
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The biological chemistry of the transition metal “transportome” of Cupriavidus metallidurans
Dietrich H.
Nies
Molecular Microbiology, Institute for Biology/Microbiology, Martin-Luther-University Halle-Wittenberg, Germany. E-mail: d.nies@mikrobiologie.uni-halle.de
First published on 21st March 2016
Abstract
This review tries to illuminate how the bacterium Cupriavidus metallidurans CH34 is able to allocate essential transition metal cations to their target proteins although these metals have similar charge-to-surface ratios and chemical features, exert toxic effects, compete with each other, and occur in the bacterial environment over a huge range of concentrations and speciations. Central to this ability is the “transportome”, the totality of all interacting metal import and export systems, which, as an emergent feature, transforms the environmental metal content and speciation into the cellular metal mélange. In a kinetic flow equilibrium resulting from controlled uptake and efflux reactions, the periplasmic and cytoplasmic metal content is adjusted in a way that minimizes toxic effects. A central core function of the transportome is to shape the metal ion composition using high-rate and low-specificity reactions to avoid time and/or energy-requiring metal discrimination reactions. This core is augmented by metal-specific channels that may even deliver metals all the way from outside of the cell to the cytoplasm. This review begins with a description of the basic chemical features of transition metal cations and the biochemical consequences of these attributes, and which transition metals are available to C. metallidurans. It then illustrates how the environment influences the metal content and speciation, and how the transportome adjusts this metal content. It concludes with an outlook on the fate of metals in the cytoplasm. By generalization, insights coming from C. metallidurans shed light on multiple transition metal homoeostatic mechanisms in all kinds of bacteria including pathogenic species, where the “battle” for metals is an important part of the host–pathogen interaction.
 Dietrich H. Nies | Dietrich H. Nies has been full Professor of Molecular Microbiology at the Martin-Luther-University of Halle, Germany, since 1993. He began his academic career in Göttingen as a student of Hans H. Schlegel, carrying out his postdoc in Berlin with Bärbel Friedrich and at the University of Illinois at Chicago with Simon Silver. Professor Nies's research interest is in the interaction of bacteria with transition metals. |
Introduction: Cupriavidus metallidurans CH34
C. metallidurans CH34 was isolated from a zinc decantation tank as a metal-resistant, hydrogen-oxidizing “Pseudomonas” and its name has changed several times in the last few decades: Alcaligenes eutrophus, Ralstonia sp., Ralstonia metallidurans, Wautersia metallidurans and finally C. metallidurans.1–8 Its genome contains 6717 protein-encoding genes located on a main chromosome, a second chromosome or “chromid”, and two large plasmids named pMOL28 and pMOL30 to honor the laboratory in Belgium that isolated this strain.3 More than half of the predicted proteins were quantified and identified in a bottom-up proteomic approach.9C. metallidurans is widespread in nature. Its niches are metal-containing soils, e.g. zinc deserts, serpentine soils in New Caledonia and auriferous soils in Australia.10–13 The genes on the two plasmids and on genomic islands were probably horizontally acquired5,14–16 and indicate that C. metallidurans has obtained during its evolution the ability to (i) resist high concentrations of transition metals,2,4,17–20 (ii) oxidize molecular hydrogen subsequently allowing chemolithoautotrophic growth,4,14 and (iii) degrade a variety of aromatic and unusual organic compounds.2,21,22 Since weathering of metal-containing minerals produces both molecular hydrogen and soluble transition metal ions12,23–26C. metallidurans can be nicely pictured as living in biofilms in between the hydrogen- and metal-generating weathering minerals on the one side and molecular oxygen plus an additional supply of organic compounds on the other side. This would also explain the presence of the outstanding and highly sophisticated metal resistance determinants in the genome of this bacterium, and why it is a “hydrogen-oxidizing, metal-resistant” bacterium.27
Taxonomically, C. metallidurans CH34 belongs to the phylum Proteobacteria, class Betaproteobacteria, order Burkholderiales, and to one of the five families within this order, the Burkholderiaceae.6,7 Proteobacteria are Gram-negative bacteria enveloped by two biological membranes, the outer and the inner membrane, with a periplasmic space in-between. This defines a second cellular compartment in addition to the cytoplasmic space. All substrates have to pass the outer membrane before transport across the inner membrane can be undertaken.
While C. metallidurans CH34 is a harmless bacterium unable to grow at 37 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and to produce toxins, some bacterial strains described under the same species name might have some pathogenic potential29,30 although further research is needed to actually demonstrate that these strains are indeed able to reproducibly infect and cause disease symptoms in healthy individuals. Such results should prompt description of those strains as a new bacterial species. The genus Cupriavidus currently comprises 14 bacterial species, including the close relatives C. necator H16 (synonym C. eutrophus H16, Ralstonia eutropha H16)31 and C. pinatubonensis JMP134 (synonym Ralstonia eutropha JMP134).32,33 Strain H16 is a well investigated bacterium able to grow facultatively as a chemolithoautotroph. It oxidizes molecular hydrogen with molecular oxygen and uses this energy to assimilate carbon dioxide via the Calvin cycle.31 Strain JMP134 was isolated as a xenobiotic-degrading bacterium able to degrade the agent orange component 2,4-dichlorphenoxyacetic acid (2,4-D) with the help of plasmid pJ4.32,33
28 and to produce toxins, some bacterial strains described under the same species name might have some pathogenic potential29,30 although further research is needed to actually demonstrate that these strains are indeed able to reproducibly infect and cause disease symptoms in healthy individuals. Such results should prompt description of those strains as a new bacterial species. The genus Cupriavidus currently comprises 14 bacterial species, including the close relatives C. necator H16 (synonym C. eutrophus H16, Ralstonia eutropha H16)31 and C. pinatubonensis JMP134 (synonym Ralstonia eutropha JMP134).32,33 Strain H16 is a well investigated bacterium able to grow facultatively as a chemolithoautotroph. It oxidizes molecular hydrogen with molecular oxygen and uses this energy to assimilate carbon dioxide via the Calvin cycle.31 Strain JMP134 was isolated as a xenobiotic-degrading bacterium able to degrade the agent orange component 2,4-dichlorphenoxyacetic acid (2,4-D) with the help of plasmid pJ4.32,33
The family Burkholderiaceae is composed of 12 genera with the genus Burkholderia containing important pathogens such as B. mallei, B. pseudomallei and the B. cepacia complex, as well as the plant pathogen Ralstonia solanacearum. The close phylogenetic relationship of C. metallidurans to pathogens, the important function of transition metals in pathogenicity and the fact that the metal homoeostasis systems of other bacteria are usually simpler versions of the C. metallidurans system indicate that insights coming from C. metallidurans are useful to understand the physiology of other free-living and pathogenic bacteria.
Transition metals
Transition metals and heavy metals
Transition metals, which belong to groups 3 to 12 of the periodic system of elements (Fig. 1), fill their 3d, 4d, 5d or 6d orbitals during the first, second, third or fourth transition period, which are part of the general periods 4, 5, 6, and 7, respectively.34 All transition metals have an electron pair in the outermost 4s, 5s, 6s or 7s orbital, empty p orbitals with the same principal quantum number 4 to 7, and empty (Sc to Zn, Y to Cd, La, Ac) or completely filled f-orbitals (all others). They differ only in the number of d electrons. With the exception of Sc, Ti and Y, the metallic forms of these elements have densities >5 g cm−3, so that most transition metals can be considered as “heavy metals”. Some metals, and even metalloids of the p group (groups 13 to 18, Fig. 1) and all f group elements (not shown in Fig. 1), are also “heavy”, so that the sets of transition and heavy metals overlap but not completely (for a detailed treatment of how the periodic system of elements results from the Schrödinger equation and why the chemical elements are arranged in the system shown in Fig. 1, please refer to the freely available companion website of ref. 35 at http://media.wiley.com/product_ancillary/07/35273165/DOWNLOAD/Website_Chapter1.pdf).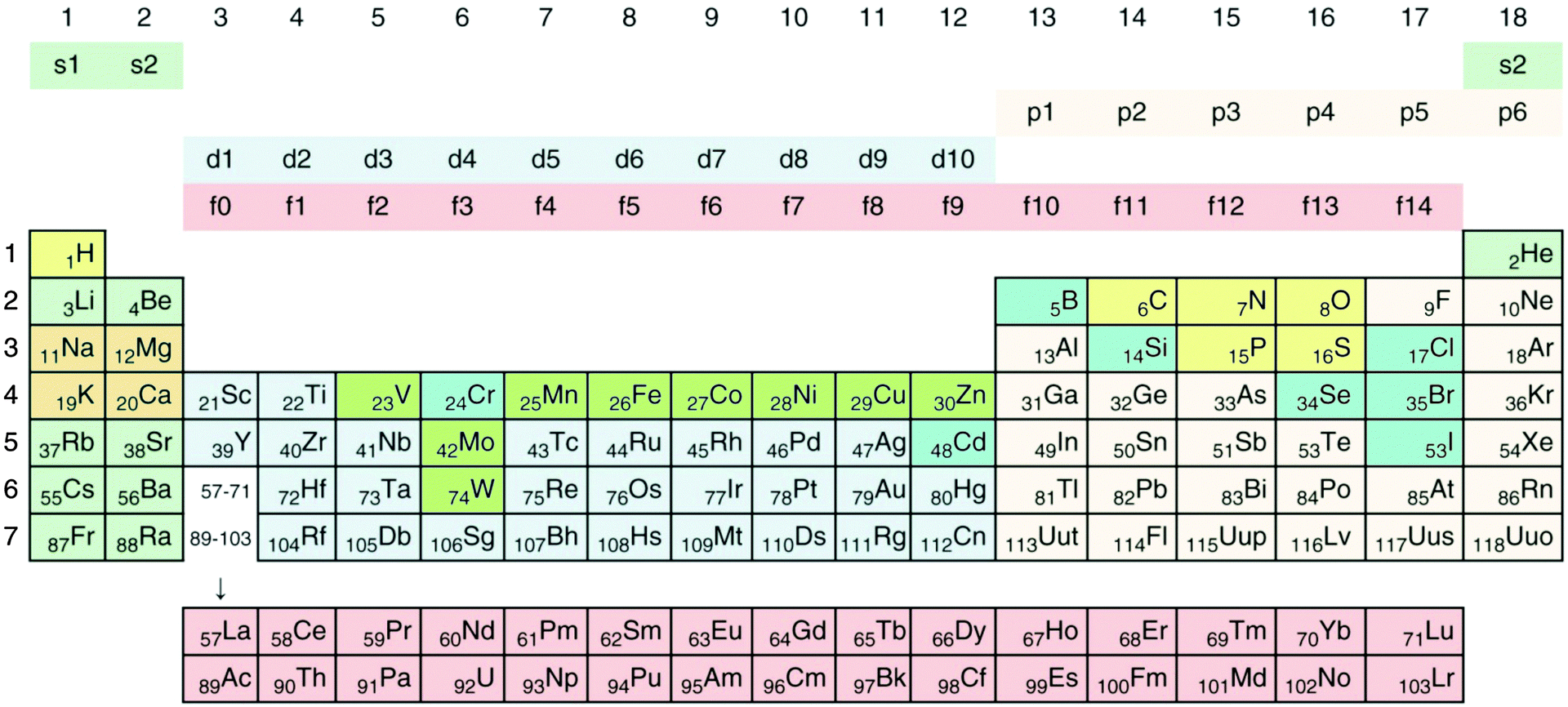 | ||
| Fig. 1 Periodic system of elements. The periods are defined by the principal quantum number n of the valence electrons. Elements of groups 1 and 2 fill up their respective s orbitals and elements of groups 13 to 18 their p-orbitals. The first period contains H and He, in the following periods groups 1 and 2 the alkali and alkaline earth metals. In-between are the transition metals, which fill up the d-orbitals, plus the lanthanides (rare earth elements) and actinides, which fill up f-orbitals. No elements are known that fill up the 5g or 6g orbitals. Such a process should start with elements of period 8; however, the element with the highest atomic number and the highest atomic mass discovered so far is the artificially synthesized and very unstable ununoctium (118-Uuo) of group 18 and period 7. Its half-life is below 1 millisecond. The ten major bio-elements are on a yellow (non-metals) and orange field (metals), and additional minor or trace elements on a green (transition metals) and blue (Se, Cr, Cd, B, Cl, Br, J) field. Cd and Cr are not known to be trace elements in bacteria. Reproduced with permission from ref. 35. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. | ||
As all transition metals have two valence electrons in an s orbital plus a different number of electrons in their d orbital, they tend to form divalent cations in aqueous solution when the valence electrons are lost, or cations of a higher order when additional d electrons are gone. At oxidation states of +5 or more, these cations react with water and form oxyanions, which can even polymerize. The electron configuration of the three noble elements Cu, Ag and Au is not d9 s2 but d10 (=filled) s1, because a half or completely occupied group of electrons with the same principal and azimuthal quantum number represents a state of low energy. Consequently, noble metals may also occur in the +1 oxidation state, in addition to +2 (Cu) and +3 (Au).34
Due to the fact that divalent transition metal cations have similar empty s valence orbitals, their ionic radius is also in the same range, in the first transition metal period from 690 pm for Cu(II) via 720 pm for Ni(II), 740 pm for Co(II) or Zn(II) and 760 pm for Fe(II) to 800 pm for Mn(II).36 Because of shielding effects caused by the innermost electrons, this value increases only slightly with the principal quantum number, e.g. from 740 pm for Zn(II) with 3d10via 970 pm for Cd(II) with 4d10 to 1.1 nm for Hg(II) with 5d10. Consequently, transition metals adjacent to each other in the same transition period or group have not only similar chemical features but also similar ionic radii and similar values of Linus Paulings' electronegativity.34 Due to increased shielding by the d electrons, this value increases within the first transition period from 1.3 (Ac) to 1.9 (Cu) and to the highest value among all transition metals of 2.4 for Au. The difference in electronegativity between two elements in a chemical bond is a measure of the percent ionic and covalent character of this bond. Derived from this, the Au–S or Au–C bonds are approximately 99.5% covalent and even the Au–O bond has only 22% ionic character. All bonds of transition metals with non-metals have a large percentage of a covalent character, especially the noble metals in group 11 but also their neighbors in groups 8, 9, 10 and 12 (Fig. 1). This is the reason why transition metals of these groups are described as “soft” metals or “borderline” metals while alkali, alkaline earth and transition metals with a low number of d electrons are considered as “hard” metals: “soft” metal cations form strong, nearly covalent bonds with sulfur while “hard” metals prefer ionic interactions with oxygen.37
Oxidation state, redox potential, solubility and bio-availability of transition metals
With the exception of hydrogen and a minor amount of helium and lithium, all chemical elements in this universe are produced by nuclear fusion reactions in stars and released by supernova explosions when the respective star dies. The fusion runs from H to He, subsequently to C, N, O, and further on via Si to elements with higher atomic number up to Fe.38 The Fe nucleus is that with the highest atomic number that is generated in an exergonic nuclear fusion reaction; synthesis of all elements with higher atomic number is endergonic.39 Consequently, the element distribution in this part of the universe decreases hyper-exponentially from hydrogen to elements with high atomic numbers, with a comparable high amount of the atoms of Fe and its neighbors, and a very low amount of Li, Be, and B, which are only transient products of the second fusion step from He to C, N, O.From these “ashes of a long-dead star” the earth was formed and re-modeled by cataclysmic events such as the moon-forming impact of another small planet on the proto-earth, which transformed the element distribution of the universe into that of the earth's crust.40 Since life as we understand it41 depends on water, elements have to be released from the minerals in the earth's crust into water to be bio-available. After the oceans had been formed on the earth,42 the average metal content in sea water reached an equilibrium of input and precipitation reactions, so that this average metal content may be assumed as the average metal content of all global ecosystems. Derived from this, the ratio of metal concentration in g per kg seawater to metal concentration in g per kg earth's crust may give a measure of the overall mobility of an individual metal (Fig. 2).
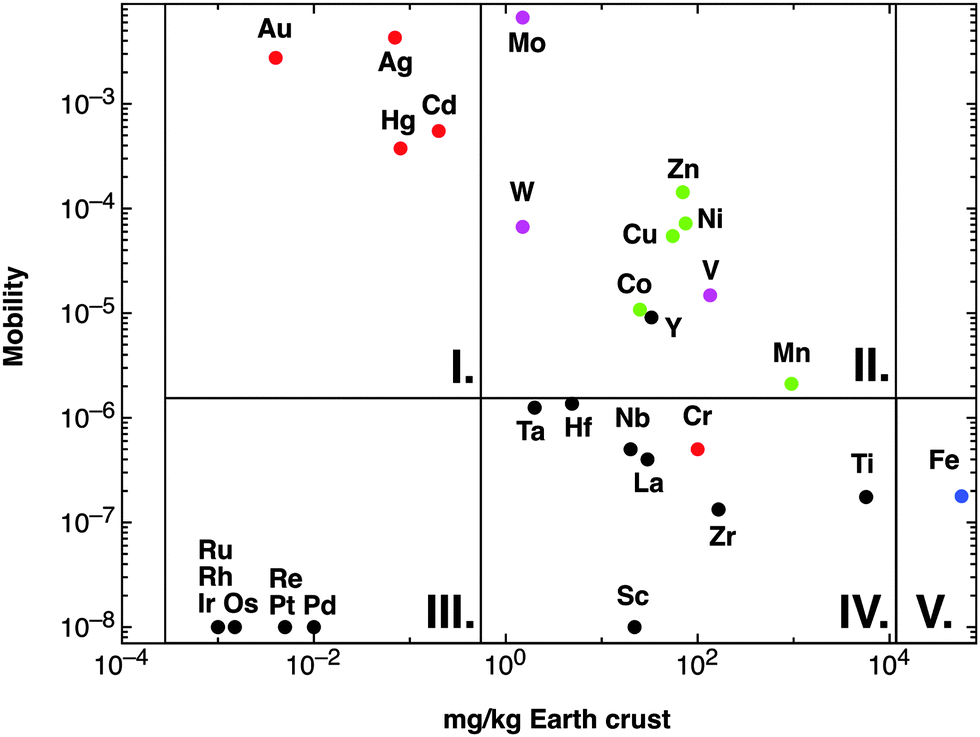 | ||
| Fig. 2 Availability of transition metals. The figure plots the content of a transition metal against the mobility, calculated as sea water content of this metal divided by the content of the earth's crust.36 This clearly defines five sets of transition metals: (I) those with high mobility despite a low content in the earth's crust; (II) with high availability; (III) with extremely limited availability; (IV) with a high content in the earth's crust but low availability; and (V) iron. Group II contains all essential oxy-anions (purple), essential-but-toxic cations (green) plus Y but except iron (blue) and group I the toxic-only transition metals (red). With the exception of Cr, the metals in groups III and IV are of no relevance for C. metallidurans. | ||
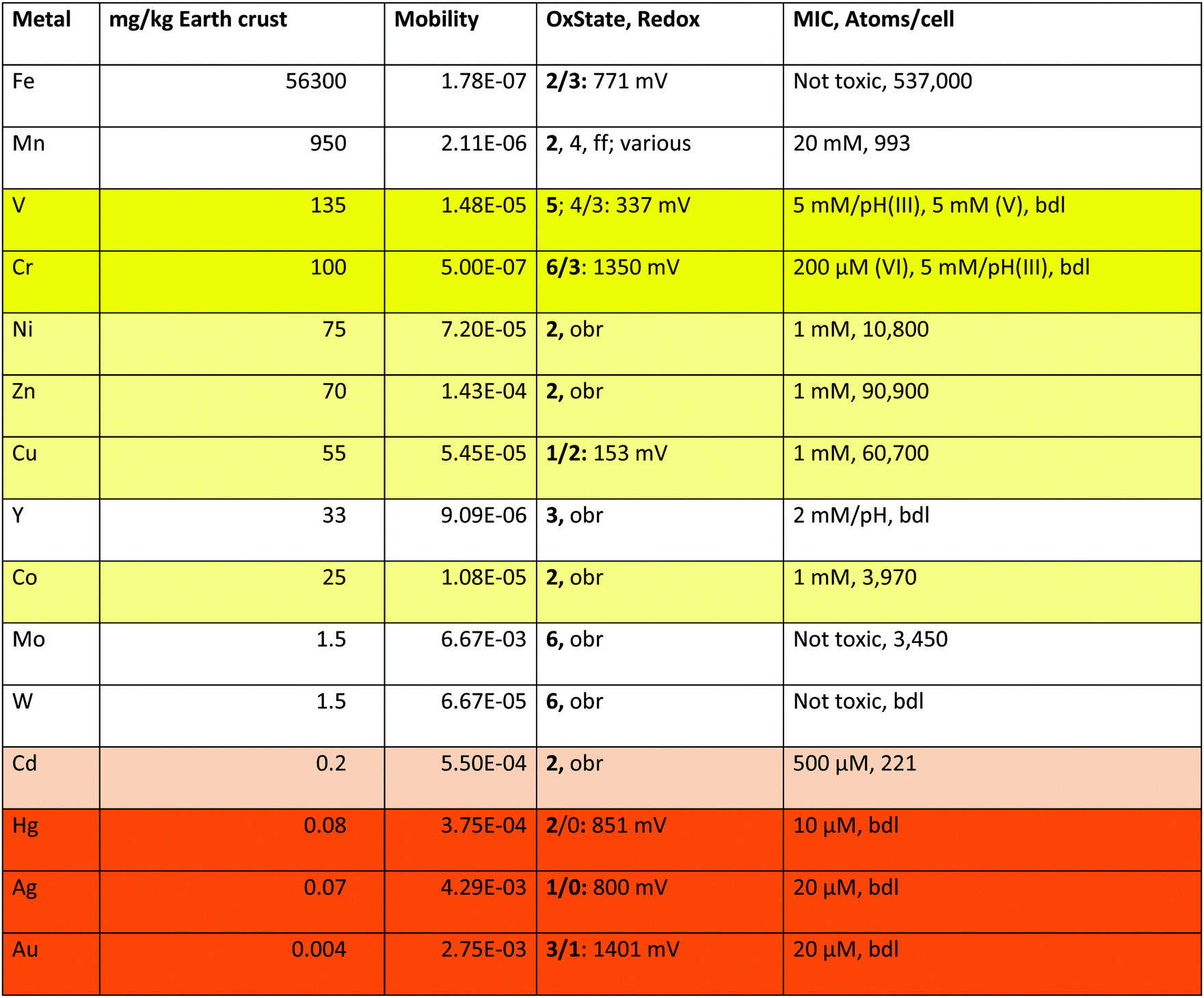
Of the 40 transition metals (Fig. 1), the ten 6d elements plus Tc of 4d elements do not contain stable isotopes, are not naturally occurring and were only artificially produced. For the remaining 29 transition metals plotting the mobility against the content of the earth's crust defines five sets of transition metals (Fig. 2): (I) four have high mobility despite a low content in the earth's crust; (II) nine have high bio-availability; (III) seven are not bio-available metals; (IV) eight have a high content in the earth's crust but have low mobility; and (V) iron. With the exception of Cr and Y, these groups neatly define the toxic-only transition metal cations (group I), essential trace elements that may be toxic nevertheless (group II), Fe as an exception (group V), and transition metals without biological importance (groups III and IV). All elements in group IV plus Y exist as trivalent, tetravalent or pentavalent cations that form insoluble hydroxides in water, which is the reason for their low bio-availability at neutral pH values. Another exception concerns Cr, which exists as an insoluble trivalent Cr(III) cation but also as Cr(VI) in chromate or bi-chromate depending on the pH value and both forms are toxic to C. metallidurans (Table 1).
| a These are the values plotted in Fig. 2 for the elements in groups I, II, and V plus Cr. The mobility is the quotient of the content of a metal in standard sea water in g kg−1 divided by the content of the earth's crust in g kg−1.36 OxState gives the oxidation states of the metals listed with the most frequently occurring states in bold type, and the half-cell redox potential E0 for the indicated states;36 obr, outside of the biological range for the cations in water; ff, and others. MIC is the minimal inhibitory concentration as determined for 5 days at 30 °C on Tris-buffered mineral salts medium with gluconate as the carbon source (TMM).4,43,44 The number of metal atoms per cell was determined by ICP-MS in TMM-grown cells;45 bdl, below detection limit. “pH” indicates that the growth medium was strongly acidified by the added metal but the pH value could not be adjusted because otherwise metal hydroxide complexes precipitated. Fields: toxic oxyanions yellow, essential-but-toxic cations light yellow, toxic-only cadmium light orange, toxic cations orange. |
|---|
|
The transition metals in groups I, II and IV, which are of consequence for C. metallidurans and its metal transportome, are also listed in Table 1. This table additionally gives the oxidation states of these metals in water, plus the redox potential under standard conditions. Water defines the borders of redox reactions in living cells, because all compounds with a redox potential more negative than that of molecular hydrogen (E0 = 0 V per definition) should reduce protons and thus be unstable. Likewise, all compounds with a redox potential higher than that of water (E0 = +1229 mV) should oxidize water and be unstable too.34 Consequently, some transition metals form cations that are redox inactive in biological systems (Zn), except in complexes (Ni, Co, Mo, W) while the redox potential of others may vary (Fe, Mn, V, Cr, Cu, Hg, Ag and Au).
Fe has a special position in group V. It exists as Fe(III) under oxic conditions, which precipitates as insoluble hydroxide.34 This would mean that iron should have low bio-availability and not be widely used as a bio-element. The contrary is, however, the case. Fe is the transition metal with the highest number of atoms in the C. metallidurans cell (Table 1).45 This has “historical” reasons: before molecular oxygen became available on earth, that is before the first great oxygenation event 2.4 billion years ago,46 Fe existed as Fe(II) that was largely bio-available and used in early life forms.47 After this event, cells were confronted with a massive iron starvation condition due to Fe(III) hydroxide precipitation, which was solved by the evolution of siderophore-dependent Fe(III) import pathways.48,49 In anoxic environments, Fe(II) is still the predominant and readily available form of iron. This is the reason for the observed discrepancy that Fe is indicated as of low availability in Fig. 2 but as an important bio-element in Table 1.
Thus, the role that any transition metal is able to play in C. metallidurans and other bacteria is simply the result of differences in the creation of these elements in ancient stars, formation of the planet earth, and their solubility. Only the transition metals in groups I and II (Fig. 2) are interesting for bacterial cells, with three exceptions: Cr belongs to group I because it is able to form the highly soluble chromate oxyanion, Y to group IV because it precipitates as a hydroxide complex similar to other trivalent metal cations, and Fe has its special role because of the outlined “historical” reasons.
Metal complexes
The high electronegativity of transition metals and the resulting covalent character of transition metal–non-metal bonds have another important consequence: transition metal cations are able to form complex compounds by accepting a free electron pair from non-metal atoms such as C, O, and N as ligands. Using the empty s and p orbitals, four ligands can be accommodated in a tetrahedral complex by the central metal cation. Some transition metal cations may harbor six ligands in an octahedral complex. The additional two metal orbitals that are needed to accept two additional ligands are recruited from the five d orbitals. Using two “binding” metal d orbitals for a bond with a non-metallic ligand produces two “anti-binding” d-orbitals because the total number of orbitals in a system has to remain constant (Fig. 3). The remaining three d orbitals are not involved in the formation of the metal complex, they are “non-binding”.34 Depending on the individual metal in the center, the complex can be redox-active by accepting electrons during the course of a biochemical reaction or by altering the oxidation state of the central metal ion.50 This allows transition metal complexes to be powerful biochemical catalysts and explains why transition metals are essential trace elements.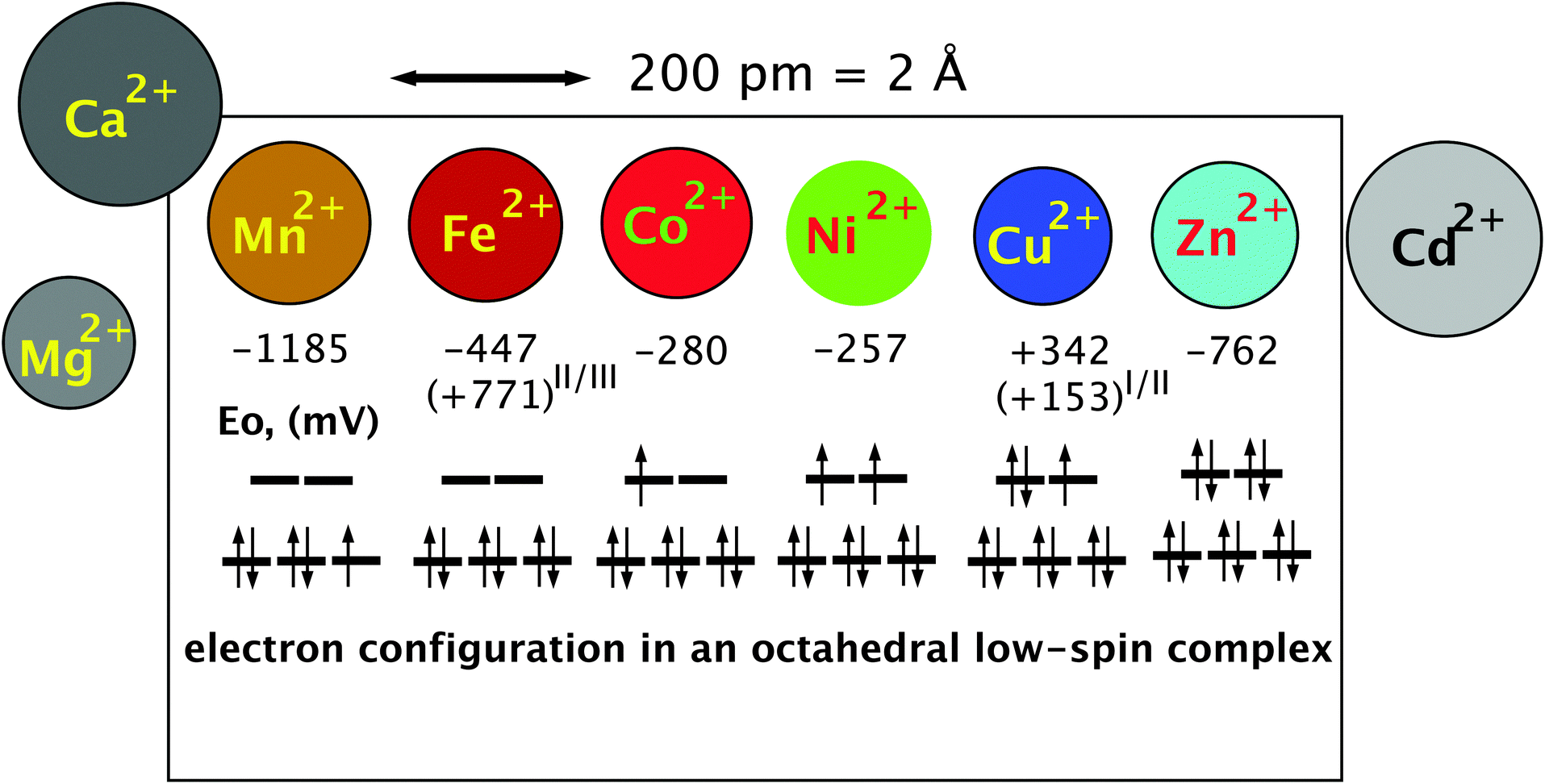 | ||
| Fig. 3 The essential transition metal cations of the first transition period. The essential divalent transition metal cations all have similar radii and can only be discriminated if variations in the complex-forming and redox abilities are considered. The diameter is indicated by the size of the circles that represent the metal. The half-cell redox potential E0 and electron configuration of a theoretical octahedral low-spin complex are indicated below these symbols. An electron spin of +1/2 is indicated by an arrow pointing up, and in a −1/2 spin the arrow points down. The three lines in the lower part of the electron configuration represent the non-binding d orbitals, and the two lines above represent the anti-binding d orbitals. During the formation of the six ligand bonds in this example, six free electron pairs residing in six different ligand orbitals recruit six metal orbitals, namely the empty s valence orbital, the three p orbitals with the same principal quantum number and the two binding orbitals. This forms six new complex orbitals as bonds between ligands and the metal cation. Because the total number of orbitals in a system has to remain constant, a consequence of the first law of thermodynamics (energy conservation), six anti-binding complex orbitals come into existence to compensate for the six complex bonds, one s*, three p* of very high energy (not shown) plus the two anti-binding d orbitals. Reproduced with permission from ref. 51 (Copyright © 2012, American Society for Microbiology) and outlined in detail in the companion website of ref. 35 at http://media.wiley.com/product_ancillary/07/35273165/DOWNLOAD/Website_Chapter1.pdf. | ||
The remaining d electrons of a central transition metal cation in octahedral complex compounds reside in the three non-binding or the two anti-binding orbitals that have a higher energy than the non-binding orbitals. The number of these d electrons increases along the first transition period from the left to the right starting from 0 electrons in the case of Sc(III), Ti(IV), V(V), Cr(VI), and Mn(VII), and ending with 10 electrons in Cu(I) and Zn(II) (Fig. 3). Up to three d electrons in a metal complex all reside in the non-binding orbitals, all with a +1/2 spin. Starting with electron number four, the electron may have a −1/2 spin and pair with another electron in one of the non-binding d orbitals or use an anti-binding orbital. In the first case, the two remaining “lone” electrons can be measured by electron spin resonance and the complex is in a low-spin triplet state, and in the second case, there are four electrons with a +1/2 spin and the complex is in a high-spin state.34 The electron present in the anti-binding d-orbital here weakens the bond to one of the ligands, which remains only in a half-bond. The individual central metal cation, the ligands and their distance from the metal determine whether the complex is in a low- or high-spin state, enabling proteins to change the state of a bound metal cofactor when the conformation is changed and subsequently the distance of the atoms of amino acyl residues that function as ligands.
With electron number five in the important Mn(II) and Fe(III) oxidation states, again a low-spin complex may be formed with one electron remaining unpaired in the non-binding d-orbitals {duplet low-spin state, see Fig. 3 for Mn(II)}. Alternatively, all five electrons remain unpaired and occupy the three non-binding and the two anti-binding orbitals in a high-spin state, which weakens the bond to two of the six ligands. With d-electron number 6 as in Fe(II) and Co(III), either the non-binding orbitals are completely filled, which represents a state of low energy, or again two electrons reside in the anti-binding and four in the non-binding d-orbitals. The state of low energy is the reason why Co(III) complexes as in cobalamin derivatives are kinetically very stable. Electron number seven as in Co(II) has no choice but to enter one of the anti-binding complexes (Fig. 3) so that Co(II) cannot establish a stable octahedral complex; one ligand is always bound only half. With Ni(II), electron number eight also enters an anti-binding d-orbital, either the second with a spin of +1/2 following Hund’s rule of the largest multiplicity (Fig. 3) or, in an exited state, it may be able to revert its spin and pair with the other electron in the first anti-binding d-orbitals. Octahedral Ni(II) complexes contain two ligands bound only half or the complex is a degenerated octahedral complex, containing only five ligands. With nine d electrons a stable octahedral complex is not possible, only a tetrahedral complex or an enlarged tetrahedral complex with an additional half-bound partner. Finally, ten d-electrons as in the case of Zn(II) allow only tetrahedral complexes because the two electron pairs of ligand number five and six cannot be accepted; all d-orbitals are already occupied by metal electrons (Fig. 3).34
“Essential-but-toxic” trace elements and “toxic-only” elements
The ability to form different complex compounds, which may be redox active or not, is the reason for the important role of the transition metal cations of the first transition period in cellular biochemistry: Mn complexes serve as electron buffers, e.g. in the water-splitting complex of photosystem II in cyanobacteria; Fe in redox-active iron sulfur clusters and heme compounds; Co in B12 and other cobalamin derivatives to re-arrange C–H and C–C bonds; Ni is needed to split and form covalent bonds such as in molecular hydrogen or urea; Cu to react with molecular oxygen; and Zn as a non-redox active counterpart of Fe to accept ligands as Lewis acid and to tie polypeptide chains in protein domains into a more rigid conformation.52,53 Without these transition metal cations, a sophisticated cellular biochemistry is not possible. As a consequence, transition metal cations have to be imported into the bacterial cytoplasm.The comparably high electronegativity of the transition metal cations that allows formation of complex compounds also contributes to most of these cations being considered as “soft” or borderline metals,37 because they bind to the sulfur of thiol groups efficiently and with a highly covalent character. Since the cytoplasm of bacteria is kept in a reduced redox state,54,55 the enzymes residing here may contain thiol groups at their surface. Binding of metal cations to these thiol groups may change the conformation of the respective enzymes. Bridging two or more thiol groups in undesired metal complexes may inactivate and precipitate cytoplasmic proteins.56,57 Moreover, transition metal–bis-thiol complexes may inhibit glutathione reductase, leading to a diminished ability of the cell to eliminate reactive oxygen species because reduced glutathione cannot be recycled.58 As demonstrated for cadmium and E. coli57 this thiol-binding activity of transition metal cations with the resulting oxidative stress is one of the reasons for the toxicity of all transition metal cations, especially for the “soft” metal cations of the second and third transition period plus Cu(I). It defines the roles of the bio-available “soft” cations of the metals in group I (Fig. 2) as toxic-only metals, which are too toxic to be of any beneficial use in bacteria, and indeed, none of the metals in group I is known as a trace element in bacteria. The exception is Cd(II), which functions in a carbo-anhydrase in eukaryotic algae living in zinc-deprived marine environments.
Two additional effects contribute to the toxicity of transition metal cations. Fe(II) can reduce H2O2 in a Fenton-type reaction to a hydroxide OH− and the extremely dangerous hydroxyl radical OH˙, which rapidly oxidizes and damages all kinds of macromolecules.59 Since the resulting Fe(III) is reduced again by the superoxide radical or by glutathione, which is present in high concentrations in proteobacteria,56,60 this Haber–Weiss cycle catalyzes the formation of hydroxyl radicals from the already very toxic reactive oxygen species hydrogen peroxide and superoxide radicals. These compounds are produced by the respiratory chain and reduced flavin compounds61–63 and therefore are always present in aerobic cells, if they are not removed fast enough by superoxide dismutases and catalases. In the presence of glutathione, Cu(I) is also able to produce hydroxyl radicals in a similar fashion,64 which is the second reason for strict control of this metal in the cytoplasm. Moreover, free iron also should not be allowed in the cytoplasm. The other essential metal cations of the first transition period are not known to perform a Fenton-type reaction, either because they are not able to change their oxidation state as “free” ions (Ni, Co, Zn) or they prefer two-electron transfer reactions (Mn). Mn complexes are even able to function as superoxide dismutase so that Mn can be addressed as an “anti-Fenton” metal, which explains its low toxicity to C. metallidurans and its protective role in cells under oxidative stress65,66 (Table 1).
The third reason for the toxicity of transition metal cations results from their similar size-to-charge ratios and chemical features, contrasted by different affinities to ligands in metal complexes. This leads to a competition of metals for prospective metal complexes or binding sites in proteins. This was addressed in the well-known Irving–Williams67 series and can be visualized by a simple plot of the affinity to “hard” ligands and “soft” ligands as judged by the solubility constants of metal hydroxide versus metal sulfide compounds (Fig. 4). The borderline metals form a ranking order Zn > Co, Ni > Fe > Mn, while “soft” Cd and Cu are on a second line due to their higher affinity to sulfur compared to oxygen as the first-shell ligand. This agrees with the Irving–Williams series67 and indicates that Cu should efficiently remove other transition metal cations from their complexes, Zn should remove all other cations except Cu and so on.
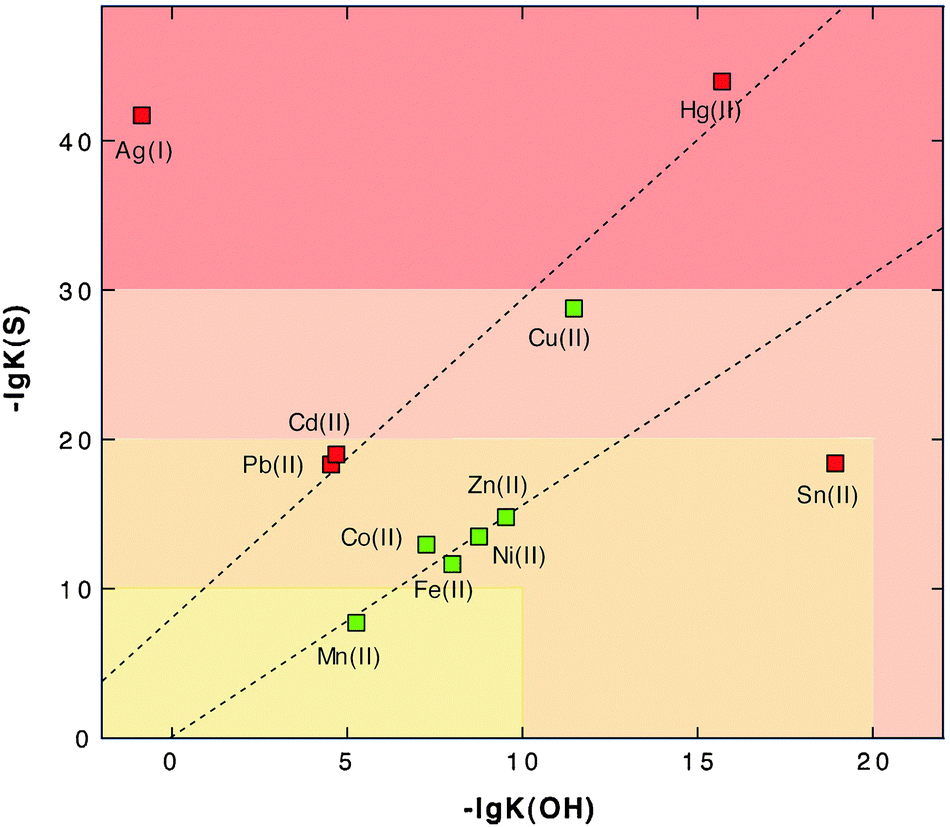 | ||
| Fig. 4 Solubility product of metal sulfides and metal hydroxides as a measure of affinity to sulfur and oxygen, assigning them to the groups of soft or borderline metals. The negative logarithm of the solubility products of divalent heavy metal cations plus Ag(I)36 was plotted against each other to illustrate the Irving–Williams series of the order of stability of metal complexes “Cu > Ni > Co > Zn > Cd > Fe > Mn”.67 Essential trace elements are green and toxic elements red. With the exception of tin and the monovalent cation Ag(I), the data points are located on two lines. The essential elements except copper are on one line, and the “toxic-only” elements due to their high affinity to sulfur are on the other line. Reproduced from ref. 53 with permission. Copyright © 2007, Springer. | ||
This interaction, however, is also influenced by the number of ligands a metal can accommodate so that Zn, which forms only tetrahedral complexes, is not able to remove Fe from octahedral complexes because the binding energy coming from two ligands would be lost. Nevertheless, competition of metals for binding sites is the third reason for the toxicity of transition metal cations. This has been clearly documented for Cd, Zn, Co and Ni.57,68–73 It is one task of the metal transportome to maintain all these metals at an appropriate level that minimizes these negative effects resulting from competition along the Irving–Williams series. Consequently, the number of copper ions has to be controlled most tightly, followed by Zn(II), Ni(II) and Co(II), and of iron due to possible Fenton-type reactions.
Chromate is a special case. The CrO42− oxyanion resembles sulfate SO42− and is imported into bacterial cells by sulfate uptake systems.74,75 Within the cytoplasm, chromate is reduced in a one- or a two-electron step via Cr(V) or Cr(IV) to Cr(III),76 which may form insoluble hydroxides at the slightly basic pH value of the cytoplasm. Therefore, chromate may be toxic because of its interaction with sulfate, the generation of radicals during its reduction, and perhaps because of the chemistry of Cr(III). Interestingly, Cr(III) is a trace element in humans and is involved in the action of insulin.77
In a similar fashion, the heavy arsenate As(V) metalloid oxyanion is imported by phosphate uptake systems and interacts with phosphate metabolism but reduction occurs in a two-electron step to As(III), a “soft” heavy metalloid cation, which can bind in its cationic form to thiol groups.78 A similar toxic action can be expected for V(V), which has been known as an inhibitor of ATPases for a long time.79
Based on the arguments elucidated above, both the beneficial and the toxic effects of transition metals and their bio-availability can be explained. In order for both effects to manifest themselves, however, these metals have to be imported into C. metallidurans or other bacterial cells. The transportome is responsible for this process, which does not act only on an aqueous solution of a single metal cation. Instead, a mixture of metal oxyanions, cations and non-metallic compounds is imported. These compounds may bind or sequester transition metal cations, influencing their availability. To understand the function of the transportome, the environment in which it is acting should be first considered.
The environment of a bacterial cell
General considerations
Transition metal cations and protons are Lewis acids, which interact with Lewis bases.34 These are compounds containing a negatively charged, or partially negatively charged, non-metal atom with a free-electron pair that is not involved in a chemical bond. This free-electron pair is able to bind a proton or a transition metal cation to sequester the metal or even precipitate it. Examples of biologically important Lewis bases are the hydroxide anion OH−, carbonate or bi-carbonate, sulfate, phosphate, thiol groups, carboxyl groups of organic acids, the imidazole group of the amino acid histidine, halogenides, amino groups and last but not least water. The protonated forms of all these Lewis bases act as Brønsted acids, strong acids such as HCl that are partially or completely de-protonated in water or weak acids that are only de-protonated above a certain pH value. The strength of an acid is given by the pKa value, which is the pH value where half of the acid molecules are de-protonated and half remain protonated. Following the Henderson–Hasselbalch equation,80,81 which describes the protonation–deprotonation ratio in the context of the pH value and vice versa, 90% of the groups are de-protonated at pH = pKa + 1, and 90% protonated at pH = pKa − 1. At pKa ± 2, these values are 99%, at pKa ± 3 99.9% and so on.The strength of an acid and that of the corresponding de-protonated Lewis base are inversely related: the weaker the acid the stronger the Lewis base and vice versa.34 Weak acids are therefore strong reaction partners of transition metal cations but strong acids are not. On the other hand, metal cations must compete with protons. At a low pH value, a weak acid may be completely protonated so that this compound is unable to interact with a metal cation. Consequently, sequestration and binding of metal cations by weak acids strictly depends on the pH value of the environment. Moreover, one Lewis acid, the hydroxide anion, is always present in all aqueous solutions, with concentrations between 10 fM at pH = 0 and 1 M at pH = 14. From the point of view of the metal, the electronegativity, and consequently the percent covalent character, influences the interaction with the Lewis base such that transition metal cations, especially the “soft” cations Cu(I) and the toxic-only members of the second and third transition period, interact more strongly with Lewis bases than the divalent alkaline earth metal cations. The latter interaction is in turn stronger than the interaction of Lewis bases with the monovalent alkali metal cations, with a few exceptions.
Taken together, availability and speciation of a transition metal cation depend on (i) its absolute concentration; (ii) the redox potential (oxic, anoxic) that determines the oxidation state of some metals such as copper and iron; (iii) the absolute concentrations of all available Lewis bases in the environment; (iv) the pH value that influences the competition of the metal cations with protons, or, in other words, the availability of a Lewis base for interaction with a metal cation; and (v) the competition of the metal cations for available Lewis bases. Furthermore, in soils, sediments and in the presence of not entirely dissolved particles, additional negatively charged chemical groups within these solid state components may interact with metal cations in a kind of ion-exchange process.
Mineral salts media
The Tris-buffered mineral salts medium TMM used to cultivate C. metallidurans and to test its metal resistance contains 50 mM Tris, 120 mM chloride, 3 mM sulfate, 642 μM phosphate, 100 nM hydroxide (at pH 7), the carbon source gluconate (0.2% w/v = 9.2 mM), the respiration product carbonate and perhaps some excreted organic compounds as possible metal-chelating Lewis bases.4 Since strong acids are usually weak Lewis bases,34 chloride and sulfate should not be able to compete with the weaker acids for divalent metal cations. Indeed, the decadic logarithm lgK of the complexes of Mg(II), Ca(II) and the transition metal cations from Mn(II) to Zn(II) is between 2.2 and 2.4 for sulfate.82 The respective value for Zn(II) gluconate is 1.782 and that of divalent metal complexes of Tris between 5 and 6,83 so that sequestration of the metal cations by Tris should prevent their binding by gluconate, chloride or sulfate.
To calculate the lgKapp for the Zn(II) phosphate complex, regulation of expression of the gene for the zinc importer ZupT, which depends on available zinc in the cytoplasm as measured by the zinc uptake regulator Zur,84 can be used. Regulation of a zupT-lacZ reporter gene fusion with increasing phosphate concentration is compared to that of the metal-complexing compound ethylene-diamine-tetraacetate (EDTA). At pH 7, 762 μM phosphate decreases the availability of 200 nM Zn(II) to a similar extent as 28.9 μM EDTA with a lgKapp of 13.1,45,82 leading to a lgK of 11.6 for the Zn(II) phosphate complex and a ranking of the Zn(II) complex stability of EDTA > phosphate > Tris > sulfate > gluconate.
Complex stability constants for other metal cations and Lewis bases are difficult to obtain from the literature. Since formation of a metal complex might be the cause of precipitation of metal-containing compounds, the solubility product constants −lgL may serve as a proxy to compare the affinities of metal cations to Lewis bases.36,85 With respect to Zn(II), the −lgL ranks the affinities as phosphate (32) > sulfide (24.7) > hydroxide (16.9) > carbonate (10.8) (Table 2). Other divalent transition metal cations form similar ranks and the metal phosphate values are very similar for Cd(II), Co(II), Cu(II), Ni(II) and Zn(II), with those for Ca(II), Mg(II) and Mn(II) being lower (Table 2). Divalent metal cations should be available in a high ratio as metal:phosphate complexes in TMM. TMM is in fact a compromise to allow growth of C. metallidurans with added mM concentrations of heavy metals.4 The phosphate content is too low to allow precipitation of metal phosphate complexes but contains sufficient phosphate for growth of the cells; at 1/3 of the phosphate concentration used in TMM the cells become phosphate-starved at the end of the exponential phase of growth.86
| Metal | Phosphate | Sulfide | Hydroxide | Carbonate |
|---|---|---|---|---|
| a The decadic logarithm of the solubility constants is given. The values were compiled from various sources, http://www.csudh.edu/oliver/chemdata/data-ksp.htm, http://bilbo.chm.uri.edu/CHM112/tables/KspTable.htm.36,82,85 | ||||
| Cd(II) | 32.6 | 27.1 | 13.6 | 11.3 |
| Ca(II) | 28.7 | 7.2 | 5.3 | 8.5 |
| Co(II) | 34.7 | 20.4 | 14.8 | 12.8 |
| Cu(II) | 37.1 | 36.2 | 19.7 | 9.8 |
| Fe(II) | n.a. | 18.2 | 15.1 | 10.5 |
| Fe(III) | 21.9 | n.a. | 37.4 | n.a. |
| Mg(II) | 25.0 | n.a. | 10.7 | 7.5 |
| Mn(II) | 14.5 | 13.5 | 12.7 | 10.7 |
| Ni(II) | 31.3 | 18.5 | 14.7 | 8.2 |
| Zn(II) | 32.0 | 24.7 | 16.9 | 10.8 |
Another growth medium used to cultivate C. metallidurans under chemolithoautotrophic conditions is the phosphate-buffered SGK medium87 that contains 36 mM phosphate and concentrations of divalent metal cations similar to TMM. In this medium, all divalent metal cations should be complexed by phosphate; however, this should occupy only about 1/30 of the phosphate moieties while the remaining 29/30 should be present at pH values around seven as a H2PO4− and HPO42− mixture. This difference in the speciation of the minor and major bioelements should influence the metabolism of a bacterium growing in these media.
This was indeed observed in C. metallidurans. In SGK medium, C. metallidurans CH34 wild type and its plasmid-free, metal-sensitive derivative AE104 were able to produce the key enzymes for chemolithoautotrophic growth as a hydrogen-oxidizing bacterium, including two different nickel-containing hydrogenases and the enzymes of the Calvin cycle needed for CO2-assimilation, even under heterotrophic conditions with gluconate as the carbon source.4 In contrast, strain AE104 was partially zinc-stressed already when 200 nM Zn(II) was present in the TMM medium and silenced many genomic islands on its chromosomes.14 Among those were two islands that contained the hydrogenase and Calvin enzyme genes so that strain AE104 no longer produced all these proteins while CH34 wild type continued to do so. When the gene for the zinc importer ZupT was deleted in strain AE104, zinc stress was turned into zinc starvation, and one of the two hydrogenases was produced again due to an “un-silencing” of the respective genomic islands.14 The phosphate content strongly influences the availability of transition metal cations in mineral salts media, indicating a strong contribution of the metal:phosphate importer PitA to metal and phosphate supply in bacteria, as will be discussed below.
Complex media
Complex media contain hydrolyzed organic compounds, exclusively or together with a mineral salts medium, and these organic compounds also interfere with the metal availability. Important complex media are LB and NB.88,89LB comprises 10 g per L tryptone and 5 g yeast extract, and NB comprises 5 g per L peptone and 3 g yeast extract. In each case, proteins from casein or other sources were enzymatically hydrolyzed, releasing single amino acids and oligopeptides. This adds the important Lewis bases cysteine and histidine to the growth medium, which are derived from the hydrolysates.90 The tri-peptide glutathione is also present and descends from yeast extract. Based on the content of the individual casein species in milk and a glutathione content of 5 mM in yeast cells, this adds 22 μmol cysteine and 198 μmol histidine per g casein and 23 μmol glutathione per g yeast extract. Thus, LB contains 450 μM cysteine88 including the cysteine residue of glutathione, and NB contains 179 μM. From the metal binding properties of cysteine (Table 3), it can be concluded that even at μM concentrations all transition metal cations may reside in metal–bis-cysteinato complexes in both media, even in the presence of phosphate or phosphate-containing compounds. Complexing of metal cations by other amino acids is negligible compared to cysteine. However, ternary complexes of transition metal cations with other amino acids may even be more stable, but the nature of such complexes is difficult to predict using current speciation models.
| Amino acid | Mg(II) < | Mn(II) < | Fe(II) < | Co(II) < | Ni(II) < | Cu(II) > | Zn(II) |
|---|---|---|---|---|---|---|---|
| a The log10(K1·K2) for the formation of a metal–bis-ligand complex is shown.82,93 Values in parentheses give only the log10(K1) value. The metals are oriented in the Irving–Williams-series; n.v., no value available. | |||||||
| Cysteine | (<4) | (4.1) | 11.8 | 16.9 | 19.3 | 18.3 | 18.7 |
| Histidine | n.v. | 7.7 | 9.3 | 13.9 | 15.9 | 16.0 | 11.8 |
| Aspartic acid | 2.4 | (3.7) | 8.5 | 10.2 | 12.4 | 15.4 | 10.2 |
| Glutamic acid | 1.9 | (3.3) | (4.6) | 8.1 | 10.3 | 14.6 | 9.5 |
| Tyrosine | 2 | (2.4) | 7.1 | 8.1 | 10.1 | 14.9 | 8.1 |
| Methionine | 4.7 | n.v. | 6.7 | 7.9 | 10.3 | 14.8 | 8.5 |
| Alanine | 2.0 | 6.1 | 7.3 | 8.6 | 10.7 | 15.4 | 9.4 |
Naturally occurring environments
Organic and inorganic particles present in natural environments also sequester metal cations. Organic compounds such as humic acids present in natural environments complex Zn(II) with lgK values between 2.3 and 5.191 and thus are between Tris and sulfate; they should not be able to compete with phosphate for zinc at pH 7 if sufficient phosphate is available. In the presence of weathering minerals, bacterial cells may be confronted by high concentrations of released transition metals that are not sequestered by phosphate or amino acids.2,10,23,26 In decomposing plant material and inside the gut in mammals such as humans, the transition metal mixture usually has been adjusted by the plant or organisms serving as food, respectively, and the organic substances present are binding partners of the transition metal cations residing there. Bacteria able to infect the remainder of the organism are confronted with an actively decreased metal availability as part of the host response to the infection.92
In complex growth media, transition metal cations will be sequestered by cysteine, cysteine-containing peptides or ternary complexes composed of other amino acyl residues while in mineral salts media, metal:phosphate complexes will be important. In soils and sediments the metal concentration may vary over a wide range. In hosts or decomposing organic material, the transition metal mixture is adjusted or had been adjusted by the respective host or previously living organism. Therefore, in different environments, not only the absolute content of a metal may be different but also its speciation and the composition of the local transition metal cation melánge. In oxic environments, Fe(III) and Cu(II) should be the dominant species. In anoxic environments, these should be Fe(II) and Cu(I), which may be sequestered and precipitated by sulfide. Finally, the pH value and the resulting availability of Lewis bases plus that of the hydroxide anion strongly influence metal availability and speciation.
The transition metal transportome: general considerations
Selectivity and affinity versus import rate
Since transition metal cations or oxyanions cannot pass the hydrophobic core of biological membranes, diffusion of these metals is only possible in small molecules with nearly covalent metal/non-metal bonds, such as in organic mercury compounds. Charged ions can be transported by (i) facilitated diffusion across membranes devoid of any charge gradient such as the outer membrane of Gram-negative bacteria; (ii) primary, gradient-forming transport processes, e.g. ATP-driven transporters; or (iii) secondary, gradient-using transport processes. Cytoplasmic or inner membranes of living bacteria have a proton gradient (and/or sodium gradient). This proton motive force (pmf) is composed of the charge gradient (Δ¥) plus the chemical potential formed by the difference in proton concentration (ΔpH) outside and inside of the membrane, pmf: ΔμH+ = Δ¥ + Z·ΔpH, which is about 150 mV to 200 mV in most respiring bacteria with Δ¥ contributing the largest part in mesophilic bacteria at pH = 7 {for more details and reasons please refer to the freely available companion website of ref. 35 available under http://media.wiley.com/product_ancillary/07/35273165/DOWNLOAD/Website_Chapter1.pdf}.Because the positive charges are outside, any import of a cation and any export of an anion are driven by the pmf in a secondary uniport reaction. Cation-importing reaction may be additionally energized as cation–proton symport to increase the theoretically possible accumulation inside/outside. Import of anions by secondary transport systems can be organized as anion–proton symport. If a net charge of zero is imported, the Z·ΔpH portion of the pmf remains as the sole driving force. To increase accumulation, the negative charge has to be overcompensated by the sum of the charge of the protons. Alternatively, cations can be exported using cation–proton–antiport reactions. Again, if the net charge transfer is zero, only Z·ΔpH drives this reaction and an overcompensation (more proton charges imported than cation charges exported) can enhance the accumulation process. In general, secondary metal transport reactions using a pmf of 180 mV may yield a maximum accumulation factor of 100-fold to 100000-fold. In contrast, transport of a neutral component that is driven by ATP hydrolysis can accumulate nearly half a billion-fold. Nevertheless, also in the case of ATP-driven transport reactions of charged substrate molecules, the influence of the pmf should also be taken into consideration.
In addition to thermodynamics, which rules the maximum accumulation factor obtainable, five other factors are important to understand transition metal homoeostasis in bacteria: (i) number of metals needed per cell; (ii) absolute content of a metal in the environment; (iii) speciation; (iv) kinetics of transport; and (v) metal cation sorting.
C. metallidurans contains for instance 90900 Zn (=151 zmol) per cell (Table 1), corresponding to a quota of 265 μM at a cell volume of 0.57 fL.6 Neglecting any sequestration of a metal within a cell and assuming a cell volume of 0.57 fL, a simple uniport reaction could provide sufficient Zn(II) to the cell if the environmental concentration is above 27 nM. Accordingly, the required concentration for the other divalent cations is between 1.2 nM for cobalt and 159 nM for iron. Under these circumstances the minimum magnesium content for successful uniport is above 3.3 μM.
Second, to produce a cell density of 1012 cells per L containing 90900 Zn per cell, a concentration of 151 nM Zn(II) is required. The minimum zinc content needed by C. metallidurans is 20000 Zn per cell9,94 corresponding to an absolute minimum of 33 nM in the growth medium. TMM contains 35.2 nM zinc added,4 close to the absolute minimum, but due to contamination from water and the salts of the major bioelements it is actually 200 nM.9,14,94 For the other transition metals, the absolute minimum number per cell has not been evaluated. For the standard cell composition (Table 1), the required medium composition is between 6.6 nM for cobalt, 0.9 μM for iron, 18 μM for Mg and 200 μM for phosphate, which corresponds with published data, e.g. complete incorporation of 214 μM phosphate by C. metallidurans grown in TMM with 2 g L−1 sodium gluconate as the carbon source.86
Third, taking now also complexation into consideration, tetra-aquo zinc complexes that are usually referred to as “Zn2+” or hex-aquo complexes of other divalent transition metals occur neither within the cell nor in the environment. As outlined above, transition metal cations may be bound and/or precipitated by Lewis bases in the environment such as hydroxide, carbonate, or phosphate as the most important Lewis bases in mineral salts media and cysteine residues in complex growth media. Within the cell, the totality of the zinc-binding sites of all zinc-binding proteins, the zinc repository,9 and the tripeptide glutathione or similar compounds in other bacteria56,57,60,95 may sequester transition metals. Consequently, the actual thermodynamic equilibrium is not between “Zn2+” outside and inside the cell but in fact between the different speciations of this metal in the growth medium and inside the cell. Binding in the cytoplasm, by four ligands in the zinc repository or two cysteine residues in glutathione, is at least as tight as binding in the cellular environment to phosphate or in bis-cysteinato complexes. The energy released by tight binding in the cytoplasm is higher than that needed to free the metal in the environment, so that this energy difference should be added to the energy used for transport. On the other hand, the transition state resulting from the release of a cation from its Lewis base in the environment might heavily influence the kinetics of metal cation import. Due to the higher affinity to all kinds of Lewis bases, this poses an especial problem for the import of transition metal cations.
Consequently, to import cations sequestered in the environment, the overall metal-Lewis base compound can be imported, e.g. zinc phosphate or nickel complexes, or the affinity of the substrate-binding site of the metal uptake system must be higher than that of the sequestering compound to release it. This is indeed the case; zinc is imported into C. metallidurans by the metal:phosphate importer PitA and the zinc importer ZupT.45 ZupT is essential at medium range EDTA concentrations and therefore able to obtain the metal out of zinc:EDTA complexes {lgKapp = 13.182}, so that zinc phosphate {lgKapp = 11.6, see above} should not be a point of concern for the general function of ZupT but for the turnover number of ZupT-dependent zinc uptake in the environment.
To understand the constraints for kinetics of transition metal transport, a general limitation of all protein-catalyzed biochemical reactions is important. In these reactions, a substrate is bound by an enzyme or transporter in an enzyme–substrate ES complex, which represents a state of low energy if binding is driven by thermodynamics in the simplest case. For the actual reaction, the enzyme–product complex is changed by a chemical transformation or a transport process. This requires a transition compound (TC) of high energy, and the difference between the energy of the TC and that of the ES complex determines the rate of this reaction. Consequently, binding a substrate with high affinity and selectivity means a low energy of the ES complex, a large difference in energy ES/TC, resulting in a low reaction rate (Fig. 5). On the other hand, low substrate affinity and selectivity allows a high reaction rate. Finally, using additional energy sources for the reaction such as ATP hydrolysis could “tunnel” the energy of the TC and allow high selectivity with a high reaction rate, albeit at additional energetic costs.
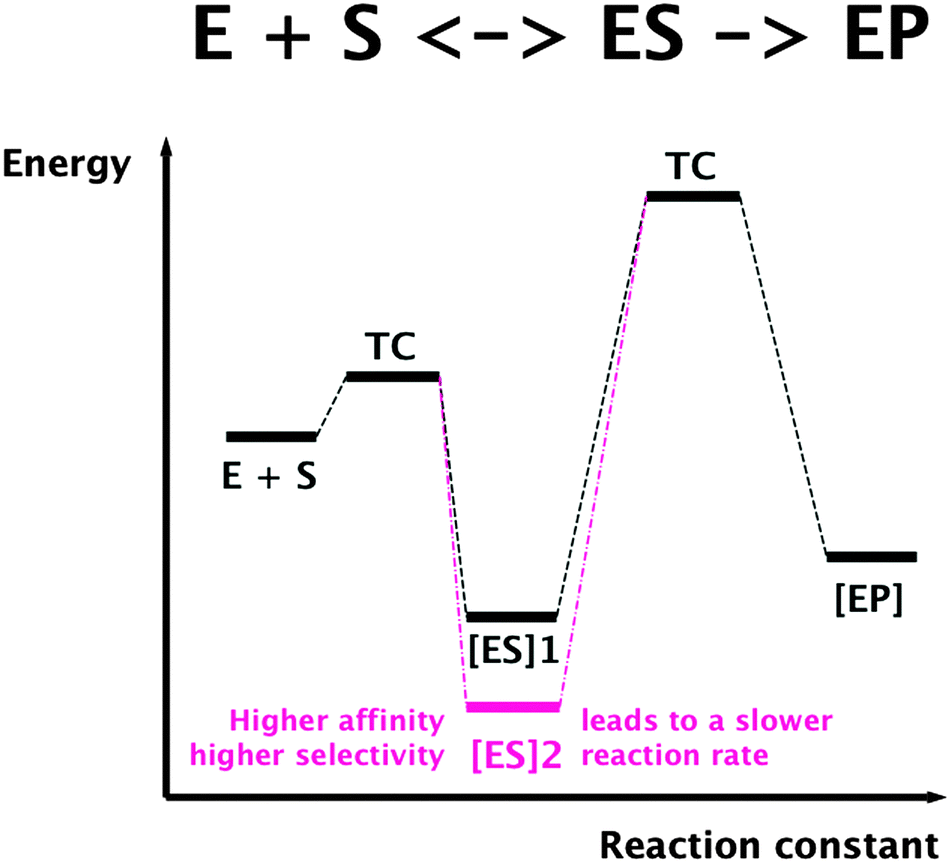 | ||
| Fig. 5 Selectivity and substrate affinity of biochemical reactions are antagonists of the reaction rate. TC, transition compound; ES, enzyme–substrate complex; EP, enzyme–product complex. | ||
This leaves two options for transition metal cation transport reactions. Due to the similar charge to surface ratios of these metals (Fig. 3) they can be transported by secondary systems with a low substrate specificity and selectivity but a comparatively high transport rate and a low energetic cost of transport. Metal cation importers could be uniporters or proton–cation symporters, while exporters can be proton–cation or cation–cation antiporters. Alternatively, a highly specific transport could be performed by primary transporters, e.g. driven by ATP hydrolysis, and the discrimination between transition metals should use the differences in their number of occupied d-orbitals by formation of metal complexes during transport, or differences in the redox potential (Fig. 3).
These two options for transport also connect the metal-sorting process to transport kinetics. If an individual metal concentration is too low, additional primary import systems may be activated in the cell to increase the content of this metal. Alternatively, surplus metals may be removed by efflux systems. The entropy generated by the futile cycle composed of import and export reactions is needed to adjust and maintain the metal composition, which minimalizes negative interferences between the metals when in a correct range. So, a high-rate coupled with a low-specificity transport process should be at the core of the transition metal transportome, which is supplemented by inducible primary high-specificity import systems or inducible efflux systems.
Such an arrangement may work surprisingly well and with a high energy efficiency. Fig. 6 compares the elemental composition of the C. metallidurans cells with the elemental content of standard sea water (OSW). With the exception of most major bio-elements (S, K, Mg, Na, K), the elemental composition is a double logarithmic function of the sea water content. Import of all of the required transition metal cations by uniport that uses a Δ¥ of 93 mV supplies these metals in the required metal concentration and composition. At higher Δ¥ values or metal concentrations, flux control of the importers may limit uptake and/or efflux systems may remove individual cations or groups thereof. Please note also the position of phosphate in this picture (Fig. 6), since it points out again the importance of metal:phosphate complexes as resources.
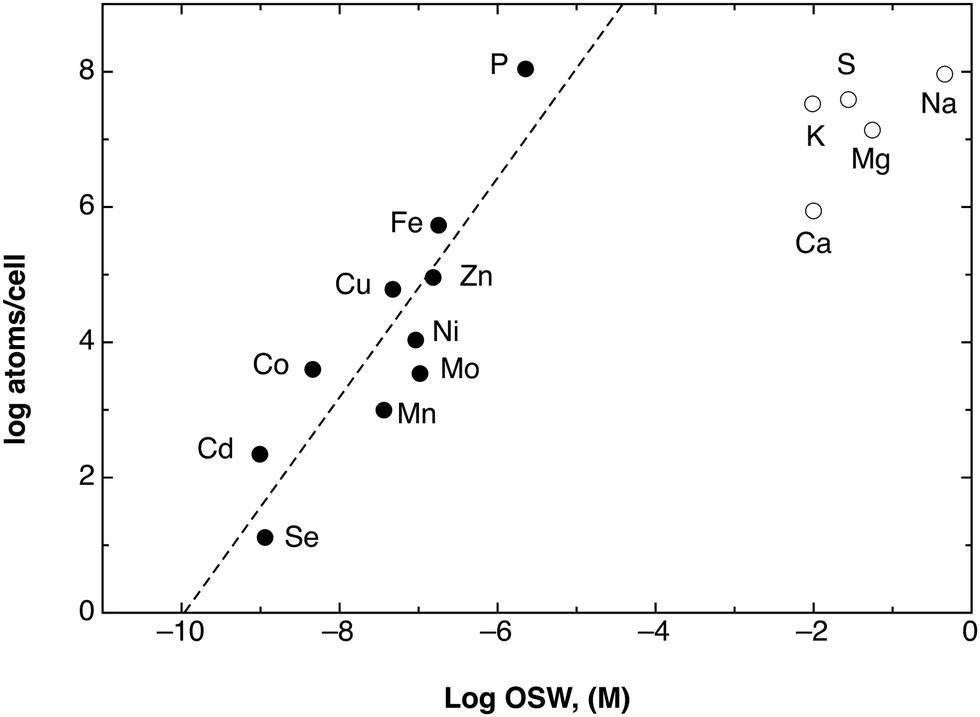 | ||
| Fig. 6 Plot of the elements per cell against the OSW. A double logarithmic plot of the number of atoms in the cell of the bacterium C. metallidurans as determined by ICP-MS45 against their occurrence in sea water (OSW) as a model of a standard environment.36 Please note that the transition metals plus phosphate and selenium (black dots) reside on a line (dashed). Assuming a cell without internal metal-chelating capacity with a cellular volume of 0.57 fL,6 this line would represent accumulation of the shown metal cations with an energy of 93 ± 20 mV across the cytoplasmic membrane. Reproduced from ref. 51 with permission. Copyright © 2012, American Society for Microbiology. | ||
Task of the transition metal transportome
The transition metal transportome transforms the environmental metal content into the cytoplasmic transition metal content. These metals are needed because of their ability to form complex compounds, making sophisticated biochemical reactions possible. The cations in group II of Fig. 2 are available in the environment for these functions and are not too toxic. Due to their similar surface-to-charge ratios, it should be efficient to import them by low-specificity, high-rate uptake systems, complemented by inducible efflux systems or high-specificity uptake systems if the environmental concentrations are too high or too low, respectively. The high-rate, low-specificity import systems allow an efficient supply of metals. But additional uptake of toxic-only metal ions is the price to be paid.Since the environmental content of individual metals may vary between nM and mM concentrations, more than a million-fold difference, this transformation of the environmental metal concentration into that of the cytoplasm is performed in two steps in Gram-negative bacteria. First, the metal content in the periplasm is pre-adjusted so that the metal uptake and efflux systems of the inner membrane alone do not have to cover for this huge, potentially million-fold difference in concentration. So, there is a periplasmic and a cytoplasmic metal transportome in Gram-negative bacteria, and for each compartment there is a high-rate, low-specificity import supplemented by inducible high-specificity uptake or efflux reactions.
The transition metal transportome for the periplasm
Adjusting periplasmic availability: import across the outer membrane
The first barrier that transition metal ions have to pass in Gram-negative bacteria is the outer membrane. Outer membrane porins form more or less ion-specific pores through this barricade, allowing facilitated diffusion of small hydrophilic and charged ions or molecules from the outside into the periplasm.96–98 These porins form the high-rate, low-specificity core of the periplasmic metal import transportome.C. metallidurans possesses the genes for about 40 outer membrane porins (Fig. 7). In comparison with the most important porins from E. coli, five of the E. coli porins,98 with the general porins OmpC and OmpF among them, are grouped in a tight cluster with only one porin from C. metallidurans. Three C. metallidurans porins are related to OmpG from E. coli, two to MotB, two to OmpA and three to OmpA and MotB (Fig. 7). Most of the C. metallidurans porins, however, form an extra cluster separated from the cluster of E. coli porins. This indicates that the import of substances into the periplasm may follow different rules in E. coli and C. metallidurans.
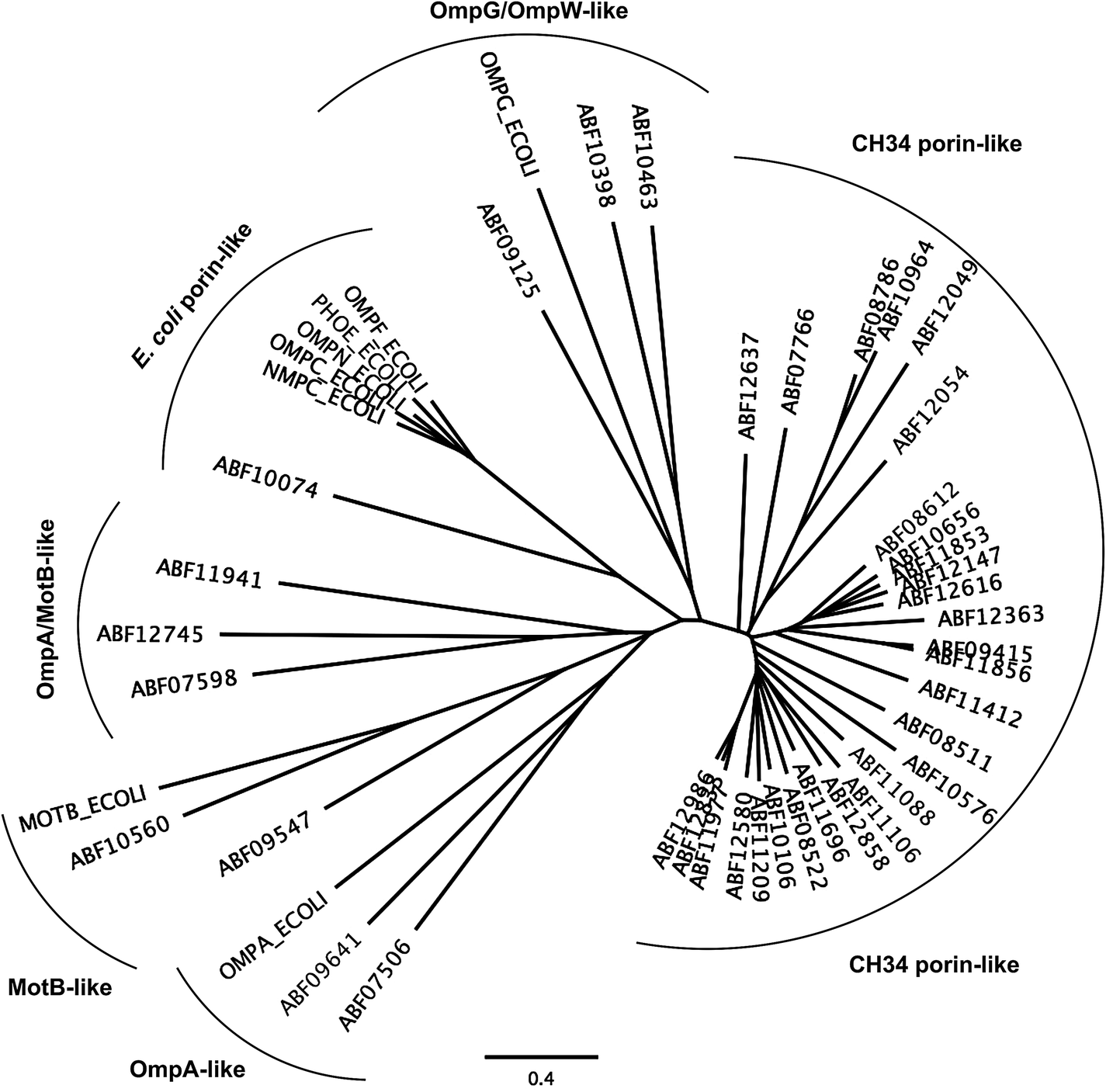 | ||
| Fig. 7 Comparison of the predicted outer membrane porins from C. metallidurans with porins from E. coli. The comparison was performed by multiple alignment using Geneious 6.1.6 (http://www.gneious.com). The scale bar and data bank entries are indicated. | ||
Synthesis of most of the C. metallidurans porins could not be confirmed using a bottom-up proteomic approach.9 One OmpA-, one MotB-, one E. coli porin-related, and three MotB/OmpA-related proteins were identified with Rmet_0712 being the porin with the highest detected copy number in C. metallidurans. OmpA in E. coli plays a structural role in the integrity of the bacterial cell surface and occurs at about 100000 copies per cell.98 MotB is not a protein of the outer membrane but of the inner membrane and acts together with MotA as a torque-generating motor protein complex of the bacterial flagellum.99 The outer membrane protein OmpA and the inner membrane protein MotB, however, share a conserved protein domain (Pfam PF00691), so that predicted proteins are sometimes annotated as “OmpA/MotB protein”. Rmet_2674 is closest to MotB from E. coli; however, none of the OmpA/MotB proteins have their genes in the vicinity of chemotaxis or flagellum determinants. The number of all porins in the C. metallidurans cell is lower than 100000 copies per cell but membrane proteins were quantified to be five times lower than soluble proteins.9 When this fact is taken into account, the total number of these porins in C. metallidurans AE104 would be 62000, which might as well indicate a lower general permeability of the outer membrane of the metal-resistant C. metallidurans that also grows 3.5-times slower than E. coli.
The ΔzupT mutant of C. metallidurans AE104, which suffers from decreased zinc import and disturbed zinc allocation, nevertheless diminished the number of proteins involved in motility and transport,9 including porins (Table 4). In each cluster of porins specific to C. metallidurans, one or two proteins could be identified in the proteome and most of them were down-regulated in the ΔzupT strain (Fig. 8). It is unclear why a metal-starved cell should decrease its import capacity. In general, an outer membrane porin different from that of E. coli plus a decreased number of porins could just as easily contribute to metal resistance in C. metallidurans.
| Rmet | Accession | Number per cell | |
|---|---|---|---|
| AE104 | ΔzupT | ||
| a The Rmet and accession numbers used in Fig. 7 are indicated. The numbers of proteins in the cell of the plasmid-free C. metallidurans strain AE104 and its ΔzupT mutant are from Ref. 9. The numbers in green indicate up-regulation in AE104 compared to CH34, or in ΔzupT compared to AE104, and the numbers in red indicate down-regulation. Bold-faced numbers are significantly different; the numbers in italics indicate detection and quantification only in one out of three biological reproductions. NF, not found in one strain. Porins that have not been found so far: CH34 porins: Rmet_0880 (ABF07766), Rmet_1628 (ABF08511), Rmet_1639 (ABF08522), Rmet_1733 (ABF08612), Rmet_1907 (ABF08786), Rmet_3786 (ABF10656), Rmet_4097 (ABF10964), Rmet_4239 (ABF11106), Rmet_4344 (ABF11209), Rmet_4994 (ABF11856), Rmet_5195 (ABF12054), Rmet_5721 (ABF12580), Rmet_5757 (ABF12616), Rmet_5778 (ABF12637), Rmet_5974 (ABF12833), Rmet_5999 (ABF12858), Rmet_6127 (ABF12986). MotB-like porin Rmet_3688 (ABF10560), OmpA-like porin Rmet_0620 (ABF07506), and OmpW/OmpG-like porins Rmet_3526 (ABF10398), Rmet_3591 (ABF10463), Rmet_2246 (ABF09125). | |||
| Rmet_2538 | ABF09415 |

|

|
| Rmet_3234 | ABF10106 | 792 |

|
| Rmet_3704 | ABF10576 |

|

|
| Rmet_4221 | ABF11088 |
|
20 |
| Rmet_4547 | ABF11412 |

|

|
| Rmet_4834 | ABF11696 | 61 | 61 |
| Rmet_4991 | ABF11853 |
|

|
| Rmet_5118 | ABF11977 | 88 | 85 |
| Rmet_5190 | ABF12049 |
|

|
| Rmet_5288 | ABF12147 |
|
135 |
| Rmet_5504 | ABF12363 | 92 | 124 |
| OmpA/MotB | |||
| Rmet_0712 | ABF07598 | 4696 | 2666 |
| Rmet_5082 | ABF11941 | NF |
|
| Rmet_5886 | ABF12745 |
|

|
| MotB | |||
| Rmet_2674 | ABF09547 |
|
1461 |
| OmpA | |||
| Rmet_2768 | ABF09641 | 1856 | 930 |
| Others | |||
| Rmet_3202 | ABF10074 | 292 | 195 |
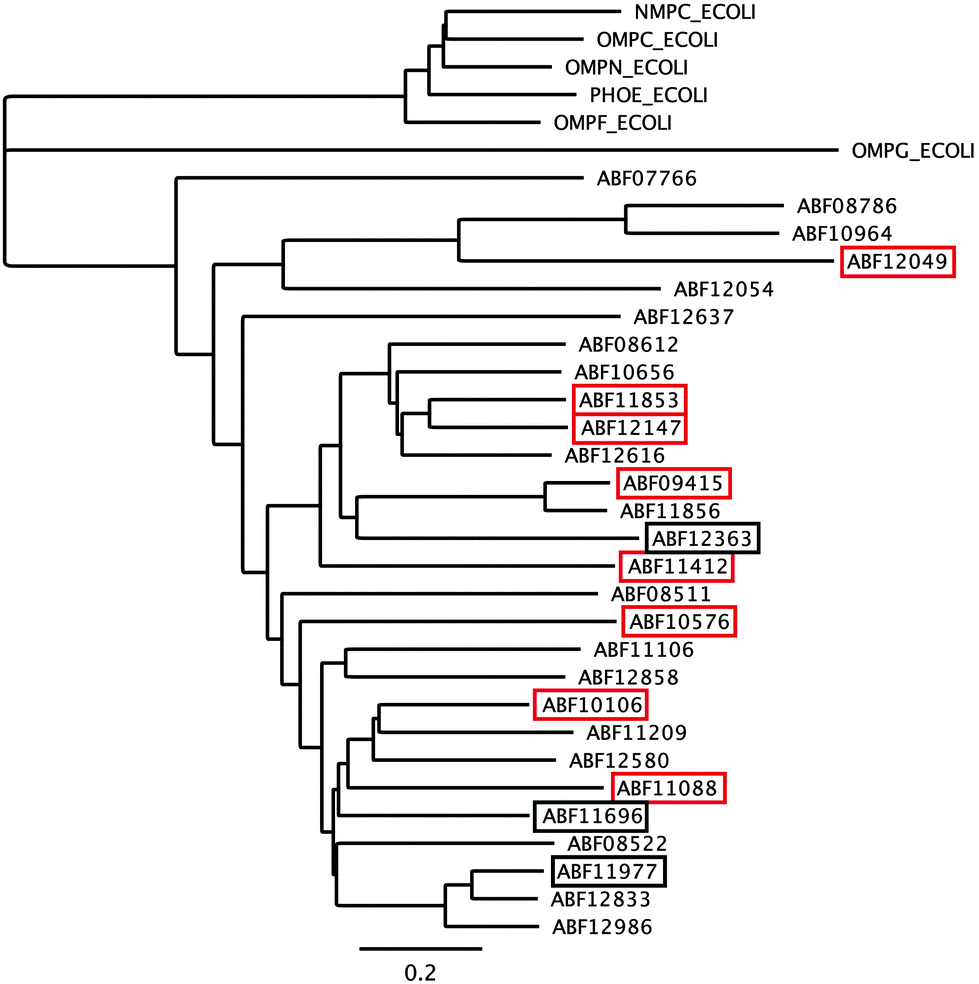 | ||
| Fig. 8 Porins specific to C. metallidurans were compared to E. coli. Only the porins of the CH34 porin-like group and the E. coli porins (Fig. 7) were compared using Geneious. Boxes indicate porins found in the proteome; red boxes indicate down-regulation in the ΔzupT mutant compared to its parent strain AE104 9. | ||
Similar to E. coli and other bacteria, C. metallidurans possesses also a number of TonB-dependent outer membrane proteins, which are able to import actively substances into the periplasm that are rare in the environment or too large to pass through the other porins, complementing the high-rate, low-specificity transport by the other porins with a substrate-specific import reaction. Since the outer membrane does not have a chemiosmotic gradient like the pmf and because energy-rich compounds are not readily available in the periplasm, this active transport process is fueled by the pmf across the inner membrane. Energy is transmitted to TonB-dependent outer membrane proteins as conformational energy by the ExbB–ExbD–TonB protein complex.100–102 Upon binding of an external substrate to such a TonB-dependent outer membrane protein, the energy conserved by the rigid conformation of TonB is used to move a plug out of the center of the TonB-dependent outer membrane protein to allow passage of the respective substrate.48,103
The best characterized TonB-dependent importers are involved in the uptake of Fe(III) siderophores.48,100,104C. metallidurans produces one siderophore, staphyloferrin B, while E. coli synthesizes enterobactin.105–107 Most bacteria are able to import also iron complexes with siderophores produced by other organisms, which can be a symbiotic or parasitic arrangement. Bacteria that try to populate a human host are usually confronted by a host response that decreases the amount of available iron by sequestration to iron-binding proteins, such as transferrin.108 Active pathogens respond by excretion of exotoxins or delivery of such substances into the host cell to kill it and release for instance heme compounds, which can be readily imported. Alternatively, transferrin is directly attacked or iron is released from it by a siderophore with an even higher affinity for ferric iron.109
C. metallidurans possesses the genes for 18 TonB-dependent outer membrane proteins, six of which were found in the proteome.9 Three of these six proteins were again down-regulated in the ΔzupT mutant of strain AE104 (Table 5) but up-regulated in strain AE104 under conditions of metal starvation:14 Rmet_0837, Rmet_1118, and Rmet_5373 encoding Fiu-, YncD- and FepA-like proteins, respectively. Three more genes for TonB-dependent proteins were also up-regulated in strain AE104, namely Rmet_0123, Rmet_1819 and Rmet_4617. The genetic environment and the relationship to TonB-dependent proteins of E. coli assigned to Rmet_0123 and Rmet_1104 roles in zinc transport, while Rmet_1118 is suggested to be involved in ferric staphyloferrin B, Rmet_2789 in cobalamin/B12 and Rmet_5373 in the import of unknown metal complexes.
| Rmet | Accession | Gene | Q | AE104 | ΔzupT | Remark |
|---|---|---|---|---|---|---|
| a The Rmet and accession numbers used in Fig. 7 are indicated. The numbers of proteins in the cell of the plasmid-free C. metallidurans strain AE104 and its ΔzupT mutant are from ref. 9. Q gives the up-regulation of the respective gene in AE104 treated with EDTA in comparison with zinc.14 The subsequent rows are again the number of proteins in the cell of AE104 and its ΔzupT mutant. The numbers in green indicate up-regulation in AE104 compared to CH34, or in ΔzupT compared to AE104, and the numbers in red indicate down-regulation. Bold-faced numbers are significantly different; the numbers in italics indicate detection and quantification only in one out of three biological reproductions. NF, not found in one strain; NeF, never found in the proteome of any C. metallidurans strain up to now. The remark concerns the genetic environment and relationship to TonB-dependent proteins from E. coli. | ||||||
| Rmet_0123 | ABF07009 | cirA |
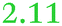
|
NeF | NeF | zur, cobW2, cobW3 cluster, related to CirA_Ecoli and BtuB_Ecoli |
| Rmet_0352 | ABF07238 | oprB | 0.79 | NeF | NeF | Single gene |
| Rmet_0837 | ABF07723 | fiu |
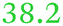
|

|

|
Part of a three-gene cluster up-regulated under metal starvation, related to Fiu_Ecoli |
| Rmet_1104 | ABF07990 | fhuE | 1.89 | NeF | NeF | Adjacent to the cobW1 zinc starvation cluster, minor relationship to FhuE_Ecoli |
| Rmet_1108 | ABF07994 | fcuA |
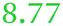
|
NeF | NeF | Adjacent to the staphyloferrin B biosynthesis cluster, minor relationship to FhuE_Ecoli |
| Rmet_1118 | ABF08004 | yncD |
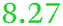
|
209 |

|
Adjacent to the staphyloferrin B biosynthesis cluster, upstream of rpoI, related to YncD_Ecoli |
| Rmet_1819 | ABF08698 | fhuE |
|
NeF | NeF | Part of a two-gene cluster up-regulated under metal starvation, minor relationship to FhuE_Ecoli |
| Rmet_2789 | ABF09662 | btuB | 0.62 |
|
|
Part of the cobalamin biosynthesis and uptake cluster |
| Rmet_3077 | ABF09949 | fiu | 0.89 | NeF | NeF | No information, never noticeable, related to Fiu_Ecoli |
| Rmet_3999 | ABF10867 | fhuE | 1.15 | NF | NF | Downstream of rpoK, minor relationship to FhuE_Ecoli |
| Rmet_4496 | ABF11361 | fhuE | 1.12 | NeF | NeF | Downstream of rpoJ, minor relationship to FhuE_Ecoli |
| Rmet_4497 | ABF11632 | fhuE | 1.07 | NeF | NeF | Downstream of rpoJ, minor relationship to FhuE_Ecoli |
| Rmet_4565 | ABF11427 | cirA | 1.51 |

|
50 | Downstream of highly expressed gene for B-12 independent methionine synthase |
| Rmet_4607 | ABF11469 | oprC | 0.75 | NeF | NeF | Some proximity to the ancient, interrupted czcI2C2B2′ region |
| Rmet_4617 | ABF11479 | fiu |
|
NF | NF/ | Up-regulated under metal starvation, related to Fiu_Ecoli |
| Rmet_5373 | ABF12232 | fepA |
|

|

|
Part of the hmu “hemin uptake” region strongly up-regulated under metal starvation |
| Rmet_5806 | ABF12665 | fecA | 1.36 |
|
|
In a two-gene region with the gene for Rmet_5807, related to FecA_Ecoli |
| Rmet_5807 | ABF12666 | fecA | 1.20 | NeF | NeF | In a two-gene region with the gene for Rmet_5806, related to FecA_Ecoli |
TonB-dependent import processes are also described for zinc and nickel.110–113 Copper can be complexed by a chalkophore,114,115 which is subsequently delivered to the bacterial cell. Cobalamin derivatives can be assumed to be cobalt–cobaltophore” complexes, that are never unloaded due to the stability of the central cobalt cation as described above. Import is again by TonB-dependent uptake systems. Finally, it seems possible that siderophores can also be recruited to form complexes and import other transition metal cations such as Zn(II).116
Adjusting periplasmic availability: active efflux across the outer membrane
TonB-dependent outer membrane proteins as part of the periplasmic metal uptake transportome complement the periplasmic core transportome and provide metal complexes and/or metals with low external availability to this cellular compartment. Metal cations are not only actively imported across the outer membrane, but they are also actively exported. This adjusts the periplasmic content of metal ions and other substances in a controlled fashion as the result of a kinetic flow equilibrium, with free energy transformed into negative entropy (negentropy, order),41 in this case the composition of the periplasmic metabolome. Again, transport is driven by the pmf of the inner membrane. RND-driven tripartite transenvelope efflux systems are responsible for this process in the case of metal cations.117,118 Again, the pmf is transformed into conformational energy. This engine part is performed by the trimeric RND protein of the inner membrane, which possesses a “jellyfish”-like head that extends into the periplasm.119–123 In a peristaltic pump mechanism, periplasmic metal cations are bound to the substrate-binding site of one monomer while the occupied substrate-binding site of the second monomer closes and that of the third monomer delivers the cation further through the complex. Driven by proton import,124 the three sites rotate between the three states in an ordered mechanism.The second component is the tube-like trimeric outer membrane factor OMF,125 which spans the outer membrane, extends into the periplasm and contacts the head of the RND trimer. RND and OMF trimers are connected by the hexameric membrane fusion protein MFP.120,126,127 Since some RND–MFP protein complexes might share a common OMF, the MFPs were also designated “adapter proteins”.128In vitro, membrane-reconstituted RND proteins or RND-containing protein complexes are also able to transport their substrates across the inner membrane119,124 but physiological evidence indicates that in vivo transport for detoxification is from the periplasm to the outside.117,129–131 Nevertheless, transport from the cytoplasm to the periplasm could play a role in the flux-control of the activity of the efflux system to prevent too strong export,132 which might lead to starvation conditions. In addition to this hypothetical flux-control of RND exporters by cytoplasmic ions, other control mechanisms seem to exist: the CzcA subunit of the cobalt–zinc–cadmium RND-driven transenvelope system CzcCBA is not translated or degraded under conditions of zinc starvation.94
C. metallidurans contains the genes for 12 putative metal-exporting RND-driven systems (Table 6).117,133 Two of these are predicted to be Cu(I)/Ag(I) exporters: the SilCBA pump is constitutively expressed from genes located in the large copper resistance region of plasmid pMOL30 while CusCBA systems are chromosomal products and expression of the genes is up-regulated by Au(III) and Ag(I). The other ten systems are likely to be exporters of divalent transition metal cations but only three or four systems can be produced: CzcCBA from pMOL30, CnrCBA from pMOL28 plus the chromosomal ZniCBA and perhaps the chromosomal ZneCBA systems. The other gene regions are silent and/or re-arranged, interrupted or otherwise inactivated (Table 6).
| Group | Name | Rmet | Gene region | Remark |
|---|---|---|---|---|
| a Data from ref. 133 and 117. | ||||
| HME1 | CzcA | Rmet_5980 | czcNICBADRSE | Inducible, pMOL30 |
| HME1 | HmuA | Rmet_4488 | zntA<>czcICB′//hmuB′A | Silent and defect |
| HME2 | CnrA | Rmet_6210 | cnrYXHCBA-cnrT | Inducible, pMOL28 |
| HME2 | NccA | Rmet_6145 | nccCBA | Silent, pMOL30 |
| HME3a | ZniA | Rmet_5319 | zniCBA | Inducible |
| HME3a | ZneA | Rmet_5329 | zneCBA | Silent |
| HME3a | HmvA | Rmet_3838//9 | hmvCBA | Constitutive, nonsense mutation |
| HME3b | NimA | Rmet_5678-Tn-Rmet_5681 | nimBA | Inducible, transposon insertion |
| HME3b | HmzA | Rmet_3011 | hmzCBA | Silent |
| HME3c | HmyA | Rmet_4123 | hmyA | Silent? |
| HME4 | CusA | Rmet_5033 | cusCBA | Inducible |
| HME4 | SilA | Rmet_6136 | silCBA | Constitutive, pMOL30 |
The czc determinant on plasmid pMOL3018,19 carries czcCBA134,135 plus a variety of other genes for additional transporters, regulators and possible periplasmic metal-binding proteins.1 It is involved in resistance to cobalt–zinc–cadmium with Zn(II) being the prime substrate. Plasmid pMOL28 carries the cnr determinant for cobalt–nickel-resistance, composed of the cnrYXH regulatory gene region, cnrCBA plus cnrT for a nickel exporter of the inner membrane.136 The chromosomal zni and zne determinants are much more simple but in the vicinity of each other in four directly adjacent divergons: zniBA<>zniC, zniS<>zniR, Rmet_5324<>Rmet_5325-zneR1S1, zneCBA<> zneR2S2. All three response regulators, ZniR, ZneR1, ZneR2, were found in the proteome of C. metallidurans and also the two histidine kinases ZneS1 and ZneS2. Interestingly, these components of three two-component regulatory systems are not closely related to typical two-component regulatory systems that regulate periplasmic metal homoeostasis processes, namely (i) CzcS is involved in the regulation of the czc region on plasmid pMOL30; (ii) HmuS is encoded as part of the ancient, interrupted czcICB′//czcB′′A (=hmuB′A) region on a chromosome;5 (iii and iv) the two copper resistance-regulating histidine kinases CopS1 and CopS2; and two other putative histidine kinases that seem not to be synthesized under standard conditions. Instead, they are more closely related to (i) BasS, which is involved in iron homoeostasis, (ii) BaeS responding to unknown environmental stress signals,137 and (iii) QseC being involved in motility regulation and global resistance to cobalt, copper, nickel, ruthenium and cesium cations.138 This indicates that the Zni–Zne systems are involved in a global resistance to environmental stress rather than in a specific response to transition metals. In agreement with this, they do not contribute much to metal resistance in C. metallidurans.129,133
Interestingly, the cnr operon is not controlled by a two-component regulatory system but by a sigma factor of the extracytoplasmic function (ECF) family,139,140 CnrH, and a nickel-sensing, membrane-bound anti-sigma factor complex, CnrYX.141–143 This may allow nickel-dependent control of cnrCBA expression despite the cross-talk “noise” of the two-component regulatory systems. C. metallidurans contains 11 sigma factors of the ECF family144 and many of them might be involved in the control of metal homoeostasis.145 RpoI is essential for production of the iron siderophore of C. metallidurans, staphyloferrin B.145 Its ortholog FecI from E. coli is released from its membrane-bound anti-sigma factor and loaded into the RNA polymerase only when the substrate ferric citrate is bound to the TonB-dependent outer membrane protein FecA.146,147 The genes for the two closest related paralogs of RpoI in C. metallidurans, RpoJ and RpoK,145 are in the vicinity of genes for TonB-dependent protein (Table 5), indicating that a similar regulatory process might be involved in the control of these sigma factors too.
CnrX binds Ni(II) in a specific, degenerated octahedral complex and binds Co(II) with lower affinity but not Zn(II).148–151 The CnrCBA efflux complex is produced with a half-maximum rate at an external concentration of about 50 μM Ni(II) and in the presence of Co(II) at a lower level. It is this nickel-specific expression of cnrCBA that makes CnrCBA a nickel exporter.141,142 When produced in an uncontrolled fashion, e.g. in mutant strains without the anti-sigma factor, CnrCBA also mediates zinc resistance.152 Similarly, the substrate specificity of CzcCBA also includes three cations, Co(II), Zn(II) and Cd(II), and here zinc is the predominant regulator with some activity with Co(II) > Cd(II).153–155 CnrCBA and CzcCBA both interact in mediating cobalt resistance. At concentrations higher than 0.75 mM, both systems are needed, but at lower concentrations, the presence of cnr prevents up-regulation of czc by cobalt.133
The RND-driven transenvelope efflux systems of C. metallidurans are all regulated by two-component regulatory systems or ECF sigma factors, which again agrees with the hypothesis that these efflux systems export periplasmic metal cations. The metal specificity of gene regulation rather than the metal specificity of the transport process assigns the particular function to these systems. This solves a dilemma: an efflux system should function rapidly enough to compensate for the transport rate of the corresponding uptake systems, in the case of the RND-driven systems by facilitated diffusion through outer membrane porins. The resulting high-rate export, however, does not allow a high substrate specificity. On the other hand, the regulator is not required to perform a high-rate reaction so that a sufficiently high substrate affinity is allowed. In the case of Ni(II), discrimination of the metal by CnrX uses the occupation state of its d-orbitals, which allows for the specific formation of a degenerated octahedral complex only in the case of nickel ions, but not iron or zinc or copper.148–151 Consequently, CnrCBA is only produced when the nickel concentration in the environment is high. The CnrCBA complex also exports other ions such as Co(II) and Zn(II) but since these ions compete with a high nickel concentration in the periplasm, in C. metallidurans CnrCBA exports in vivo only Ni(II) and to a certain level Co(II) but not Zn(II).
The question is whether RND-driven efflux systems are the second step in the removal of surplus transition metal cations from the cytoplasm via the periplasm to the outside, or whether these systems pre-adjust the periplasmic metal mixture so that the subsequently working metal import systems are enabled to supply these metals in the desired fashion to the cytoplasm. Probably both views are the flipsides of the same coin. Of the cations available to C. metallidurans, grouped in panels I and II of Fig. 2, only for Hg(II), Y(III) and Mn(II) is evidence lacking for the involvement of an RND-driven efflux system in homoeostasis and detoxification. The increase in metal resistance mediated by RND systems is huge, about 20-fold for zinc, 300-fold for cobalt, and 4-fold for cadmium and nickel so that this pre-adjustment or second step efflux is at the core of the outstanding metal resistance of C. metallidurans.4,129
Summary: the periplasmic transition metal transportome
For many transition metal cations that are toxic and/or essential for C. metallidurans, there is a rapid, low-specificity import by facilitated diffusion through outer membrane porins at the core of the periplasmic metal transportome (Table 4). RND-driven transenvelope efflux systems can be produced that actively remove surplus periplasmic cations back to the outside (Table 6). While the transport process is also of low specificity, probably to keep the reaction rate high to cope with the high transport rates of the outer membrane porins, regulation of synthesis of these protein complexes by two-component regulatory systems or ECF sigma factors is highly specific, and thus assigns the function to the individual efflux system. RND-driven efflux systems in C. metallidurans can export Zn(II), Co(II), Cd(II), Ni(II), Cu(I), Ag(I) and maybe Au(I) so that all cations in the fields I and II of Fig. 2 can be removed when in surplus, except Mn(II), Y(III) and Hg(II). Since some of these cations are also trace elements, mechanisms seem to exist that prevent the activity of these exporters during simultaneously existing metal starvation. On the other hand, when the periplasmic content of a metal is too low, TonB-dependent uptake systems ensure an increase in the periplasmic content of the desired metal or metal complex (Table 5).Redox reactions in the periplasm
Of the transition metals C. metallidurans may be confronted with (Table 2), Fe, Mn, V, Cr, Cu, Ag, Au and Hg may occur in ionic oxidation states that are redox active under physiological conditions. In fact, several anaerobically respiring bacteria may use Fe(III), Mn(IV), vanadate or chromate as electron acceptors while several chemolithoautotrophs use Fe(II), Mn(II), V(III) or Cr(III) as electron donors.156C. metallidurans, however, does not possess the required systems for these reactions and uses only molecular oxygen or nitrate as electron acceptors and molecular hydrogen, formiate or organic substances as electron donors.4E. coli is also able to change the oxidation state of iron ions in the periplasm, followed by import of the resulting ferric or ferrous iron, by using the EfeU or FetM iron import pathways.157–159 Again, these systems are not available to C. metallidurans and the only homolog of the FetM or EfeU inner membrane transport systems is PbrT, which is involved in lead resistance.160 Consequently, the redox states of Fe, Mn, V and Cr compounds are not changed in the periplasm of C. metallidurans. In contrast, Au in Au(I) or Au(III) complexes is taken up by C. metallidurans and deposited as periplasmic metallic gold particles.13,161Copper is mainly present as Cu(II) under oxic conditions. Upon contact with the respiratory chain, most likely through reduced quinone compounds,60,162 Cu(II) can be reduced to Cu(I). This ion is much more rapidly imported into the cytoplasm than Cu(II),163 probably by using Na(I)-dependent or K(I) transporters as unspecific substrates.60,164 This makes Cu(I) much more toxic to bacteria than Cu(II). C. metallidurans, E. coli and many other bacteria are able to solve this problem by (i) sequestration of periplasmic copper ions, e.g. by CusF from E. coli,165,166 (ii) export of Cu(I) by the RND-driven CusCBA efflux systems back to the outside,164,166–168 or (iii) oxidation of Cu(I) to Cu(II) using molecular oxygen as an electron acceptor.169 The last reaction functions consequently only under aerobic conditions.164 In E. coli, the respective oxidase is CueO.130,170,171 The two homologs of this protein in C. metallidurans are CopA1 and CopA2, one chromosomally encoded and one by plasmid pMOL30,1,18,19 and both are homologs of the plasmid-encoded E. coli protein PcoA.172,173 Unfortunately, the copper-exporting P-type ATPase of E. coli was named “CopA”,174,175 which results in some confusion between the E. coli P-type ATPase CopA and the copper oxidases CopA from C. metallidurans and Pseudomonas.176,177 The genes for the periplasmic copper oxidases copA/pcoA are usually in a gene region together with genes for proteins that provide copper to the active center of CopA during assembly in the cytoplasm as well as with genes for a two-component regulatory system.169 Cus-like efflux systems also export Ag(I).167,178,179 Together, of the transition metals in group I and II of Fig. 2, C. metallidurans should reduce in the periplasm complexes of Au(III) to Au(I) complexes and complexes of Cu(II) to Cu(I).
Sequestration in the periplasm counterbalances the transportome
Periplasmic metal binding chaperones may serve as a buffer and as an interaction partner of transport systems and client proteins to diminish these toxic effects and to increase the efficiency of import. Highly substrate-specific import systems of the ABC protein superfamilies {transporter classification180 database TC#3.A.1} usually interact with specific periplasmic metal-binding proteins or, in the case of “ECF”-ABC systems181 (which has nothing to do with ECF sigma factors, TC#2.A.88) possess substrate binding sites with a high affinity. Periplasmic metal-binding proteins that deliver their substrate to an ABC importer are known for molybdate, tungstate, Fe(III) siderophore complexes, Co-containing cobalamin derivatives, Fe(II)/Mn(II), Zn(II) and Ni(II),182 which sums up all essential transition metals except copper. C. metallidurans does not possess a ZnuABC183 or a NikABC184,185 ortholog for zinc and nickel, respectively, but it does have a ModABC186,187 importer for molybdate/tungstate (Rmet_0569, 70, 71) and one for the staphyloferrin B-type siderophore, probably HmuTUV (Rmet_5376, 77, 78). In the case of small substrates that were previously imported by TonB-dependent processes, it would be useful if the periplasmic binding protein for the subsequent import into the cytoplasm by an ABC importer would interact directly with the TonB-dependent protein to prevent loss by diffusion through the outer membrane porins. This dangerous situation does not exist for metal complexes that are too big to pass through the porins so that such a protein–protein interaction is less likely to occur.The extremely toxic mercury cation Hg(II) is sequestered in the periplasm by the MerP protein to prevent damage, and subsequently passed on to Hg(II)-specific uptake systems such as MerT, MerC or MerF, and from there further on to the NADPH-dependent mercury reductase MerA.169 This transforms the cation into the volatile metallic mercury that diffuses rapidly out of the cell and its environment. C. metallidurans contains three active mercury resistance determinants both chromosomally encoded and encoded by each of the plasmids.188,189
There are only a few examples of copper-dependent proteins that need to be loaded with this cofactor in the cytoplasm. These are the periplasmic copper-dependent Cu(I) oxidases CopA1, CopA2 in C. metallidurans and PcoA, CueO in E. coli.169 The cop and pco determinants also encode proteins that may import copper into the cytoplasm for delivery to these client proteins. Export of the resulting oxidase is subsequently achieved by the TAT (twin-arginine transport) pathway, which is able to export folded proteins to the periplasm.190 Other copper-dependent proteins are transported to this compartment by the Sec general secretion pathway,191 which transports unfolded proteins. These cases, e.g. the periplasmic Cu–Zn superoxide dismutase SodC and the copper sites in the cytochrome c oxidase, the respiratory chain component IV, comprise the main copper-dependent cellular proteins. Copper needs to be loaded into the client proteins in the periplasm and this is accomplished by periplasmic copper-binding chaperones.192,193
Theoretically, this should make it unnecessary to import copper into the cytoplasm at all, except for delivery to CopA-like proteins that are produced when the cells are exposed to copper. Interestingly, C. metallidurans and other bacteria carry the genes for two types of copper-exporting P-type ATPases.194,195 One group of proteins has the main task of removing toxic copper from the cytoplasm (CupA and CopF in C. metallidurans), while the other group seems to deliver copper into the periplasm for allocation to the periplasmic copper-binding sites of copper-dependent proteins (Rmet_2046, Rmet_2379) such as SodC.193
Delivery of Cu(I) from P-type ATPases to periplasmic copper chaperones is likely and has been demonstrated for CusF.196 CusF subsequently hands the ion over to the CusB MFP component of the CusCBA transenvelope efflux system.197,198Via this mechanism, and to prevent import into the cytoplasm, Cu(I) is (i) oxidized by CopA/CueO/PcoA to the less toxic Cu(II); (ii) exported by CusCBA systems; (iii) sequestered by CusF and related proteins or accepted by these proteins from copper-exporters and passed on to CusCBA systems; (iv) sequestered/accepted by other periplasmic copper chaperones for delivery as cofactor to client proteins.
Interestingly, synthesis of the Cu–Zn SodC in C. metallidurans also requires prior import of the zinc into the cytoplasm to deliver this ion efficiently to this periplasmic protein.9,94C. metallidurans contains a variety of possible metal-binding chaperones as part of the Czc system, e.g. CzcI, CzcE, and CzcJ. CzcE might be involved in zinc and/or copper homoeostasis.153,199 Since many czc-like determinants in other bacteria and the (now interrupted and inverted) ancient czc determinant on the chromosome of C. metallidurans are in fact czcICBA regions, in many cases with the gene for a zinc-exporting P-type ATPase in a divergon situation,5 CzcI rather like CusF might accept Zn(II) from the P-type ATPase for delivery to the CzcCBA system. Alternatively, CzcI and the other possible periplasmic zinc chaperones may store periplasmic zinc, protect it from export by the CzcCBA system, and deliver it to import systems. Such a periplasmic zinc storage activity has been described for ZinT from E. coli.200,201
Silver is in the same group of the periodic system as copper, but the only stable ionic oxidation state is Ag(I). This ion is on the one hand less available to cells compared to copper ions because it is readily precipitated by chloride, binds strongly to thiol compounds, or is reduced to metallic silver, especially in the presence of light. On the other hand, as a “soft” thiol-loving ion, Ag(I) is inferior in toxicity only to Hg(II). Detoxification of Ag(I) is accomplished by a process parallel to Cu(I) detoxification, by specific silver resistance systems related to copper resistance determinants, or by such systems accepting both ions as substrates. Export to the periplasm is via P-type ATPases and CusF-like chaperones may take over to export it further on to the outside by CusCBA/SilCBA-like systems.178,179 Since Ag(II) does not exist, CopA-like proteins are not involved in silver detoxification. However, the SilE protein might serve as a periplasmic “silver sponge” that binds and stores the toxic ion.
Gold can exist as Au(III) and Au(I) but only in complexes. The cations are immediately reduced to metallic gold by reduction by water. Due to the extremely high electronegativity of gold, the highest of all transition metals, Au–C, Au–S and even Au–halogenide bonds are almost covalent so that Au(III) can exist in water as an Au(III)Cl4− complex, which is taken up by C. metallidurans into the periplasm and cytoplasm, and reduced during this process to Au(I) complexes. These are finally reduced further to metallic gold nanoparticles in the periplasm,13,161 and, interestingly, the copper oxidase CopA1 is involved in this process.202
Taken together, periplasmic metal chaperones counterbalance the activity of the transport systems that introduce or remove transition metals into or out of the periplasm. They play a role in homoeostasis of nearly all ions in groups I, II and V in Fig. 2. Iron (group V) can be reduced or oxidized and bound for further import into the cytoplasm. This is similar for iron siderophore complexes. For the ions in group II, periplasmic chaperones together with importers exist for Mo, W, V, Zn, Ni, Mn and B12. Chaperones for delivery of Cu to exporters or client proteins also exist, leaving only Y, which is in any case misplaced in this group. Among group I metals, Ag(I) is sequestered by SilE and CusF-like proteins and Hg(II) by MerP for further uptake to the cytoplasm and subsequent reduction. Cadmium can be bound by ZinT-like periplasmic zinc chaperones but this process does not contribute to cadmium resistance.200 Gold has the lowest content in the earth's crust and consequently has the least chance of occurrence in an ecosystem of all group I, II, V elements (Fig. 2). It is only present in complexes with a strong covalent character of the bond so that transient binding by a periplasmic protein for further delivery may not be an efficient process.
The metal transportome for the cytoplasm
Import across the inner membrane
The events that shape the periplasmic transition metal content are mirrored by parallel events adjusting the cytoplasmic transition concentration and composition. Again, there is (i) high-rate, low-specificity import for an efficient supply with a mixture of transition metal cations; (ii) high-rate, low-specificity export that is able to compensate for the high import rate of these systems; (iii) metal-specific control of synthesis of the efflux systems plus flux control; (iv) redox reactions in the case of a few metal cations; (v) metal-binding events that counterbalance the kinetic flow equilibrium, which is the result of the export and import reactions; and (vi) highly specific but low-rate import of transition metals that are under-represented in the actual cytoplasmic metal mixture, which is performed by the ABC-type importers described above.In C. metallidurans a battery of redundant secondary import systems with low substrate specificity supply ions to the cytoplasm (Table 7). In some cases the primary substrate is a major bioelement, which is needed by the cells in a much higher amount than the minor transition metal bioelements, e.g. 14 million Mg compared to 90 thousand Zn45,94 (Table 7). Consequently, high import rates are needed, which lowers the substrate specificity, so that other ions are usually additional substrates of these importers.
| Rmet | Protein | Family | TC number | Substrates |
|---|---|---|---|---|
| a The proteins were assigned to the transporter protein family and TC number using the transporter classification database.5,180,210 Substrate assignment due to published45,94,160,169,203–205,207–209 and unpublished data (Herzberg and Nies et al., unpublished). | ||||
| Rmet_3052 | CorA1 | MIT | 1.A.35 | Mg, Co, Ni, Zn203 |
| Rmet_0036 | CorA2 | MIT | 1.A.35 | Mg, Co, Ni, Zn203 |
| Rmet_3287 | CorA3 | MIT | 1.A.35 | Mg, Co, Ni, Zn203 |
| Rmet_0549 | ZntB | MIT | 1.A.35 | Zn and others203 |
| Rmet_1973 | PitA | PiT | 2.A.20 | Metal:phosphate203 |
| Remt_2621 | ZupT | ZIP | 2.A.5 | Zn and others203 |
| Rmet_1533 | HoxN | NiCoT | 2.A.52 | Ni, Co, Zn203 |
| None | MgtE | MgtE | 9.A.19 | Absent in CH34 |
| None | MntH | NRAMP | 2.A.55 | Absent in CH34 |
| Rmet_5936 | MgtA | P-type | 3.A.3 | Mg, Zn, others204,205 |
| Rmet_2211 | MgtB | P-type | 3.A.3 | Mg, Zn, others204,205 |
| Rmet_5890 | FeoB | FeoB | 9.A.8 | Fe207,208 |
| Rmet_5945 | PbrT | ILT | 2.A.108 | Pb?160 |
| Rmet_6115 | CopD1 | CopD | 9.B.82 | Cu169 |
| Rmet_5668 | CopD2 | CopD | 9.B.82 | Cu169 |
| Rmet_2369 | Not named | MFS | 2.A.1 | Cu209 |
| Rmet_3466 | Not named | MFS | 2.A.1 | Cu209 |
Sulfate permeases (candidates Rmet_0606, 1188, 3720, 4472, 4600) import sulfate but also the structural analogs molybdate and chromate, while phosphate importers (PitA) transport metal phosphate complexes, vanadate and arsenate. Proton symport with a net neutral charge is sufficient to drive this import by using the Z·ΔpH portion of the pmf. Known uptake systems for metal cations are the magnesium importers of the CorA (MIT family) or the MgtE (MgtE family), the zinc/iron-, iron/manganese-, and nickel/cobalt-importing ZIP, NRAMP and NiCoT proteins, respectively, and metal-importing P-type ATPases. C. metallidurans possesses four MIT proteins (CorA1, CorA2, CorA3, ZntB), one ZIP (ZupT), one NiCoT (HoxN), no MgtE, no NRAMP but two Mg/Ca-transporting P-type ATPases (Table 7).45,210
The structure of a MgtE and a CorA protein from other bacteria indicates that the activity of these unrelated importers can be flux-controlled.211–215 Import activity of the pentameric CorA5 complex from T. maritima is locked in a transport-incompetent conformation by allosteric, sequential binding of cytoplasmic Mg(II) cations to the five protomers.216 Since the amino acids required for transport and flux control by gating are all conserved within the MIT protein family, eukaryotic homologs included, this flux-controlled uptake of magnesium seems to be a general mechanism.212 Binding of cytoplasmic Mg(II) to regulatory sites shuts down the import so that the cells cannot be overloaded with magnesium. Most bacteria possess either a CorA or a MgtE ortholog.
All these secondary and primary importers have a broad substrate specificity and transport a variety of other divalent metal cations in addition to their primary substrates. The three CorAs in C. metallidurans are involved in Zn(II) and probably Mg(II) import, CorA1 and CorA2 are also involved in nickel transport and CorA1 and CorA3 have something to do with cobalt toxicity, although CorA123 together function as cobalt importers.203 These CorA substrates were already identified a long time ago in E. coli and Aerobacter (=Enterobacter) aerogenes,217 and nickel uptake by a magnesium uptake system in Cupriavidus necator (=Ralstonia eutropha, Alcaligenes eutrophus)218 and C. metallidurans.75,86
Since CorA-like importers can be switched off at high cytoplasmic magnesium concentrations by flux control, additional uptake systems for divalent transition metal cations must exist. The ZupT protein of the ZIP family is not essential for net zinc import45 but deletion of ΔzupT causes multiple pleiotropic effects so that zinc import by ZupT is connected to zinc allocation in the cytoplasm.94 The cells might contain a sufficient number of zinc atoms, for instance 120000 Zn(II) per cell at an outside concentration of 100 μM; nevertheless, allocation of this metal to the important zinc-dependent RpoC or beta-prime subunit of the RNA polymerase is not efficient. Even supply of zinc to the periplasmic Zn–Cu superoxide dismutase SodC, which is not exported as a folded protein by the TAT system but as an unfolded polypeptide by the general secretion system, is hampered, indicating that zinc allocation to SodC may need uptake into the cytoplasm by ZupT and re-export to the periplasm.9,94
Due to a potential sequestration of zinc and other metals by phosphate in TMM, the metal:phosphate uptake system PitA is also an important source for transition metals in the cytoplasm.203Table 2 predicts that in oxic environments, in the absence of sulfide, all divalent transition metal cations with the exception of Mn(II), which is out-competed by Mg(II), should be present as a metal:phosphate complex. Indeed, involvement of PitA in the import of Zn(II) and other transition metal cations, including Cu(II) and Fe(II), can be demonstrated.203 In E. coli, PitA also seems to be involved in zinc uptake219 and some strains of this bacterium even contain a paralog of PitA, called PitB.220 Consequently, mutations in pitA cause zinc resistance.221 PitA also imports phosphate complexes with magnesium and calcium, and Zn(II) competes with Mg(II) for import by PitA.221 This highlights an important connection between phosphate and metal import in all kinds of mineral salts media or natural environments, which are poor in sulfide or cysteine-containing compounds. It also explains the long standing observation that phosphate and magnesium homeostases are closely linked.
In enterobacteria, a two-component regulatory system composed of the membrane-bound histidine kinase sensor PhoR (C. metallidurans ortholog Rmet_2179) and the response regulator PhoB senses low phosphate concentrations on the inside of the cells. This leads to PhoB-phosphate-dependent up-regulation of a variety of operons including phoBR itself, pstSCAB-phoU for the phosphate-specific ABC importer, and, interestingly, phoQ.222,223 PhoU is a dimer that binds Mn(II) and Mg(II) and interacts with PhoR and the component of the high-affinity phosphate uptake system PstABC, PstB.224 This leads to the formation of a signaling complex that responds to low environmental phosphate levels by the scanning activity of the PstABC uptake system, and may integrate this information with the cytoplasmic magnesium content. The PstABC importer transports orthophosphate only without bound metal cations.225,226
PhoQ (C. metallidurans ortholog Rmet_5129) is another membrane-bound histidine kinase sensor. It senses low magnesium concentrations outside the cells, which leads to phosphorylation of its response regulator PhoP.227,228 The PhoPQ two-component regulatory system is up-regulated at low magnesium concentrations, acidic pH values, and in the presence of antimicrobial peptides in Salmonella.229 When this system is activated at low magnesium concentrations, expression of the genes responding to this signal needs the magnesium-transporting P-type ATPase MgtA, while activation of another part of the PhoR regulon by acidic stress or antimicrobial peptides does not.230 At acidic pH values, the PhoPQ system also promotes uptake of Fe(II) by the Feo iron uptake system via the response regulator RstA.229 At low magnesium concentrations, a variety of operons are up-regulated, including the genes for the magnesium-importing P-type ATPases MgtA in E. coli, or MgtA and MgtB in Salmonella enterica. In addition to transcription initiation control, transcription elongation into the coding regions of these genes is regulated by a riboswitch, and is only allowed at low cytosolic magnesium concentrations.231 This highlights the important role of phosphate in metal homoeostasis.
If the five systems CorA1, CorA2, CorA3, ZupT, and PitA are removed from C. metallidurans, net import of all metals is still possible.203 The transporters involved in this uptake process are the fourth MIT protein, ZntB, HoxN and the two P-type ATPases MgtA and MgtB. Remarkably, further deletion of the genes for these four importers does not abolish net metal import, although some multiple deletion mutants show a clearly diminished fitness. All these nine systems are important for metal homeostasis, but additional, currently unknown metal importers clearly must exist in C. metallidurans.206
Import of iron into C. metallidurans occurs as the Fe(III) staphyloferrin B complex or by the FeoAB system (Rmet_5891/90), which is not an ABC system but may be a GTPase.207,208 Iron and copper can also be imported by PitA as a phosphate complex.203 Additionally, copper import by CopD-like systems could be needed to supply this ion to the periplasmic copper-dependent copper oxidase CopA before export to the periplasm by the twin-arginine transport system.169,238 In Rhodobacter capsulatus,209 copper transfer to periplasmic copper-dependent proteins first requires uptake of copper via importers of the major facilitator superfamily (MFS), followed by export by specific anabolic P-type ATPases194–196 and subsequent transfer to periplasmic copper chaperones. C. metallidurans contains two candidates for anabolic P-type ATPases encoded close to cytochrome oxidase biosynthesis gene clusters, RdxI and CtpA (Table 8). In the vicinity of the genes for each exporter are genes encoding two putative copper-importing MFS proteins (Table 7). Why the lead-resistance determinant on plasmid pMOL30 contains the gene for a possible lead importer PbrT160 has not yet been established.
| Rmet | Protein | Family | TC number | Substrates |
|---|---|---|---|---|
| a The proteins were assigned to the transporter protein family and TC number using the transporter classification database.5,180,210 Substrate assignment was based on published data as indicated. | ||||
| Rmet_4594 | ZntA | PIB2-type | 3.A.3 | Zn, Cd129,232 |
| Rmet_2303 | CadA | PIB2-type | 3.A.3 | Zn, Cd129,232 |
| Rmet_5947 | PbrA | PIB2-type | 3.A.3 | Pb, Zn, Cd129,160 |
| Rmet_5970 | CzcP | PIB4-type | 3.A.3 | Zn, Cd, Co129 |
| Rmet_3524 | CupA | PIB1-type | 3.A.3 | Cu, Ag202 |
| Rmet_6119 | CopF | PIB1-type | 3.A.3 | Cu202 |
| Rmet_2046 | RdxI | PIB1-type | 3.A.3 | Cu anabolic194,195 |
| Rmet_2379 | CtpA | PIB1-type | 3.A.3 | Cu anabolic194,195 |
| Rmet_5979 | CzcD | CDF | 2.A.4 | Zn, Cd, Co129,233,234 |
| Rmet_0198 | DmeF | CDF | 2.A.4 | Co, Ni129,235 |
| Rmet_3406 | FieF | CDF | 2.A.4 | Fe235,236 |
| Rmet_6211 | CnrT | DMT | 2.A.7 | Ni5,210 |
| Rmet_6144 | NreB | MFS | 2.A.1 | Not expressed |
| Rmet_0391 | AtmA | ABC | 3.A.1 | Ni237 |
| Rmet_6202 | ChrA1 | CHR | 2.A.51 | Chromate133 |
| Rmet_3865 | ChrA2 | CHR | 2.A.51 | Chromate133 |
| Rmet_4831 | ChrA3 | CHR | 2.A.51 | Unknown133 |
| Rmet_2517/8 | ChrA4a/b | CHR | 2.A.51 | Unknown133 |
Due to the fact that during the formation of metal:phosphate complexes Mn(II) should be out-competed by Mg(II) most bacteria need to carry a manganese–proton symporter of the NRAMP protein family or ABC importers for this metal,239e.g. to metallate client proteins with manganese under conditions of redox stress.65,66C. metallidurans, however, does not contain such a manganese importer to decrease the import of Cd(II)45 and consequently seems not to use this metal, which explains the low cellular content of manganese in C. metallidurans45 and the absence of a Mn-type superoxide dismutase.240 An iron import pathway via an NRAMP system is therefore not available in C. metallidurans.
The battery of redundant metal importers supplies the cytoplasm of C. metallidurans with the overall mixture of transition metals available in the periplasm, at a high rate, but with a low substrate-specificity and comparably low energy. In ecosystems and media comparable to sea water the mineral composition is sufficient to warrant a balanced supply with these ions (Fig. 6), but in others, single metals may be over- or under-represented. As outlined above, most bacteria are able to produce highly specific metal import channels for under-represented metals, which can be composed of the following modules allowing (i) excretion of metal-chelating compounds; (ii) TonB-dependent uptake into the periplasm; (iii) binding by periplasmic metal-binding proteins; and (iv) further import into the cytoplasm by ABC transport systems such as ZnuABC for zinc, NikABCD for nickel, and SitABC for manganese and iron. C. metallidurans, however, does not possess orthologous ABC importers, and instead relies mostly on its battery of redundant uptake systems (Table 7).
Efflux back into the periplasm
C. metallidurans is adapted to environments with a high metal content and lacks orthologs for these uptake systems. Instead, this bacterium possesses a broad repertoire of efflux systems that are able to move surplus ions back to the periplasm for further export to the outside by the RND-driven systems. These efflux systems are P-type ATPases or secondary transport systems.C. metallidurans contains eight P-type ATPases of the PIB protein family (Table 8), which are efflux systems for heavy metal cations. Four members of the PIB1 subgroup241 mentioned above export Cu(I) and contribute to Ag(I), but not to gold, resistance.202 The three PIB2 subgroup members ZntA, CadA and PbrA export Zn(II), Cd(II) and Pb(II). Again, all three proteins transport Zn(II) and Cd(II) with similar affinities and turnover rates.129 Once more it is the specificity of their production that assigns the particular function to the respective protein. As in the case of the RND transporters, a certain low substrate specificity might be required to allow a sufficient transport rate whereas the regulatory process is not hampered by this constraint.
PIB-type ATPases contain a conserved cysteine residue in the transmembrane part of the protein with a proline residue adjacent to it.241 In PIB1 and PIB2 ATPases, a second cysteine residue is present and form a “CPC” motif, which is part of the substrate-binding site of these exporters. Additional metal-binding sites in various arrangements are present in the cytoplasmic domains of these proteins. These sites have an important function in flux control and/or acquisition of the substrate cation from the cytoplasm for delivery to the “CPC” site and subsequent export.242
C. metallidurans contains an additional zinc-exporting P-type ATPase of the PIB4 subgroup, CzcP, which is encoded by plasmid pMOL30 as part of the czc metal resistance determinant. CzcP enhances zinc resistance but is not able to mediate a notable zinc resistance without one of the three PIB2-type ATPases.129 This indicates that CzcP is only able to reach some cytoplasmic zinc speciations or zinc pools while the PIB2-type ATPases such as ZntA are able to reach all zinc species for removal and consequently detoxification. Since it is important for efficient zinc supply to zinc-dependent proteins if the ion was imported by ZupT or other importers, this indicates the presence of at least two, or perhaps three, zinc pools or speciations in the cytoplasm: loosely bound zinc ions that are imported by the other transport systems and can be removed by CzcP, and more tightly bound zinc imported by ZupT, which may need ZntA for export from the cytoplasm.9,94,129
Although CzcP is not able to reach all cytoplasmic zinc species, it is able to export its substrates with a higher rate than the PIB2-type ATPases such as ZntA. CzcP also has a broader substrate specificity, which may include Co(II) under certain conditions. The metal resistance gained by CzcP is similar to that mediated by secondary metal efflux systems, which belong in C. metallidurans to the CDF or other protein families.129 The three CDF proteins in C. metallidurans are the Co(II)/Zn(II)/Cd(II) exporter CzcD, the Co(II)/Ni(II) exporter DmeF and the Fe(II) exporter FieF. Flux control has also been described for a CDF metal exporter from E. coli.243,244 Members of other protein families are the nickel exporter CnrT, the two chromate exporters ChrA1 and ChrA2 and two paralogs of the CHR protein family with unknown function (Table 8).
Fate of transition metal cations in C. metallidurans
The following is the fate of the transition metal cations (from the upper left to the lower right corner of Fig. 2) when they reach C. metallidurans cells:(1) Au(III) complexes are rapidly reduced to Au(I) complexes, which reach the cytoplasm. Re-export to the periplasm does not involve the PIB1-type ATPase CupA,202 although other bacteria (Salmonella) are able to remove Au(I) from their periplasm with a CupA-homologous system.245 In the periplasm, Au(I) is further reduced to metallic gold nanoparticles, which is the first step in the formation of gold nuggets by this bacterium.13,202
(2) Ag(I) can be removed from the cytoplasm by CupA202 and from there to the outside by CusCBA and SilCBA.178
(3) Cd(II) enters as a phosphate complex through PitA and other transporters of the battery but accumulation of cadmium is low in C. metallidurans compared to E. coli because an NRAMP importer for Mn(II), Fe(II) and Cd(II) is missing.45 Export from the cytoplasm is at low cadmium concentrations by the inducible P-type ATPase CadA, at high concentrations additionally by ZntA and CzcD, and from the periplasm to the outside by CzcCBA.129,232
(4) Hg(II) is sequestered in the periplasm by MerP proteins, it is imported to the cytoplasm by MerT proteins, reduced to metallic mercury by MerA proteins, and leaves the cell by diffusion.188,189,246
(5) Molybdate and tungstate can be imported by an ABC import system when needed.247
(6) Zn(II) is imported by at least nine members of the battery of import systems.203,206 At low concentrations, it is exported by ZntA, at higher concentrations by ZntA, CadA, CzcP and CzcD to the periplasm and from there by CzcCBA back to the outside.129
(7) Cu(II) can be imported as a phosphate complex by PitA, by other systems of the redundant import battery, by CopD- and MFS-like specific importers (Table 8), or rapidly by Na(I)- or K(I)-utilizing importers after reduction to Cu(I) by respiratory chain components, most likely reduced quinones.60,162 Imported Cu(II) is also reduced immediately to Cu(I) upon contact with glutathione.248 Cytoplasmic Cu(I) is sequestered by the cytoplasmic copper chaperone CupC and exported by CupA and other PIB1-type ATPases for detoxification or allocation to periplasmic copper-requiring proteins. It is highly likely that periplasmic copper chaperones are involved in this process.193 Surplus Cu(I) in the periplasm is bound by CusF165,196,197 and exported to the outside by CusCBA and SilCBA, or oxidized by CopAs back to the less toxic Cu(II).169
(8) Ni(II) is imported by members of the redundant battery of importers and is used for synthesis of hydrogenase and urease. Surplus nickel is removed by DmeF at low,235 and additionally by CnrT at high, concentrations to the periplasm and from there by CnrCBA back to the outside. Another transporter involved in nickel resistance is the AtmA efflux system of the ABC protein family.237
(9) Co(II) is imported in a fashion similar to Ni(II) and is used for the synthesis of cobalamin compounds such as B12. It is removed by DmeF when in surplus to the periplasm,235 and from there by CzcCBA134,135,249 and CnrCBA136 to the outside.
(10) Nothing is known about vanadate and Y(III) transport. Mn(II) is not very toxic to C. metallidurans. It is not used for instance as a cofactor in a Mn-superoxide dismutase and not taken up by a NRAMP importer.45
(11) Chromate is imported by sulfate uptake systems and exported by ChrA1 and ChrA2 efflux systems.250–252 Reduction to Cr(III) might occur in the cytoplasm but the fate of this ion is not known.
(12) Finally, iron can be imported as a Fe(III) staphyloferrin B106,107 complex and possibly as an ion by FeoAB, PitA or other members of the redundant importer battery. If in surplus, it can be removed from the cytoplasm by FieF.235,236
In summary, the concentration of a metal cation can be pre-adjusted in the periplasm but following that, the whole mixture of metals is imported into the cytoplasm by the battery of redundant low-specificity, high-rate transport systems. Surplus cations are removed by efficient efflux systems to adjust and fulfill the cytoplasmic metal cation requirement.
Outlook: fate of metal cations in the cytoplasm
Cytoplasmic Fe(III) will be rapidly reduced to Fe(II) by the NADP-dependent Fe(III)-siderophore reductase ViuB (Rmet_4199) or upon contact with glutathione, GSH. Similarly, Cu(II) will be reduced to cytoplasmic Cu(I),248 which is sequestered by the cytoplasmic copper chaperone CupC. Theoretically, the divalent transition metal cations should bind immediately to thiol groups, e.g. cysteine groups at the outside of proteins. The importance of GSH or other cellular small thiol peptides in bacteria may be to keep transition metal cations from reaching these thiol groups or to remove them as metal–bis-glutathionato complexes GS–Me(II)–SG.Removal of transition metal cations from the metal–bis-glutathionato complexes may be catalyzed by PIB-type ATPases in the case of Cu(I), Zn(II) and Cd(II), and in the case of Cu(I) by binding to CupC. For zinc, this cannot be the only route because the metal is needed as a cofactor. The number of zinc atoms per C. metallidurans cell is at least 20000 as a minimum number and increases with the external zinc concentration to 120000.9,94 At this point, the efflux systems prevent a further increase in content. These cells also contain 120000 zinc-binding proteins. Since these may contain more than one zinc-binding site per protein, there are actually more zinc-binding sites in the cytoplasmic proteins of C. metallidurans than zinc atoms per cell.9 The totality of these zinc binding sites may form a zinc repository that accepts incoming zinc ions, prevents them from binding to glutathione or other thiols, and stores them for allocation or for export of surplus zinc ions. Most of these sites are located in ribosomal proteins and those subunits of the RNA polymerase that are not zinc-dependent proteins (RpoA, RpoB).9 It could be the task of all these sites to provide zinc ions to newly translated, folding proteins, especially the zinc-dependent RpoC subunit of the RNA polymerase.
When all zinc efflux systems are removed, C. metallidurans contains about 200000 Zn(II) per cell already at an external concentration of 10 μM zinc chloride,9,94 which is close to the inhibitory concentration of this zinc-sensitive mutant strain.129 This number could correspond to the actual size of the zinc repository. Interestingly, the cell-bound number of other transition metals, including even gold, also reaches a similar number of 200000 to 300000 atoms per cell when present if high external concentrations exist (Wiesemann, Reith and Nies et al., unpublished results).
This leaves a number of fascinating questions concerning the cytoplasmic counterbalance of the transition metal transportome:
(i) Do other transition metal cations use the zinc repository too, and if they do, how are they removed from it?
(ii) What is the interaction between the zinc repository and glutathione?
(iii) How is zinc bound in the repository or bound to glutathione related to the pools of loosely and more firmly bound zinc that were identified in the ZupT and CzcP experiments?
(iv) How is iron kept out of the zinc repository?
Conclusion
Metal resistance is simply the ability to maintain homeostasis of multiple metals at high external metal concentrations. Excretion of metal-sequestering compounds may contribute to resistance, e.g. of charged polysaccharides or carbonate, but since several frequently occurring transition metal cations are also essential, they have to be imported into the cell, which leads to toxic effects. The metal transportome is central to an efficient multiple metal homeostasis. The first barrier in proteobacteria is the periplasm, and the metal content and composition can be pre-adjusted by (i) TonB-dependent import of scarce metals; (ii) export of surplus metals by inducible and flux-controlled RND-driven transenvelope efflux complexes; (iii) sequestration of periplasmic metal cations for delivery to client proteins, uptake systems, or efflux systems even immediately after export from the cytoplasm; (iv) oxidation to less toxic compounds as in the case of Cu(I); (v) selective import for immediate reduction to a less toxic form as in the case of Hg(II).Starting from the pre-adjusted metal content and composition in the periplasm, this metal mixture is imported into the cytoplasm by low-specificity, high-rate import systems with overlapping substrate specificities, which supply metal cations to the cell with high energy efficiency. Surplus cations are removed by efficient efflux systems, e.g. PIB-type ATPases or CDF proteins, which are also of a low-substrate specificity to allow an export rate rapid enough to cope with the rate of the import systems, and which are assigned their task by control of gene expression plus flux control. Limiting transition metal ions are imported by specifically induced importers, in most cases of the ABC protein superfamily. The zinc repository and cellular small thiol-containing polypeptides such as glutathione possibly counterbalance the metal transportome. One remaining question is how cytoplasmic components integrate into this homeostasis mechanism and how they help to specifically allocate a transition metal to its client protein.253
Acknowledgements
I thank Gary Sawers for critically reading the manuscript.References
- P. J. Janssen, R. Van Houdt, H. Moors, P. Monsieurs, N. Morin, A. Michaux, M. A. Benotmane, N. Leys, T. Vallaeys, A. Lapidus, S. Monchy, C. Medigue, S. Taghavi, S. McCorkle, J. Dunn, D. van der Lelie and M. Mergeay, PLoS One, 2010, 5, e10433 Search PubMed.
- M. Mergeay, in Bacterial stress responses, ed. G. Storz and R. Hengge-Aronis, ASM Press, Washington, DC, 2000, pp. 403–414 Search PubMed.
- M. Mergeay, C. Houba and J. Gerits, Arch. Int. Physiol. Biochim., 1978, 86, 440–441 CAS.
- M. Mergeay, D. Nies, H. G. Schlegel, J. Gerits, P. Charles and F. van Gijsegem, J. Bacteriol., 1985, 162, 328–334 CAS.
- T. von Rozycki and D. H. Nies, Antonie van Leeuwenhoek, 2009, 96, 115–139 CrossRef CAS PubMed.
- J. Goris, P. De Vos, T. Coenye, B. Hoste, D. Janssens, H. Brim, L. Diels, M. Mergeay, K. Kersters and P. Vandamme, Int. J. Syst. Evol. Microbiol., 2001, 51, 1773–1782 CrossRef CAS PubMed.
- P. Vandamme and T. Coenye, Int. J. Syst. Evol. Microbiol., 2004, 54, 2285–2289 CrossRef PubMed.
- M. Vaneechoutte, P. Kämpfer, T. De Baere, E. Falsen and G. Verschraegen, Int. J. Syst. Evol. Microbiol., 2004, 54, 317–327 CrossRef PubMed.
- M. Herzberg, D. Dobritzsch, S. Helm, S. Baginski and D. H. Nies, Metallomics, 2014, 6, 2157–2165 RSC.
- L. Diels and M. Mergeay, Appl. Environ. Microbiol., 1990, 56, 1485–1491 CAS.
- H. Brim, M. Heyndrickx, P. De Vos, A. Wilmotte, D. Springael, H. G. Schlegel and M. Mergeay, Syst. Appl. Microbiol., 1999, 22, 258–268 CrossRef CAS PubMed.
- F. Reith, M. F. Lengke, D. Falconer, D. Craw and G. Southam, ISME J., 2007, 1, 567–584 CrossRef CAS PubMed.
- F. Reith, S. L. Rogers, D. C. McPhail and D. Webb, Science, 2006, 313, 233–236 CrossRef CAS PubMed.
- M. Herzberg, M. Schüttau, M. Reimers, C. Grosse, H. G. Schlegel and D. H. Nies, Metallomics, 2015, 7, 632–649 RSC.
- R. Van Houdt, S. Monchy, N. Leys and M. Mergeay, Antonie van Leeuwenhoek, 2009, 96, 205–226 CrossRef CAS PubMed.
- R. Van Houdt, P. Monsieurs, K. Mijnendonckx, A. Provoost, A. Janssen, M. Mergeay and N. Leys, BMC Genomics, 2012, 13, 111 CrossRef CAS PubMed.
- M. Mergeay, S. Monchy, T. Vallaeys, V. Auquier, A. Benotmane, P. Bertin, S. Taghavi, J. Dunn, D. van der Lelie and R. Wattiez, FEMS Microbiol. Rev., 2003, 27, 385–410 CrossRef CAS PubMed.
- S. Monchy, M. A. Benotmane, P. Janssen, T. Vallaeys, S. Taghavi, D. van der Lelie and M. Mergeay, J. Bacteriol., 2007, 189, 7417–7425 CrossRef CAS PubMed.
- S. Monchy, M. A. Benotmane, R. Wattiez, S. van Aelst, V. Auquier, B. Borremans, M. Mergeay, S. Taghavi, D. van der Lelie and T. Vallaeys, Microbiology, 2006, 152, 1765–1776 CrossRef CAS PubMed.
- S. Taghavi, C. Lesaulnier, S. Monchy, R. Wattiez, M. Mergeay and D. van der Lelie, Antonie van Leeuwenhoek, 2009, 96, 171–182 CrossRef CAS PubMed.
- D. Springael, L. Diels, L. Hooyberghs, S. Kreps and M. Mergeay, Appl. Environ. Microbiol., 1993, 59, 334–339 CAS.
- C. Miyake-Nakayama, H. Ikatsu, M. Kashihara, M. Tanaka, M. Arita, S. Miyoshi and S. Shinoda, Appl. Microbiol. Biotechnol., 2006, 70, 625–630 CrossRef CAS PubMed.
- Y. Ohara, M. K. Reagan, K. Fujikura, H. Watanabe, K. Michibayashi, T. Ishii, R. J. Stern, I. Pujana, F. Martinez, G. Girard, J. Ribeiro, M. Brounce, N. Komori and M. Kino, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2831–2835 CrossRef CAS PubMed.
- C. Oze, L. C. Jones, J. I. Goldsmith and R. J. Rosenbauer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9750–9754 CrossRef CAS PubMed.
- B. N. Orcutt, J. B. Sylvan, N. J. Knab and K. J. Edwards, Microbiol. Mol. Biol. Rev., 2011, 75, 361–422 CrossRef CAS PubMed.
- F. Reith, J. Brugger, C. M. Zammit, A. L. Gregg, K. C. Goldfarb, G. L. Andersen, T. Z. DeSantis, Y. M. Piceno, E. L. Brodie, Z. Lu, Z. He, J. Zhou and S. A. Wakelin, ISME J., 2012, 6, 2107–2118 CrossRef CAS PubMed.
- D. Nies, Dissertation, Georg-August-Universität Göttingen, 1985.
- K. Mijnendonckx, A. Provoost, C. M. Ott, K. Venkateswaran, J. Mahillon, N. Leys and R. Van Houdt, Microb. Ecol., 2013, 65, 347–360 CrossRef CAS PubMed.
- T. D'Inzeo, R. Santangelo, B. Fiori, G. De Angelis, V. Conte, A. Giaquinto, I. Palucci, G. Scoppettuolo, V. Di Florio, T. Giani, M. Sanguinetti, G. M. Rossolini and T. Spanu, Diagn. Microbiol. Infect. Dis., 2015, 81, 9–12 CrossRef PubMed.
- S. Langevin, J. Vincelette, S. Bekal and C. Gaudreau, J. Clin. Microbiol., 2011, 49, 744–745 CrossRef PubMed.
- A. Pohlmann, W. F. Fricke, F. Reinecke, B. Kusian, H. Liesegang, R. Cramm, T. Eitinger, C. Ewering, M. Potter, E. Schwartz, A. Strittmatter, I. Voss, G. Gottschalk, A. Steinbüchel, B. Friedrich and B. Bowien, Nat. Biotechnol., 2006, 24, 1257–1262 CrossRef PubMed.
- R. A. Don and J. M. Pemberton, J. Bacteriol., 1981, 145, 681–686 CAS.
- B. F. Johnson and R. Y. Stanier, J. Bacteriol., 1971, 107, 468–475 CAS.
- C. E. Housecroft and E. C. Constable, Chemistry, Pearson Education Limited, Essex, England, 3rd edn, 2006 Search PubMed.
- G. J. Krauss and D. H. Nies, Ecological biochemistry – environmental and interspecies interactions, Wiley-VCH, Weinheim, 2014 Search PubMed.
- R. C. Weast, CRC handbook of chemistry and physics, CRC Press, Inc., Boca Raton, Florida, USA, 64 edn, 1984 Search PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- J. Köppen and N. Arimoto, Astrophys. Space Sci., 1989, 156, 47–50 CrossRef.
- P. Neyskens, S. Van Eck, A. Jorissen, S. Goriely, L. Siess and B. Plez, Nature, 2015, 517, 174–176 CrossRef CAS PubMed.
- S. A. Jacobson, A. Morbidelli, S. N. Raymond, D. P. O'Brien, K. J. Walsh and D. C. Rubie, Nature, 2014, 508, 84–87 CrossRef CAS PubMed.
- E. Schrödinger, What is life?: the pysical aspect of the living cell, Cambridge University Press, Cambridge, New York, 1992, first published 1944 Search PubMed.
- A. R. Sarafian, S. G. Nielsen, H. R. Marschall, F. M. McCubbin and B. D. Monteleone, Science, 2014, 346, 623–626 CrossRef CAS PubMed.
- D. H. Nies, Appl. Microbiol. Biotechnol., 1999, 51, 730–750 CrossRef CAS PubMed.
- D. H. Nies, Extremophiles, 2000, 4, 77–82 CrossRef CAS PubMed.
- A. Kirsten, M. Herzberg, A. Voigt, J. Seravalli, G. Grass, J. Scherer and D. H. Nies, J. Bacteriol., 2011, 193, 4652–4663 CrossRef CAS PubMed.
- T. W. Lyons, C. T. Reinhard and N. J. Planavsky, Nature, 2014, 506, 307–315 CrossRef CAS PubMed.
- H. J. W. de Baar and J. La Roche, Marine Science Frontiers for Europe, 2003, pp. 79–105 Search PubMed.
- V. Braun and M. Braun, Curr. Opin. Microbiol., 2002, 5, 194–201 CrossRef CAS PubMed.
- V. Braun and K. Hantke, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin-Heidelberg, 2007, vol. 6, pp. 189–217 Search PubMed.
- S. C. Rasmussen, ChemTexts, 2015, 1, 1–9 CrossRef.
- D. H. Nies, J. Bacteriol., 2012, 194, 2407–2412 CrossRef CAS PubMed.
- D. H. Nies, in Metals and their compounds in the environment, ed. K. Anke, M. Ihnat and M. Stoeppler, Wiley-VCH, Weinheim, 2004, part II.1 Search PubMed.
- D. H. Nies, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 118–142 Search PubMed.
- W. G. Kirlin, J. Cai, S. A. Thompson, D. Diaz, T. J. Kavanagh and D. P. Jones, Free Radical Biol. Med., 1999, 27, 1208–1218 CrossRef CAS PubMed.
- N. S. Kosower and E. M. Kosower, Int. Rev. Cytol., 1978, 54, 109–160 CAS.
- K. Helbig, C. Bleuel, G. J. Krauss and D. H. Nies, J. Bacteriol., 2008, 190, 5431–5438 CrossRef CAS PubMed.
- K. Helbig, C. Grosse and D. H. Nies, J. Bacteriol., 2008, 190, 5439–5454 CrossRef CAS PubMed.
- G. M. Bishop, R. Dringen and S. R. Robinson, Free Radical Biol. Med., 2007, 42, 1222–1230 Search PubMed.
- F. Haber and J. Weiss, Naturwissenschaften, 1932, 20, 948–950 CrossRef CAS.
- C. Große, G. Schleuder, C. Schmole and D. H. Nies, Appl. Environ. Microbiol., 2014, 80, 7071–7078 CrossRef PubMed.
- D. Ballou, G. Palmer and V. Massey, Biochem. Biophys. Res. Commun., 1969, 36, 898–904 CrossRef CAS PubMed.
- V. Massey, S. Strickland, S. G. Mayhew, L. G. Howell, P. C. Engel, R. G. Matthews, M. Schuman and P. A. Sullivan, Biochem. Biophys. Res. Commun., 1969, 36, 891–897 CrossRef CAS PubMed.
- A. Boveris, N. Oshino and B. Chance, Biochem. J., 1972, 128, 617–630 CrossRef CAS PubMed.
- M. E. Aliaga, C. Lopez-Alarcon, L. Garcia-Rio, M. Martin-Pastor and H. Speisky, Bioorg. Med. Chem., 2012, 20, 2869–2876 CrossRef CAS PubMed.
- A. Anjem, S. Varghese and J. A. Imlay, Mol. Microbiol., 2009, 72, 844–858 CrossRef CAS PubMed.
- J. A. Imlay, J. Biol. Chem., 2014, 289, 28121–28128 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef CAS.
- L. Macomber, S. P. Elsey and R. P. Hausinger, Mol. Microbiol., 2011, 82, 1291–1300 CrossRef CAS PubMed.
- J. R. Fantino, B. Py, M. Fontecave and F. Barras, Environ. Microbiol., 2010, 12, 2846–2857 CAS.
- C. Ranquet, S. Ollagnier-de-Choudens, L. Loiseau, F. Barras and M. Fontecave, J. Biol. Chem., 2007, 282, 30442–30451 CrossRef CAS PubMed.
- G. A. Hunter and G. C. Ferreira, J. Biol. Chem., 2010, 285, 41836–41842 CrossRef CAS PubMed.
- M. Gu and J. A. Imlay, Mol. Microbiol., 2013, 89, 123–134 CrossRef CAS PubMed.
- S. L. Begg, B. A. Eijkelkamp, Z. Luo, R. M. Counago, J. R. Morey, M. J. Maher, C. L. Ong, A. G. McEwan, B. Kobe, M. L. O'Mara, J. C. Paton and C. A. McDevitt, Nat. Commun., 2015, 6, 6418 CrossRef CAS PubMed.
- H. Ohtake, C. Cervantes and S. Silver, J. Bacteriol., 1987, 169, 3853–3856 CAS.
- D. H. Nies and S. Silver, J. Bacteriol., 1989, 171, 4073–4075 CAS.
- P. Arslan, M. Beltrame and A. Tomasi, Biochim. Biophys. Acta, 1987, 931, 10–15 CrossRef CAS.
- C. M. Davis and J. B. Vincent, Biochemistry, 1997, 36, 4382–4385 CrossRef CAS PubMed.
- H. Bhattacharjee and B. P. Rosen, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 371–406 Search PubMed.
- L. Josephson and L. C. Cantley, Jr., Biochemistry, 1977, 16, 4572–4578 CrossRef CAS PubMed.
- L. J. Henderson, Am. J. Physiol., 1908, 21, 173–179 CAS.
- K. A. Hasselbalch, Biochem. Z., 1917, 78, 112–144 Search PubMed.
- R. M. C. Dawson, D. C. Elliott, W. H. Elliott and K. M. Jones, Data for biochemical research, At The Clarendon Press, Oxford, 2nd edn, 1969 Search PubMed.
- P. J. Brignac and C. Mo, Anal. Chem., 1975, 47, 1465–1466 CrossRef CAS.
- C. Schmidt, C. Schwarzenberger, C. Grosse and D. H. Nies, J. Bacteriol., 2014, 196, 3461–3471 CrossRef PubMed.
- H. L. Clever, M. E. Derrick and S. A. Johnson, J. Phys. Chem. Ref. Data, 1992, 21, 941–966 CrossRef CAS.
- A. Legatzki, S. Franke, S. Lucke, T. Hoffmann, A. Anton, D. Neumann and D. H. Nies, Biodegradation, 2003, 14, 153–168 CrossRef CAS PubMed.
- H. G. Schlegel, G. Gottschalk and H. Kaltwasser, Arch. Microbiol., 1961, 38, 209–222 CAS.
- G. Sezonov, D. Joseleau-Petit and R. D'Ari, J. Bacteriol., 2007, 189, 8746–8749 CrossRef CAS PubMed.
- H. Nikaido, in Small things considered, ed. M. Schaechter, American Society for Microbiology, Washington, 2009, vol. 2012 Search PubMed.
- D. H. Nies and M. Herzberg, Mol. Microbiol., 2013, 87, 447–454 CrossRef CAS PubMed.
- R. F. C. Mantoura and J. P. Riley, Anal. Chim. Acta, 1975, 78, 193–200 CrossRef CAS.
- K. Y. Djoko, C. L. Y. Ong, M. J. Walker and A. G. McEwan, J. Biol. Chem., 2015, 290, 18954–18961 CrossRef CAS PubMed.
- G. Berton, Pure Appl. Chem., 1995, 67, 1117–1240 Search PubMed.
- M. Herzberg, L. Bauer and D. H. Nies, Metallomics, 2014, 6, 421–436 RSC.
- Z. Ma, P. Chandrangsu, T. C. Helmann, A. Romsang, A. Gaballa and J. D. Helmann, Mol. Microbiol., 2014, 94, 756–770 CrossRef CAS PubMed.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- H. Nikaido and M. Vaara, Microbiol. Rev., 1985, 49, 1–32 CAS.
- R. Koebnik, K. P. Locher and P. Van Gelder, Mol. Microbiol., 2000, 37, 239–253 CrossRef CAS PubMed.
- H. C. Berg, Annu. Rev. Biochem., 2003, 72, 19–54 CrossRef CAS PubMed.
- V. Braun, FEMS Microbiol. Rev., 1995, 16, 295–307 CrossRef CAS PubMed.
- P. I. Higgs, P. S. Myers and K. Postle, J. Bacteriol., 1998, 180, 6031–6038 CAS.
- N. Noinaj, M. Guillier, T. J. Barnard and S. K. Buchanan, Annu. Rev. Microbiol., 2010, 64, 43–60 CrossRef CAS PubMed.
- M. Sauer, K. Hantke and V. Braun, Mol. Microbiol., 1990, 4, 427–437 CrossRef CAS PubMed.
- R. A. Kingsley, R. Reissbrodt, W. Rabsch, J. M. Ketley, R. M. Tsolis, P. Everest, G. Dougan, A. J. Baumler, M. Roberts and P. H. Williams, Appl. Environ. Microbiol., 1999, 65, 1610–1618 CAS.
- M. Munzinger, K. Taraz and H. Budzikiewicz, Z. Naturforsch., C: J. Biosci., 1999, 54, 867–875 CAS.
- A. Gilis, P. Corbisier, W. Baeyens, S. Taghavi, M. Mergeay and D. van der Lelie, J. Ind. Microbiol. Biotechnol., 1998, 20, 61–68 CrossRef CAS PubMed.
- A. Gilis, A. M. Khan, P. Cornelis, J. M. Meyer, M. Mergeay and D. van der Lelie, J. Bacteriol., 1996, 178, 5499–5507 CAS.
- M. F. Barber and N. C. Elde, Science, 2014, 346, 1362–1366 CrossRef CAS PubMed.
- S. Subashchandrabose and H. L. T. Mobley, Metallomics, 2015, 7, 935–942 RSC.
- M. Napolitano, M. A. Rubio, J. Santamaria-Gomez, E. Olmedo-Verd, N. J. Robinson and I. Luque, J. Bacteriol., 2012, 194, 2426–2436 CrossRef CAS PubMed.
- M. Stork, M. P. Bos, I. Jongerius, N. de Kok, I. Schilders, V. E. Weynants, J. T. Poolman and J. Tommassen, PLoS Pathog., 2010, 6, e1000969 Search PubMed.
- K. Schauer, B. Gouget, M. Carriere, A. Labigne and H. de Reuse, Mol. Microbiol., 2007, 63, 1054–1068 CrossRef CAS PubMed.
- K. Schauer, D. A. Rodionov and H. de Reuse, Trends Biochem. Sci., 2008, 33, 330–338 CrossRef CAS PubMed.
- J. D. Semrau, A. A. DiSpirito and S. Yoon, FEMS Microbiol. Rev., 2010, 34, 496–531 CrossRef CAS PubMed.
- H. J. Kim, D. W. Graham, A. A. DiSpirito, M. A. Alterman, N. Galeva, C. K. Larive, D. Asunskis and P. M. Sherwood, Science, 2004, 305, 1612–1615 CrossRef CAS PubMed.
- A. G. Bobrov, O. Kirillina, J. D. Fetherston, M. C. Miller, J. A. Burlison and R. D. Perry, Mol. Microbiol., 2014, 93, 759–775 CrossRef CAS PubMed.
- D. H. Nies, in Microbial efflux pumps: current research, ed. E. W. Yu, Q. Zhang and M. H. Brown, Caister Academic Press, Norfolk, UK, 2013, pp. 79–122 Search PubMed.
- T.-T. Tseng, K. S. Gratwick, J. Kollman, D. Park, D. H. Nies, A. Goffeau and M. H. J. Saier, J. Mol. Microbiol. Biotechnol., 1999, 1, 107–125 CAS.
- F. Long, C. C. Su, M. T. Zimmermann, S. E. Boyken, K. R. Rajashankar, R. L. Jernigan and E. W. Yu, Nature, 2010, 467, 484–488 CrossRef CAS PubMed.
- C. C. Su, F. Long, M. T. Zimmermann, K. R. Rajashankar, R. L. Jernigan and E. W. Yu, Nature, 2011, 470, 558–563 CrossRef CAS PubMed.
- S. Murakami, R. Nakashima, R. Yamashita and A. Yamaguchi, Nature, 2002, 419, 587–593 CrossRef CAS PubMed.
- M. A. Seeger, A. Schiefner, T. Eicher, F. Verrey, K. Diederichs and K. M. Pos, Science, 2006, 313, 1295–1298 CrossRef CAS PubMed.
- Y. Takatsuka and H. Nikaido, J. Bacteriol., 2009, 191, 1729–1737 CrossRef CAS PubMed.
- M. Goldberg, T. Pribyl, S. Juhnke and D. H. Nies, J. Biol. Chem., 1999, 274, 26065–26070 CrossRef CAS PubMed.
- V. Koronakis, A. Sharff, E. Koronakis, B. Luisi and C. Hughes, Nature, 2000, 405, 914–919 CrossRef CAS PubMed.
- M. F. Symmons, E. Bokma, E. Koronakis, C. Hughes and V. Koronakis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7173–7178 CrossRef CAS PubMed.
- M. K. Higgins, E. Bokma, E. Koronakis, C. Hughes and V. Koronakis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9994–9999 CrossRef CAS PubMed.
- D. Nehme and K. Poole, J. Bacteriol., 2007, 189, 6118–6127 CrossRef CAS PubMed.
- J. Scherer and D. H. Nies, Mol. Microbiol., 2009, 73, 601–621 CrossRef CAS PubMed.
- G. Grass and C. Rensing, Biochem. Biophys. Res. Commun., 2001, 286, 902–908 CrossRef CAS PubMed.
- G. Grass and C. Rensing, J. Bacteriol., 2001, 183, 2145–2147 CrossRef CAS PubMed.
- E.-H. Kim, D. H. Nies, M. McEvoy and C. Rensing, J. Bacteriol., 2011, 193, 2381–2387 CrossRef CAS PubMed.
- D. H. Nies, G. Rehbein, T. Hoffmann, C. Baumann and C. Grosse, J. Mol. Microbiol. Biotechnol., 2006, 11, 82–93 CrossRef CAS PubMed.
- D. Nies, M. Mergeay, B. Friedrich and H. G. Schlegel, J. Bacteriol., 1987, 169, 4865–4868 CAS.
- D. H. Nies, A. Nies, L. Chu and S. Silver, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7351–7355 CrossRef CAS.
- H. Liesegang, K. Lemke, R. A. Siddiqui and H.-G. Schlegel, J. Bacteriol., 1993, 175, 767–778 CAS.
- K. Yamamoto, K. Hirao, T. Oshima, H. Aiba, R. Utsumi and A. Ishihama, J. Biol. Chem., 2005, 280, 1448–1456 CrossRef CAS PubMed.
- L. Zhou, X. H. Lei, B. R. Bochner and B. L. Wanner, J. Bacteriol., 2003, 185, 4956–4972 CrossRef CAS PubMed.
- D. Missiakas and S. Raina, Mol. Microbiol., 1998, 28, 1059–1066 CrossRef CAS PubMed.
- J. D. Helmann, Adv. Microb. Physiol., 2002, 46, 47–110 CrossRef CAS PubMed.
- G. Grass, B. Fricke and D. H. Nies, BioMetals, 2005, 18, 437–448 CrossRef CAS PubMed.
- G. Grass, C. Große and D. H. Nies, J. Bacteriol., 2000, 182, 1390–1398 CrossRef CAS PubMed.
- C. Tibazarwa, S. Wuertz, M. Mergeay, L. Wyns and D. van der Lelie, J. Bacteriol., 2000, 182, 1399–1409 CrossRef CAS PubMed.
- D. H. Nies, Arch. Microbiol., 2004, 181, 255–268 CrossRef CAS PubMed.
- C. Große, S. Friedrich and D. H. Nies, J. Mol. Microbiol. Biotechnol., 2007, 12, 227–240 CrossRef PubMed.
- S. Mahren and V. Braun, J. Bacteriol., 2003, 185, 1796–1802 CrossRef CAS PubMed.
- W. Wagegg and V. Braun, J. Bacteriol., 1981, 145, 156–163 CAS.
- A. P. Maillard, E. Girard, W. Ziani, I. Petit-Hartlein, R. Kahn and J. Coves, J. Mol. Biol., 2014, 426, 2313–2327 CrossRef CAS PubMed.
- G. Pompidor, A. P. Maillard, E. Girard, S. Gambarelli, R. Kahn and J. Coves, FEBS Lett., 2008, 582, 3954–3958 CrossRef CAS PubMed.
- J. Trepreau, E. Girard, A. P. Maillard, E. de Rosny, I. Petit-Haertlein, R. Kahn and J. Coves, J. Mol. Biol., 2011, 408, 766–779 CrossRef CAS PubMed.
- J. Trepreau, C. Grosse, J.-M. Mouesca, G. Sarret, E. Girard, I. Petit-Hartlein, S. Kuennemann, C. Desbourdes, E. de Rosny, A. P. Maillard, D. H. Nies and J. Covès, Metallomics, 2014, 6, 263–273 RSC.
- J.-M. Collard, A. Provoost, S. Taghavi and M. Mergeay, J. Bacteriol., 1993, 175, 779–784 CAS.
- C. Große, A. Anton, T. Hoffmann, S. Franke, G. Schleuder and D. H. Nies, Arch. Microbiol., 2004, 182, 109–118 CrossRef PubMed.
- C. Große, G. Grass, A. Anton, S. Franke, A. Navarrete Santos, B. Lawley, N. L. Brown and D. H. Nies, J. Bacteriol., 1999, 181, 2385–2393 Search PubMed.
- D. van der Lelie, T. Schwuchow, U. Schwidetzky, S. Wuertz, W. Baeyens, M. Mergeay and D. H. Nies, Mol. Microbiol., 1997, 23, 493–503 CrossRef CAS PubMed.
- D. H. Nies, in Ecological Biochemistry: Environmental and Interspecies Interactions, ed. G. J. Krauss and D. H. Nies, Wiley-VCH, Weinheim, 2014 Search PubMed.
- D. Koch, A. C. Chan, M. E. Murphy, H. Lilie, G. Grass and D. H. Nies, J. Biol. Chem., 2011, 286, 25317–25330 CrossRef CAS PubMed.
- C. Grosse, J. Scherer, D. Koch, M. Otto, N. Taudte and G. Grass, Mol. Microbiol., 2006, 62, 120–131 CrossRef CAS PubMed.
- J. Cao, M. R. Woodhall, J. Alvarez, M. L. Cartron and S. C. Andrews, Mol. Microbiol., 2007, 65, 857–875 CrossRef CAS PubMed.
- B. Borremans, J. L. Hobman, A. Provoost, N. L. Brown and D. Van der Lelie, J. Bacteriol., 2001, 183, 5651–5658 CrossRef CAS PubMed.
- L. Fairbrother, B. Etschmann, J. Brugger, J. Shapter, G. Southam and F. Reith, Environ. Sci. Technol., 2013, 47, 2628–2635 CrossRef CAS PubMed.
- S. I. Volentini, R. N. Farias, L. Rodriguez-Montelongo and V. A. Rapisarda, BioMetals, 2011, 24, 827–835 CrossRef CAS PubMed.
- D. Thieme, P. Neubauer, D. H. Nies and G. Grass, Appl. Environ. Microbiol., 2008, 74, 7463–7470 CrossRef CAS PubMed.
- F. W. Outten, D. L. Huffman, J. A. Hale and T. V. O'Halloran, J. Biol. Chem., 2001, 276, 30670–30677 CrossRef CAS PubMed.
- I. R. Loftin, S. Franke, S. A. Roberts, A. Weichsel, A. Heroux, W. R. Montfort, C. Rensing and M. M. McEvoy, Biochemistry, 2005, 44, 10533–10540 CrossRef CAS PubMed.
- S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2003, 185, 3804–3812 CrossRef PubMed.
- S. Franke, G. Grass and D. H. Nies, Microbiology, 2001, 147, 965–972 CrossRef CAS PubMed.
- F. W. Outten, C. E. Outten, J. Hale and T. V. O'Halloran, J. Biol. Chem., 2000, 275, 31024–31029 CrossRef CAS PubMed.
- D. Magnani and M. Solioz, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 259–285 Search PubMed.
- S. A. Roberts, A. Weichsel, G. Grass, K. Thakali, J. T. Hazzard, G. Tollin, C. Rensing and W. R. Montfort, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2766–2771 CrossRef CAS PubMed.
- S. K. Singh, G. Grass, C. Rensing and W. R. Montfort, J. Bacteriol., 2004, 186, 7815–7817 CrossRef CAS PubMed.
- T. J. Tetaz and R. K. J. Luke, J. Bacteriol., 1983, 154, 1263–1268 CAS.
- N. L. Brown, S. R. Barrett, J. Camakaris, B. T. Lee and D. A. Rouch, Mol. Microbiol., 1995, 17, 1153–1166 CAS.
- C. Rensing, B. Fan, R. Sharma, B. Mitra and B. P. Rosen, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 652–656 CrossRef CAS.
- C. Rensing, M. Ghosh and B. P. Rosen, J. Bacteriol., 1999, 181, 5891–5897 CAS.
- D. A. Cooksey, Appl. Environ. Microbiol., 1987, 53, 454–456 CAS.
- M. A. Mellano and D. A. Cooksey, J. Bacteriol., 1988, 170, 2879–2883 CAS.
- S. Silver, FEMS Microbiol. Rev., 2003, 27, 341–353 CrossRef CAS PubMed.
- S. Franke, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 343–355 Search PubMed.
- M. H. J. Saier, C. V. Tran and R. D. Barabote, Nucleic Acids Res., 2006, 34, D181–D186 CrossRef CAS PubMed.
- S. Siche, O. Neubauer, P. Hebbeln and T. Eitinger, Res. Microbiol., 2010, 161, 824–829 CrossRef CAS PubMed.
- A. J. Rice, A. Park and H. W. Pinkett, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 426–437 CrossRef CAS PubMed.
- S. I. Patzer and K. Hantke, Mol. Microbiol., 1998, 28, 1199–1210 CrossRef CAS PubMed.
- C. Navarro, L. F. Wu and M. A. Mandrand-Berthelot, Mol. Microbiol., 1993, 9, 1181–1191 CrossRef CAS PubMed.
- L. F. Wu, C. Navarro and M. A. Mandrand Berthelot, Gene, 1991, 107, 37–42 CrossRef CAS PubMed.
- A. J. Rice, A. Harrison, F. J. Alvarez, A. L. Davidson and H. W. Pinkett, J. Biol. Chem., 2014, 289, 15005–15013 CrossRef CAS PubMed.
- G. Schwarz, P.-L. Hagedoorn and K. Fischer, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 421–451 Search PubMed.
- L. Diels, M. Faelen, M. Mergeay and D. Nies, Arch. Int. Physiol. Biochim., 1985, 93, B27–B28 Search PubMed.
- C. Dressler, U. Kües, D. H. Nies and B. Friedrich, Appl. Environ. Microbiol., 1991, 57, 3079–3085 CAS.
- M. Müller and R. B. Klösgen, Mol. Membr. Biol., 2005, 22, 113–121 CrossRef.
- E. Vrontou and A. Economou, Biochim. Biophys. Acta, 2004, 1694, 67–80 CrossRef CAS PubMed.
- T. V. O'Halloran and V. C. Culotta, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef PubMed.
- D. Osman, C. J. Patterson, K. Bailey, K. Fisher, N. J. Robinson, S. E. J. Rigby and J. S. Cavet, Mol. Microbiol., 2013, 87, 466–477 CrossRef CAS PubMed.
- M. Gonzalez-Guerrero, D. Raimunda, X. Cheng and J. M. Argüello, Mol. Microbiol., 2010, 78, 1246–1258 CrossRef CAS PubMed.
- D. Raimunda, M. Gonzalez-Guerrero, B. W. Leeber and J. M. Argüello, BioMetals, 2011, 24, 467–475 CrossRef CAS PubMed.
- T. Padilla-Benavides, A. M. G. Thompson, M. M. McEvoy and J. M. Argüello, J. Biol. Chem., 2014, 289, 20492–20501 CrossRef CAS PubMed.
- M. N. Ucisik, D. K. Chakravorty and K. M. Merz, Biochemistry, 2015, 54, 4226–4235 CrossRef CAS PubMed.
- I. Bagai, C. Rensing, N. J. Blackburn and M. M. McEvoy, Biochemistry, 2008, 47, 11408–11414 CrossRef CAS PubMed.
- A. Zoropogui, S. Gambarelli and J. Coves, Biochem. Biophys. Res. Commun., 2008, 365, 735–739 CrossRef CAS PubMed.
- C. J. Kershaw, N. L. Brown and J. L. Hobman, Biochem. Biophys. Res. Commun., 2007, 364, 66–71 CrossRef CAS PubMed.
- P. Petrarca, S. Ammendola, P. Pasquali and A. Battistoni, J. Bacteriol., 2010, 192, 1553–1564 CrossRef CAS PubMed.
- N. Wiesemann, J. Mohr, C. Grosse, M. Herzberg, G. Hause, F. Reith and D. H. Nies, J. Bacteriol., 2013, 195, 2298–2308 CrossRef CAS PubMed.
- M. Herzberg, L. Bauer, A. Kirsten and D. H. Nies, Metallomics, 2016, 8, 313–326 RSC.
- M. D. Snavely, J. B. Florer, C. G. Miller and M. E. Maguire, J. Bacteriol., 1989, 171, 4761–4766 CAS.
- M. D. Snavely, C. G. Miller and M. E. Maguire, J. Biol. Chem., 1991, 266, 815–823 CAS.
- C. Grosse, M. Herzberg, M. Schüttau and D. H. Nies, mSystems, 2016, 1, e00004 Search PubMed.
- M. L. Cartron, S. Maddocks, P. Gillingham, C. J. Craven and S. C. Andrews, BioMetals, 2006, 19, 143–157 CrossRef CAS PubMed.
- T. C. Marlovits, W. Haase, C. Herrmann, S. G. Aller and V. M. Unger, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16243–16248 CrossRef CAS PubMed.
- S. Ekici, H. Yang, H. G. Koch and F. Daldal, mBio, 2012, 3, e00293-11 CrossRef PubMed.
- T. von Rozycki, D. H. Nies and M. H. J. Saier, Comp. Funct. Genomics, 2005, 6, 17–56 CrossRef CAS PubMed.
- S. Eshaghi, D. Niegowski, A. Kohl, D. M. Molina, S. A. Lesley and P. Nordlund, Science, 2006, 313, 354–357 CrossRef CAS PubMed.
- A. Guskov, N. Nordin, A. Reynaud, H. Engman, A. K. Lundback, A. J. O. Jong, T. Cornvik, T. Phua and S. Eshaghi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18459–18464 CrossRef CAS PubMed.
- V. Lunin, E. Dobrovetsky, G. Khutoreskaya, R. Zhang, A. Joachimiak, D. A. Doyle, A. Bochkarev, M. E. Maguire, A. M. Edwards and C. M. Koth, Nature, 2006, 440, 833–837 CrossRef CAS PubMed.
- J. Payandeh, C. Li, M. Ramjeesingh, E. Poduch, C. E. Bear and E. F. Pai, J. Biol. Chem., 2008, 283, 11721–11733 CrossRef CAS PubMed.
- M. Hattori, Y. Tanaka, S. Fukai, R. Ishitani and O. Nureki, Nature, 2007, 448, 1072–1076 CrossRef CAS PubMed.
- R. Pfoh, A. Li, N. Chakrabarti, J. Payandeh, R. Pomes and E. F. Pai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18809–18814 CrossRef CAS PubMed.
- M. Webb, Biochim. Biophys. Acta, 1970, 222, 428–439 CrossRef CAS.
- M. Lohmeyer and C. G. Friedrich, Arch. Microbiol., 1987, 149, 130–135 CrossRef CAS.
- S. J. Beard, R. Hashim, G. H. Wu, M. R. B. Binet, M. N. Hughes and R. K. Poole, FEMS Microbiol. Lett., 2000, 184, 231–235 CrossRef CAS PubMed.
- R. M. Harris, D. C. Webb, S. M. Howitt and G. B. Cox, J. Bacteriol., 2001, 183, 5008–5014 CrossRef CAS PubMed.
- R. J. Jackson, M. R. B. Binet, L. J. Lee, R. Ma, A. I. Graham, C. W. McLeod and R. K. Poole, FEMS Microbiol. Lett., 2008, 289, 219–224 CrossRef CAS PubMed.
- R. A. van Bogelen, E. R. Olson, B. L. Wanner and F. C. Neidhardt, J. Bacteriol., 1996, 178, 4344–4366 CAS.
- L. W. Marzan, C. M. M. Hasan and K. Shimizu, Arch. Microbiol., 2013, 195, 161–171 CrossRef CAS PubMed.
- S. G. Gardner, K. D. Johns, R. Tanner and W. R. McCleary, J. Bacteriol., 2014, 196, 1741–1752 CrossRef PubMed.
- Z. M. Wang, A. Choudhary, P. S. Ledvina and F. A. Quiocho, J. Biol. Chem., 1994, 269, 25091–25094 CAS.
- H. Rosenberg, R. G. Gerdes and K. Chegwidden, J. Bacteriol., 1977, 131, 505–511 CAS.
- F. C. Soncini, E. G. Vescovi, F. Solomon and E. A. Groisman, J. Bacteriol., 1996, 178, 5092–5099 CAS.
- E. G. Vescovi, F. C. Soncini and E. A. Groisman, Cell, 1996, 84, 165–174 CrossRef CAS.
- E. Choi, E. A. Groisman and D. Shin, J. Bacteriol., 2009, 191, 7174–7181 CrossRef CAS PubMed.
- S. Y. Park and E. A. Groisman, Mol. Microbiol., 2014, 91, 135–144 CrossRef CAS PubMed.
- M. J. Cromie, Y. X. Shi, T. Latifi and E. A. Groisman, Cell, 2006, 125, 71–84 CrossRef CAS PubMed.
- A. Legatzki, A. Anton, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2003, 185, 435404361 CrossRef.
- A. Anton, C. Große, J. Reißman, T. Pribyl and D. H. Nies, J. Bacteriol., 1999, 181, 6876–6881 CAS.
- A. Anton, A. Weltrowski, J. H. Haney, S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2004, 186, 7499–7507 CrossRef CAS PubMed.
- D. Munkelt, G. Grass and D. H. Nies, J. Bacteriol., 2004, 186, 8036–8043 CrossRef CAS PubMed.
- G. Grass, M. Otto, B. Fricke, C. J. Haney, C. Rensing, D. H. Nies and D. Munkelt, Arch. Microbiol., 2005, 183, 9–18 CrossRef CAS PubMed.
- A. Mikolay and D. H. Nies, Antonie van Leeuwenhoek, 2009, 96, 183–191 CrossRef CAS PubMed.
- O. Barre, F. Mourlane and M. Solioz, J. Bacteriol., 2007, 189, 5947–5954 CrossRef CAS PubMed.
- K. M. Papp-Wallace, A. S. Moomaw and M. E. Maguire, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2005, vol. 6, pp. 235–257 Search PubMed.
- M. Roux and J. Coves, FEMS Microbiol. Lett., 2002, 210, 129–133 CrossRef CAS PubMed.
- J. M. Argüello, E. Eren and M. Gonzalez-Guerrero, BioMetals, 2007, 20, 233–248 CrossRef PubMed.
- S. L. Drees, D. F. Beyer, C. Lenders-Lomscher and M. Lübben, Mol. Microbiol., 2015, 97, 423–438 CrossRef CAS PubMed.
- M. Lu, J. Chai and D. Fu, Nat. Struct. Mol. Biol., 2009, 16, 1063–1068 CAS.
- M. Lu and D. Fu, Science, 2007, 317, 1746–1748 CrossRef CAS PubMed.
- L. B. Pontel, M. E. Audero, M. Espariz, S. K. Checa and F. C. Soncini, Mol. Microbiol., 2007, 66, 814–825 CrossRef CAS PubMed.
- S. Silver and J. L. Hobman, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer, Berlin, 2007, vol. 6, pp. 357–370 Search PubMed.
- G. Schwarz, P.-L. Hagedoorn and K. Fischer, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer, Berlin, 2007, vol. 6 Search PubMed.
- J. H. Freedman, M. R. Ciriolo and J. Peisach, J. Biol. Chem., 1989, 264, 5598–5605 CAS.
- D. H. Nies and S. Silver, J. Bacteriol., 1989, 171, 896–900 CAS.
- S. Juhnke, N. Peitzsch, N. Hübener, C. Große and D. H. Nies, Arch. Microbiol., 2002, 179, 15–25 CrossRef CAS PubMed.
- A. Nies, D. H. Nies and S. Silver, J. Bacteriol., 1989, 171, 5065–5070 CAS.
- A. Nies, D. H. Nies and S. Silver, J. Biol. Chem., 1990, 265, 5648–5653 CAS.
- S. Tottey, D. R. Harvie and N. J. Robinson, in Molecular microbiology of heavy metals, ed. D. H. Nies and S. Silver, Springer-Verlag, Berlin, 2007, vol. 6, pp. 3–36 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |