
Porous silicon–graphene oxide core–shell nanoparticles for targeted delivery of siRNA to the injured brain†
Jinmyoung
Joo‡
 a,
Ester J.
Kwon
b,
Jinyoung
Kang
c,
Matthew
Skalak
b,
Emily J.
Anglin
a,
Aman P.
Mann
d,
Erkki
Ruoslahti
de,
Sangeeta N.
Bhatia
bfghij and
Michael J.
Sailor
*ac
a,
Ester J.
Kwon
b,
Jinyoung
Kang
c,
Matthew
Skalak
b,
Emily J.
Anglin
a,
Aman P.
Mann
d,
Erkki
Ruoslahti
de,
Sangeeta N.
Bhatia
bfghij and
Michael J.
Sailor
*ac
aDepartment of Chemistry, University of California, San Diego, La Jolla, CA 92093, USA. E-mail: msailor@ucsd.edu
bHarvard-MIT Division of Health Science and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Nanoengineering, University of California, San Diego, La Jolla, CA 92093, USA
dCancer Research Center, Sanford-Burnham-Prebys Medical Discovery Institute, La Jolla, CA 92037, USA
eCenter for Nanomedicine and Department of Molecular, Cellular and Developmental Biology, University of California, Santa Barbara, Santa Barbara, CA 93106, USA
fDepartment of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
gDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
hDepartment of Medicine, Brigham and Women's Hospital, Boston, MA 02115, USA
iHoward Hughes Medical Institute, Chevy Chase, MD 20815, USA
jBroad Institute of Harvard and MIT, Cambridge, MA 02142, USA
First published on 14th June 2016
Abstract
We report the synthesis, characterization, and assessment of a nanoparticle-based RNAi delivery platform that protects siRNA payloads against nuclease-induced degradation and efficiently delivers them to target cells. The nanocarrier is based on biodegradable mesoporous silicon nanoparticles (pSiNPs), where the voids of the nanoparticles are loaded with siRNA and the nanoparticles are encapsulated with graphene oxide nanosheets (GO–pSiNPs). The graphene oxide encapsulant delays release of the oligonucleotide payloads in vitro by a factor of 3. When conjugated to a targeting peptide derived from the rabies virus glycoprotein (RVG), the nanoparticles show 2-fold greater cellular uptake and gene silencing. Intravenous administration of the nanoparticles into brain-injured mice results in substantial accumulation specifically at the site of injury.
Conceptual insightsGene silencing by RNA interference (RNAi) is typically limited by extracellular nucleolytic degradation and by inefficient delivery into the cytoplasm of target cells. We employ porous silicon nanoparticles as a therapeutic nanocarrier platform that can protect siRNA payloads against degradation while enabling release of siRNA in target cells. This work presents the first use of graphene oxide nanosheets as a protective “shell” for rational design of “hollow” nanoparticle-based RNAi therapeutics. The porous nanocarrier allows a high level of siRNA loading and protects the oligonucleotide from nucleolytic degradation, while the nanosheets slow the release of the drug payload to a therapeutically useful timescale. In order to localize and penetrate into the intended cellular targets, we deploy a targeting peptide conjugated to the exterior surface of the nanoparticles. The targeted construct displays enhanced cellular uptake and gene silencing. The intravenously injected nano-constructs are found to target brain injury in a mouse model, and the nanostructures exhibit remarkable in vivo delivery efficiency. The data demonstrate the potential of this biodegradable delivery system for RNAi-based therapeutics that safely transports the drug payload through the bloodstream and delivers it to the target cells. |
Since the discovery of RNA interference (RNAi) as a modulator of eukaryotic gene expression, it has attracted attention as a tool for manipulation of cellular pathways that regulate fundamental biological processes and as a potential therapeutic to treat genetic diseases with high specificity.1–4 Clinical translation of RNAi-based therapies such as siRNA have been hampered by poor cellular delivery, due in part to the large size and negative charge of the oligonucleotides.5 Even if the cellular membrane penetration problem could be solved, systemic administration of naked siRNA is limited by immune system activation, enzymatic degradation, rapid renal clearance, and poor accumulation at target sites.5,6
Efforts to overcome these obstacles have resulted in a number of siRNA delivery strategies.7–9 A variety of approaches to increase stability and evade immune system activation have been pursued by using viral- or non-viral nanocarrier-enabled delivery systems. Viral vectors delivering siRNA in the form of a viral genome have been shown to efficiently achieve gene silencing, but challenges of scale-up, low loading capacity and safety concerns such as mutagenesis or immunogenicity have so far limited clinical translation of these constructs.10,11 Non-viral nanoparticle delivery systems include lipid-based complexes,12,13 cationic polymers,14,15 inorganic nanoparticles,16–18 and their hybrid systems.19–21 Inorganic nanoparticles such as gold,22 iron oxide,23 semiconductor quantum dots,24 carbon nanotubes,25 and silica26 have all been used for gene delivery.
For solid nanoparticles, oligonucleotide payloads conjugated or adsorbed onto the surface are readily subjected to degradation by nucleases,27 whereas hollow structures have the potential to sequester and protect the siRNA payload from enzymatic degradation until it reaches its target. In this work we employed a hollow nanoparticle system based on porous Si nanoparticles (pSiNPs), which have been shown to be effective in delivering oligonucleotide and other therapeutic payloads in vivo.28,29
The pSiNPs show promise as a non-biological carrier for siRNA payloads because they exhibit high drug loading capacity, easy surface functionalization, biocompatibility, low toxicity, and intrinsic photoluminescence that provides a theranostic imaging capability.30–33 In particular, a pSiNP-based nanocarrier is a candidate therapeutic vehicle for targeting brain tissues due to its biodegradable nature, which is expected to minimize harmful side effects caused by residual components. Herein, we aimed to improve in vivo delivery of siRNA by coating the pSiNP carrier with a protective “shell” of graphene oxide nanosheets. Graphene oxide has been explored as a potential in vivo imaging agent, and its biocompatibility and biodegradability in vivo has been described.34–36 We find that graphene oxide nanosheets effectively wrap pSiNPs, protecting the siRNA payload contained within from nucleolytic degradation and slowing release of active siRNA. Furthermore, we show that attaching a peptide derived from the rabies virus glycoprotein (RVG) improves targeting of the nano-construct to neuronal cells in vitro, and to injured regions of the mouse brain in vivo.
The pSiNPs were prepared by electrochemical etching of single-crystalline silicon wafers in ethanolic HF solution, followed by lift-off of the porous film and ultrasonic fracture into nanometer sized particles as previously described.37 The nanoparticle surface was then oxidized in aqueous sodium borate solution, generating a thin surface oxide layer that activates photoluminescence from the quantum-confined silicon domains.38 At this stage the particles were negatively charged, with a zeta potential −28.4 ± 3 mV (measured by dynamic light scattering, DLS, Fig. 1). In order to load the negatively charged siRNA payload more effectively, the surface charge of the pSiNPs was made positive by reaction with (3-aminopropyl)-dimethyl-ethoxysilane (zeta potential +41.9 ± 4 mV). The mean hydrodynamic diameter of the pSiNPs (measured by DLS) was ∼170 nm, consistent with the transmission electron microscope (TEM) images (Fig. 1). The pSiNPs contained ∼10 nm-size pores, and the porosity of the particles was 52% (measured using the spectroscopic liquid infiltration method on the intact porous film, prior to nanoparticle generation by ultrasonic fracture).39 The porous nanostructure appeared evenly distributed throughout the nanoparticles. Nitrogen adsorption–desorption isotherm analysis yielded a Brunauer–Emmett–Teller (BET) surface area of ∼450 m2 g−1 and Barrett–Joyner–Halenda (BJH) average pore size of 10 nm, consistent with the scanning electron microscope (SEM) image (Fig. S1 in ESI†).
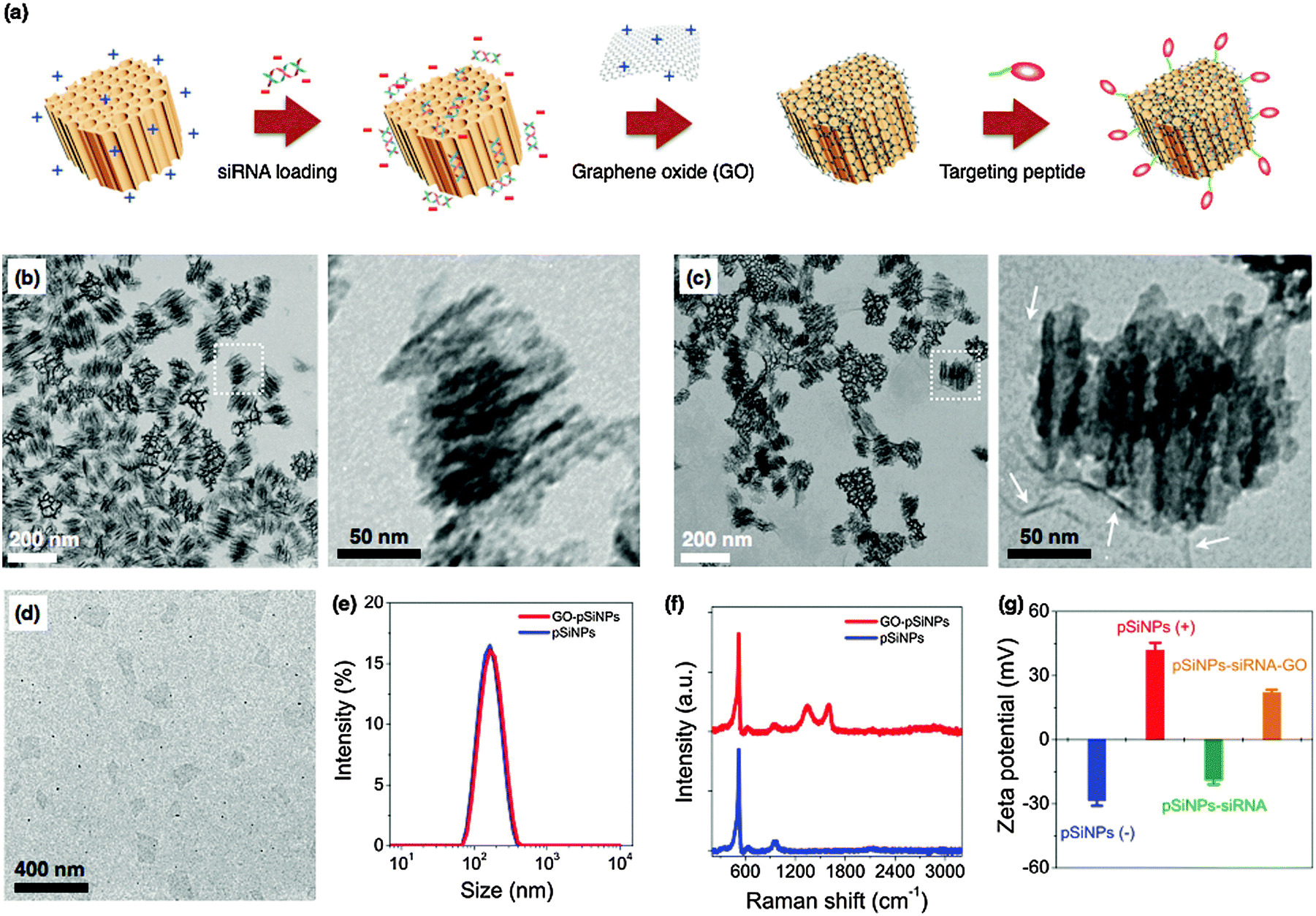 | ||
| Fig. 1 (a) Schematic illustration depicting preparation of siRNA-loaded, porous silicon–graphene oxide core–shell nanoparticles. (b) Transmission electron microscope (TEM) image of porous Si nanoparticles (pSiNPs). Enlargement of the indicated region is shown on the right. (c) TEM image of graphene oxide-wrapped pSiNPs (GO–pSiNPs). Enlargement of the indicated region is shown on the right. White arrows indicate graphene oxide. (d) TEM image of individual graphene oxide (GO) nanosheets. (e) Intensity-weighted size distribution (from DLS) of pSiNP (blue) and GO–pSiNP (red) formulations. (f) Raman spectra of pSiNPs (blue) and GO–pSiNPs (red). (g) Zeta potential of pSiNPs during the course of siRNA loading and GO wrapping (pSiNPs(−): as-oxidized pSiNPs (blue); pSiNPs(+): amine-terminated pSiNPs (red); pSiNPs–siRNA: siRNA-loaded pSiNPs (green); pSiNPs–siRNA–GO: siRNA-loaded pSiNPs after the GO wrapping process (orange)). As-oxidized pSiNPs are negatively charged, and the net surface charge becomes positive upon conjugation of (3-aminopropyl)-dimethyl-ethoxysilane. Loading of siRNA into this structure then converts the net surface charge to negative. Finally, wrapping of aminated GO around the nano-constructs transforms the net charge to positive. Note that GO was chemically modified with amine groups as described in the experimental section. | ||
The siRNA payload was loaded into the cationic (amine-terminated) pSiNPs by electrostatic means. Mixing of the pSiNPs in a solution of siRNA led to 4% mass loading within the first 30 min, and a total of 7% of siRNA was loaded after 24 h (Fig. S2a in ESI†). The mass loading of siRNA was optimized by systematically varying the siRNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pSiNP ratio (Fig. S2b in ESI†). A maximum loading capacity of 12% by mass was achieved with a siRNA:pSiNPs mixing ratio of 2, corresponding to 2.6 × 104 siRNA molecules per nanoparticle. This loading capacity compares favorably with other non-porous nanoparticles or polyplex systems,40–42 and is attributed to the relatively high open porosity (52%) of the pSiNPs and the high positive charge on the interior pore walls imparted by the amination chemistry. The electrostatic loading of negatively charged siRNA to the pSiNPs was confirmed by measured changes in surface charge of the nanocarriers (Fig. 1g), and the results are consistent with the above interpretation.
:pSiNP ratio (Fig. S2b in ESI†). A maximum loading capacity of 12% by mass was achieved with a siRNA:pSiNPs mixing ratio of 2, corresponding to 2.6 × 104 siRNA molecules per nanoparticle. This loading capacity compares favorably with other non-porous nanoparticles or polyplex systems,40–42 and is attributed to the relatively high open porosity (52%) of the pSiNPs and the high positive charge on the interior pore walls imparted by the amination chemistry. The electrostatic loading of negatively charged siRNA to the pSiNPs was confirmed by measured changes in surface charge of the nanocarriers (Fig. 1g), and the results are consistent with the above interpretation.
For therapeutics destined for in vivo applications, it is preferable to achieve delayed release of siRNA payloads in order to avoid unwanted loss in the bloodstream before reaching the designated tissues. To slow release of the siRNA payload, we coated the nanoparticles with graphene oxide (GO). GO has attracted increased attention for biological applications in sensing, imaging, and drug delivery due to its colloidal stability, its biocompatibility, and its reported low toxicity,43–47 though it has seen limited use as an encapsulant of other nanoparticles. Suspensions of the graphene oxide (GO) sheets were subjected to ultrasonication to obtain the desired lateral dimensions of ∼200 nm (Fig. 1d). The GO nanosheets at this point displayed a negative surface charge indicative of carboxylate and other negatively charged moieties. The surface charge was changed to positive (zeta potential +23.4 ± 4 mV) by grafting of amine groups (NH2) through the EDC-mediated reaction with ethylenediamine.48 The positively charged GO nanosheets were then added to the siRNA-loaded pSiNPs, and the mixture was maintained at 4 °C for 1 h to generate the GO-wrapped pSiNPs (GO–pSiNPs).
We expect the wrapping process for GO–pSiNP formation was driven by electrostatic interaction between the positively charged GO nanosheets and the negatively charged siRNA-loaded pSiNPs (zeta potential −18.7 ± 3 mV). The GO–pSiNP product exhibited a positive zeta potential of +22.1 ± 2 mV (Fig. 1g), consistent with a structure in which the GO nanosheets lie on the exterior. TEM images of GO–pSiNPs revealed crinkled textures on the exterior, ascribed to thin, layered structures of the GO nanosheets (Fig. 1c). Although isolated two-dimensional GO nanosheets are normally planar (Fig. 1d), they have been observed to take on a curved texture when wrapping around nanoparticles.49,50 The encapsulation of individual nanoparticles did not lead to aggregation or excessive layering. After wrapping with GO nanosheets, the pSiNPs exhibited only a slight increase in average hydrodynamic diameter (Fig. 1e). The Raman spectra further support the presence of GO sheets on the pSiNPs (Fig. 1f), exhibiting strong peaks characteristic of GO (D band at 1342 cm−1; G band at 1594 cm−1) in addition to the 520 cm−1 band corresponding to the phonon band of crystalline silicon.
The GO coating delayed release of the siRNA payload in vitro. When incubated in phosphate-buffered saline (PBS, pH 7.4, 37 °C), the uncoated siRNA-loaded pSiNPs exhibited a half-life for release of the siRNA payload of 1.3 h, whereas the GO coating extended the half-life of siRNA release to 4.0 h (Fig. 2a). The degradation of the pSiNPs, as measured by the loss of intrinsic photoluminescence from the Si nanostructures,33 was also substantially slower for the GO-coated pSiNPs (Fig. 2b). The GO nanosheets thus slowed both pSiNP degradation and release of the siRNA payload. The leakage of siRNA payload correlated with degradation of the nanocarrier, and the loss in photoluminescence intensity from the pSiNP skeleton showed a linear correlation with siRNA release for both the uncoated and the GO-coated nanoparticles (Fig. 2c). This result demonstrates a potentially useful self-reporting drug delivery characteristic of these materials.
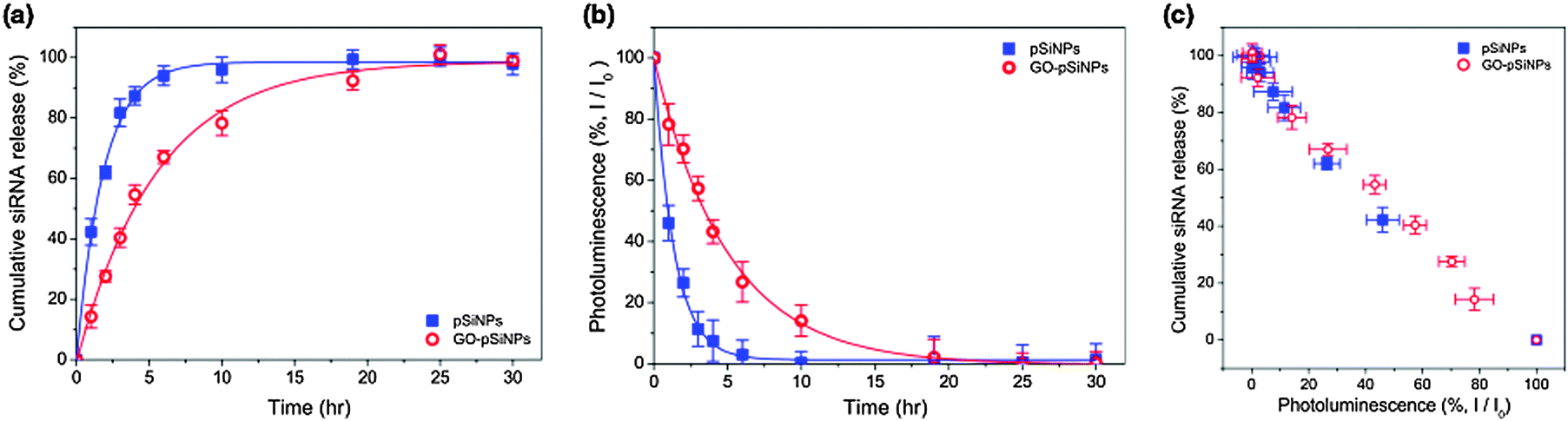 | ||
| Fig. 2 (a) Cumulative release profile of siRNA payloads from the nanocarriers incubated in phosphate-buffered saline (PBS) at 37 °C (blue: pSiNPs, red: GO–pSiNPs). Dy677-labeled siRNA was loaded to the nanocarriers, and supernatant was separated by centrifugation to measure fluorescence intensity (λex: 670 nm/λem: 700 nm). (b) Integrated PL intensity (λex: 365 nm/λem: 550–950 nm) of pSiNPs (blue) and GO–pSiNP (red) incubated in PBS at 37 °C as a function of time. (c) Correlation of cumulative siRNA release with the photoluminescence of pSiNPs during incubation in PBS at 37 °C (re-plotted from (a) and (b)). | ||
We then modified the nanocarrier with a targeting peptide to allow cell-specific homing. Selective binding and efficient internalization into target cells are critical factors to achieve significant gene silencing both in vitro and in vivo. We used a peptide derived from rabies virus glycoprotein (RVG), which has been employed for targeting neuronal cells in the central nervous system.51 The RVG peptide binds specifically to the acetylcholine receptor on neuronal cells to enable entry by receptor-mediated endocytosis.52 For these experiments the GO–pSiNPs were loaded with Dy547-labeled siRNA to allow tracking of the oligonucleotide, and RVG was grafted to the nanoparticle constructs via a PEG linker (Mw, 5000 Da) to reduce nonspecific binding. After peptide conjugation, the nanocarriers were still stably dispersed in aqueous solution without any aggregation. Grafting of the PEG linkers and targeting peptides slightly increased the mean hydrodynamic diameter of the nanocarriers to ∼190 nm (measured by DLS, polydispersity index <0.2).53 When incubated with Neuro-2a cells, confocal microscopy indicated significant uptake of the RVG peptide-conjugated pSiNPs (Fig. 3a). The confocal images of the RVG-targeted constructs showed coincident signals for the intrinsic photoluminescence from the pSiNPs and the fluorescence from the dye-labeled siRNA, which were well localized in the cytosol of the cells. Negligible uptake was observed for free siRNA, typical of the low cell permeability usually observed with free oligonucleotides due to their strong electrostatic repulsion from the anionic cell membrane surface.20
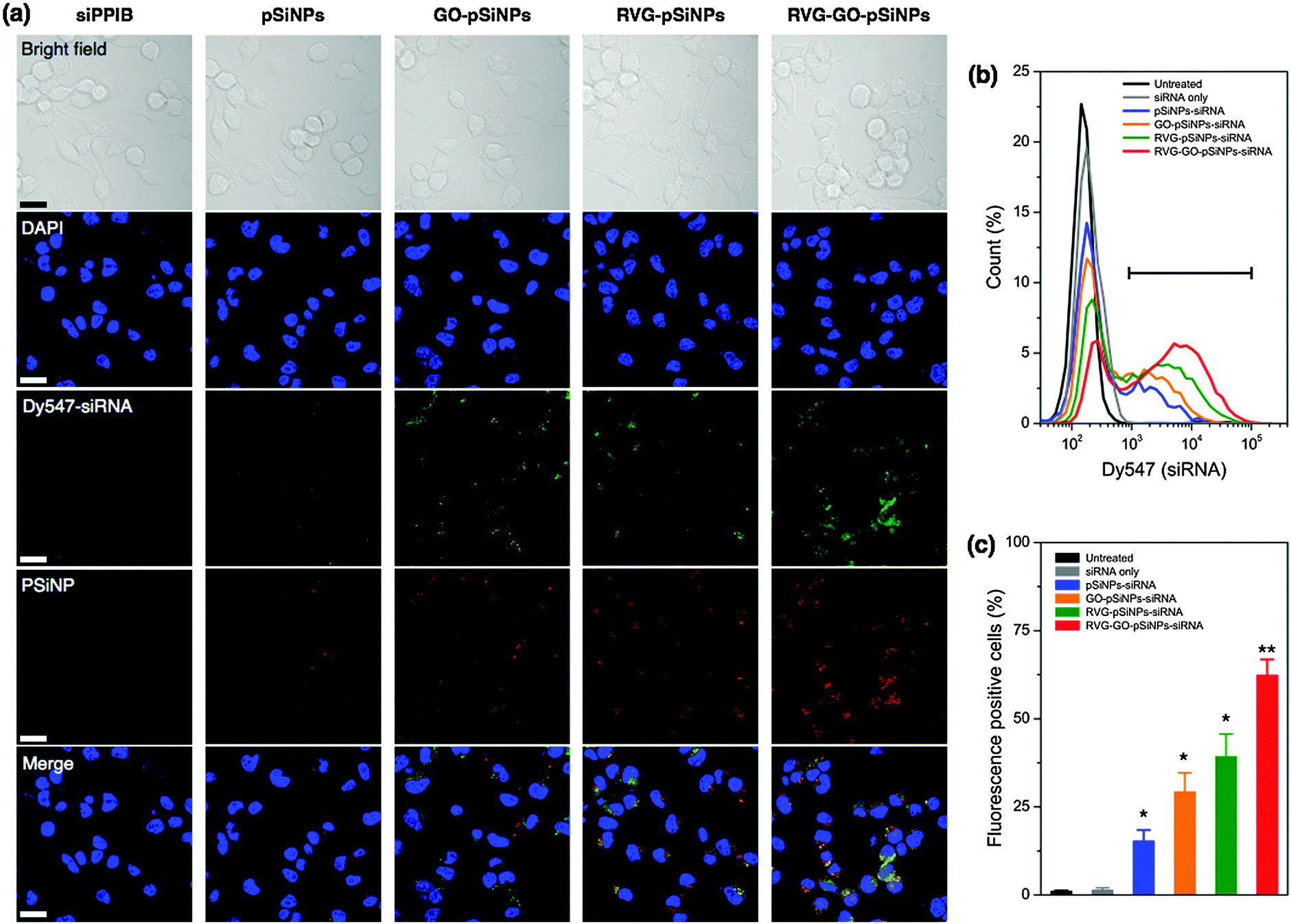 | ||
| Fig. 3 Targeting of Neuro-2a cells with nanoparticles containing siRNA against the PPIB gene (siPPIB). (a) Confocal fluorescence microscope images of cells treated for 2 h with: free (control) siPPIB, pSiNPs, GO–pSiNPs, RVG–pSiNPs, and RVG–GO–pSiNPs, as indicated. All nanoparticle and control formulations contained comparable quantities of siPPIB, which was labeled with the fluorescent dye Dy547 to enable imaging. Blue channel represents the DAPI nuclear stain, green represents the Dy547 label conjugated to the siRNA, and red channel represents the signal from intrinsic photoluminescence of the pSiNPs. Scale bar: 20 μm. (b) Flow cytometry histograms of Neuro-2a cells after incubation with nanoparticle formulations of Dy547-labeled siPPIB. (c) Percentage of fluorescence-positive cells appearing in the gate indicated in (b). Statistics: n = 3, * is p < 0.05 and ** is p < 0.005 against the untreated control. | ||
The confocal microscopy results were confirmed by flow cytometry (Fig. 3b and c). When Neuro-2a cells were incubated with RVG-targeted, siRNA-loaded pSiNPs (either the pSiNP or GO–pSiNP constructs), the cell population showed significantly increased fluorescence signal compared with those incubated with siRNA-loaded pSiNPs containing no RVG targeting peptide (p < 0.02). Notably, a significant extent of intracellular uptake of siRNA was observed for GO–pSiNPs compared to pSiNPs containing no GO nanosheet coating (p < 0.04, Fig. 3c). This is attributed to the improved retention of the siRNA payload by GO–pSiNPs discussed above (Fig. 2). RVG–GO–pSiNPs exhibited the strongest association and the highest percentage of siRNA-positive cells compared to other groups. Whereas commercially available transfection agents, such as Lipofectamine (Life Technologies), have shown impressive cellular uptake efficiency, they are only useful for in vitro experiments due to short circulation times. In contrast, the porous silicon-based nanocarriers are promising formulations for in vivo administration of potential therapeutic agents (see below).23,54
In order to evaluate the ability of GO–pSiNPs to mediate gene-specific knockdown, this study used siRNA against peptidylprolyl isomerase B (PPIB), an endogenously expressed gene with validated siRNA sequences. The levels of PPIB mRNA expression in Neuro-2a cells were quantitatively determined by reverse transcription polymerase chain reaction (qRT-PCR) after 48 h of incubation with the nanocarriers (Fig. 4a). To eliminate the possibility that gene silencing was caused by toxicity of the nanocarriers, the same formulations without siRNA payloads or with control sequences against luciferase (siLuc) were also tested, and they showed no significant cytotoxicity and gene silencing compared to untreated cells. Controls using free siRNA against PPIB also showed negligible knockdown. By contrast, RVG–GO–pSiNPs demonstrated significant knockdown efficiency (∼65%) of PPIB mRNA levels. Taken with the results of Fig. 3, these results clearly demonstrate RVG-mediated uptake and target gene knockdown in vitro. As additional evidence supporting the importance of the cellular targeting feature of the nanoparticles, substantially less knockdown in PPIB expression was observed if the RVG targeting moiety was omitted from the nano-constructs. In the case of siRNA-loaded pSiNPs lacking both the RVG peptide and the protective GO shell, negligible knockdown was observed (blue bar in Fig. 4a, “pSiNPs–siPPIB”) and in the case of siRNA-loaded pSiNPs lacking the RVG peptide but containing the protective GO shell, only ∼33% knockdown of PPIB expression was observed (orange bar in Fig. 4a, “GO–pSiNPs–siPPIB”). It is likely the positive charge on the GO nanosheets contributes to non-specific cellular binding and cellular uptake of the GO–pSiNP nanocarriers, similar to the behavior of the commonly used gene delivery vectors poly-L-lysine or Lipofectamine. Higher gene knockdown could be achieved with Lipofectamine, however, the use of such transfection agents is generally limited to in vitro experiments due to the low circulation time and toxicity associated with such cationic materials in vivo.55,56
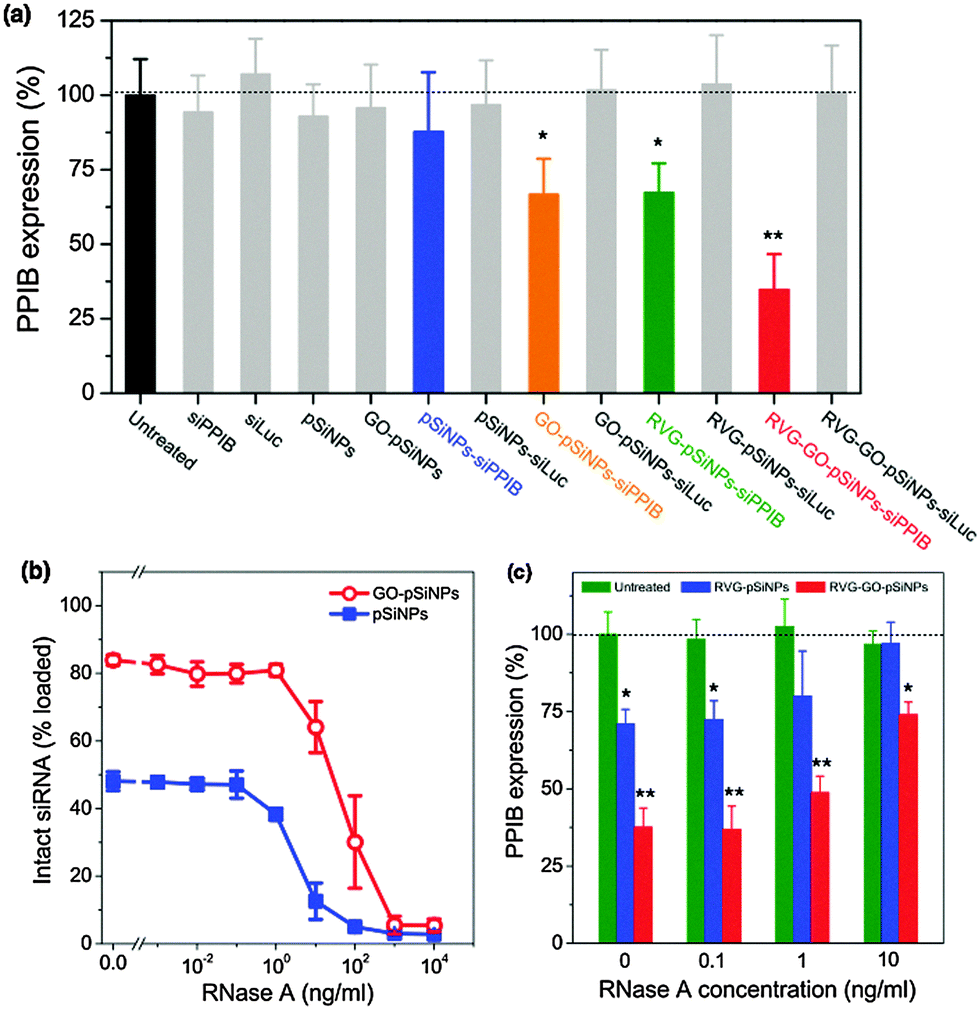 | ||
| Fig. 4 (a) Relative PPIB gene silencing in Neuro-2a cells after treatment with the nanocarrier formulation or free siPPIB, as indicated, in serum-free transfection media (Opti-MEM). Gray-colored bars show negative controls including a negative control siRNA against luciferase (siLuc)-loaded nanocarriers and the empty nanocarriers without siRNA (n = 5; * is p < 0.05 and ** is p < 0.01 against the untreated control). (b and c) The porous Si nanoparticles protect siRNA payloads from nuclease-induced degradation. (b) Percent of siRNA loaded in pSiNPs (blue solid squares) or GO–pSiNPs (red open circles) that is still active after exposure to the indicated concentrations of RNase A. Nanoparticles containing siRNA were incubated in RNase A for 1 h, isolated, and the remaining siRNA payload was assayed for intact RNA. Under these conditions, free siRNA was completely degraded by RNase A concentrations of 10−2 ng mL−1 or greater. However, siRNA contained in the nanocarriers retained detectable activity after exposure to RNase A concentrations of up to 100 ng mL−1. The quantity of intact siRNA detected is comparable to the quantity retained in the nanoparticles during the experiment. (c) Relative PPIB gene silencing in Neuro-2a cells after treatment with the indicated nanocarrier formulations in RNase A-containing serum media (n = 3; *p < 0.05 and **p < 0.01 against the untreated/no RNase control). | ||
In addition to inhibiting prompt release of siRNA, it is critical that a carrier designed for in vivo RNAi-based therapy protect the oligonucleotide payload against nucleolytic degradation when circulating in the bloodstream. We thus assessed if the siRNA payload remained intact when incubated with ribonuclease (RNase) in serum-containing media (Fig. 4b). Whereas exposure of free siRNA to RNase resulted in complete enzymatic degradation at RNase concentrations as low as 10−2 ng mL−1, degradation of siRNA contained in uncoated pSiNPs was not detectable at RNase concentrations less than 10−1 ng mL−1, and in GO-coated pSiNPs, fragmentation of siRNA was not detectable at RNase concentrations less than 1 ng mL−1. Complete degradation of siRNA in either of the nanoparticle types was not achieved until RNase concentrations reached ∼103 ng mL−1 (Fig. 4b). Thus the GO-coated pSiNPs imparted 10-fold greater stability to the siRNA payload than the uncoated pSiNPs, and 100-fold greater stability relative to free siRNA in the presence of ribonuclease. Under the conditions of these experiments, approximately 50% or 80% of the siRNA originally loaded into the nanocarriers was retained in the pSiNPs or GO–pSiNPs, respectively (Fig. 2a), while the rest was released into solution and degraded on the timescale of the assay. Thus the quantity of intact siRNA detected was comparable to the quantity that was retained in the nanoparticles during the experiment.
In order to test the extent of protection from nucleolytic degradation that was afforded by the pSiNP or the GO–pSiNP carriers, PPIB gene knockdown experiments were also run in culture media containing RNase and fetal bovine serum (Fig. 4c). Significant PPIB knockdown was achieved with the siRNA-containing RVG–GO–pSiNP construct in the presence of as much as 10 ng mL−1 of RNase. Consistent with the siRNA fragmentation assay discussed above (Fig. 4b), the GO–pSiNPs showed substantially greater gene knockdown than the uncoated pSiNPs, and both formulations performed better than free siRNA, which was ineffective in the presence of 10−1 ng mL−1 or higher concentrations of RNase. The demonstration of gene silencing in the presence of added RNase and serum proteins is a considerable improvement over cationic lipid-based vectors whose gene silencing performance is impeded by serum co-incubation.57
We next tested whether the nanocarriers could specifically deliver siRNA to a target tissue via systemic administration in a mouse model. The in vivo mouse model involved a penetrating brain injury on the right hemisphere, which generates localized inflammation and immune deficiencies that can impede systemic delivery of diagnostic and therapeutic agents. The nanocarriers were administered intravenously 6 h post-injury via a tail-vein injection, and the brains were harvested and imaged 2 h later. For the imaging, we employed a time-gated method (GLISiN, Gated Luminescence Imaging of Silicon Nanoparticles) because it highlights the long-lived excited state of pSiNPs and suppresses the strong autofluorescent background of the brain tissues.53 When administered by intravenous injection, time-gated luminescence images indicated that the RVG-targeted nanocarriers homed to the injured site (right hemisphere) of the brain while non-targeted nanocarriers nonspecifically spread throughout the entire brain (Fig. 5a). Moreover, the amount of RVG-targeted pSiNPs accumulated at the injured area was significantly larger than that in the uninjured (healthy) areas (Fig. 5b).
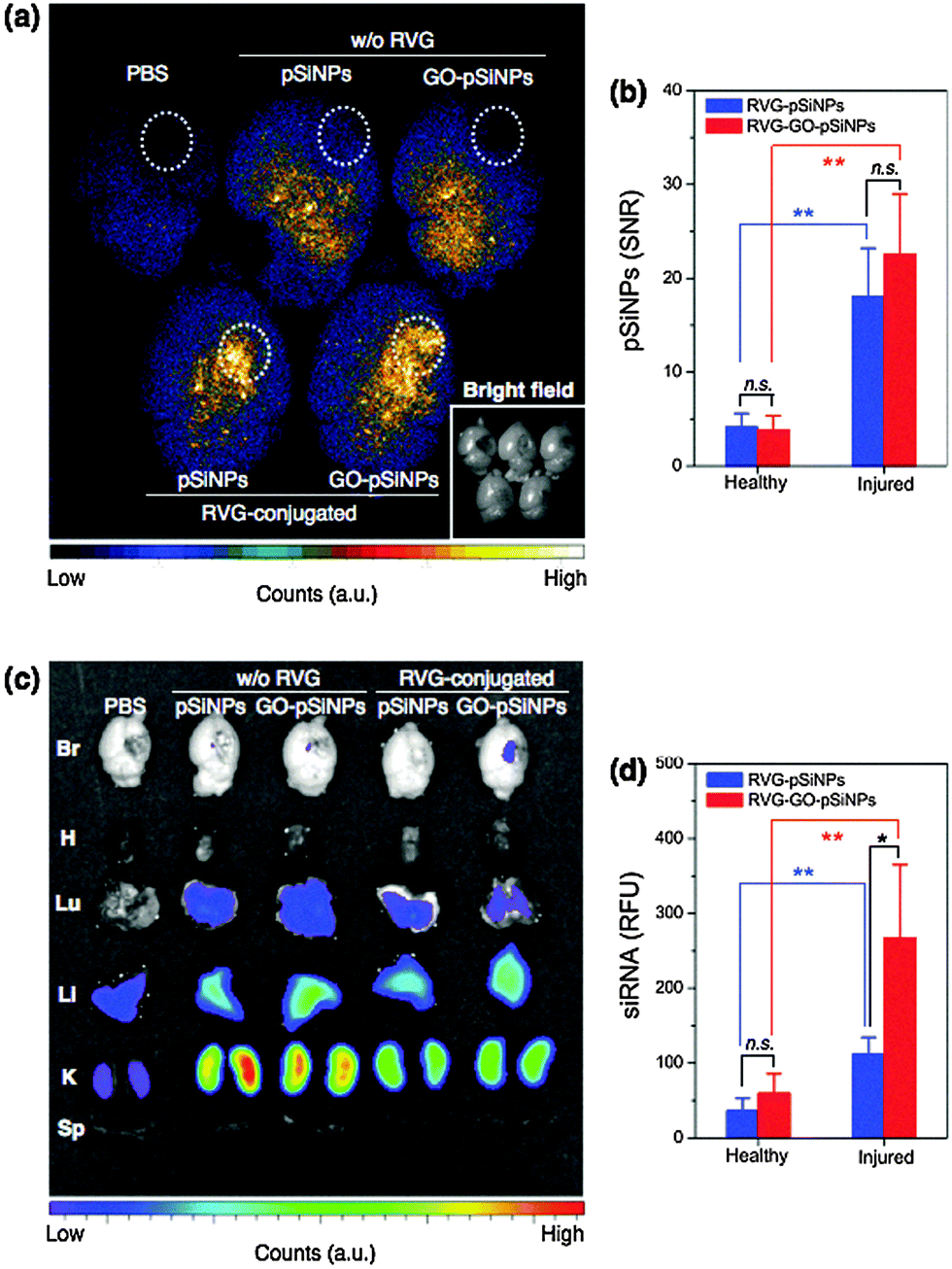 | ||
| Fig. 5 Luminescence images testing specific targeting of pSiNPs and GO-coated pSiNPs to injured mouse brain. (a) Time-gated luminescence image of injured mouse brains (λex: 365 nm). Dashed white circles indicate region of penetrating brain injury. Targeted (“RVG-conjugated”) and non-targeted (“w/o RVG”) nanoparticles are compared. Inset: Bright field image (in gray scale) under ambient light. (b) Signal-to-noise ratio (SNR) calculated for luminescent pSiNPs accumulated at healthy (left hemisphere) or injured (right hemisphere) region of the brain tissues. (c) Fluorescence image of Dy677-labeled siRNA accumulated in mouse organs obtained from IVIS 200 imaging system (λex: 670 nm, λem: 700 nm). (d) Relative fluorescence intensity of Dy677-labeled siRNA at healthy (left hemisphere) or injured (right hemisphere) region of the brain tissues. For (b) and (d), * is p < 0.05; ** is p < 0.01; n.s. is not significant, from two-tailed Student's t test. Data are presented as means ± SD (n = 3). | ||
The RVG peptide has been reported to cross the blood–brain barrier (BBB) and penetrate into neuronal cells.51 Our results show that the RVG peptide can be employed as a targeting ligand for brain injury. The ability of the peptide to cross the BBB may not be relevant in this case because the BBB is compromised in brain injury. However, we can expect a greater number of neuronal cells to become exposed at the damaged site. Consistent with this, the amount of RVG-targeted pSiNPs accumulated at the injured area was significantly larger than that of the healthy area (Fig. 5b). The greater accumulation seen in the damaged region of the mouse brain is attributed to specific interaction of the RVG peptide with acetylcholine receptor expressed on neuronal cells or macrophages exposed at the site of injury.52,58,59
Fluorescence microscope images configured to monitor the emission from a Dy677 tag attached to the siRNA payload showed that the RVG–GO–pSiNP construct delivered a substantial quantity of siRNA to the injured site (Fig. 5c). Delivery of siRNA by RVG–GO–pSiNPs was 2.5-fold greater (p < 0.05) than with the RVG–pSiNP construct that did not contain a GO coating (Fig. 5d). This finding illustrates a potential advantage of the GO coating for delivery of siRNA, as the quantity of nanoparticles that accumulated at the injured site was not affected significantly by the GO-coating (p ∼ 0.2 for RVG–GO–pSiNPs vs. RVG–pSiNPs). Thus the RVG–GO–pSiNPs delivered a larger dose of siRNA, which is attributed to the ability of the GO coating to slow release of the siRNA payload (Fig. 2) or to inhibit its degradation (Fig. 4). For all the pSiNP formulations, the labeled siRNA was also detected in the kidney, liver, and (to a lesser extent) the lung.
Conclusions
This work demonstrates a targeted gene delivery platform based on biodegradable porous silicon nanoparticles containing a cell-specific targeting moiety, a therapeutic siRNA payload, and a graphene oxide coating. The graphene oxide coating slowed release and enzymatic degradation of the siRNA payload, and it also slowed dissolution of the porous Si nanoparticle host. Multivalent functionalization of the exterior of the nanoparticle with a neuronal cell-specific targeting peptide facilitated homing and delivery of siRNA, and effective gene silencing was achieved in a mouse neuroblastoma cell line in vitro, even in the presence of extracellular nucleases. The in vitro and in vivo studies demonstrated high specificity toward neuronal cells and brain injury, respectively, and targeting played an essential role in efficient cellular uptake and subsequent gene silencing. Although the scope of the current study was limited to targeting of neuronal cells, the platform could be adapted for other cellular targets based on available homing molecules.60 The high loading capacity, delayed release and protection of siRNA combined with the silencing activity and in vivo localization of systemically delivered nanocarriers demonstrates the potential for GO–pSiNPs to serve as a delivery platform for RNAi-based therapeutics.Acknowledgements
The authors thank Dr Sang Woo Seo (University of California, San Diego) for helpful discussions. This work supported by the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement HR0011-13-2-0017. The content of the information within this document does not necessarily reflect the position or the policy of the Government. This work also supported in part by the Marie-D. & Pierre Casimir-Lambert Fund (MIT) and by internal funds from the Sanford-Burnham-Prebys Medical Discovery Institute. E. J. Kwon acknowledges support from the Ruth L. Kirschstein National Research Service Award (1F32CA177094-01). S. Bhatia is an HHMI Investigator.References
- T. M. Rana, Nat. Rev. Mol. Cell Biol., 2007, 8, 23–36 CrossRef CAS PubMed.
- A. de Fougerolles, H. P. Vornlocher, J. Maraganore and J. Lieberman, Nat. Rev. Drug Discovery, 2007, 6, 443–453 CrossRef CAS PubMed.
- B. L. Davidson and P. B. McCray, Nat. Rev. Genet., 2011, 12, 329–340 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS PubMed.
- C. V. Pecot, G. A. Calin, R. L. Coleman, G. Lopez-Berestein and A. K. Sood, Nat. Rev. Cancer, 2011, 11, 59–67 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- Y. Ren, H. W. Cheung, G. von Maltzhan, A. Agrawal, G. S. Cowley, B. A. Weir, J. S. Boehm, P. Tamayo, A. M. Karst, J. F. Liu, M. S. Hirsch, J. P. Mesirov, R. Drapkin, D. E. Root, J. Lo, V. Fogal, E. Ruoslahti, W. C. Hahn and S. N. Bhatia, Sci. Transl. Med., 2012, 4, 147ra112 Search PubMed.
- A. Daka and D. Peer, Adv. Drug Delivery Rev., 2012, 64, 1508–1521 CrossRef CAS PubMed.
- Y. K. Oh and T. G. Park, Adv. Drug Delivery Rev., 2009, 61, 850–862 CrossRef CAS PubMed.
- R. Wadhwa, S. C. Kaul, M. Miyagishi and K. Taira, Curr. Opin. Mol. Ther., 2004, 6, 367–372 CAS.
- S. Sabbioni, E. Callegari, M. Manservigi, R. Argnani, A. Corallini, M. Negrini and R. Manservigi, Gene Ther., 2007, 14, 459–464 CrossRef CAS PubMed.
- S. B. Zhang, B. Zhao, H. M. Jiang, B. Wang and B. C. Ma, J. Controlled Release, 2007, 123, 1–10 CrossRef CAS PubMed.
- T. Lobovkina, G. B. Jacobson, E. Gonzalez-Gonzalez, R. P. Hickerson, D. Leake, R. L. Kaspar, C. H. Contag and R. N. Zare, ACS Nano, 2011, 5, 9977–9983 CrossRef CAS PubMed.
- M. L. Chen, S. Gao, M. D. Dong, J. Song, C. X. Yang, K. A. Howard, J. Kjems and F. Besenbacher, ACS Nano, 2012, 6, 4835–4844 CrossRef CAS PubMed.
- J. G. Li, X. S. Yu, Y. Wang, Y. Y. Yuan, H. Xiao, D. Cheng and X. T. Shuai, Adv. Mater., 2014, 26, 8217–8224 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, A. E. Prigodich, P. C. Patel and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 2072–2073 CrossRef CAS PubMed.
- J. S. Lee, J. J. Green, K. T. Love, J. Sunshine, R. Langer and D. G. Anderson, Nano Lett., 2009, 9, 2402–2406 CrossRef CAS PubMed.
- F. Pittella, M. Z. Zhang, Y. Lee, H. J. Kim, T. Tockary, K. Osada, T. Ishii, K. Miyata, N. Nishiyama and K. Kataoka, Biomaterials, 2011, 32, 3106–3114 CrossRef CAS PubMed.
- L. C. Yin, Z. Y. Song, K. H. Kim, N. Zheng, N. P. Gabrielson and J. J. Cheng, Adv. Mater., 2013, 25, 3063–3070 CrossRef CAS PubMed.
- L. Han, J. Zhao, X. Zhang, W. P. Cao, X. X. Hu, G. Z. Zou, X. L. Duan and X. J. Liang, ACS Nano, 2012, 6, 7340–7351 CrossRef CAS PubMed.
- T. A. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS PubMed.
- D. Zheng, D. A. Giljohann, D. L. Chen, M. D. Massich, X. Q. Wang, H. Iordanov, C. A. Mirkin and A. S. Paller, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11975–11980 CrossRef CAS PubMed.
- S. Jiang, A. A. Eltoukhy, K. T. Love, R. Langer and D. G. Anderson, Nano Lett., 2013, 13, 1059–1064 CrossRef CAS PubMed.
- A. M. Derfus, A. A. Chen, D. H. Min, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2007, 18, 1391–1396 CrossRef CAS PubMed.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J. P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Y. Zhang, M. Xue, T. Xia, S. J. Lin, X. Wang, Y. Zhao, Z. X. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- M. H. Kim, H. K. Na, Y. K. Kim, S. R. Ryoo, H. S. Cho, K. E. Lee, H. Jeon, R. Ryoo and D. H. Min, ACS Nano, 2011, 5, 3568–3576 CrossRef CAS PubMed.
- T. Tanaka, L. S. Mangala, P. E. Vivas-Mejia, R. Nieves-Alicea, A. P. Mann, E. Mora, H. D. Han, M. M. K. Shahzad, X. W. Liu, R. Bhavane, J. H. Gu, J. R. Fakhoury, C. Chiappini, C. H. Lu, K. Matsuo, B. Godin, R. L. Stone, A. M. Nick, G. Lopez-Berestein, A. K. Sood and M. Ferrari, Cancer Res., 2010, 70, 3687–3696 CrossRef CAS PubMed.
- L. T. Canham, in Porous Silicon for Biomedical Applications, ed. H. A. Santos, 2014, pp. 3–20 DOI:10.1533/9780857097156.1.3.
- B. Godin, C. Chiappini, S. Srinivasan, J. F. Alexander, K. Yokoi, M. Ferrari, P. Decuzzi and X. Liu, Adv. Funct. Mater., 2012, 22, 4225–4235 CrossRef CAS PubMed.
- J. Salonen, in Handbook of Porous Silicon, ed. L. T. Canham, Springer, Switzerland, 2014, p. 909 Search PubMed.
- L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326 Search PubMed.
- J.-H. Park, L. Gu, G. v. Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- Z. Qin, J. Joo, L. Gu and M. J. Sailor, Part. Part. Syst. Charact., 2014, 31, 252–256 CrossRef CAS.
- J. Joo, J. F. Cruz, S. Vijayakumar, J. Grondek and M. J. Sailor, Adv. Funct. Mater., 2014, 24, 5688–5694 CrossRef CAS.
- E. Segal, L. A. Perelman, F. Cunin, F. D. Renzo, J.-M. Devoisselle, Y. Y. Li and M. J. Sailor, Adv. Funct. Mater., 2007, 17, 1153–1162 CrossRef CAS.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, ACS Nano, 2013, 7, 9571–9584 CrossRef CAS PubMed.
- A. Elbakry, A. Zaky, R. Liebkl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. M. Tian, M. E. Napier, P. D. Pohlhaus, J. P. Rolland and J. M. DeSimone, Nano Lett., 2012, 12, 287–292 CrossRef CAS PubMed.
- H. Kim and W. J. Kim, Small, 2014, 10, 117–126 CrossRef CAS PubMed.
- M. Nurunnabi, Z. Khatun, K. M. Huh, S. Y. Park, D. Y. Lee, K. J. Cho and Y. K. Lee, ACS Nano, 2013, 7, 6858–6867 CrossRef CAS PubMed.
- M. L. Chen, Y. J. He, X. W. Chen and J. H. Wang, Bioconjugate Chem., 2013, 24, 387–397 CrossRef CAS PubMed.
- S. Y. Kwak, J. K. Yang, S. J. Jeon, H. I. Kim, J. Yim, H. Kang, S. Kyeong, Y. S. Lee and J. H. Kim, Adv. Funct. Mater., 2014, 24, 5119–5128 CrossRef CAS.
- H. K. Na, M. H. Kim, J. Lee, Y. K. Kim, H. Jang, K. E. Lee, H. Park, W. D. Heo, H. Jeon, I. S. Choi, Y. Lee and D. H. Min, Nanoscale, 2013, 5, 1669–1677 RSC.
- W. Choi, J. Choi, J. Bang and J. H. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 12510–12519 Search PubMed.
- S. B. Yang, X. L. Feng, S. Ivanovici and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS PubMed.
- S. Sreejith, X. Ma and Y. L. Zhao, J. Am. Chem. Soc., 2012, 134, 17346–17349 CrossRef CAS PubMed.
- P. Kumar, H. Q. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–43 CrossRef CAS PubMed.
- J. Y. Kim, W. I. Choi, Y. H. Kim and G. Tae, Biomaterials, 2013, 34, 1170–1178 CrossRef CAS PubMed.
- J. Joo, X. Liu, V. R. Kotamraju, E. Ruoslahti, Y. Nam and M. J. Sailor, ACS Nano, 2015, 9, 6233–6241 CrossRef CAS PubMed.
- C. Zheng, M. Zheng, P. Gong, J. Deng, H. Yi, P. Zhang, Y. Zhang, P. Liu, Y. Ma and L. Cai, Biomaterials, 2013, 34, 3431–3438 CrossRef CAS PubMed.
- L. Parhamifar, A. K. Larsen, A. C. Hunter, T. L. Andresen and S. M. Moghimi, Soft Matter, 2010, 6, 4001–4009 RSC.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- M. Yan, M. Liang, J. Wen, Y. Liu, Y. F. Lu and I. S. Y. Chen, J. Am. Chem. Soc., 2012, 134, 13542–13545 CrossRef CAS PubMed.
- Y. K. Gao, Z. Y. Wang, J. H. Zhang, Y. X. Zhang, H. Huo, T. Y. Wang, T. Y. Jiang and S. L. Wang, Biomacromolecules, 2014, 15, 1010–1018 CrossRef CAS PubMed.
- S. S. Kim, C. T. Ye, P. Kumar, I. Chiu, S. Subramanya, H. Q. Wu, P. Shankar and N. Manjunath, Mol. Ther., 2010, 18, 993–1001 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Mater., 2012, 24, 3747–3756 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on experimental information. See DOI: 10.1039/c6nh00082g |
| ‡ Current address: Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2016 |