 5% CO2 for 32 h followed by a 1 h metal treatment of ZnSO4 at IC50 (240 μM). The confocal imaging was then performed in the same way as described above.
Metal treatment of CK2 knockout mutants of the model organism S. cerevisiae
Single colonies of the wild type (BY4743) and the CK2 knockout mutants (cka1Δ, cka2Δ, ckb1Δ and ckb2Δ) were inoculated into minimal medium (pH 4.3) (20 g L−1D-glucose, 1.7 g L−1 yeast nitrogen base without ammonium sulphate, 5 g L−1 ammonium sulphate and 0.79 g L−1 complete supplement mixture which contained 10 mg L−1 adenine, 50 mg L−1L-arginine, 80 mg L−1L-aspartic acid, 20 mg L−1L-histidine HCl, 50 mg L−1L-isoleucine, 100 mg L−1L-leucine, 50 mg L−1L-lysine HCl, 20 mg L−1L-methionine, 50 mg L−1L-phenylalanine, 100 mg L−1L-threonine, 50 mg L−1L-tryptophan, 50 mg L−1L-tyrosine, 140 mg L−1L-valine, 20 mg L−1 uracil) and incubated at 30 °C, with shaking at 150 rpm in an orbital mixer incubator (Ratek) until the cultures reached optical density 600 nm (OD600) of approximately 1.0. Cultures were then diluted in minimal medium to OD600 0.05 and transferred into 96-well plates. The plated mutants and wild type were then treated with 5 metals at varying concentrations, including CaCl2 (0, 40, 80, 160, 320 and 640 mM), ZnSO4 (0, 4, 8, 16, 25 and 32 mM), CoCl2 (0, 0.4, 0.8, 1.6, 3.2 and 6.4 mM), Al2(SO4)3 (0, 0.4, 0.8, 1.6, 2.5 and 3.2 mM), NaAsO2 (0, 0.2, 0.4, 0.8, 1.6 and 3.2 mM) and CrO3 (0, 0.05, 0.1, 0.2, 0.4 and 0.8 mM).The OD600 at the time of treatment (T0) was measured using a 96-well plate reader (Multiskan EX, Thermo Electron, USA) then the cultures were incubated at 30 °C while shaking at 750 rpm (Ika MTS 2/4 digital microtiter shaker). OD600 was subsequently measured every 4 h for a period of 24 h.
Statistical analysis
The data were analysed with one-way ANOVA in SPSS statistical software. Analysis and quantification of confocal images was achieved through Image-J with Fiji attachment software.Results
Determination of IC50 of metal ions
Inhibitory concentrations (IC50) of the metal ions for the viability of Neuro-2a cells were determined (Fig. 1). The IC50 of ZnSO4 is 240 μM, CoCl2 310 μM, Al(mal)3 150 μM, NaAsO2 15 μM and CrO3 13 μM. Ca(II) showed no inhibition to the cell's growth up to 2 mM CaCl2.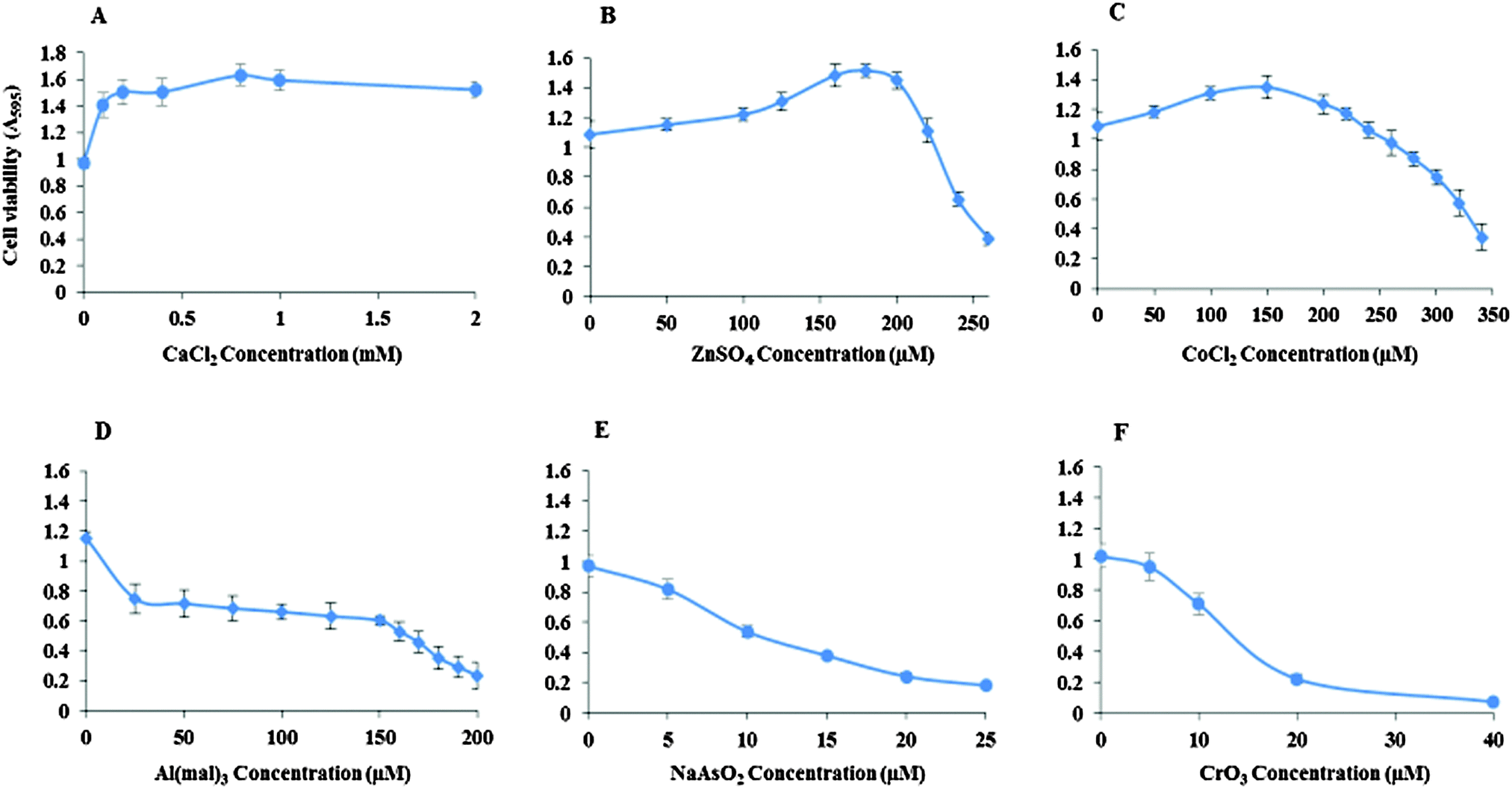 | ||
| Fig. 1 Inhibitory concentration (IC50) of the metal ions. Neuro-2a cells were cultured in the presence of CaCl2 (A), ZnSO4 (B), CoCl2 (C), Al(mal)3 (D), NaAsO2 (E) and CrO3 (F). No inhibition was exhibited by CaCl2 up to 2 mM. IC50 for ZnSO4 was found to be 240 μM, CoCl2 310 μM, Al(mal)3 150 μM, NaAsO2 15 μM and CrO3 13 μM. Error bars denote standard deviation of 8 replicates for each concentration. | ||
Effect of CK2 inhibition by TBB and quinalizarin on metal toxicity
Inhibition of CK2 by TBB or quinalizarin at 20 μM demonstrated a significant reduction in toxicity of Zn(II), Al(III), As(III) and Cr(VI) compared to control (Fig. 2). Both of the inhibitors have a similar effect, although the effect of quinalizarin was comparatively more pronounced. As expected, calcium showed no toxicity and inhibition of CK2 had no effect on the cell under Ca(II) exposure.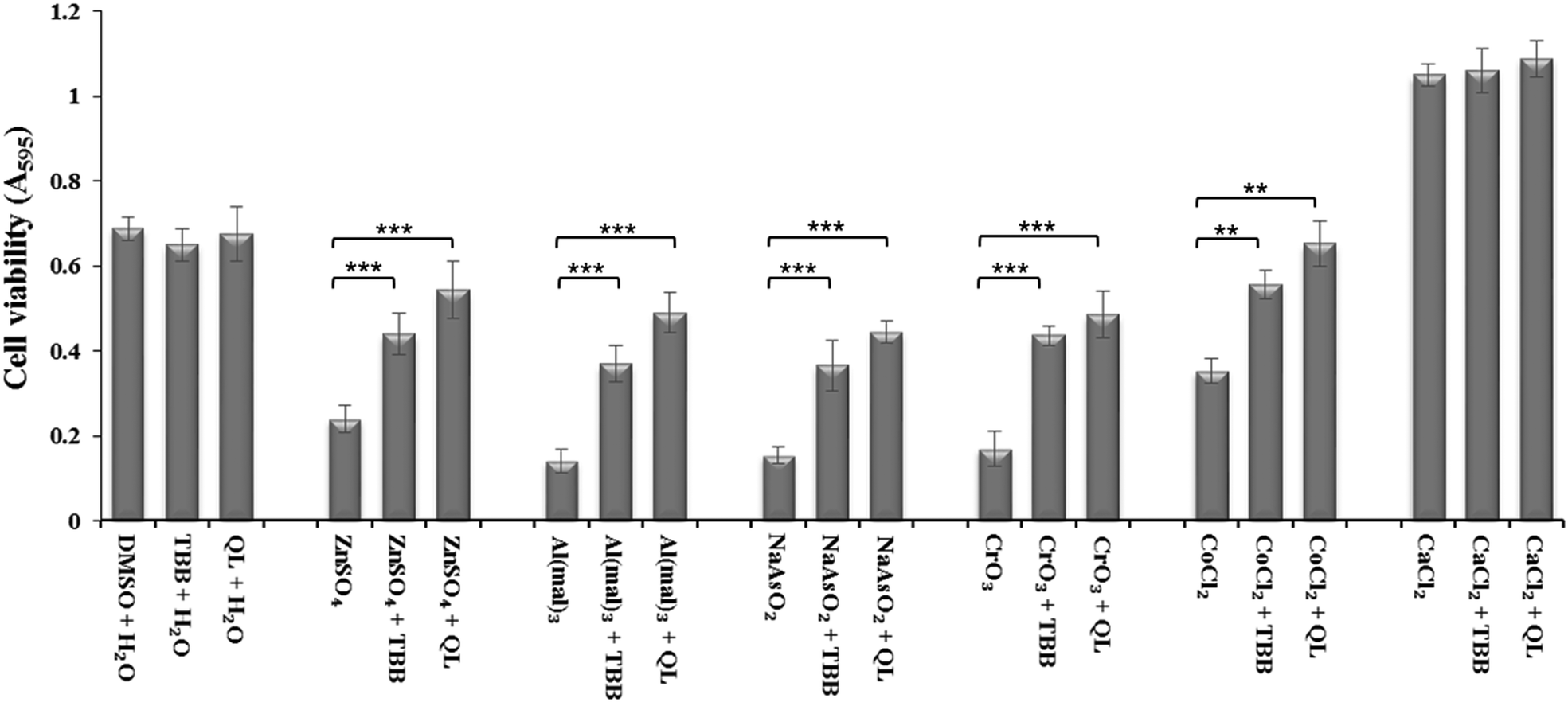 | ||
| Fig. 2 Inhibition of CK2 by TBB and quinalizarin reduces metal toxicity. Neuro-2a cells were grown for 32 h followed by 3 h treatment of 20 μM inhibitor and 5 h incubation with IC50 treatment of ZnSO4 (240 μM), Al(mal)3 (150 μM), NaAsO2 (15 μM), CrO3 (13 μM), CoCl2 (310 μM) and 2 mM CaCl2. The metal toxicity was then measure by MTT assay. P values were calculated by one-way anova. ** denotes P < 0.01 and *** P < 0.001. Error bars represent standard deviation of 8 replicates for each treatment. | ||
Effect of CK2 subunit knockdown via siRNA on metal toxicity
siRNA knockdown of CK2 subunits α, α′ and β exposed individual roles of CK2 subunits in regulating metal ion toxicity (Fig. 3). Mock transfection plus metal treatments showed clear toxicity except CaCl2, by displaying lower cell viability as compared to CK2 subunit knockdowns. Interestingly, the MTT assay demonstrated that CK2α′ is the prominent subunit in regulating metal toxicity against ZnSO4, Al(mal)3, NaAsO2, CrO3 and CoCl2. Little effect of CK2β knockdown was revealed against Zn(II), Cr(VI) and Co(II).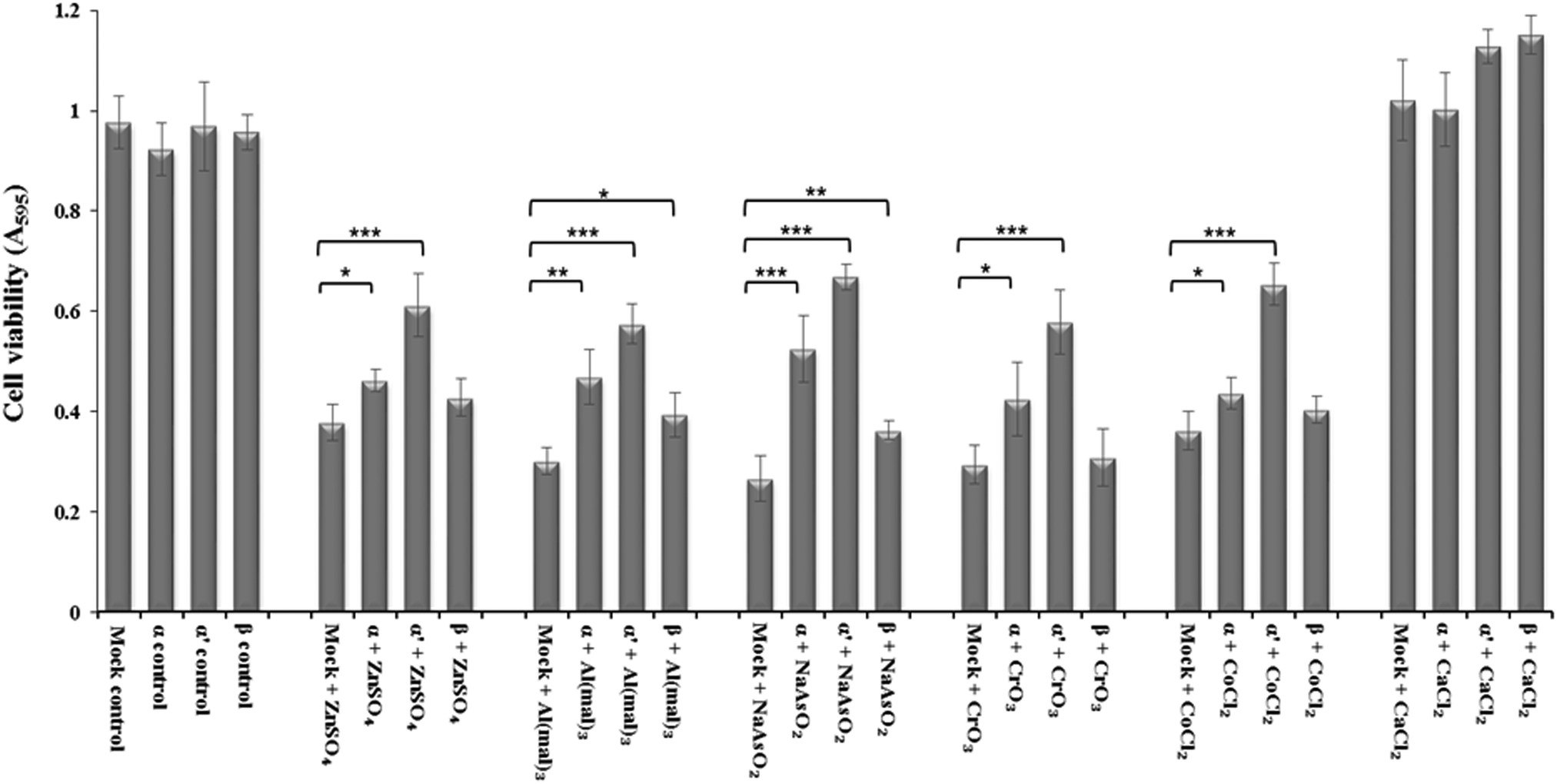 | ||
| Fig. 3 siRNA knockdown of CK2 subunits decreases metal toxicity. Transfection of siRNA was followed by a 32 h incubation of Neuro-2a cells, 5 h treatment of metal ions and then an MTT assay to quantify cell viability. No siRNA mock treatment served as the control. * denotes P < 0.05, ** P < 0.01 and *** P < 0.001. Error bars represent standard deviation of at least 8 replicates for each treatment. | ||
Imaging of intracellular zinc and calcium by confocal microscopy
Confocal imaging for intracellular zinc and calcium was carried out using specific fluorophores for zinc (FluoZin-3 AM) and calcium (Fluo-4 AM) in Neuro-2a cells. A marked reduction of intracellular Zn(II) in the cells pre-treated with CK2 inhibitors TBB or quinalizarin, as compared to cells treated with ZnSO4 IC50 (240 μM) alone was demonstrated (Fig. 4A–E). Intriguingly, it was found that intracellular Ca(II) concentration was increased by ZnSO4 IC50 treatment, but this was prevented by inhibitor pre-treatment (Fig. 4F–J). Neuro-2a cells treated with ZnSO4 IC50 (240 μM) (Fig. 4B and G) lost the typical dendrite structure observed in control cells (Fig. 4A and F), whereas cells pre-treated with CK2 inhibitors (Fig. 4C, D, H and I) maintained the neuronal morphology.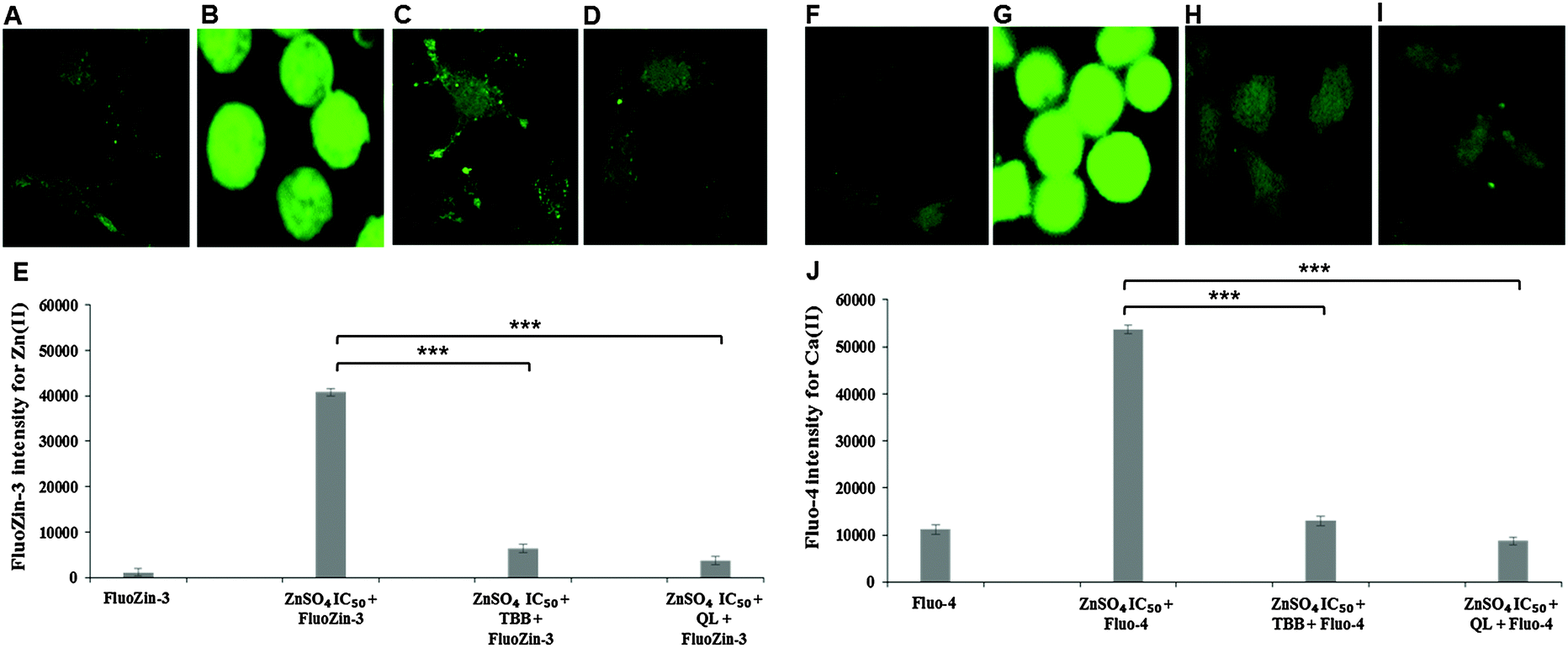 | ||
| Fig. 4 Confocal imaging of CK2 inhibition through TBB and quinalizarin. Neuro-2a cells were grown in confocal imaging culture dishes for 32 h then treated with either TBB or quinalizarin (QL) for 3 h followed by ZnSO4 IC50 (240 μM) treatment and incubation with fluorescent probe FluoZin-3 AM or Fluo-4 AM. The morphological effect was observed by images at 63× magnification (A–D, F–I) while fluorescence intensity (E and J) was quantified with confocal images at 40× magnifications. (A) FluoZin-3 only, (B) ZnSO4 IC50 plus FluoZin-3, (C) TBB plus ZnSO4 IC50 and FluoZin-3, (D) quinalizarin plus ZnSO4 IC50 and FluoZin-3 and (E) quantification of FluoZin-3 fluorescence intensity. (F) Fluo-4 only, (G) ZnSO4 IC50 plus Fluo-4, (H) TBB plus ZnSO4 IC50 and Fluo-4, (I) quinalizarin plus ZnSO4 IC50 and Fluo-4 and (J) quantification of Fluo-4 fluorescence intensity. *** P < 0.001 respectively. Error bars represent standard deviation of 45 cells for each treatment. | ||
Similarly to inhibitor treatment, the siRNA knockdown of CK2 subunits α, α′ and β led to a significant reduction in metal toxicity against ZnSO4 IC50 (240 μM) (Fig. 5). A marked reduction of intracellular Zn(II) in the cells treated with CK2 siRNA compared to mock siRNA treatment was demonstrated in cells incubated with ZnSO4 IC50 (Fig. 5A–F), with CK2α′ knockdown having the most prominent effect. The increase in intracellular Ca(II) concentration observed with ZnSO4 IC50 treatment was also reduced by CK2 knockdown (Fig. 5G–L). Neuro-2a cells treated with CK2 siRNA prior to ZnSO4 IC50 (240 μM) exposure retained an apparent healthy morphology (Fig. 5C–E and I–K) similar to mock siRNA control cells not exposed to ZnSO4 IC50 (A and G) This rescue from the morphological changes and cell death observed with exposure of control cells to ZnSO4 IC50 (Fig. 5B and H) is in agreement with the toxicity observed in the MTT assay (Fig. 3). These results confirm that CK2α′ is the prominent subunit involved in regulating Zn(II) toxicity. Additionally, confocal imaging confirmed that CK2 subunit knockdowns, particularly CK2α′, significantly decrease intracellular Ca(II). Therefore, taken together Fig. 4 and 5, the data demonstrate CK2 regulates the transport of both Zn(II) and Ca(II).
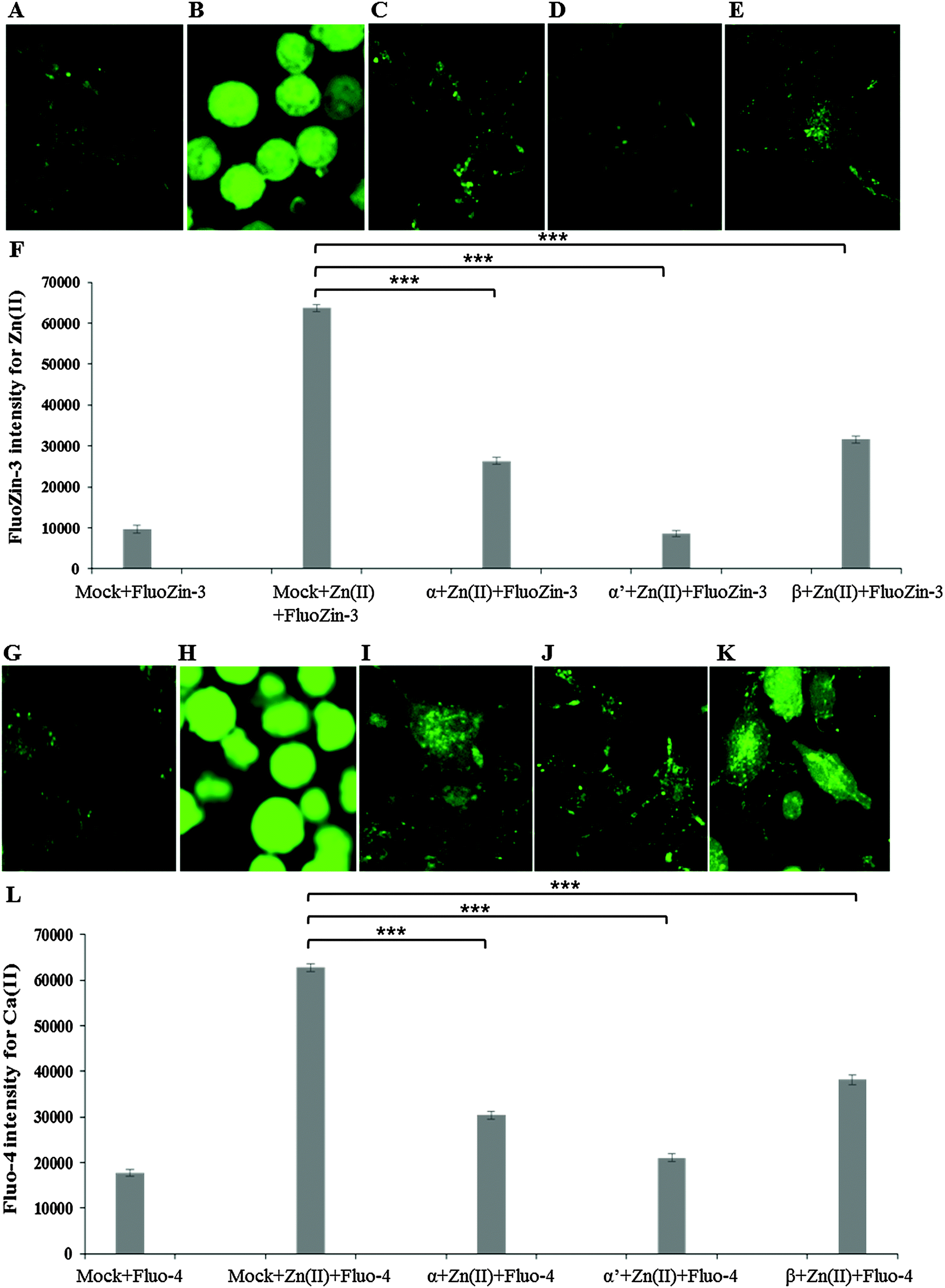 | ||
| Fig. 5 Effect of siRNA against CK2 observed by confocal imaging. Neuro-2a cells were transfected with siRNA to knockdown CK2 subunits α, α′, β and grown in confocal imaging plates for 32 h prior to ZnSO4 IC50 (240 μM) treatment and incubation with the fluorescent probe FluoZin-3 AM or Fluo-4 AM. (A) Mock transfection plus FluoZin-3, (B) Mock transfection plus ZnSO4 IC50 and FluoZin-3, (C) CK2α siRNA plus ZnSO4 IC50 and FluoZin-3, (D) CK2α′ siRNA plus ZnSO4 IC50 and FluoZin-3, (E) CK2β siRNA plus ZnSO4 IC50 and FluoZin-3, (F) quantification of fluorescence intensity of FluoZin-3. (G) Mock transfection plus Fluo-4, (H) Mock transfection plus ZnSO4 IC50 and Fluo-4, (I) CK2α siRNA plus ZnSO4 IC50 and Fluo-4, (J) CK2α′ siRNA plus ZnSO4 IC50 and Fluo-4, (K) CK2β siRNA plus ZnSO4 IC50 and Fluo-4. *** denotes P < 0.001. Error bars represent standard deviation of 45 cells for each treatment. | ||
Conservation of CK2's role in metal toxicity in S. cerevisiae
The complete knockout mutants for individual CK2 subunits in S. cerevisiae (cka1Δ, cka2Δ, ckb1Δ and ckb2Δ) were screened against the panel of metal ions used in Neuro-2a cells. The findings, as shown in Fig. 6, are strikingly similar to what were uncovered in Neuro-2a cells. CKA2 (analogous to CKα′ in mammalian cells) was found to be the prominent subunit involved in metal toxicity such as in response to Zn(II) and Al(III). Interestingly deletion of one of the two yeast regulatory subunits, CKB2, conferred the greatest resistance to As(III) insult, while deletion of the other (CKB1) led to the highest resistance to Cr(VI) toxicity. That these two mutants differ in phenotype under different stressors, and based on the accepted fact that in yeast both regulatory subunits are required for tetramer formation,27 suggests that these regulatory subunits have roles in regulating metal toxicity as monomers.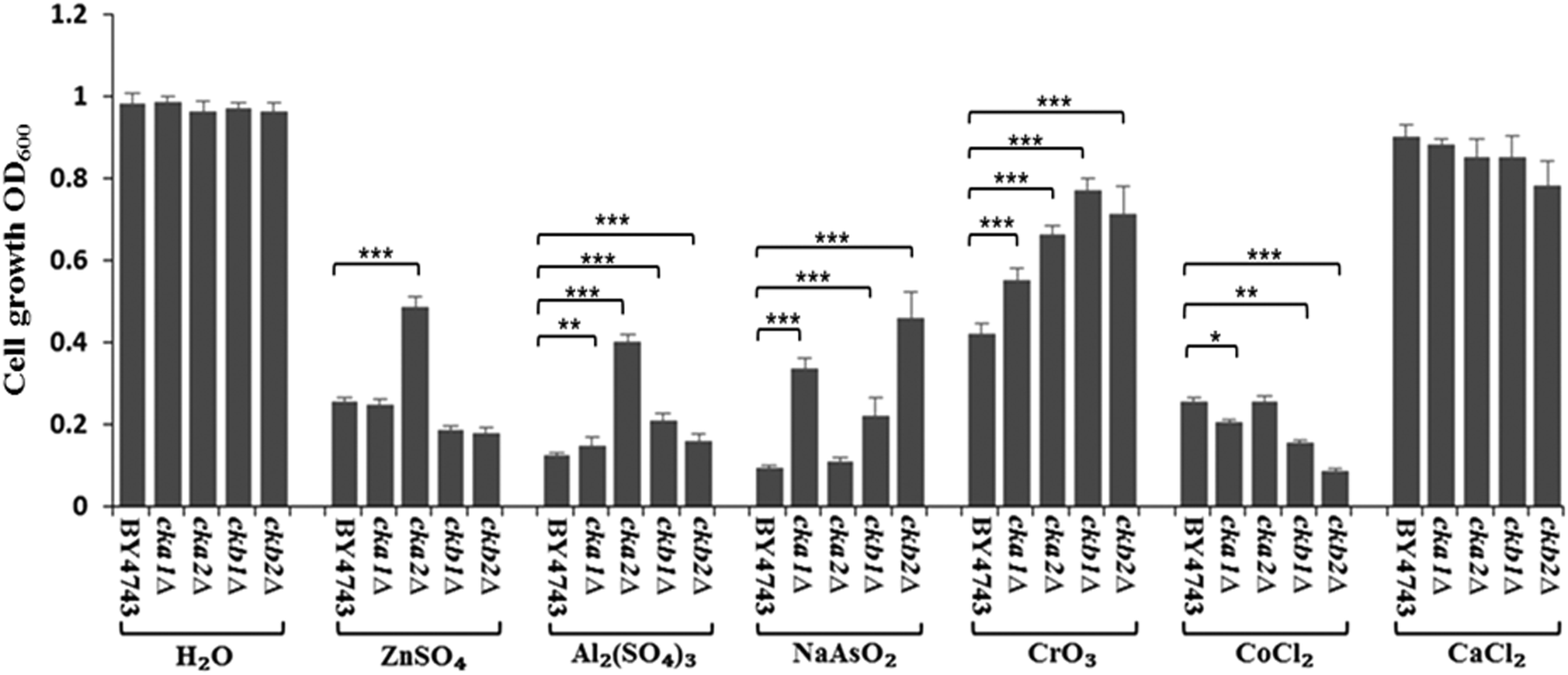 | ||
| Fig. 6 Validation of CK2's role in metal toxicity by the model organism S. cerevisiae. Wild type S. cerevisiae (BY4743) and the CK2 deletion mutants (cka1Δ, cka2Δ, ckb1Δ and ckb2Δ) were screened against H2O (control), ZnSO4 (25 mM), Al2(SO4)3 (2.5 mM), NaAsO2 (1.6 mM), CrO3 (0.4 mM), CoCl2 (3.2 mM) and CaCl2 (80 mM). * denotes P < 0.05, ** P < 0.01 and *** P < 0.001. Error bars represent standard error of 15 replicates for each treatment. | ||
Discussion
By three lines of evidence from the specific inhibitors, confocal imaging and siRNA knockdown, this study demonstrates clearly that protein kinase CK2 is involved in metal toxicity in neuronal cells. Heretofore, such a role for CK2 has never been reported. Given that metal ions can be the contributing factor for certain health problems such as Alzheimer's disease,17–19 the identification of CK2's role in regulating metal toxicity is therefore relevant to the understanding of human disorders in which metal ions play a part.Under the toxic dosage of metal ions, CK2 activity is inversely related to the cell viability. As previous studies demonstrate, CK2 plays a key role in promoting cell proliferation and supressing apoptosis.28 Such effect is due to its constitutive basal activity as well as additional activation by cellular stimuli such as growth factors. The aberrant and prolonged activation of CK2 may lead to the development of diseases such as certain cancers. However, the knowledge about the acute regulation of CK2 in response to sudden insults such as toxic metal ion Al(III) or high dose of nutrient metal ion Zn(II) is scarce. In this study, we are dealing with the acute role of CK2 in response to metal toxicity. We firstly show that inhibition of CK2 activity by TBB and quinalizarin led to increased cell viability under the metal exposure at IC50 or IC25, suggesting a negative relationship between CK2 activity and cell viability under excess metal ions. Because the degree of metal toxicity is dictated by metal uptake, sequestration into subcellular compartments, and metal efflux, further results demonstrate the decrease of zinc toxicity is due to the reduction of Zn(II) uptake, as illustrated by FluoZin-3 staining (Fig. 4). Since the toxicity levels of Zn(II), Al(III), As(III), Cr(VI) and Co(II) were all markedly reduced by the inhibition of CK2, it is conceivable that there might be a shared pathway regulating these metals' uptake. Considering these findings together with the fact that CK2 is overexpressed in cancer cells, we therefore reason that the cancer cells would be more sensitive to metal toxicity than normal cells. The intriguing question is: can metal compounds be used as therapeutic agents for treating cancers? This is indeed answered by the reality that metal complexes containing arsenic and platinum have already been used in medicine for a long time.29 Thus, the finding here provides a likely molecular basis for the application of metal for cancer therapy.
Furthermore, the findings of the Zn(II) and Ca(II) imaging revealed a clear link between zinc and calcium uptake via CK2. Inhibition of CK2 in the Neuro-2a cells by its inhibitors, either TBB or quinalizarin, resulted in reduction of Zn(II) uptake and the concurrent reduction of Ca(II), as compared to the high intracellular concentrations of Zn(II) and Ca(II) in the cells exposed to IC50 Zn(II) (240 μM) but not treated with the CK2 inhibitors (Fig. 4). Two fundamental questions are how CK2 regulates zinc uptake across plasma membrane and how zinc influx triggers the increase of intracellular calcium. One past study sheds light on these questions, which demonstrated that extracellular Zn(II) can be sensed by the cell, leading to the release of intracellular Ca(II).20 In the context of excessive metal ions as applied in this study, acute toxicity of free zinc probably involves zinc influx, and subsequent increase of intracellular calcium could be due to Ca(II) influx through transporters such as L-type calcium channels or release from intracellular Ca(II) stores.30 An increase of intracellular calcium upon exposure to the toxic metal cadmium was observed in neuroblastoma cells and zinc could block this action.31 Such findings support the likelihood that there is a link between zinc and calcium homeostasis in Neuro-2a cells. Ionotropic NMDA (N-methyl-D-aspartate), glutamate receptors and voltage-gated calcium channels might also serve as the routes for zinc uptake. So far, there is no report about CK2 regulation of these channels. According to the current knowledge, numerous protein transporters are involved in zinc import (ZIP transporters) and zinc efflux (Zn-T).32 Physiologically, there is little amount of free zinc available in neuronal cells although zinc is the most abundant trace element in the brain. Apart from the zinc ions that are bound to enzymes as a cofactor and other proteins as structural components such as the zinc ions in CK2β homodimer, the free zinc ions are sequestered in subcellular compartments. The excess zinc condition used in this study is relevant to the findings that excess zinc can cause neurodegenerative disorders such as AD and neuronal death after transient global ischemia. Therefore, the findings here suggests that CK2 may be a therapeutic target for treating disorders related to excesses of metal ions such as zinc.
Although the substrate repertoire of CK2 is increasing to the thousands,2 only about three membrane-bound ion transporters have been found to be regulated by CK2, including the zinc channel (ZIP7) in the membrane of endoplasmic reticulum,12 CFTR (cystic fibrosis transmembrane conductance regulator) which is essentially a chloride channel,33,34 as well as the epithelial Na+ channel.35 The regulation of CFTR by CK2 revealed the fine molecular details of the interaction between the kinase and the channel. Inhibition of CK2 closes CFTR wild type but not the cystic fibrosis mutant channel ΔF508-CFTR.36 The deletion of phenylalanine (F) of the 508th residue in CFTR abolishes the interaction of CK2 with ΔF508-CFTR, suggesting phenylalanine residue serves as a docking site in the wild type for CK2 regulation. The data here suggests that it is likely that CK2 regulates a plasma membrane-bound zinc importer for its uptake. Future investigation will be carried out to identify the zinc channel.
The siRNA knockdown results suggest it is CK2α′ that plays the prominent role in regulating metal toxicity. This finding highlights the specific functionality of individual subunits of CK2, a notion well described in the past studies.3,37 Both independent CK2α′ or the quaternary complex (α′α′ββ) could be involved in regulating the metal uptake. However, the small effect shown by the siRNA against CK2β as compared to the control indicates quaternary CK2 does not play a major role in this, because CK2β knockdown abolishes the formation of tetrameric holoenzyme CK2. As the CK2 holoenzymes are seemingly responsible for a lion's share of CK2 duties such as in cell proliferation,38 the significant involvement of CK2α′ monomer in metal toxicity is a novel finding itself, and also demonstrates the notion that individual CK2 subunits can regulate different cellular processes. The phenotypes of complete knockout mutants for individual CK2 subunits in the model organism, Saccharomyces cerevisiae, further support the finding. Using the complete deletion mutants of CK2, we demonstrate that the role of CK2 in metal toxicity is conserved between mammalian cells and S. cerevisiae. CK2 is involved in metal toxicity in S. cerevisiae for Zn(II), Al(III), As(III), Cr(VI) and Co(II) (Fig. 6). This further shows that the yeast is a useful model organism for eukaryotic cells.39 Historically, its quintessential contributions to the understanding of mammalian cell biology include the pioneering works on cell cycle control of S. cerevisiae by Hartwell40 and Nurse,41 and the discovery of the key signalling protein, TOR (target of rapamycin) serine/threonine kinases.42,43 The results here with yeast deletion mutants of CK2 demonstrate unequivocally that the independent CK2 subunits function distinctly (Fig. 6), with CKA2 (CK2α′ in the mammalian cells) being the prominent subunit. This agrees with the findings in Neuro-2a (Fig. 3).
The findings of this study highlight the role of CK2 in regulating metal toxicity. We showed that, under excessive metal conditions, CK2 activity seems not required for the cell's survival. In fact, its inhibition and expression knockdown by the specific inhibitors and siRNA respectively are beneficial. This notion appears contrary with the current general description of CK2 functionality as an essential regulator for cell proliferation. However, the discovery here reveals a new facet of CK2's multi-personalities. The diversity of CK2's functions, we believe, is at least partly due to the multiple entities of the CK2 subunits, existing either in quaternary holoenzymes or discrete monomers, which would have different biochemical roles in maintaining the cell's proliferation and response to environmental challenges such as excess of metal ions. The subcellular localisation of different formats of CK2 is also critical to understanding the intricacy of CK2. Therefore, further study is needed to dissect the molecular functions for individual monomers and the holoenzyme of CK2 under varying conditions. Moreover, the findings of this study provide a basis for further identification of plasma membrane-bound ion transporters and their regulation details by CK2.
Acknowledgements
The authors are grateful to Ms Karen Ballantyne, Dr Francesca Charlton and Ms Lindy Villanueva for their technical support.References
- G. Burnett and E. P. Kennedy, J. Biol. Chem., 1954, 211, 969–980 CAS.
- F. Meggio and L. A. Pinna, FASEB J., 2003, 17, 349–368 CrossRef CAS PubMed.
- A. C. Bibby and D. W. Litchfield, Int. J. Biol. Sci., 2005, 1, 67–79 CrossRef CAS PubMed.
- B. Guerra and O. G. Issinger, Electrophoresis, 1999, 20, 391–408 CrossRef CAS PubMed.
- L. A. Pinna, Acc. Chem. Res., 2003, 36, 378–384 CrossRef CAS PubMed.
- M. Sayed, S. O. Kim, B. S. Salh, O. G. Issinger and S. L. Pelech, J. Biol. Chem., 2000, 275, 16569–16573 CrossRef CAS PubMed.
- M. Daya-Makin, J. S. Sanghera, T. L. Mogentale, M. Lipp, J. Parchomchuk, J. C. Hogg and S. L. Pelech, Cancer Res., 1994, 54, 2262–2268 CAS.
- J. H. Trembley, Z. Chen, G. Unger, J. Slaton, B. T. Kren, C. Van Waes and K. Ahmed, BioFactors, 2010, 36, 187–195 CrossRef CAS PubMed.
- M. Kalathur, A. Toso, J. Chen, A. Revandkar, C. Danzer-Baltzer, I. Guccini, A. Alajati, M. Sarti, S. Pinton, L. Brambilla, D. Di Mitri, G. Carbone, R. Garcia-Escudero, A. Padova, L. Magnoni, A. Tarditi, L. Maccari, F. Malusa, R. K. R. Kalathur, L. A. Pinna, G. Cozza, M. Ruzzene, N. Delaleu, C. V. Catapano, I. J. Frew and A. Alimonti, Nat. Commun., 2015, 6, 7227 CrossRef CAS PubMed.
- N. Tun, P. O'Doherty, Z. Chen, X. Wu, T. Bailey, C. Kersaitis and M. Wu, Metallomics, 2014, 6, 1558–1564 RSC.
- Y. Jin, P. Dunlap, S. McBride, H. Al-Refai, P. Bushel and J. Freedman, PLoS Genet., 2008, 4, e1000053 CrossRef PubMed.
- K. M. Taylor, S. Hiscox, R. I. Nicholson, C. Hogstrand and P. Kille, Sci. Signaling, 2012, 5, ra11 Search PubMed.
- D. Y. Lou, I. Dominguez, P. Toselli, E. Landesman-Bollag, C. O'Brien and D. C. Seldin, Mol. Cell. Biol., 2008, 28, 131–139 CrossRef CAS PubMed.
- P. R. Blanquet, Prog. Neurobiol., 2000, 60, 211–246 CrossRef CAS PubMed.
- D. I. Perez, C. Gil and A. Martinez, Med. Res. Rev., 2010, 31, 924–954 CrossRef PubMed.
- M. V. Aksenova, G. S. Burbaeva, K. V. Kandror, D. V. Kapkov and A. S. Stepanov, FEBS Lett., 1991, 279, 55–57 CrossRef CAS PubMed.
- S. Bolognin, L. Messori, D. Drago, C. Gabbiani, L. Cendron and P. Zatta, Int. J. Biochem. Cell Biol., 2011, 43, 877–885 CrossRef CAS PubMed.
- L. Tomljenovic, J. Alzheimer's Dis., 2011, 23, 567–598 CAS.
- A. I. Bush, Curr. Opin. Chem. Biol., 2000, 4, 184–191 CrossRef CAS PubMed.
- M. Hershfinkel, A. Moran, N. Grossman and I. Sekler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11749–11754 CrossRef CAS PubMed.
- S. Yamasaki, K. Sakata-Sogawa, A. Hasegawa, T. Suzuki, K. Kabu, E. Sato, T. Kurosaki, S. Yamashita, M. Tokunaga, K. Nishida and T. Hirano, J. Cell Biol., 2007, 177, 637–645 CrossRef CAS PubMed.
- C. Grienberger and A. Konnerth, Neuron, 2012, 73, 862–885 CrossRef CAS PubMed.
- B. Scharf, C. C. Clement, V. Zolla, G. Perino, B. Yan, S. G. Elci, E. Purdue, S. Goldring, F. Macaluso, N. Cobelli, R. W. Vachet and L. Santambrogio, Sci. Rep., 2014, 4, 5729 CrossRef CAS PubMed.
- A. W. Schaffer, A. Schaffer, A. Pilger, C. Engelhardt, K. Zweymueller and H. W. Ruediger, Clin. Toxicol., 1999, 37, 839–844 CAS.
- F. Zwicker, M. Ebert, P. E. Huber, J. Debus and K.-J. Weber, Radiat. Oncol., 2011, 6, 15 CrossRef CAS PubMed.
- G. Cozza, M. Mazzorana, E. Papinutto, J. Bain, M. Elliott, G. Di Maira, A. Gianoncelli, M. Pagano, S. Sarno and M. Ruzzene, Biochem. J., 2009, 421, 387–395 CrossRef CAS PubMed.
- K. Kubiński, K. Domańska, E. Sajnaga, E. Mazur, R. Zieliński and R. Szyszka, Mol. Cell. Biochem., 2007, 295, 229–236 CrossRef PubMed.
- D. W. Litchfield, Biochem. J., 2003, 369, 1–15 CrossRef CAS PubMed.
- B. Desoize, Anticancer Res., 2004, 24, 1529–1544 CAS.
- J. Y. Koh and D. W. Choi, Neuroscience, 1994, 60, 1049–1057 CrossRef CAS PubMed.
- J. Benters, U. Flögel, T. Schäfer, D. Leibfritz, S. Hechtenberg and D. Beyersmann, Biochem. J., 1997, 322, 793–799 CrossRef CAS PubMed.
- R. A. Colvin, C. P. Fontaine, M. Laskowski and D. Thomas, Eur. J. Pharmacol., 2003, 479, 171–185 CrossRef CAS PubMed.
- D. De Stefano, V. Villella, S. Esposito, A. Tosco, A. Sepe, F. De Gregorio, L. Salvadori, R. Grassia, C. Leone, G. De Rosa, M. Maiuri, M. Pettoello-Mantovani, S. Guido, A. Bossi, A. Zolin, A. Venerando, L. Pinna, A. Mehta, G. Bona, G. Kroemer, L. Maiuri and V. Raia, Autophagy, 2014, 10, 2053–2074 CrossRef PubMed.
- A. Venerando, M. A. Pagano, K. Tosoni, F. Meggio, D. Cassidy, M. Stobbart, L. A. Pinna and A. Mehta, Naunyn-Schmiedeberg's Arch. Pharmacol., 2011, 384, 473–488 CrossRef CAS PubMed.
- T. Bachhuber, J. Almaça, F. Aldehni, A. Mehta, M. D. Amaral, R. Schreiber and K. Kunzelmann, J. Biol. Chem., 2008, 283, 13225–13232 CrossRef CAS PubMed.
- K. J. Treharne, Z. Xu, J.-H. Chen, O. G. Best, D. M. Cassidy, D. C. Gruenert, P. Hegyi, M. A. Gray, D. N. Sheppard, K. Kunzelmann and A. Mehta, Cell. Physiol. Biochem., 2009, 24, 347–360 CrossRef CAS PubMed.
- O. Filhol, A. Nueda, V. Martel, D. Gerber-Scokaert, M. J. Benitez, C. Souchie, Y. Saoudi and C. Cochet, Mol. Cell. Biol., 2003, 23, 975–987 CrossRef CAS PubMed.
- O. Filhol, J.-L. Martiel and C. Cochet, EMBO Rep., 2004, 5, 351–355 CrossRef CAS PubMed.
- D. Botstein and G. R. Fink, Genetics, 2011, 189, 695–704 CrossRef CAS PubMed.
- L. H. Hartwell, R. K. Mortimer, J. Culotti and M. Culotti, Genetics, 1973, 74, 267–286 CAS.
- M. G. Lee and P. Nurse, Nature, 1987, 327, 31–35 CrossRef CAS PubMed.
- R. Loewith, Biochem. Soc. Trans., 2011, 39, 437–442 CrossRef CAS PubMed.
- J. Heitman, N. Movva and M. Hall, Science, 1991, 253, 905–909 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |