
PdII-mediated integration of isocyanides and azide ions might proceed via formal 1,3-dipolar cycloaddition between RNC ligands and uncomplexed azide†
Mikhail A.
Kinzhalov
a,
Alexander S.
Novikov
a,
Konstantin V.
Luzyanin
ab,
Matti
Haukka
c,
Armando J. L.
Pombeiro
d and
Vadim Yu.
Kukushkin
*a
aSt. Petersburg State University, 7-9, Universitetskaya nab., St. Petersburg, 199034, Russia. E-mail: v.kukushkin@spbu.ru
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
cDepartment of Chemistry, University of Jyväskylä, P.O. Box 35, FI-40014, Finland
dCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
First published on 6th November 2015
Abstract
Reaction between equimolar amounts of trans-[PdCl(PPh3)2(CNR)][BF4] (R = t-Bu 1, Xyl 2) and diisopropylammonium azide 3 gives the tetrazolate trans-[PdCl(PPh3)2(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) N4t-Bu)] (67%, 4) or trans-[PdCl(PPh3)2(N4Xyl)] (72%, 5) complexes. 4 and 5 were characterized by elemental analyses (C, H, N), HRESI+-MS, 1H and 13C{1H} NMR spectroscopies. In addition, the structure of 4 was elucidated by a single-crystal X-ray diffraction. DFT calculations showed that the mechanism for the formal cycloaddition (CA) of N3− to trans-[PdCl(PH3)2(CNMe)]+ is stepwise. The process is both kinetically and thermodynamically favorable and occurs via the formation of an acyclic NNNCN-intermediate. The second step of the formal CA, i.e. cyclization, is rate limiting. Despite the fact that the substitution of CNMe by the N3− ligand is slightly thermodynamically favorable, we were unable to find paths on the potential energy surface for hypothetical CA between uncomplexed isocyanide and palladium-bound azide. Thus, we believe that the experimentally observed palladium tetrazolate complexes are, in fact, generated from the negatively charged uncomplexed azide and the positively charged metal-bound isocyanide species, and this reaction path is favorable from the viewpoint of Coulomb attraction.
N4t-Bu)] (67%, 4) or trans-[PdCl(PPh3)2(N4Xyl)] (72%, 5) complexes. 4 and 5 were characterized by elemental analyses (C, H, N), HRESI+-MS, 1H and 13C{1H} NMR spectroscopies. In addition, the structure of 4 was elucidated by a single-crystal X-ray diffraction. DFT calculations showed that the mechanism for the formal cycloaddition (CA) of N3− to trans-[PdCl(PH3)2(CNMe)]+ is stepwise. The process is both kinetically and thermodynamically favorable and occurs via the formation of an acyclic NNNCN-intermediate. The second step of the formal CA, i.e. cyclization, is rate limiting. Despite the fact that the substitution of CNMe by the N3− ligand is slightly thermodynamically favorable, we were unable to find paths on the potential energy surface for hypothetical CA between uncomplexed isocyanide and palladium-bound azide. Thus, we believe that the experimentally observed palladium tetrazolate complexes are, in fact, generated from the negatively charged uncomplexed azide and the positively charged metal-bound isocyanide species, and this reaction path is favorable from the viewpoint of Coulomb attraction.
Introduction
Being one of the most studied ligands in contemporary organometallic chemistry and catalysis, N-heterocyclic carbenes (NHCs) continue to attract a lot of attention due to their valuable characteristics, e.g., high chemical and thermal stability, low toxicity, and variability of donor and steric properties.1 The preparation of metal complexes with NHC ligands can be achieved via either the direct coordination of pre-prepared (or generated in situ) free NHCs to the metal center,1 or through diverse cyclizations of metal-bound isocyanides and this subject was repeatedly reviewed including recent surveys by some of us.2Firstly (Scheme 1, route A), cyclic carbenes are prepared via two-step cascade synthesis that includes a nucleophilic attack of an amino or alcohol function of X(H)CH2CH2Br (XH = OH, NH2) on a coordinated isocyanide followed by ring closure.2b,d
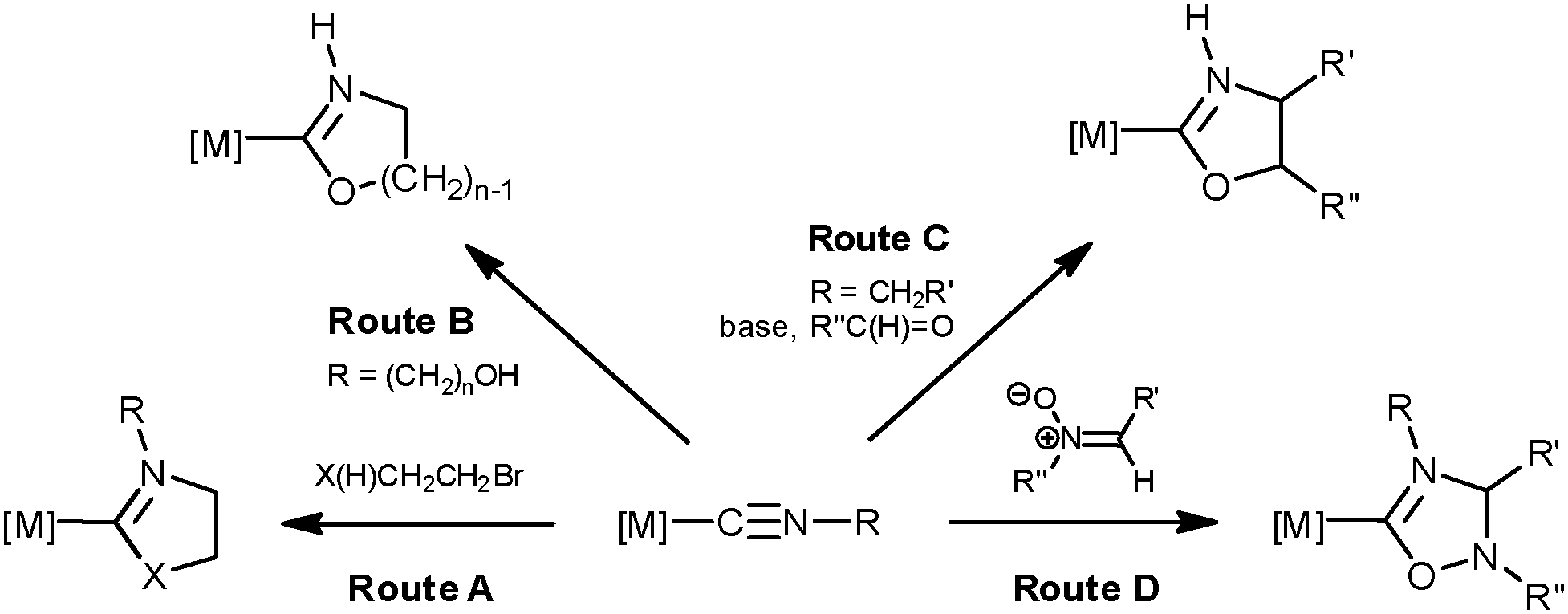 | ||
| Scheme 1 Conversion of ligated isocyanides into N-heterocyclic carbene complexes. | ||
Secondly (route B), metal–NHCs are generated via a spontaneous or base-promoted intramolecular cyclization of complexes featuring functionalized isocyanides, e.g. M–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–(CH2)n–OH.2d Thirdly (route C), isocyanides bearing acidic C–H bonds in the α-position to the CN group, e.g. M–CN–CH2EWG (where EWG stands for an electron withdrawing moiety), being ionized with a base, react with polar double bonds (e.g. with RC(H)
N–(CH2)n–OH.2d Thirdly (route C), isocyanides bearing acidic C–H bonds in the α-position to the CN group, e.g. M–CN–CH2EWG (where EWG stands for an electron withdrawing moiety), being ionized with a base, react with polar double bonds (e.g. with RC(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as a 1,3-dipolarophile) furnishing heterocycles.3
O as a 1,3-dipolarophile) furnishing heterocycles.3
Finally, complexes with NHC ligands can be generated via a metal-mediated 1,3-dipolar cycloaddition (CA) of some dipoles to isocyanides (route D). Examples of the latter route are so far limited to CA of nitrile imines [accomplishing carbene {C(NR′′NCaR′)NbR(Ca–Nb)}],4 nitrile ylides4 [yielding the {C(CHR′′NCaR′)NbR(Ca–Nb)} functionality] and, as we previously observed, acyclic and cyclic nitrones [giving the {C(CHR′′NCaR′)NbR(Ca–Nb)} moiety] to coordinated RNCs,5 and the metal-mediated interplay between RNC and azide leading to complexes with C-bound tetrazolate ligands, i.e. {C(NNNa)NbR(Na–Nb)}.2d,6 The mechanisms of these processes can be concerted or stepwise; in the latter case the formal CA is completely asynchronous and occurs via the formation of an acyclic intermediate followed by ring-closure.
Inspection of the literature relevant to metal-involving CA between azides and isocyanides6,7 indicated that up to now a substantial number of reports were devoted to reactions between uncomplexed isocyanides and metal-bound azides at palladium(II),8 platinum(II),8a,d–f,j,9 gold(I),10 gold(III),8c,11 and nickel(II)8d,f,12 centers (Scheme 2, route E). In the vast majority of cases,8b–e,h–j,9a,10,12 it is believed that CA proceeds through the attack of the free isocyanide on a complexed azide, albeit no mechanistic studies were undertaken. In one work,13 it is suggested that the reaction starts from the joint coordination of azide and isocyanide to a metal center giving an intermediate pentacoordinated species that is subsequently transformed into C-tetrazolates via an intramolecular CA (route H). However, the latter mechanism was neither supported experimentally, nor theoretically.
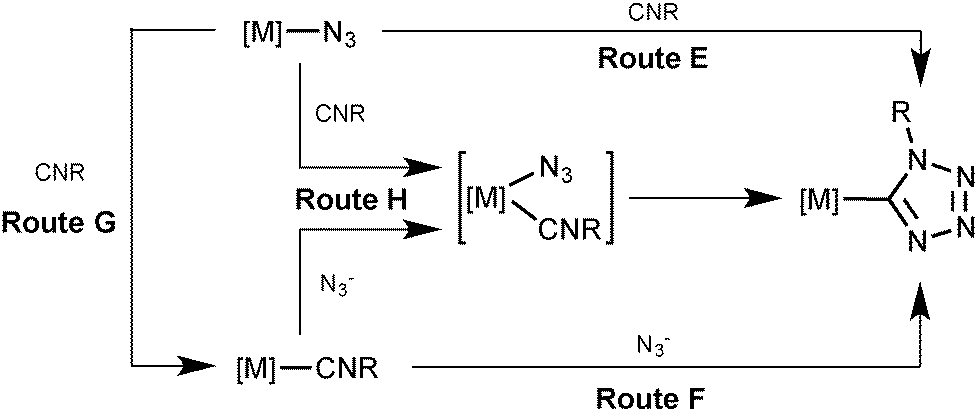 | ||
| Scheme 2 Metal-mediated generation of tetrazolate species. | ||
An alternative approach to the generation of tetrazolate complexes via CA is based on the use of metal-bound isocyanides and uncomplexed azide ions. Until now this approach was represented by a single study where CA was conducted at a platinum(II) center (route F) when the isocyanide complexes [Pt(PPh3)2(CNMe)2][BF4]2 and [Pt(diphos)(CNMe)2][PF6]2 were treated with one and two equiv. of NaN3 in water, respectively, and CA led to the C-tetrazolate species [Pt(PPh3)2(CNMe)(CN4Me)][BF4] (78%) and [Pt(diphos)(CN4Me)2] (90%), respectively.13 As believed, this CA proceeds by the attack of azide ions on the square-planar complex to give an intermediate five-coordinate species (route H), which then undergoes a facile cyclization accomplishing the tetrazolate complexes.
In principle, the mechanism of metal-mediated CA between isocyanides and azides could involve complexed azide as a dipole and uncomplexed CNR as a dipolarophile (Sustman III type of CA;14route E), metal-bound isocyanide as a dipolarophile and free azide as a dipole (Sustman I type of CA;14route F), and the joint coordination of the reactants furnishing C-tetrazolate species (route H). Alternatively the reaction between metal-azides and isocyanides may proceed via the azide substitution step followed by CA (routes G–F).
In pursuit of our previous studies on CA to metal-bound (in particular to PdII-ligated) nitriles (see our reviews,15 recent experimental,16 and also theoretical17 studies) and isocyanides (see reviews,2d experimental,5,18 and theoretical19 studies) and general interest in the metal-involving chemistry of azides (see recent review by Fehlhammer and Beck6) we decided to verify the most probable route for the generation of palladium(II)-tetrazolates from RNCs and N3−. To reach this goal we, first of all, decided to find experimental evidence that the palladium-tetrazolates could be obtained not only by the known reaction between (azide)PdII species and uncomplexed RNCs,8 but also via yet unreported integration of some (RNC)PdII precursors and uncomplexed azide. These experimental examples should form the solid background for theoretical studies – which comprise the main goal of this study – directed toward the verification of a plausible reaction mechanism that may proceed via routes E, F, H, or G–F (Scheme 2), the establishment of driving forces of the generation of (tetrazolate)PdII complexes and the identification of key intermediates of this palladium(II)-mediated integration.
Results and discussion
Cycloaddition of azide to PdII-bound isocyanides
In order to verify the reactivity of palladium-isocyanide species toward N3−, we attempted to carry out CA of diisopropylammonium azide 3 to the isocyanide ligands in trans-[PdCl(PPh3)2(CNR)][BF4] (ref. 20) (R = t-Bu 1, Xyl 2). Reaction between equimolar amounts of complex 1 (or 2) and 3 proceeds in CH2Cl2 at room temperature (RT) for ca. 1 d giving the tetrazolate trans-[PdCl(PPh3)2(N4t-Bu)] (4) or trans-[PdCl(PPh3)2(N4Xyl)] (5) (Scheme 3) species. After the evaporation of the solvent, these complexes were washed with a MeOH/Et2O mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and purified by recrystallization from a CH2Cl2/Et2O mixture giving 4 (67%) and 5 (72%) in good isolated yields.
:1, v/v) and purified by recrystallization from a CH2Cl2/Et2O mixture giving 4 (67%) and 5 (72%) in good isolated yields.
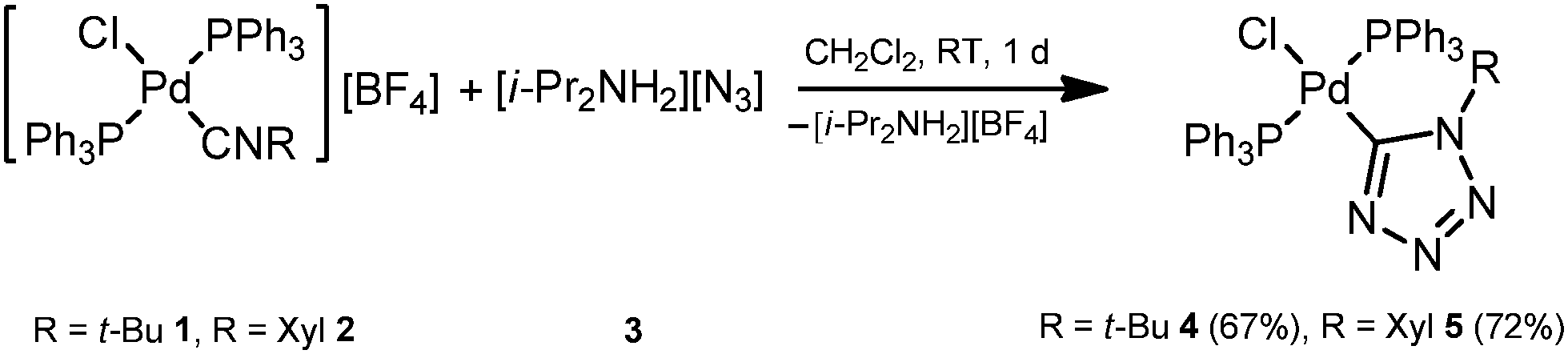 | ||
| Scheme 3 Reaction between trans-[PdCl(PPh3)2(CNR)][BF4] (R = t-Bu 1, Xyl 2) and diisopropylammonium azide (3). | ||
Complexes 4 and 5 were obtained as pale yellow solids and characterized by elemental analyses (C, H, N), HRESI+-MS, 1H and 13C{1H} NMR spectroscopies. In addition, the structure of 4 was elucidated by a single-crystal X-ray diffraction. Complexes 4 and 5 gave satisfactory C, H, and N elemental analyses, which are consistent with the proposed formulae. The ESI+ mass spectra demonstrated a fragmentation pattern corresponding to the loss of Cl from the molecular ion along with the characteristic isotopic distribution. CA of 3 to 1 or 2 is accompanied by a pronounced downfield δ13C shift of the isocyanide quaternary C atom to the range common for tetrazolate ring Pd–N4R (δC 150–165 ppm).8a–f,k,9a In 4 and 5, the N4R 13C signals were found to resonate at δC 151.5 and 161.9 ppm, respectively; that is ca. 45 ppm downfield shifted vs.1 and 2 (e.g. 115 ppm for CN in 1).
In 4 (Fig. 1), The PPh3 ligands are mutually trans (P1–Pd1–P2 167.93(2)°) and a slightly distorted square-planar environment around the metal center is completed with one tetrazolate and one chloride ligand. Bond angles around Pd as vertex are all within 3.0° of the values for an ideal square-plane. The Pd–C distance [Pd1–C1 1.989(2) Å] is comparable to those reported for the related palladium tetrazolate complex trans-[Pd(PMe3)2(N4Xyl)(NCNR)] (2.006(5) Å).8e
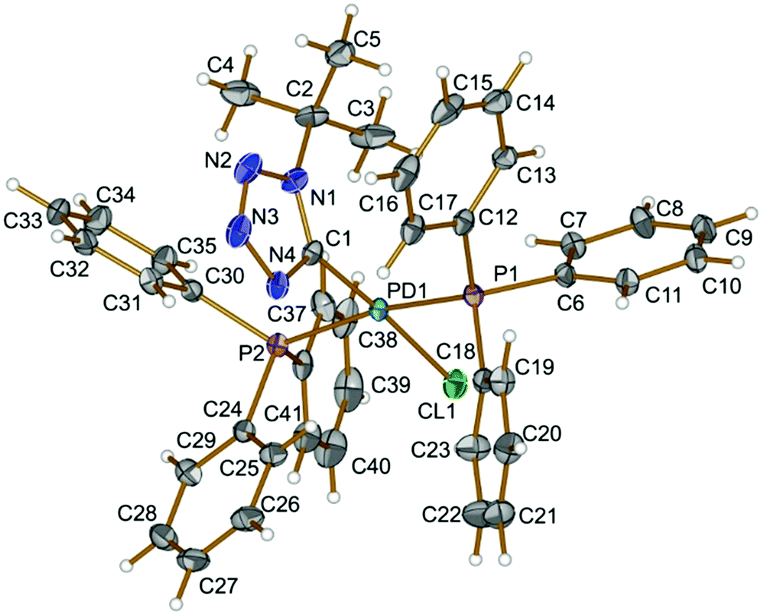 | ||
| Fig. 1 View of 4 with the atomic numbering schemes. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen labels are omitted for simplicity. | ||
Our little synthetic experiment proved that the integration between RNC and N3− species giving C-tetrazolate complexes might proceed viaroute F (Scheme 2) not only at platinum(II),13 but also at palladium(II) centers. These data form the background for a further theoretical study aimed toward the verification of a plausible mechanism of this reaction and results of the study are disclosed in the next section.
Theoretical study
In order to shed light on the mechanism of the PdII-mediated azide–isocyanide integration, a quantum chemical DFT study of this process in a model system was undertaken (CPCM-B3LYP/6-311+G(d,p)//gas-B3LYP/6-31G(d)). The route F was found to be the most preferred out of all possible reaction paths shown in Scheme 2. Discussion about this and other plausible reaction channels is given below.The mechanism of the formation of (tetrazolate)PdII complexes via formal CA of N3− to trans-[PdCl(PH3)2(CNMe)]+ complexes (Scheme 2, route F) was found to be stepwise (Scheme 4).
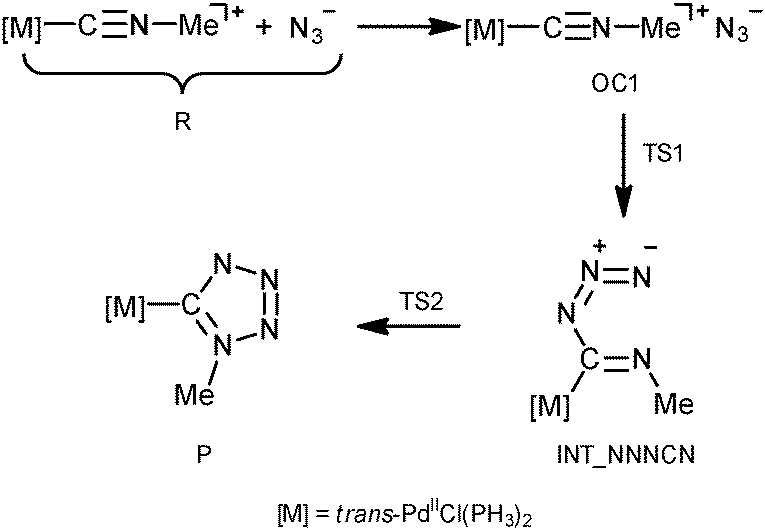 | ||
| Scheme 4 Mechanism of PdII-mediated azide–isocyanide integration. | ||
The process is initiated by the formation of the orientation complex trans-[PdCl(PH3)2(CNMe)]+·N3− (OC1), and two transition states (TSs) were located. The first one (TS1) corresponds to the formation of the C–N bond giving the acyclic NNNCN-intermediate (INT-NNNCN) and the second one (TS2) is associated with the ring closure forming the C-tetrazolate complex trans-[PdCl(PH3)2(N4Me)] (P). The second step is rate limiting (Fig. 2 and Table S3, ESI†). Noteworthy that we were unable to locate on the potential energy surface any TSs for the concerted pathway or other acyclic NNNCN-intermediates. Based upon our quantum chemical calculations one can state that the formation of the C-tetrazolate palladium complexes trans-[PdCl(PH3)2(N4Me)] from coordinated isocyanides and free azides is quite favorable from both kinetic and thermodynamic viewpoints.
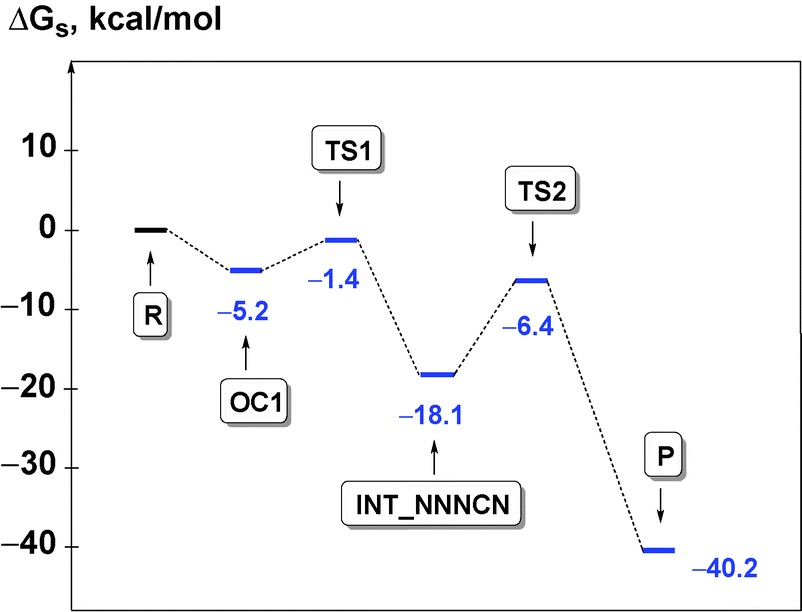 | ||
| Fig. 2 Energy profile of R → P transformation for PdII-mediated azide–isocyanide integration. | ||
We estimated the relative stability of two possible types of the orientation complexes in the model reaction system, viz. trans-[PdCl(PH3)2(CNMe)]+·N3− (OC1) versus trans-[PdCl(PH3)2(N3)]·CNMe (OC2) and found that the OC1 → OC2 transformation (Scheme 2, inverse route G) is just slightly thermodynamically favorable (ΔGs = −1.0 kcal mol−1). Taking this into account, the hypothetical reactions between uncomplexed isocyanide and metal-bound azide (Scheme 2, route E) were also inspected (Table S3, ESI†) and the following conclusions can be drawn. Firstly, we were unable to locate any TSs and intermediates for the CNMe + trans-[PdCl(PH3)2(N3)] → trans-[PdCl(PH3)2(N4Me)] processes. Secondly, we found two TSs (TS3 and TS4) for the CNMe + trans-[PdCl(PH3)2(N3)] → N2 + trans-[PdCl(PH3)2(NCNMe)] (ΔG≠s =36.2 kcal mol−1, ΔGs = −62.2 kcal mol−1) and CNMe + trans-[PdCl(PH3)2(N3)] → trans-[PdCl(PH3)2(N2)]+ + NCNMe− (ΔG≠s = 39.0 kcal mol−1, ΔGs = −9.0 kcal mol−1) concerted transformations. It is obvious that although being thermodynamically favorable these paths are not realized at room temperature due to too high activation barriers.
In order to get experimental evidence supporting the generation of (isocyanide)Pd complexes from the corresponding azide precursors, we additionally conducted IR monitoring of two ligand substitution reactions, viz. between [PdCl(N3)(PPh3)2] and CNXyl on the one hand, and between [PdCl(PPh3)2(CNXyl)][BF4] and N3− on the other hand. In the former case, immediately after mixing the two reactants we detected the appearance of a new band at 2204 cm−1 from coordinated isocyanide (in [PdCl(PPh3)2(CNXyl)][BF4] ν(CN) 2204 cm−1), whereas in the latter case, only after approximately 20 min we observed the emergence of weak absorbance from the uncomplexed isocyanide (2099 cm−1). These observations suggest that for the studied system the complexation equilibrium is shifted toward the isocyanide complex and, in turn, the isocyanide is a better ligand than azide. All these give additional (but not the final) evidence that the isocyanide–azide integration might proceed via cycloaddition of N3− to the CNXyl ligand.
In order to verify the hypothesis13 on the formation of intermediates bearing both azide and isocyanide species bound to the metal center (Scheme 2, route H), we undertook an extensive search of appropriate minima on the potential energy surface with various mutual positions of these ligands in the model starting structures. However, we were unable to locate any pentacoordinated intermediates, and all attempts to fix them led to the migration of one ligand from the coordination sphere of the metal center during the geometry optimization procedure or to the collapse of the whole structure of the palladium model complex. Hence, route H can be excluded from consideration.
The metal-free reaction N3− + CNMe → CN4Me− is thermodynamically unfavorable (ΔGs = 4.7 kcal mol−1) and we were unable to locate appropriate TSs and intermediates for both concerted and stepwise paths. However, we found TS (TS_metal-free) for the N3− + CNMe → N2 + NCNMe− concerted transformation and it is highly thermodynamically favorable (ΔGs = −55.5 kcal mol−1), but kinetically hindered (ΔG≠s = 35.2 kcal mol−1).
We checked the tendency toward the decomposition of trans-[PdCl(PH3)2(N4Me)] complexes and uncomplexed tetrazolate CN4Me−via the paths trans-[PdCl(PH3)2(N4Me)] → N2 + trans-[PdCl(PH3)2(NCNMe)] (by TS5) and CN4Me− → N2 + NCNMe− (by TS5_metal-free), respectively (Table S3, ESI†). One can conclude that the tetrazolate CN4Me− species are very susceptible to decomposition (ΔG≠s = 10.8 kcal mol−1, ΔGs = −60.2 kcal mol−1), but the palladium center is an excellent stabilizer for such anionic species (ΔG≠s = 36.5 kcal mol−1, ΔGs = −35.2 kcal mol−1).
Finally, we compared the thermodynamic stability of the tetrazolate complexes trans-[PdCl(PH3)2(CN4Me)], where the CN4Me− ligand is coordinated to the metal by different alternative sites (Fig. 3). The stability series is the following: trans-[PdCl(PH3)2(N4Me)] (0 kcal mol−1 in terms of Gibbs free energy in solution, internal standard) > A (24.4 kcal mol−1) > B (33.9 kcal mol−1) > C (42.2 kcal mol−1).
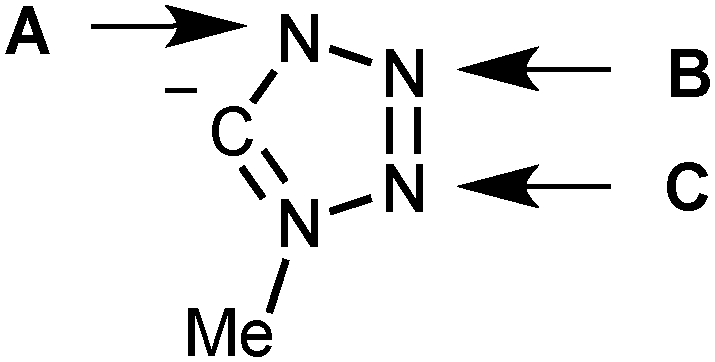 | ||
| Fig. 3 Alternative binding sites of the tetrazolate ligand. | ||
Conclusions
The results of this work could be considered from the following perspectives. Firstly, in pursuit of our studies on cycloaddition to metal-bound RNCs,5,18,19 we observed that the metal-mediated reaction between equimolar amounts of the isocyanide ligand in trans-[PdCl(PPh3)2(CNR)][BF4] (R = t-Bu, Xyl) and diisopropylammonium azide proceeds in CH2Cl2 at room temperature furnishing the C-tetrazolate complexes trans-[PdCl(PPh3)2(N4R)] (ca. 70%). The generation of C-tetrazolato ligands from metal-bound RNCs and uncomplexed N3− was previously observed only at a platinum(II) center, whereas many other synthetic routes towards C-tetrazolato metal complexes involve a reaction between metal-bound azide and uncomplexed isocyanides.
Secondly, based upon quantum chemical calculations we established that the mechanism for the formal cycloaddition of N3− to trans-[PdCl(PH3)2(CNMe)]+ is stepwise. The process is both kinetically and thermodynamically favorable and occurs via the formation of an acyclic NNNCN-intermediate. The second step of formal cycloaddition, i.e. cyclization, is rate limiting. Despite the fact that the substitution of CNMe by the N3− ligand is slightly thermodynamically favorable, we were unable to find the paths on the potential energy surface for the hypothetical cycloaddition between uncomplexed isocyanide and palladium-bound azide. Thus, we believe that the experimentally observed palladium tetrazolate complexes are, in fact, generated from the negatively charged uncomplexed azide and positively charged metal-bound isocyanide species, and this reaction path is favorable from the viewpoint of Coulomb attraction. However, the alternative mechanism that involves complexed azide and uncomplexed RNCs (Scheme 2, route E) can be feasible at other metal centers and the previously obtained synthetic results collaterally confirm this assumption. Previously published data8 for the integration between azide complexes and isocyanides may potentially require reconsideration to address the possibility that the interaction proceeds through an alternative mechanism involving the substitution of an azide ligand with isocyanide followed by cycloaddition.
Experimental section
Materials and instrumentation
Solvents and reagents were obtained from commercial sources and used as received, apart from chloroform, which was purified by the conventional distillation over calcium chloride. Complexes 1–2 were prepared by the known procedures.20 C, H, and N elemental analyses were carried out on a Euro EA 3028HT CHNS/O analyzer. Mass spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source and MeCN was used as a solvent. The instrument was operated both at positive and negative ion modes using the m/z range of 50–3000. The capillary voltage of the ion source was set at −4500 V (ESI+) or 3500 V (ESI−) and the capillary exit at ±(70–150) V. The nebulizer gas pressure was 0.4 bar and drying gas flow 4.0 L min−1. The most intensive peak in the isotopic pattern is reported. Infrared spectra (4000–400 cm−1) were recorded on a Shimadzu FTIR 8400S instrument in CHCl3 solutions. 1D (1H, 13C{1H}, DEPT) NMR spectra were acquired on a Bruker Avance 400 spectrometer at ambient temperature.Synthetic work
:Et2O (1:1, v/v) mixture at RT.
4 (80 mg, 67%). Anal. calcd for C41H39N4ClP2Pd: C, 61.21; H, 4.97; N, 7.08. Found: C, 61.44; H, 4.98; N, 7.12. HRESI+-MS, m/z: calcd for C41H39N4P2Pd+ 755.1679, found 755.1652 [M − Cl]+. 1H NMR (400.13 MHz, CDCl3, δ): 1.21 (s, 9H, CH3), 7.32 (t, 3JH,H = 7.3 Hz, 12H), 7.40 (t, 3JH,H = 7.3 Hz, 6H), 7.47 (q, 3JH,H = 6.2 Hz, 12H). 13C{1H} NMR (100.61 MHz, CDCl3, δ): 30.2 (CH3), 58.0 (![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) (CH3)3), 128.4 (t, JC,P = 5 Hz, CH, C from PPh3), 129.8 (t, JC,P = 24 Hz, C, C from PPh3), 130.6 (CH, C from PPh3), 134.5 (t, JC,P = 6 Hz, CH, C from PPh3), 151.5 (C, C from the tetrazolate ring).
(CH3)3), 128.4 (t, JC,P = 5 Hz, CH, C from PPh3), 129.8 (t, JC,P = 24 Hz, C, C from PPh3), 130.6 (CH, C from PPh3), 134.5 (t, JC,P = 6 Hz, CH, C from PPh3), 151.5 (C, C from the tetrazolate ring).
5 (88 mg, 72%). Anal. calcd. for C45H39N4ClP2Pd: C, 64.37; H, 4.68; N, 6.67. Found: C, 63.42; H, 4.64; N, 6.70. HRESI+-MS, m/z: calcd for C43H39N4P2Pd+ 803.1679, found 803.1661 [M − Cl]+. 1H NMR (400.13 MHz, CDCl3, δ): 2.22 (s, 6H, CH3), 6.96 (d, 3JH,H = 7.3 Hz, 12H), 7.12–7.72 (m, 32H). 13C{1H} NMR (100.61 MHz, CDCl3, δ): 21.0 (CH3), 128.0–136.2 (C from Xyl and PPh3), 161.9 (C, C from the tetrazolate ring).
X-ray structure determination
The crystal of 4 was immersed in cryo-oil, mounted in a MiTeGen loop, and measured at a temperature of 123 K. The X-ray diffraction data were collected on a Bruker Axs KappaCCD diffractometer using Mo Kα radiation (λ = 0.71073 Å). The Denzo-Scalepack21 program package was used for cell refinement and data reduction. The structure was solved by the charge flipping method using the Superflip22 program with the Olex223 graphical user interface. A semi-empirical absorption correction (SADABS)24 was applied to the data. Structural refinement was carried out using SHELXL-97.25 Hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–1.00 and Uiso = 1.2–1.5 Ueq (parent atom). The crystallographic details are summarized in Table S1 (see ESI†). CCDC 1414621.Computational details
The full geometry optimization of all structures and transition states was carried out at the DFT/HF hybrid level of theory using Becke's three-parameter hybrid exchange functional in combination with the gradient-corrected correlation functional of Lee, Yang and Parr (B3LYP)26 with the help of the Gaussian-0927 program package. No symmetry operations have been applied. The geometry optimization was carried out using a quasi-relativistic Stuttgart pseudopotential that described 28 core electrons and the appropriate contracted basis sets28 for the palladium atoms and the 6-31G(d) basis set for other atoms. Single-point calculations were performed on the basis of the equilibrium geometries found by using the 6-311+G(d,p) basis set for nonmetal atoms. As was shown previously,17a,b,d–f this approach is sufficiently accurate for the description of CAs to the CN triple bond providing results close to those obtained by such methods as MP2, MP4, CCSD(T), CBS-Q, and G3B3.
The Hessian matrix was calculated analytically for the optimized structures in order to prove the location of correct minima (no imaginary frequencies) or saddle points (only one imaginary frequency), and to estimate the thermodynamic parameters, the latter being calculated at 25 °C. The nature of all transition states was investigated by the analysis of vectors associated with the imaginary frequency and by the calculations of the intrinsic reaction coordinates (IRC) using the Gonzalez–Schlegel method.29
The total energies corrected for solvent effects (Es) were estimated at the single-point calculations on the basis of gas-phase geometries at the CPCM-B3LYP/6-311+G(d,p)//gas-B3LYP/6-31G(d) level of theory using the polarizable continuum model in the CPCM version30,31 with CH2Cl2 as a solvent. The UAKS model was applied for the molecular cavity and dispersion, cavitation, and repulsion terms were taken into account. The entropic term in CH2Cl2 solution (Ss) was calculated according to the procedure described by Wertz32 and Cooper and Ziegler33 (see eqn (S1)–(S4)), as well as enthalpies and Gibbs free energies in solution (Hs and Gs) using the expressions (S5) and (S6) (all equations are provided in the ESI†).
Acknowledgements
Theoretical part of this work was conducted within Russian Science Foundation project (14-43-00017), whereas the synthetic part was done under Russian Foundation for Basic Research project (14-03-31204 mol_a) and the authors are grateful to both foundations for support of their studies. MAK and ASN acknowledge the financial support from Saint Petersburg State University (travel grant 12.42.1262.2014 and postdoctoral fellowship 12.50.1190.2014, correspondingly). Physicochemical studies were performed at the Center for Magnetic Resonance and Center for Chemical Analysis and Materials Research (both belong to Saint Petersburg State University).References
- (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (c) S. T. Liddle, I. S. Edworthy and P. L. Arnold, Chem. Soc. Rev., 2007, 36, 1732–1744 RSC; (d) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC; (e) P. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS.
- (a) R. A. Michelin, A. J. L. Pombeiro and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2001, 218, 75–112 CrossRef CAS; (b) R. S. Chay and K. V. Luzyanin, Inorg. Chim. Acta, 2012, 380, 322–327 CrossRef CAS; (c) V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS; (d) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed.
- (a) W. P. Fehlhammer, K. Bartel and W. Petri, J. Organomet. Chem., 1975, 87, C34–C36 CrossRef CAS; (b) K. R. Grundy and W. R. Roper, J. Organomet. Chem., 1975, 91, C61–C64 CrossRef CAS.
- Y. Fuchita, K. Hidaka, S. Morinaga and K. Hiraki, Bull. Chem. Soc. Jpn., 1981, 54, 800 CrossRef CAS.
- K. V. Luzyanin, A. G. Tskhovrebov, M. F. C. Guedes da Silva, M. Haukka, A. J. L. Pombeiro and V. Y. Kukushkin, Chem. – Eur. J., 2009, 15, 5969–5978 CrossRef CAS PubMed.
- W. P. Fehlhammer and W. Beck, Z. Anorg. Allg. Chem., 2015, 641, 1599–1678 CrossRef CAS.
- (a) H.-W. Frühauf, Chem. Rev., 1997, 97, 523–596 CrossRef; (b) W. Beck, K. Burger and W. P. Fehlhammer, Chem. Ber., 1971, 104, 1816–1825 CrossRef CAS.
- (a) Y.-J. Kim, D.-H. Kim, J.-Y. Lee and S.-W. Lee, J. Organomet. Chem., 1997, 538, 189–197 CrossRef CAS; (b) Y.-J. Kim, Y.-S. Kwak and S.-W. Lee, J. Organomet. Chem., 2000, 603, 152–160 CrossRef CAS; (c) M. Wehlan, R. Thiel, J. Fuchs, W. Beck and W. P. Felhammer, J. Organomet. Chem., 2000, 613, 159 CrossRef CAS; (d) Y.-J. Kim, Y.-S. Joo, J.-T. Han, W. S. Han and S.-W. Lee, Dalton Trans., 2002, 3611 RSC; (e) Y.-J. Kim, Y.-S. Kwak, Y.-S. Joo and S.-W. Lee, Dalton Trans., 2002, 144–151 RSC; (f) Y.-J. Kim, J.-T. Han, S. Kang, W. S. Han and S. W. Lee, Dalton Trans., 2003, 3357–3364 RSC; (g) W. S. Han, H. K. Lee, Y.-J. Kim and S. W. Lee, Acta Crystallogr., 2004, E60, m1813–m1814 Search PubMed; (h) Y.-J. Kim, X. Chang, J.-T. Han, M. S. Lim and S. W. Lee, Dalton Trans., 2004, 3699–3708 RSC; (i) Y.-J. Kim, S.-H. Lee, S.-H. Lee, S. I. Jeon, M. S. Lim and S. W. Lee, Inorg. Chim. Acta, 2005, 358, 650–658 CrossRef CAS; (j) H. S. Huh, Y. K. Lee and S. W. Lee, J. Mol. Struct., 2006, 789, 209 CrossRef CAS; (k) Y.-J. Kim, K.-E. Lee, H.-T. Jeon, H. S. Huh and S. W. Lee, Inorg. Chim. Acta, 2008, 361, 2159–2165 CrossRef CAS.
- (a) X. Chang, K.-E. Lee, Y.-J. Kim and S. W. Lee, Inorg. Chim. Acta, 2006, 359, 4436–4440 CrossRef CAS; (b) W. S. Kim, Y.-J. Kim and S. W. Lee, Bull. Korean Chem. Soc., 2002, 23, 1177–1180 CrossRef CAS.
- W. F. Gabrielli, S. D. Nogai, J. M. McKenzie, S. Cronje and H. G. Raubenheimer, New J. Chem., 2009, 33, 2208–2218 RSC.
- (a) W. Beck and W. P. Fehlhammer, Angew. Chem., Int. Ed., 1967, 6, 169–170 CrossRef CAS; (b) W. P. Fehlhammer and L. F. Dahl, J. Am. Chem. Soc., 1972, 94, 3370–3377 CrossRef CAS.
- R. Jothibasu and H. V. Huynh, Organometallics, 2009, 28, 2505–2511 CrossRef CAS.
- P. M. Treichel, W. J. Knebel and R. W. Hess, J. Am. Chem. Soc., 1971, 93, 5424–5433 CrossRef CAS.
- R. Sustmann, Tetrahedron Lett., 1971, 12, 2717–2720 CrossRef.
- N. A. Bokach, M. L. Kuznetsov and V. Y. Kukushkin, Coord. Chem. Rev., 2011, 255, 2946–2967 CrossRef CAS.
- (a) A. S. Kritchenkov, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, Inorg. Chem., 2012, 51, 11971 CrossRef CAS PubMed; (b) A. L. Mindich, N. A. Bokach, F. M. Dolgushin, M. Haukka, L. A. Lisitsyn, A. P. Zhdanov, K. Y. Zhizhin, S. A. Miltsov, N. T. Kuznetsov and V. Y. Kukushkin, Organometallics, 2012, 31, 1716–1724 CrossRef CAS; (c) A. L. Mindich, N. A. Bokach, M. L. Kuznetsov, G. L. Starova, A. P. Zhdanov, K. Y. Zhizhin, S. A. Miltsov, N. T. Kuznetsov and V. Y. Kukushkin, Organometallics, 2013, 32, 6576–6586 CrossRef CAS; (d) A. S. Kritchenkov, N. A. Bokach, M. L. Kuznetsov, F. M. Dolgushin, T. Q. Tung, A. P. Molchanov and V. Y. Kukushkin, Organometallics, 2012, 31, 687–699 CrossRef CAS.
- (a) M. L. Kuznetsov, V. Y. Kukushkin and A. J. L. Pombeiro, J. Org. Chem., 2010, 75, 1474–1490 CrossRef CAS PubMed; (b) N. A. Bokach, M. L. Kuznetsov, M. Haukka, V. I. Ovcharenko, E. V. Tretyakov and V. Y. Kukushkin, Organometallics, 2009, 28, 1406–1413 CrossRef CAS; (c) M. L. Kuznetsov, V. Y. Kukushkin and A. J. L. Pombeiro, Dalton Trans., 2008, 1312–1322 RSC; (d) M. L. Kuznetsov, A. A. Nazarov, L. V. Kozlova and V. Y. Kukushkin, J. Org. Chem., 2007, 72, 4475–4477 CrossRef CAS PubMed; (e) M. L. Kuznetsov and V. Y. Kukushkin, J. Org. Chem., 2006, 71, 582–592 CrossRef CAS PubMed; (f) M. L. Kuznetsov, V. Y. Kukushkin, A. I. Dement'ev and A. J. L. Pombeiro, J. Phys. Chem. A, 2003, 107, 6108–6120 CrossRef CAS.
- A. S. Kritchenkov, K. V. Luzyanin, N. A. Bokach, M. L. Kuznetsov, V. V. Gurzhiy and V. Y. Kukushkin, Organometallics, 2013, 32, 1979–1987 CrossRef CAS.
- (a) A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 2874–2888 CrossRef CAS PubMed; (b) A. S. Novikov, A. I. Dement'ev and Y. N. Medvedev, Russ. J. Inorg. Chem., 2013, 58, 320–330 CrossRef CAS; (c) A. S. Novikov and M. L. Kuznetsov, Inorg. Chim. Acta, 2012, 380, 78–89 CrossRef CAS.
- R. A. Michelin, L. Zanotto, D. Braga, P. Sabatino and R. J. Angelici, Inorg. Chem., 1988, 27, 85–92 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods Enzymol., ed. J. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SADABS – Bruker AXS scaling and absorption correction, Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, B37, 785 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS; (c) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- D. H. Wertz, J. Am. Chem. Soc., 1980, 102, 5316–5322 CrossRef CAS.
- J. Cooper and T. Ziegler, Inorg. Chem., 2002, 41, 6614–6622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data, calculated total energies, enthalpies, Gibbs free energies and entropies in the gas phase and CH2Cl2 solution, calculated total activation and reaction energies, enthalpies and Gibbs free energies of activation and reaction for the gas phase and CH2Cl2 solution, Cartesian atomic coordinates of the calculated equilibrium structures. CCDC 1414621. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02564h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |