
Dereplication, sequencing and identification of peptidic natural products: from genome mining to peptidogenomics to spectral networks
Hosein
Mohimani
and
Pavel A.
Pevzner
*
Department of Computer Science and Engineering, University of California, San Diego, USA. E-mail: ppevzner@ucsd.edu
First published on 26th October 2015
Abstract
Covering: 2000 to 2015
While recent breakthroughs in the discovery of peptide antibiotics and other Peptidic Natural Products (PNPs) raise a challenge for developing new algorithms for their analyses, the computational technologies for high-throughput PNP discovery are still lacking. We discuss the computational bottlenecks in analyzing PNPs and review recent advances in genome mining, peptidogenomics, and spectral networks that are now enabling the discovery of new PNPs via mass spectrometry. We further describe the connections between these advances and the new generation of software tools for PNP dereplication, de novo sequencing, and identification.
 Hosein Mohimani | Hosein Mohimani is a project scientist in the Computer Science and Engineering department, University of California, San Diego. Dr Mohimani is leading the antibiotic sequencing research area at NIH Center for Computational Mass Spectrometry. He holds a Ph.D. (2013) from UCSD. |
 Pavel A. Pevzner | Pavel Pevzner is a Ronald R. Taylor Distinguished Professor of Computer Science and Engineering and Director of the NIH Center for Computational Mass Spectrometry at University of California, San Diego. He holds a Ph.D. (1988) from Moscow Institute of Physics and Technology, Russia. He was named Howard Hughes Medical Institute Professor in 2006. He was elected as the ACM Fellow (2010) for “contribution to algorithms for genome rearrangements, DNA sequencing, and proteomics” and ISCB Fellow (2012). He was awarded a Honoris Causa (2011) from Simon Fraser University in Vancouver. Dr Pevzner has authored the textbooks, “Computational Molecular Biology: An Algorithmic Approach” in 2000, “Introduction to Bioinformatics Algorithms” in 2004 (jointly with Neal Jones) and “Bioinformatics Algorithms: an Active Learning Approach” in 2014 (jointly with Phillip Compeau). |
1 Introduction
The golden age of antibiotics, that started in the 1940s, was followed by a decline in the pace of antibiotics discovery in the 1990s.1 However, antibiotics and other natural products (including immunosuppressive, antiproliferative, herbicidal, insecticidal, fungicidal and antiparasitic drugs) are again at the center of attention as exemplified by the recent discovery of teixobactin.2,3 The recent launch of the Global Natural Products Social (GNPS) Molecular Networking project4 (the first large community project for natural product discovery) brought together over a hundred laboratories that have already generated over a billion mass spectra of natural products. While these spectra represent a gold mine for future antibiotics discovery (over 70 million of them are publicly available), their interpretation remains a challenging computational problem.While the first computational methods for analyzing mass spectra of small molecules were developed in the 1960s,5–8 three decades before their proteomics counterparts,9,10 computational mass spectrometry of small molecules is often viewed as a more complex (and less mature!) field as compared to computational proteomics.11,12 See ref. 12–16 for recent reviews of computational approaches to analyzing small molecules. Depending on their building blocks, natural products are classified into a variety of chemical classes that include Peptidic Natural Products (PNPs), the focus of this review. Starting from penicillin, PNPs have an unparalleled track record in pharmacology: many antibiotics, antiviral and antitumor agents, immunosuppressors, and toxins are PNPs.
While recent breakthroughs in PNP discovery2,17,18 raised a challenge for developing new algorithms for dereplication, de novo sequencing and identification of PNPs, computational technologies for high-throughput PNP discovery are still lacking. The traditional process of PNP discovery is to elucidate the structure of the compound by chemical assays (such as nuclear magnetic resonance) and to associate the chemical compound to its biosynthetic gene cluster by genome manipulations. This process is time-intensive, laborious, and requires large amounts of highly purified material. Moreover, rather than discovering novel PNPs, it often rediscovers known PNPs, resulting in wasted efforts.
Recently, mass spectrometry (MS) has become a cheap, fast, and reliable complementary approach to traditional PNP discovery techniques.19,20 However, compared to traditional applications of MS in proteomics, the application of MS for PNP discovery faces additional computational challenges due to a higher complexity of the compounds and unusual fragmentation patterns. Some of these challenges are now addressed through genome mining, peptidogenomics, and spectral networks:
• Genome mining. The sequencing of many bacterial and fungal genomes in the last decade opened an era of genome mining for PNP discovery. Genome mining refers to using information about the biosynthetic genes (responsible for synthesizing a PNP) to infer information about the PNP itself. The discovery of coelichelin in Streptomyces coelicolor was one of the first successes of genome mining21,22 that was followed by the characterization of many PNPs from sequenced genomes.
• Peptidogenomics. Given a mass spectrum and a peptide database, peptide identification refers to finding a peptide in the database (or its variant) that generated the given spectrum. While peptide databases in traditional proteomics consist of known peptides, peptide databases in peptidogenomics are often dominated by putative peptides derived via genome mining. Since many PNPs are not directly encoded in genomes, genome mining often fails to generate a database of putative PNPs that contains the exact amino acid sequence of a PNP corresponding to a given spectrum. Instead, it produces a database containing an error-prone template that makes matching spectra against such a template difficult. Therefore, popular proteomics tools such as Sequest9 and Mascot10 fail to identify PNPs. Also, identification of spectra derived from PNPs is more difficult than traditional peptide identification in proteomics because many PNPs are non-linear peptides with extensive modifications that generate complex spectra (the standard proteomics tools fail to identify non-linear peptides).
• Spectral networks. Bandeira et al.23 introduced the concept of spectral networks (also known as molecular networks18) that reveal spectra of related compounds (without knowing what these compounds are) using spectral alignment algorithms.24,25 Nodes in the spectral networks correspond to spectra while edges connect spectral pairs, i.e., pairs of spectra that are generated from related peptides (e.g., peptides differing by a single mutation or a modification). Spectral networks enable the discovery of novel variants of known PNPs as well as novel PNP families. Thus, since most PNPs form families of related peptides,18 spectral networks are ideally suited for analyzing PNPs.
PNPs are produced by two types of biosynthetic machineries: Non-Ribosomal Peptide synthetase (NRP synthetase)26,27 and Ribosomally synthesized and Posttranslationally modified Peptide synthetase (RiPP synthetase).28,29 NRP and RiPP synthetases produce Non-Ribosomal Peptides (NRPs) and Ribosomally synthesized and Posttranslationally modified Peptides (RiPPs), respectively. NRPs are widely distributed and biomedically important natural products that are not directly inscribed in genomes but instead are encoded by NRP synthetases using the non-ribosomal code.30 In addition to standard amino acids, NRPs often include non-proteinogenic amino acids such as ornithine. Known NRPs include hundreds of non-proteinogenic building blocks and some NRPs like kutznerides31 are built entirely from non-proteinogenic amino acids. Since the non-ribosomal code remains poorly understood, an accurate prediction of PNPs from NRP synthetases remains challenging.
While RiPPs are encoded in the genome, the genes encoding RiPPs are often short making it difficult to annotate them (short genes often evade gene prediction methods32,33). Moreover, RiPPs often have many unusual Post Translational Modifications (PTMs) making it difficult to identify them via MS. Heavily modified peptides with more than two blind modifications often evade identification algorithms such as InsPecT25 and MODa34 designed for discovery of unexpected PTMs.
Analysis of over 1000 bacterial genomes from the Joint Genome Institute (JGI) database revealed that 71% of them harbor at least one RiPP protein family (Pfam) domain and 69% harbor at least one NRP synthetase Pfam domain.17 Recent analysis of 830 Actinobacteria genomes revealed that Actinobacteria encode thousands of potential drug leads.35 These and other studies18 suggest that we only saw a tip of the iceberg with respect to PNP discovery and raise the challenge of developing new methods for PNP discovery.
Understanding how PNP biosynthetic machineries work is a prerequisite to genome mining and peptidogenomics that involves two steps; predicting the candidate gene clusters responsible for the synthesis of a PNP and connecting them to their chemical products by MS. However, connecting biosynthetic gene clusters to their products is not a trivial task since the rules defining how a gene cluster specifies its products remain poorly understood. For example, the existing tools for predicting NRPs from NRP synthetases remain error-prone. The transition from a gene cluster to its product becomes particularly difficult in the case of modifications involved in the maturation of PNPs. For example, the gene cluster for coelichelin (NRP synthetase) was elucidated in 2000,21 but coelichelin itself (NRP) was sequenced only in 2005.22
Below we review recent advances in genome mining, peptidogenomics, and spectral networks (Section 2) and further describe PNP dereplication (Section 3), PNP sequencing (Section 4), and PNP identification (Section 5). We note that, as opposed to dereplication (that reveals known PNPs or their variants), PNP sequencing and identification may reveal previously unknown PNPs. Fig. 1 illustrates various approaches to PNP discovery.
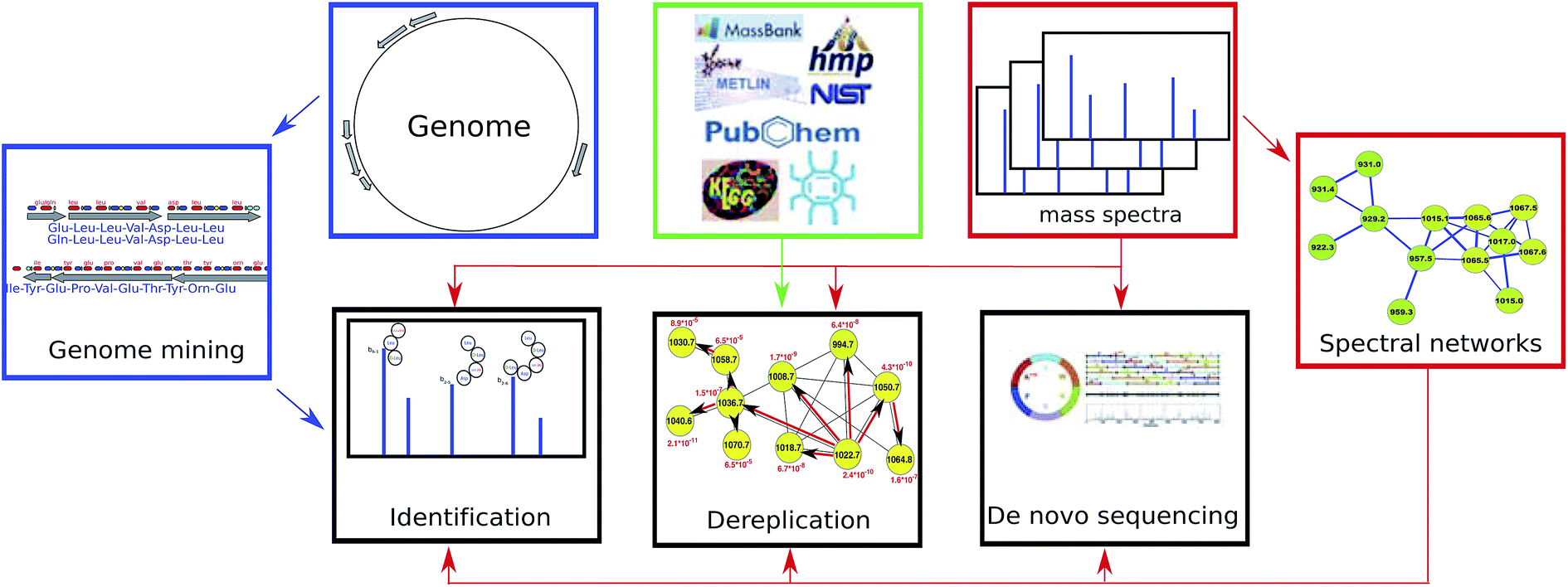 | ||
| Fig. 1 Three computational approaches to PNP discovery. | ||
2 Genome mining, peptidogenomics, and spectral networks
2.1 Genome mining for PNPs
Genome mining tools for the identification of NRP synthetase gene clusters and the prediction of the NRPs they produce include ClustScan,36 NP.searcher,37 NRPSpredictor,38 NRPSpredictor2,39 and antiSMASH.40–42 For polyketide synthetase gene cluster predictors see ref. 43–47. Fig. 2 illustrates how NRP genome mining tools work. Medema et al.48 recently developed the Pep2Path genome mining tool that works for both NRPs (NRP2Path) and RiPPs (RiPP2Path) by matching peptide sequence tags in the spectra against biosynthetic gene clusters that have the highest likelihood of generating PNPs containing these tags.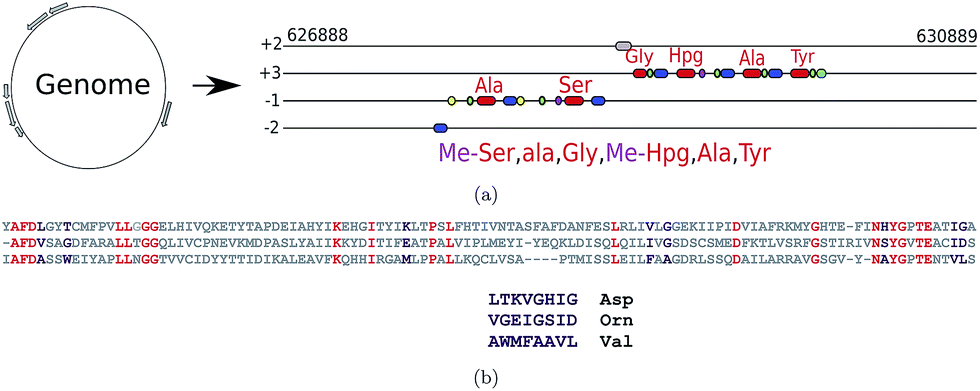 | ||
| Fig. 2 (a) Predicting NRPs based on NRP synthetase analysis using tools such as NRPSpredictor2 (ref. 39) and antiSMASH.40 The following domains are shown: A-domains (red), PCP-domains (green), C-domains (blue), methylation domains (yellow), and thioester domains (purple). Note that different modules of the same NRP synthetase can appear in different frames. (b) Extracting signature sequences (non-ribosomal code) from A-domains (only short segments of the A-domains are shown). Various A-domains have conserved residues (shown in red) that enable their accurate multiple alignment using profile HMMs. The non-ribosomal code postulates that certain amino acids in the resulting multiple alignment (shown in purple) define a single amino acid in the NRP loaded by this domain. The three A-domains shown here define 8 amino acid signatures LTKVGHIG, VGEIGSID, and WMFAAVL corresponding to the amino acids Asp, Orn, and Val, respectively. The 8 amino acid signatures shown here represent a simplified representation of the non-ribosomal code, e.g., NRPSpredictor2 uses longer signatures to predict amino acids for each A-domain. | ||
NRP synthetases are formed by an array of distinct modular sections, each of which is responsible for incorporation or modification of a single amino acid into the final NRP. A minimum of three domains are required for each NRP synthetase module, the adenylation domain (A-domain), peptidyl carrier domain (PCP-domain) and condensation domain (C-domain). The A-domain is responsible for picking the specific amino acids that will be incorporated into the NRP. Hundreds of different A-domain specificities have been classified, each one recruiting a specific amino acid. This allows us to determine the sequence of the putative NRP by looking at the order of A-domains along the assembly line and assigning a specific amino acid to each A-domain using the non-ribosomal code. However, since the non-ribosomal code is still poorly understood, the tools for defining specificities of A-domains remain error-prone. These tools often use profile Hidden Markov Models (HMMs) to align conservative amino acids within each A-domain (red amino acids in Fig. 2(b)) against previously analyzed A-domains. The constructed alignment reveals variable amino acids within A-domains (purple amino acids in Fig. 2(b)) that define the non-ribosomal code. The genome mining tools further use various machine learning techniques to derive the amino acid in the NRP defined by the non-ribosomal code.
RiPPs are classified into more than 20 classes (such as lanthibiotics, thiopeptides, cyanobactins, lasso peptides, and many others) based on structural and biosynthetic commonality.49 Various software tools for RiPP genome mining have been reviewed in ref. 50. BAGEL, a genome mining tool for bacteriocins, revealed 150 putative lanthipeptide gene clusters.51,52 ThioFinder, a genome mining tool for thiopeptides, predicted 53 novel thiopeptide producing gene clusters.53 Recent genome mining studies predicted 79 lasso peptides54 and 27 cyanobactin-producing Anabaena strains.55 The development of RiPP genome mining tools is tied to construction of databases of known RiPPs, such as Bactibase, a database of 177 bacteriocins56 or a bacteriocin database51,52 consisting of 483 bacteriocins (236 class I, 160 class II and 93 class III as of August 2015). Other examples include Thiobase, a database of 39 thiopeptides,53 and MIBiG, a natural product structure and biosynthetic gene cluster repository with over 169 RiPPs from different classes.42 Availability of these databases for diverse RiPP classes speeds up the development of novel machine learning techniques aimed at genome mining for RiPPs.50
AntiSMASH is one of the most popular genome mining tools for analyzing both NRPs and RiPPs as well as polyketides. The antiSMASH pipeline includes the following steps: (i) genes are extracted or predicted from the genome using Glimmer3,32 (ii) biosynthetic gene clusters are identified using profile HMMs, (iii) biosynthetic gene clusters are annotated, and (iv) the core chemical structure of natural products are predicted based on the annotated gene clusters. Optionally, comparative analysis of the biosynthetic gene clusters can be done using ClusterBlast.40
2.2 Peptidogenomics of PNPs
The key difficulties in peptidogenomics are that (i) many PNPs are non-linear peptides, (ii) many PNPs (all NRPs) are not directly encoded in genomes, (iii) even when a PNP is encoded in a genome (all RiPPs), they often have many modifications making it difficult to identify them using standard MS/MS searches, and (iv) many PNPs are encoded in the alphabet of 100 s building blocks rather than in the alphabet of 20 proteinogenic amino acids. Also, many PNPs fragment poorly due to multiple complex modifications and multicyclic structures. For example, spectra of RiPPs often feature very few peaks making it nearly impossible to identify them using conventional MS/MS database search tools.Kersten et al., 2011 (ref. 17) discovered many novel PNPs using a manual peptidogenomic approach for connecting PNPs to their biosynthetic genes and matching them against mass spectra. However, the manual peptidogenomic approach to PNP discovery, while useful,57 is somewhat limited in analyzing large spectral datasets (such as LC-MS/MS datasets from bacterial extracts) and complex patterns of modifications. Moreover, this approach relies on identifying long peptide sequence tags (4–5 amino acids) to reduce the search space.48 Such long tags are often not available for multicyclic peptides such as lanthipeptides or for NRPs with non-standard amino acids. Also, since the manual approach does not provide estimates of statistical significance (a pre-requisite for analyzing large spectral datasets) an automated peptidogenomics software tool is needed.
Peptidogenomics is based on the comparison of experimental spectra with the theoretical spectrum of a PNP. Various bond disconnection algorithms58–64 generate a list of bonds between atoms in a compound (excluding hydrogen) and assign them a breakage score based on the likelihood of each bond being disconnected. The theoretical spectrum is constructed from masses and breakage scores of all substructures resulting from bond disconnections. Tools such as MetFrag59 attempt to explain the peaks in the experimental spectrum using the likely substructures formed by disconnecting some bonds. Alternative machine learning approaches use large collections of MS/MS spectra for learning the rules governing the MS/MS fragmentation process.65–67 Alternative approaches to bond disconnection algorithms have also been suggested.65–68
Theoretical spectra of PNPs are formed by disconnecting only amide bonds (rather than all bonds)69,70 (see Fig. 3). Since the number of fragmented substructures grow quadratically with the PNP length (under the assumption that at most two amide bonds are disconnected), theoretical spectra of PNPs have a large number of masses making it difficult to analyze them since only a fraction of these masses have counterparts in the experimental spectra. In spite of this complication, some studies used general metabolite dereplication tools to successfully dereplicate PNPs.35
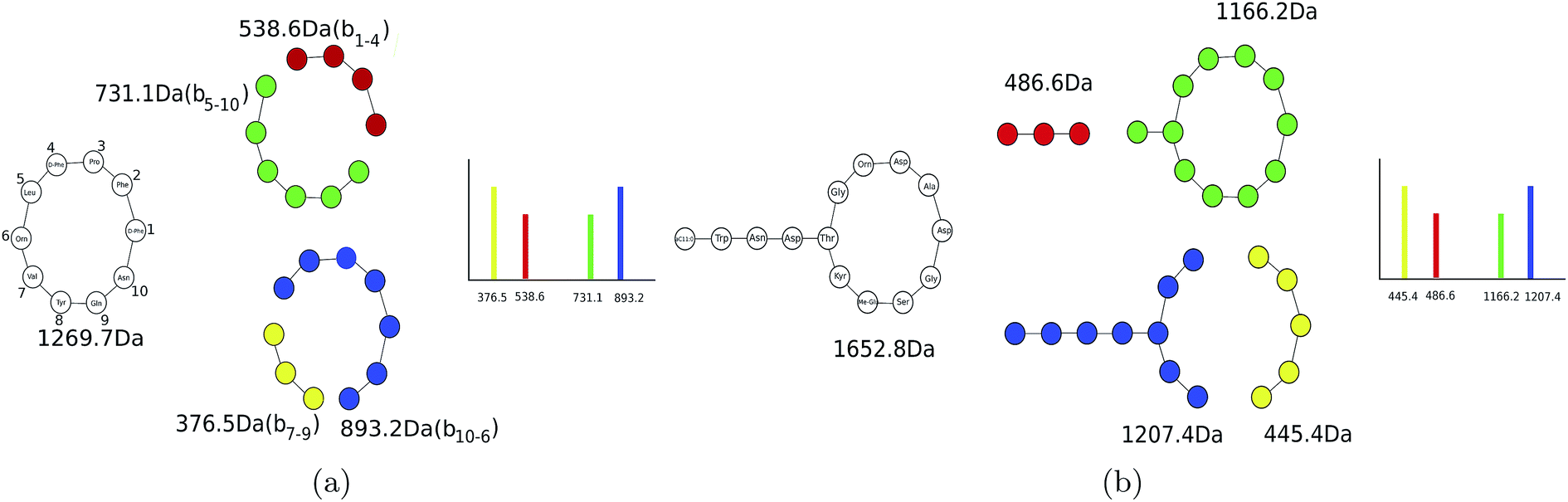 | ||
| Fig. 3 (a) Generating the theoretical spectrum of cyclic (tyrocidine) and (b) branch cyclic (daptomycin) peptides. Only four out of 9 × 10 = 90 (9 × 10 + 4 × 2 = 98) theoretical peaks in tyrocidine (daptomycin) are shown. For a cyclic PNP of length n, the theoretical spectrum contains n(n − 1) masses. For a branch cyclic PNP with a cycle length n and a branch length m, the theoretical spectrum contains n(n − 1) + 2m masses. | ||
A Peptide-Spectrum Match (PSM) is a pair of a peptide and a spectrum with the same precursor mass (up to an error δ). In the context of PNP discovery, a PSM score is often defined as the number of peaks shared between a theoretical spectrum and an experimental spectrum. Given a spectrum, a peptide that forms a PSM with the highest score against this spectrum (among all peptides in a peptide database) is reported as a potential annotation of the spectrum.
It is well known in the context of traditional proteomics that PSM scores often poorly correlate with statistical significance of PSMs such as p-values.71 This observation is greatly amplified for non-linear peptides since scoring PSMs formed by non-linear peptides is currently more primitive than scoring PSMs formed by linear peptides due to the lack of a large learning sample of PSMs formed by non-linear peptides.
To address this challenge, Ibrahim et al., 2013 (ref. 70) proposed additional statistical measures to distinguish between correct and erroneous PSMs formed by PNPs in their iSNAP approach. Mohimani et al.,72 developed the MS-DPR algorithm for computing p-values of PSMs formed by arbitrary PNPs. MS-DPR addresses the problem of deciding whether a given spectrum was generated by a linear, cyclic, or branch cyclic peptide since it enables evaluation of statistical significance of peptides with diverse structures72 (see Fig. 4).
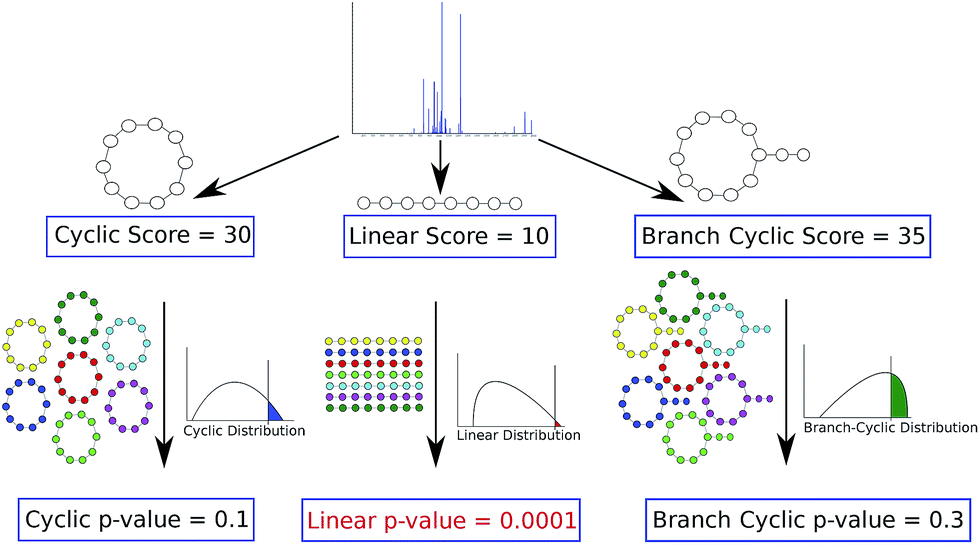 | ||
| Fig. 4 Deciding whether a peptide that produced a spectrum is linear, cyclic or branch cyclic. Given a spectrum, MS-DPR72 considers various structure assumptions for a peptide that generated the spectrum (e.g. linear, or cyclic, or branch cyclic), and derives a p-value of PSMs resulting from each such assumption. For each structure, MS-DPR explores many putative amino acid sequences (shown by different colors) to estimate the p-value. If one of the structures results in a small p-value (e.g. a linear structure with a p-value of 0.0001 shown in red), that structure is accepted as the most likely structure for a given spectrum. Note that even though the linear peptide in this example has the lowest score, it is the most statistically significant among the three structures. The figure is reproduced from ref. 72 by permission from ACS publications. | ||
2.3 Spectral networks of PNPs
Spectral networks allow one to enlarge the set of identified PNPs (and sometimes get rid of incorrectly identified PNPs) by analyzing multiple spectra to simultaneously dereplicate, sequence, or identify related unknown peptides. The advantage of this approach (as compared to analyzing individual spectra) is that finding peptides that simultaneously explain all spectra in a spectral network may result in more accurate spectral interpretations. Thus, an individual PSM deemed statistically insignificant may become reliable in the context of multiple related PSMs revealed by a spectral network (and vice versa). Since most PNPs form families of related peptides, spectral networks can be used to reveal relationships between different spectra without knowing the amino acid sequences corresponding to these spectra.Given a set of peptides P1, … Pm, their peptide network is a graph with nodes P1, … Pm, and edges connecting two peptides if they differ by a single amino acid modification. Fig. 5(a) shows the peptide network for nine variants of tyrocidine, a family of NRPs from Bacillus brevis.73 For example, peptide 1 (tyrocidine B1) in this network (red node) is connected to four peptides differing from tyrocidine B1 by a single modification: tyrocidine A1 (peptide 2), tyrocidine B (peptide 5), tyrocidine C1 (peptide 8), and a previously unreported peptide with a mass of 1338.7 (peptide 9). However, it is not connected to peptides 3, 4, 6 and 7 since they differ from peptide 1 by multiple modifications. Six of these nine tyrocidines (1, 2, 3, 5, 7, 8) are contained in the database of putative NRPs generated by NRPSpredictor2 (without modifications) and three more differ from these variants by one or two modifications/mutations.
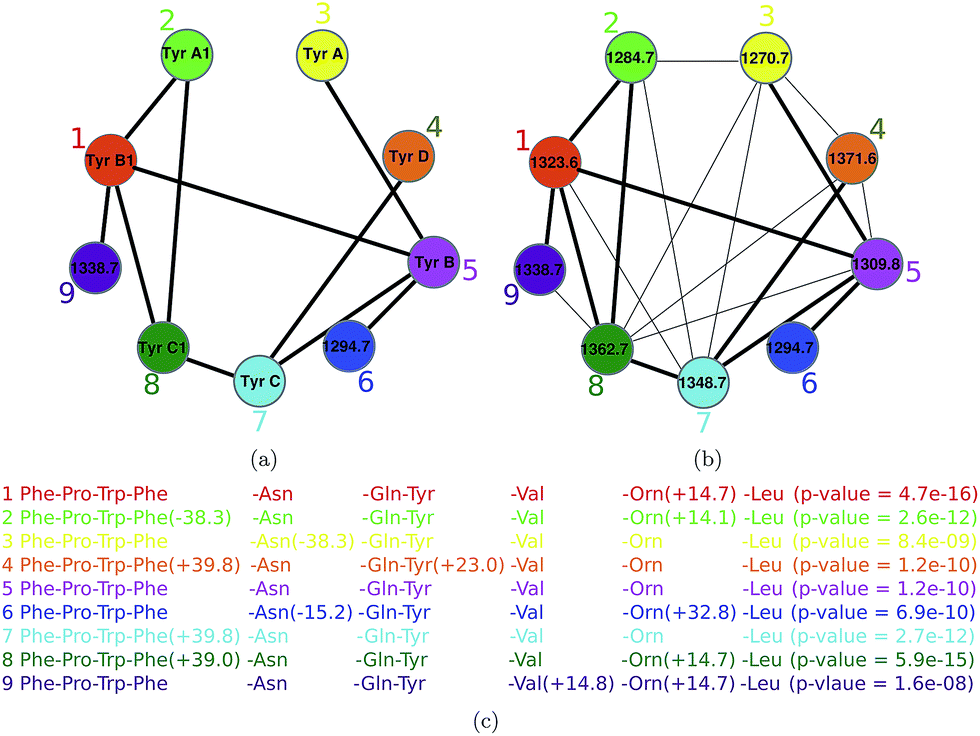 | ||
| Fig. 5 The peptide network (a) and the spectral network (b) of tyrocidines.74 The numbers within nodes represent precursor masses. Edges in the peptide network connect two peptides if they differ by a single amino acid modification. Shared edges between peptide and spectral networks are shown by thick lines. For example, peptide 1 (tyrocidine B1) in this network (red node) is connected to four peptides differing from tyrocidine B1 by a single modification: tyrocidine A1 (peptide 2), tyrocidine B (peptide 5), tyrocidine C1 (peptide 8), and a previously unreported peptide with a mass of 1338.7 (peptide 9). However, it is not connected to peptides 3, 4, 6 and 7 since they differ from peptide 1 by multiple modifications. In part (c), annotation of each node in the spectral network is shown. The spectral network revealed two novel tyrocidine variants at masses 1294.7 and 1338.7. The figure is reproduced from ref. 74 by permission from ACS publications. | ||
In reality, we are not given peptides P1, … Pm but only their spectra S1, … Sm. Nevertheless, one can approximate the peptide network by constructing the spectral network on nodes S1, … Sm where spectra Si and Sj are connected by an edge if they can be aligned against each other using a spectral alignment.23,25,75Fig. 5 shows the peptide and spectral networks of nine tyrocidines and illustrates that the spectral network captures all edges of the peptide network. While the peptide and spectral networks in Fig. 5 are not identical, their shared edges usually allow one to interpret the peptides corresponding to the nodes of the spectral network using the spectral network dereplication algorithm.76 The algorithm starts from a node with a known annotation in the spectral network, and propagates annotations from known to unknown peptides through the edges of the network.
3 PNP dereplication
PNP researchers face the challenge of maximizing the discovery of new compounds while minimizing the re-evaluation of already known PNPs. The process of using information about the chemical structure of a previously characterized compound to identify a compound in an experimental sample (without having to repeat the entire isolation and structure-determination process) is called dereplication. In many cases, a PNP in the new sample is absent in the database of known PNPs, but its variant is present in this database with a modification. Identification of a PNP from its variants is called variable dereplication.3.1 Dereplication via chemical databases
The development of chemical structure databases such as PubChem77 (≈60 million compounds, as of August 2015), ChemSpider78 (≈34 million compounds, as of August 2015), mzCloud63 (≈3 thousand compounds), KEGG79 (≈16 thousand compounds), MetaCyc80 (≈10 thousand compounds), Norine81 (≈1000 compounds), MIBiG82 (≈1200 compounds with biosynthetic gene clusters), and AntiMarin, the result of a merger between AntiBase and MarinLit databases83 (≈60 thousand compounds), paved the way for the development of bioinformatics tools for natural product dereplication. However, the number of PNPs in these databases remains limited, e.g., AntiMarin contains only 3462 compounds with more than five amide bonds.Ng et al.,69 proposed the first method for the dereplication of cyclic PNPs. Ibrahim et al.,70 proposed an alternative dereplication approach, iSNAP, that is not limited to cyclic NRPs but extends to branch cyclic and linear peptides.
iSNAP analyzes each spectrum using the following steps: (i) identify all amide bonds for each NRP in the chemical database, (ii) generate a theoretical spectrum for each NRP by cleaving at most two amide bonds at a time, (iii) generate PSMs formed by the experimental spectrum and all NRPs in the database whose mass matches the precursor mass of the spectrum, and (iv) score resulting PSMs, estimate their statistical significance, and report statistically significant PSMs.
3.2 Dereplication via spectral libraries
Since some natural products feature atypical fragmentation patterns,84 their experimental spectra have low scores against their theoretical spectra. In such cases, instead of dereplication via a search in chemical databases, researchers search spectral libraries of natural products by comparing the experimental spectrum of interest against previously identified spectra. The development of large metabolite spectral databases such as mzCloud63 (≈200 thousands spectra), NIST85 (≈120 thousand spectra), METLIN86 (≈55 thousand spectra), MassBank87 (≈36 thousand spectra), HMDB88 (≈1000 human metabolite spectra), and GNPS spectral library4 (≈1600 natural product spectra) enabled MS/MS library searches for metabolites.84,89–95While dereplication via a spectral library search is more accurate than dereplication via a search in a chemical database, the spectral libraries still contain only a fraction of PNPs present in chemical databases, e.g., as of August 2015, only 81 out of 1607 annotated spectra in GNPS Molecular Networking dataset4 represented PNPs. Therefore, the application of spectral libraries for PNP dereplication remains limited. For example, Milman and Zhurkovich96 described the dereplication of toxic NRPs based on a small spectral library consisting of only 263 spectra.
3.3 Dereplication via spectral networks
The spectral network approach to PNP dereplication analyzes connected components of a spectral network. In contrast to the traditional spectral library approaches that compare spectra with the same precursor mass, spectral networks reveal relationships between spectra with different precursor masses thus enabling the analysis of PNP variants. As long as there is at least one annotated spectrum in a connected component of a spectral network, its annotation can be propagated to all spectra of the connected components.23 Ng et al.69 and Mohimani et al.74,97 described variable PNP dereplication algorithms using spectral networks and identified many variants of previously known PNPs.Watrous et al.18 constructed spectral networks of various bacterial extracts and dereplicated many PNPs using a manual analysis of connected components in these networks. Various studies reported success in utilizing spectral networks for the discovery of natural products.4,97–108
For example, Mohimani et al.97 discovered a lanthipeptide informatipeptin, a doubly charged ion with m/z 1065.5, using the RiPPquest algorithm. This PNP belonged to a connected component of the spectral network and was connected with three doubly charged ions with m/z of 929.2, 957.5, and 1015.1, comparing the mass shifts between these ions and informatipeptin provided a hint that these peptides are N-terminal derivatives of informatipeptin. While the three resulting PSMs had borderline statistical significance and RiPPquest did not report them as significant discoveries, the fact that they clustered with informatipeptin in the spectral network provided evidence that they are indeed N-terminal derivatives of informatipeptin (Fig. 6).
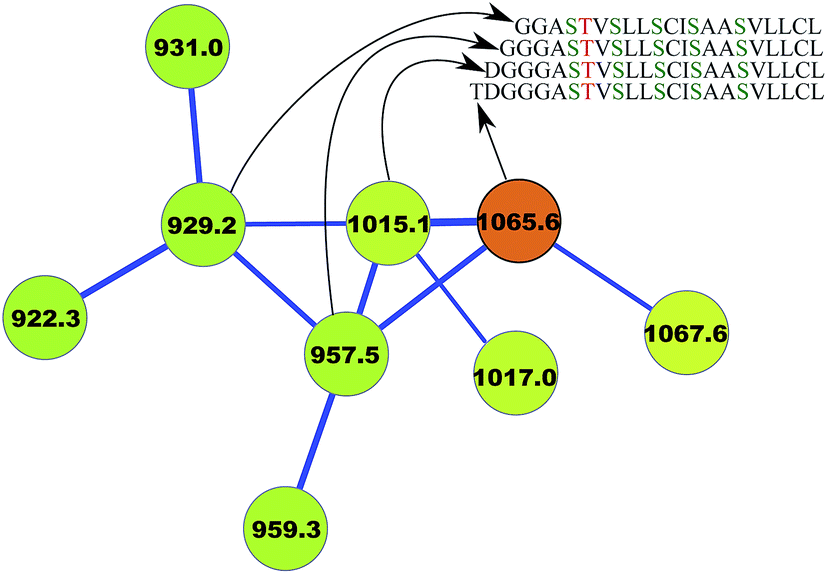 | ||
| Fig. 6 Spectral network analysis leads to variable dereplication of RiPP informatipeptin (shown in orange) into 3 variant PNPs.97 Ser → Dha and Thr → Dhb conversions in this lanthipeptide are shown in green and red, respectively. The figure is reproduced from ref. 97 by permission from ACS publications. | ||
4 PNP sequencing
While the availability of genome sequences enables PNP discovery via genome mining, many PNPs are produced by difficult-to-cultivate organisms whose genomes are still unknown. If a genome is unavailable and if the dereplication of a PNP fails, de novo sequencing69,76,109,110 remains the last resort.Allmer et al.111 recently reviewed various approaches to de novo sequencing of linear peptides. However, while dozens of tools for de novo sequencing of linear peptides have been proposed,112–115 techniques for de novo sequencing of non-linear peptides are still in the early stages of development. Ng et al.69 proposed the first algorithm for the sequencing of cyclic peptides that however works only for very well-fragmented spectra. Novak et al.116 recently developed CycloBranch that takes advantage of high resolution mass spectrometry to improve the accuracy of de novo sequencing of cyclic, branched, and branch cyclic peptides.
Mohimani et al.76 developed a multiplex de novo peptide sequencing algorithm for the case when spectra of multiple related peptides are available. Multiplex peptide sequencing starts from constructing the spectral network and identifying clusters of related compounds (connected components in the spectral network). It further attempts to sequence all compounds in each connected component (see Fig. 7). As opposed to PNP dereplication via spectral networks (when at least one spectrum in the connected component represents a known compound), de novo PNP sequencing works even when all nodes in the connected component represent unknown compounds. The advantage of spectral networks for PNP sequencing is that finding PNPs that simultaneously explain all spectra in a connected component of a spectral network results in a more accurate approach than sequencing each individual spectrum. When tandem mass spectrometry (MS2) fails to sequence a PNP, one can attempt multistage (MSn) mass spectrometry and apply the multistage de novo peptide sequencing approach109,117 (see Fig. 8).
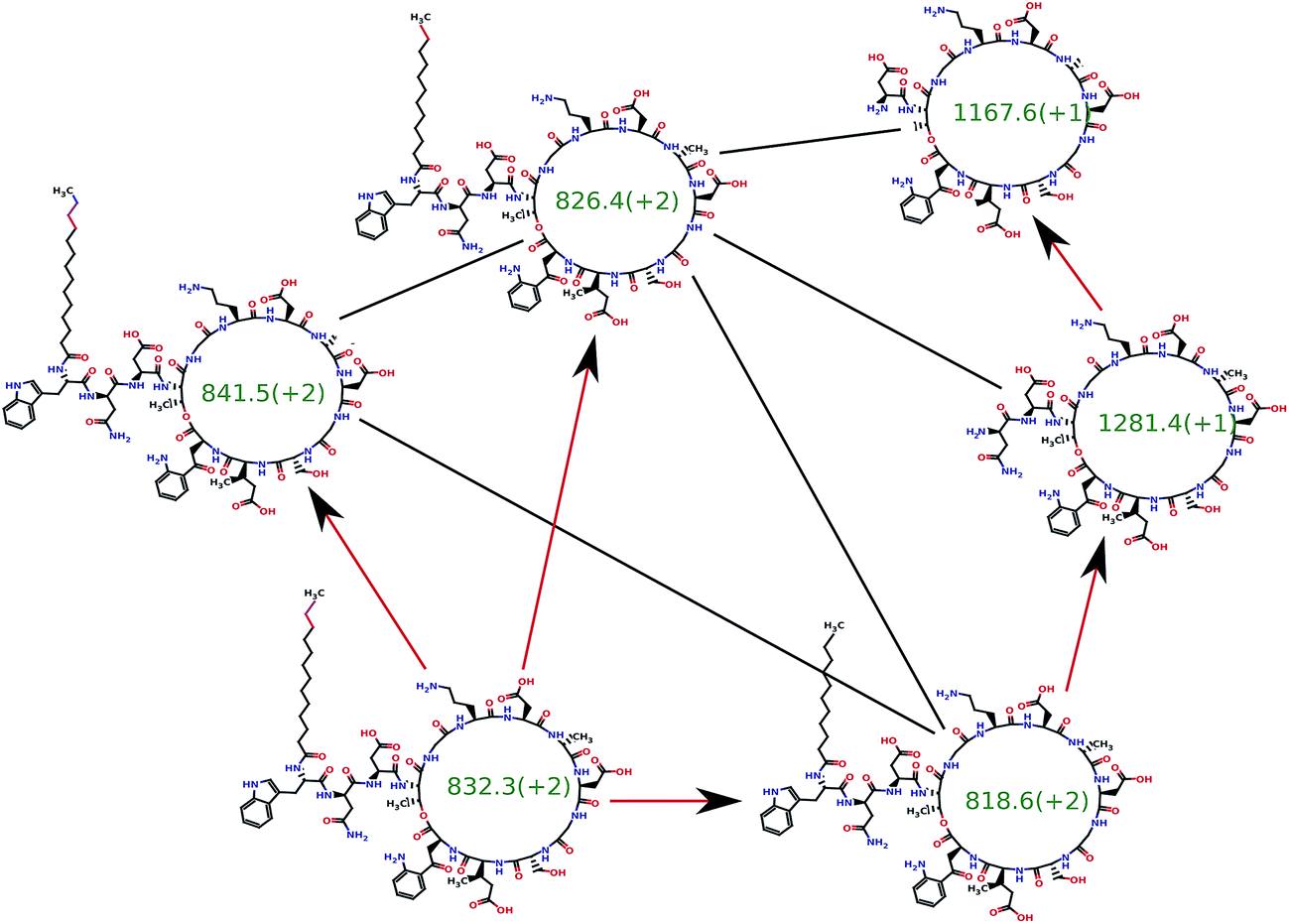 | ||
| Fig. 7 The spectral network dereplication algorithm from ref. 76 attempts to de novo sequence all spectra in a spectral network in a coordinated fashion. It starts from a putative interpretation of one of the spectra (bottom left node) and propagates this interpretation to other nodes using red edges. The propagation typically fails if the initial putative interpretation is incorrect and succeeds if it is correct. Thus, the propagation process allows one to reject the incorrect initial interpretations. The spectral network dereplication algorithm generates many putative interpretations of the spectrum and propagates them through the spectral network in an attempt to decide which one is correct. | ||
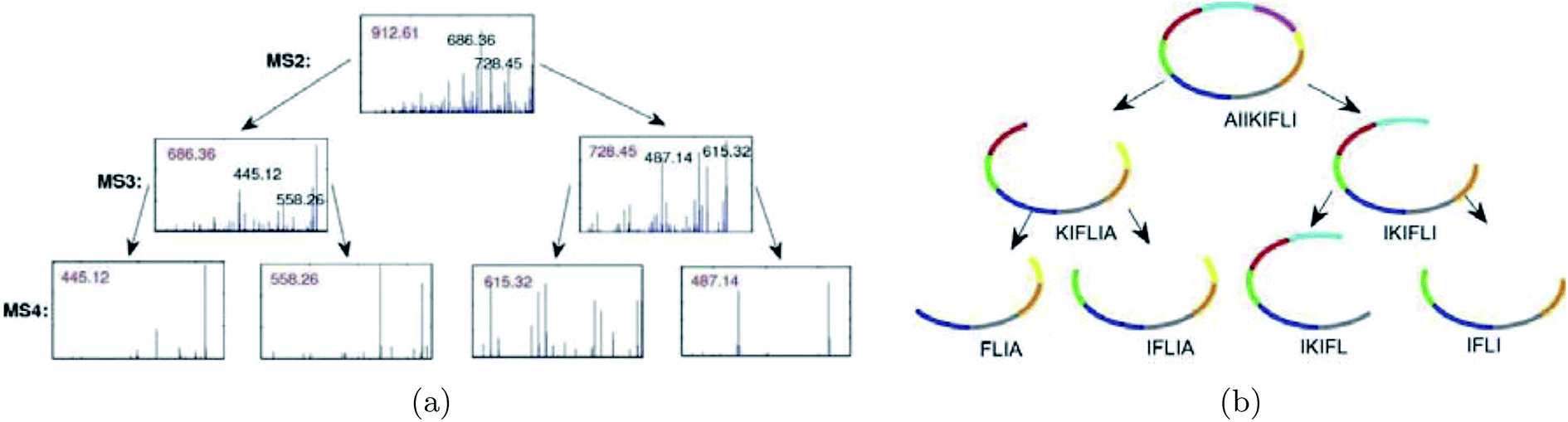 | ||
| Fig. 8 Illustration of an algorithm for peptide sequencing by multistage mass spectrometry. For each candidate peptide, the multistage peptide sequencing algorithm scores how well each subpeptide is explained by the MSn data.109 The figure is reproduced from ref. 109 by permission from Wiley publications. | ||
5 PNP identification
For both RiPPs and NRPs, the PNP identification consists of a genome mining step for detecting the biosynthetic gene clusters and their putative PNPs, and a peptidogenomics step for identifying a spectrum that matches one of the putative PNPs and finding modifications in this putative PNP. Below we describe these steps for RiPP identification and NRP identification.5.1 RiPP identification
A RiPP biosynthetic gene cluster usually includes a gene encoding a single core peptide and several genes encoding modification enzymes that are responsible for the conversion of the core peptide to a mature peptide. The standard MS/MS database search tools are limited with respect to the identification of complex RiPPs with more than two modifications. This limitation makes them inadequate for analyzing such RiPPs as lanthipeptides that often have more than five modifications. Moreover, even if these tools were able to efficiently search for peptides with more than two modifications, the resulting PSMs often would not be reported as statistically significant since many RiPPs are poorly fragmented (due to the presence of multicyclic modifications). Since the search for multiple variable modifications is statistically equivalent to the search in a huge virtual database of all modified peptides, it often results in a high false discovery rate (FDR) even for microbial organisms with small proteomes.118Even when the core RiPP sequence is known and the types of PTMs in a RiPP can be predicted, multiple possible PTM sites typically result in thousands of structures that are difficult to analyze. Due to this complication, computational approaches for RiPP identification could not keep pace with the rapid progress in RiPP discovery in recent years. Cycloquest,119 a tool for RiPP identification, is limited to cyclic peptides with very few modifications. Also, since Cycloquest does not take advantage of genome mining, it is unable to identify poorly fragmented peptides (e.g., lanthipeptides).
Genome mining is crucial for the success of RiPP identification efforts. The statistical significance (E-values) of the found PSMs deteriorates with an increase in the size of the protein database. Thus, one way to make PSMs formed by poorly fragmented spectra statistically significant is to reduce the effective size of the protein database. Fortunately, most RiPPs appear in small windows of ≈20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nucleotides around biosynthetic gene clusters, and these clusters can be identified by searching for conserved biosynthetic enzymes. Thus, limiting the search space to this small region of the genome has the potential to reduce the E-values of found PSMs by orders of magnitude thus separating them from false PSMs.
000 nucleotides around biosynthetic gene clusters, and these clusters can be identified by searching for conserved biosynthetic enzymes. Thus, limiting the search space to this small region of the genome has the potential to reduce the E-values of found PSMs by orders of magnitude thus separating them from false PSMs.
RiPPquest97 is a RiPP database search tool that addresses these complications and uses a more involved pipeline than peptide identification tools in traditional proteomics (compare Fig. 9(a) with 9(b)). While RiPPquest is currently limited to lanthipeptide analysis, it can be extended to other RiPP classes as soon as (i) it implements a genome mining rationale for a specific RiPP class, and (ii) it implements a biosynthetic rationale for transforming core peptides into mature peptides for a specific RiPP class.
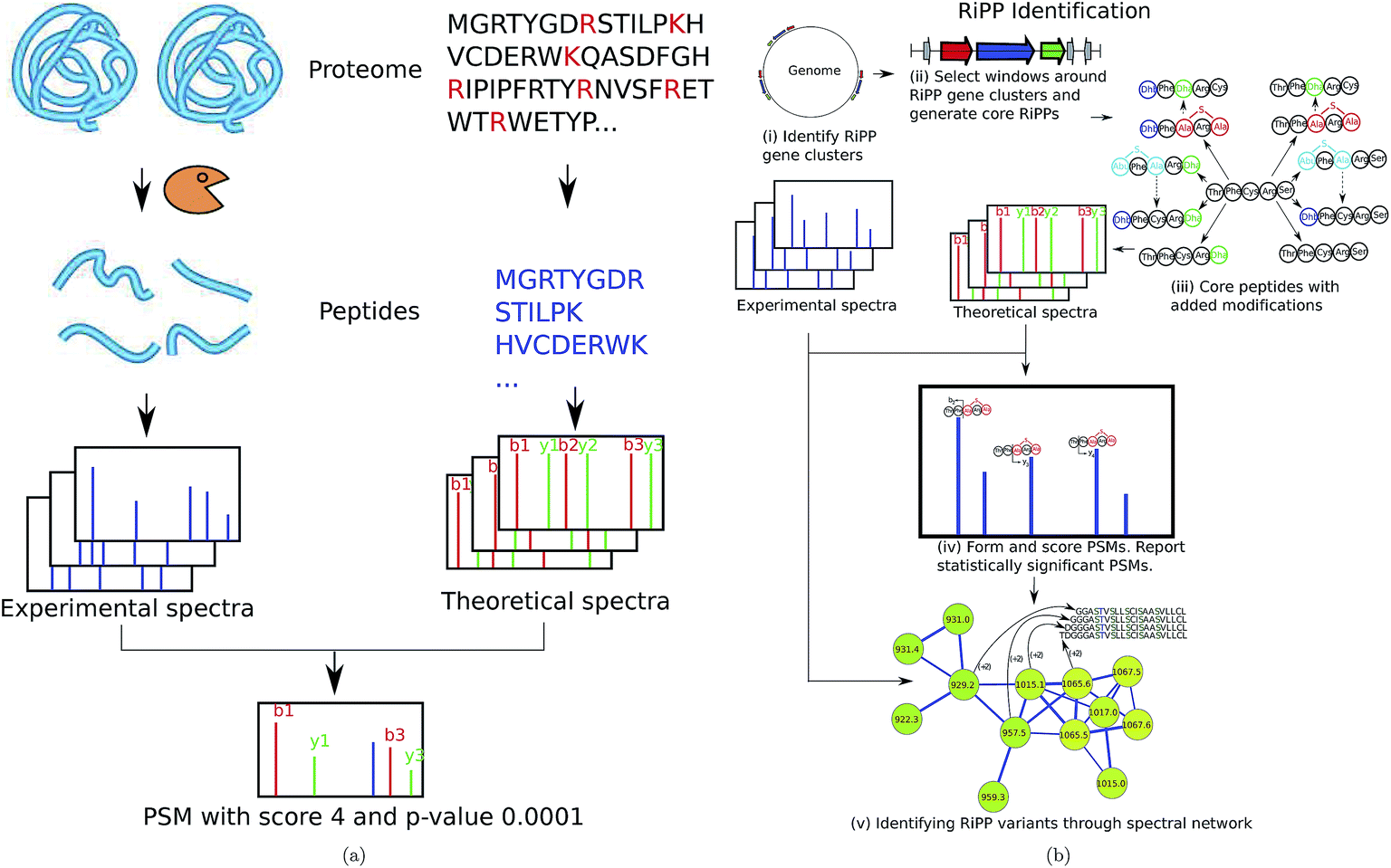 | ||
| Fig. 9 (a) Standard proteomic database search tools (e.g., Sequest9) are based on digesting proteins by an enzyme, and collecting tandem spectra of the resulting peptides. Each spectrum is matched against theoretical spectra of all peptides in a protein database and PSMs with the highest scores/lowest p-values are reported. (b) RiPPquest97 includes the following steps: (i) identifying RiPP synthetases in the genome, (ii) extracting candidate ORFs in a window around the gene cluster, (iii) adding proper modifications, (iv) matching spectra against the database of putative RiPPs and computing p-values of resulting PSMs, and (v) refining and enlarging the set of identified NRPs using spectral networks. The figure is reproduced from ref. 97 by permission from ACS publications. | ||
Zhang et al.120 recently developed the Hypothetical Structure Enumeration and Evaluation (HSEE) algorithm for RiPP identification and applied it for the identification of the lanthipeptide prochlorosin. HSEE is based on matching spectra against a collection of hypothetical structures predicted based on the biosynthetic gene cluster. HSEE generates a theoretical spectrum for each hypothetical structure and scores structures based on the shared peak count between the theoretical and experimental spectrum. The structure with the highest score is reported as a putative interpretation of an experimental spectrum.
We illustrate the PNP identification pipeline using RiPPquest97 that includes the following steps: (i) identifying RiPP synthetases in the genome, (ii) extracting candidate open reading frames (ORFs) in a window around the gene cluster, (iii) adding proper modifications, (iv) matching spectra against the database of putative RiPPs and computing p-values of resulting PSMs, and (v) refining and enlarging the set of identified RiPPs using spectral networks (Fig. 9(b)). Below is a brief description of the RiPPquest pipeline:
RiPPquest uses genome mining tools such as BAGEL,51,52 ThioFinder53, and antiSMASH40–42,121 for the identification of RiPP gene clusters. Limiting the search to small windows centered at RiPP gene clusters decreases the search space by two orders of magnitude as compared to the entire Streptomyces genome. Candidate core peptides are extracted from short ORFs found in these windows, and transformed to mature peptides according to the biosynthetic enzymes in the gene cluster. In the case of lanthipeptides, the most essential modifications are the dehydration of serine and threonine, and the formation of lanthionine and methyl-lanthionine bridges.49
Most classes of RiPPs form families of related peptides, making spectral networks helpful in RiPP analysis.18 In particular, spectral networks revealed related lanthipeptides with stepwise N-terminal leader processing and different dehydration numbers97 (see Fig. 6).
5.2 NRP identification
While genome mining techniques accurately identify NRP synthetases in the genome, accurate determination of specificities of A-domains remains difficult, especially for non-proteinogenic amino acids that are common in NRPs. While most NRPs go through modifications such as backbone macrocyclization and the addition of fatty acid chains, existing genome mining tools fail to predict most of these modifications. That is why NRP identification algorithms have to implement a blind MS/MS search that allows for multiple unexpected modifications and mutations. Blind searches refer to the case when the set of possible modifications is not restricted (different from typical searches for PTMs in traditional proteomics). This is a difficult computational problem even in the case of linear peptides,25,34 let alone non-linear peptides.NRPquest74 uses a genome sequence and a spectral dataset as an input and includes the following steps (i) identifying NRP synthetases in the genome, (ii) using the non-ribosomal code to construct a database of putative NRPs generated by each NRP synthetase, (iii) matching spectra against the database of putative NRPs and computing p-values of resulting PSMs, and (iv) refining and enlarging the set of identified NRPs using spectral networks (Fig. 10).
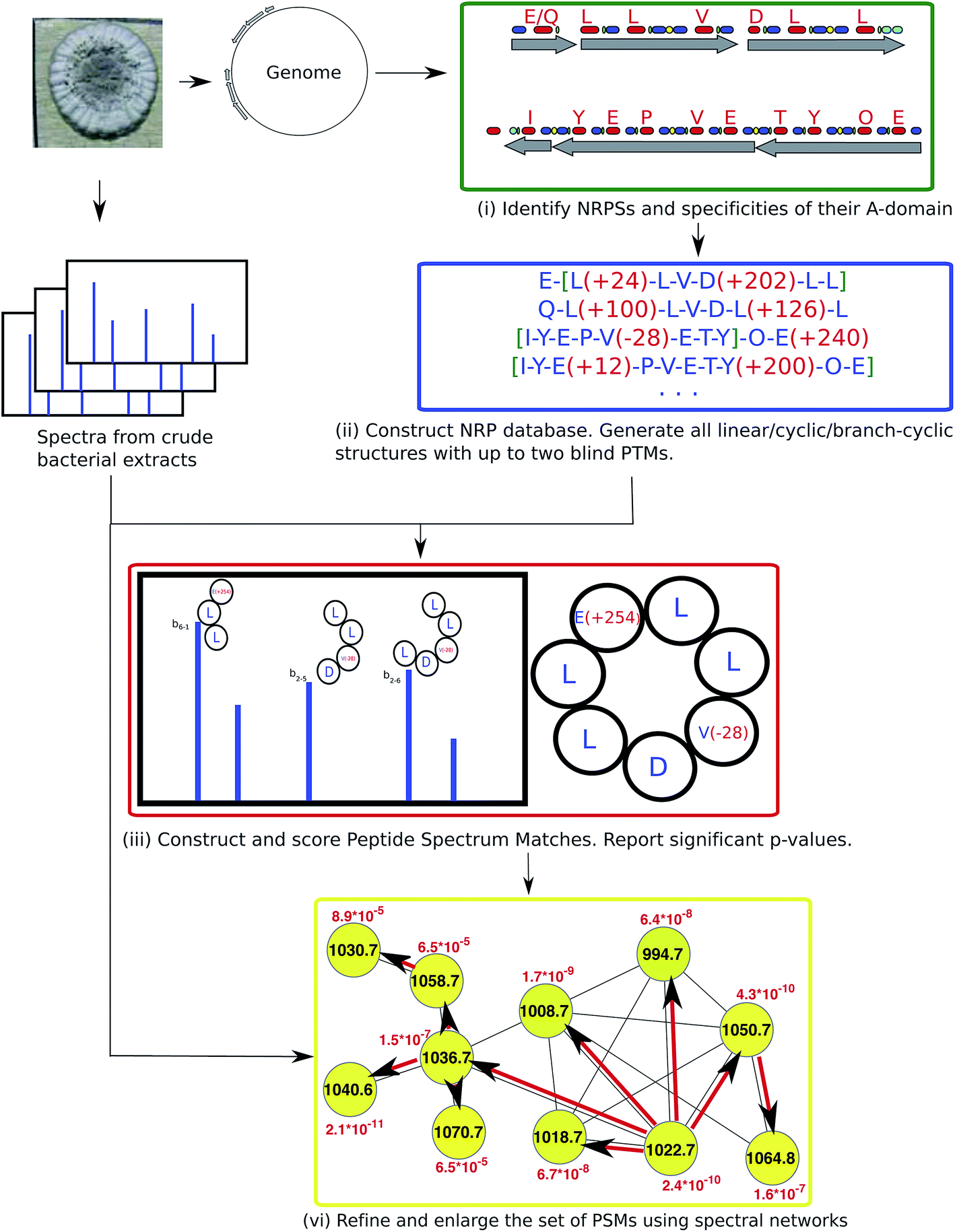 | ||
| Fig. 10 NRPquest74 pipeline includes the following steps: (i) identifying NRP synthetases in the genome, (ii) using the non-ribosomal code to construct a database of putative NRPs generated by each NRP synthetase, (iii) matching spectra against the database of putative NRPs and computing p-values of resulting PSMs, and (iv) refining and enlarging the set of identified NRPs using spectral networks. The figure is reproduced from ref. 74 by permission from ACS publications. | ||
NRPquest uses NRPSpredictor2 (ref. 39) and antiSMASH40–42 to identify NRP synthetases in the genome, and to predict the set of all possible amino acids generated by each A-domain. NRPquest further searches the genome for a methylation domain, epimerization domain, and side chain bond formation domain, and accounts for the corresponding modifications in the database of putative NRPs. Each spectrum is matched against each putative peptide in the database of putative NRPs using a brute force algorithm that allows for up to two blind modifications. These blind modifications account for potentially inaccurate predictions of specificities of A-domains that are particularly common for non-proteinogenic amino acids and modifications. Similar to RiPPquest, NRPquest scores the PSMs using the scoring function from ref. 114 and computes p-values using the MS-DPR algorithm.72
NRPquest constructs a spectral network23 to refine and enlarge the set of identified PSMs. After constructing the spectral network, its connected components are extracted and the spectral network dereplication algorithm from Mohimani et al., 2011 (ref. 76), is used for the identification of all peptides represented by spectra forming this connected component. Moreover, the peptide propagation through the spectral network23,69 allows one to identify peptides with many modifications that NRPquest missed during blind searches of individual spectra (since blind searches with more than two modifications are prohibitively time-consuming).
6 Discussion
Despite the important biomedical applications of PNPs, most pharmaceutical companies are now focusing on synthetic compounds and do not utilize the biosynthetic capacity of bacteria and fungi. However, the rise of high-throughput DNA sequencing has revealed a wealth of new PNP biosynthetic gene clusters in various genomes that exceeds previous expectations by orders of magnitude. These new discoveries suggest that there may be a reversal of focus in the pharmaceutical industry that could lead to a revival in biomedical applications of natural products. Arguably, one of the key bottlenecks for accomplishing such a transformation is the shortage of computational tools for PNP discovery. Here we have reviewed recently developed approaches for PNP discovery and computational technologies (genome mining, peptidogenomics, and spectral networks) that enable these methods. While these approaches have made a rather modest progress towards PNP discovery, further development of the algorithms for PNP discovery may enable a systematic and high-throughput exploration of PNPs.We described three approaches to PNP discovery with their own merits and limitations: PNP dereplication, PNP sequencing and PNP identification. For example, while PNP dereplication requires a chemical structure database to be available and can only identify known compounds and their variants, PNP identification requires the genome sequence to be available and can identify new compounds. The fragmentation quality of spectra required for the success of these approaches is vastly different, as they perform searches in vastly different computational spaces.
While the search space for PNP dereplication is usually small, the search space for PNP sequencing is very large since it includes all peptides with a given mass. PNP identification, for both NRPs and RiPPs, has a search space that typically includes under a million putative peptides, standing in between PNP dereplication and PNP sequencing with respect to the search space. Thus, while PNP sequencing can succeed only with extremely high quality spectra, PNP identification can succeed with a medium quality spectra, and PNP dereplication can succeed even with poorly-fragmented spectra. Since PNP sequencing using a single tandem mass spectrum rarely succeeds, researchers have tried to utilize information from multiple spectra/multistage MS to overcome this limitation.23,76,109,117,122
7 Acknowledgements
We are grateful to Alexey Gurevich and Kira Vyatkina for many helpful comments. This work was supported by the US National Institutes of Health grant 5P41GM103484 from the National Center for Research Resources. P. A. P. has an equity interest in Digital Proteomics, LLC, a company that may potentially benefit from the research results. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.8 References
- J. Li and J. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- L. Ling, T. Schneider, A. Peoples, A. Spoering, I. Engels, B. Conlon, A. Mueller, T. Schberle, D. Hughes, S. Epstein, M. Jones, L. Lazarides, V. Steadman, D. Cohen, C. Felix, K. Fetterman, W. Millett, A. Nitti, A. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- A. Harvey, R. Edrada-Ebel and R. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- M. Wang, et al. , Nat. Biotechnol., 2015 Search PubMed , accepted. See also: http://gnps.ucsd.edu.
- J. Lederberg, Proc. Natl. Acad. Sci. U. S. A., 1965, 53, 134–139 CrossRef CAS.
- J. Lederberg, ACM Conf. on the History of Medical Informatics, 1987, pp. 5–9 Search PubMed.
- I. Mun and F. Mclafferty, ACS Symp. Ser., 1981, 9, 117–124 CrossRef PubMed.
- D. Smith, N. Gray, J. Nourse and C. Crandell, Anal. Chim. Acta, 1981, 133, 471–497 CrossRef CAS.
- J. Eng, A. McCormack and J. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
- D. Perkins, D. Pappin, D. Creasy and J. Cottrell, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS.
- J. Gasteiger, W. Hanebeck and K. Schulz, J. Chem. Inf. Comput. Sci., 1992, 32, 264–271 CrossRef CAS.
- K. Scheubert, F. Hufsky and S. Bocker, J. Cheminf., 2013, 5, 12 CAS.
- S. Nuemann and S. Bocker, Anal. Bioanal. Chem., 2010, 398, 2779–2788 CrossRef PubMed.
- T. Kind and O. Fiehn, Bioanal. Rev., 2010, 2, 23–60 CrossRef PubMed.
- J. F. Xiao, B. Zhou and H. Ressom, TrAC, Trends Anal. Chem., 2012, 32, 1–14 CrossRef CAS PubMed.
- A. Vaniya and O. Fiehn, TrAC, Trends Anal. Chem., 2015, 69, 52–61 CrossRef CAS PubMed.
- R. Kersten, Y. Yang, P. Cimermancic, S. Nam, W. Fenical, M. Fischbach, B. Moore and P. Dorrestein, Nat. Chem. Biol., 2011, 7, 794–802 CrossRef CAS PubMed.
- J. Watrous, P. Roach, T. Alexandrov, B. Heath, J. Yang, R. Kersten, M. van der Voort, K. Pogliano, H. Gross, J. Raaijmakers, B. Moore, J. Laskin, N. Bandeira and P. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1743–E1752 CrossRef CAS PubMed.
- A. Bouslimani, L. Sanchez, N. Garg and P. C. Dorrestein, Nat. Prod. Rep., 2014, 31, 718–729 RSC.
- T. Ito and M. Miyako, Nat. Prod. Rep., 2014, 67, 353–360 CAS.
- G. Challis and J. Ravel, FEMS Microbiol. Lett., 2000, 187, 111–114 CrossRef CAS PubMed.
- S. Lautru, R. Deeth, L. Bailey and G. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- N. Bandeira, D. Tsur, A. Frank and P. Pevzner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6140–6145 CrossRef CAS PubMed.
- P. Pevzner, Z. Mulyukov, V. Dancik and C. Tang, Genome Res., 2001, 11, 290–299 CrossRef CAS PubMed.
- D. Tsur, S. Tanner, E. Zandi, V. Bafna and P. Pevzner, Nat. Biotechnol., 2005, 23, 1562–1567 CrossRef CAS PubMed.
- M. Marahiel, T. Stachelhaus and H. Mootz, Nat. Prod. Rep., 1997, 7, 2651–2674 Search PubMed.
- D. Schwarzer, R. Finking and M. Marahiel, Nat. Prod. Rep., 2003, 20, 275–287 RSC.
- T. Oman and W. van der Donk, Nat. Prod. Rep., 2010, 6, 9–18 CAS.
- J. McIntosh, M. Donia and E. Schmidt, Nat. Prod. Rep., 2009, 26, 537–559 RSC.
- T. Stachelhaus, H. Mootz and M. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- A. Broberg, A. Menkis and R. Vasiliauskas, J. Nat. Prod., 2006, 69, 97–102 CrossRef CAS PubMed.
- A. Delcher, D. Harmon, S. Kasif, O. White and S. Salzberg, Nucleic Acids Res., 1999, 27, 673–679 Search PubMed.
- K. Severinov, E. Semenova, A. Kazakov, T. Kazakov and M. Gelfand, Mol. Microbiol., 2007, 65, 1380–1394 CrossRef CAS PubMed.
- S. Na, N. Bandeira and E. Paek, Mol. Cell. Proteomics, 2012, 11, M111.010199 Search PubMed.
- J. Doroghazi, J. Albright, A. Goering, K. Ju, R. Haines, K. Tchalukov, D. Labeda, N. Kelleher and W. Metcalf, Nat. Chem. Biol., 2014, 10, 6963–6968 CrossRef PubMed.
- A. Starcevic, J. Zucko, J. Simunkovic, P. Long, J. Cullum and D. Hranueli, Nucleic Acids Res., 2008, 36, 6882–6892 CrossRef CAS PubMed.
- M. Li, P. Ung, J. Zajkowski, S. Garneau-Tsodikova and D. Sherman, Nucleic Acids Res., 2009, 10, 185–194 Search PubMed.
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS PubMed.
- M. Rottig, M. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W332–W337 CrossRef PubMed.
- M. Medema, K. Blin, P. Cimermancic, V. Jager, P. Zakrzewski, M. Fischbach, T. Weber, E. Takan and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- K. Blin, M. Medema, D. Kazempour, M. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. Kim, R. Bruccoleri, S. Lee, M. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef PubMed.
- S. Anand, M. Prasad, G. Yadav, N. Kumar, J. Shehara, M. Ansari and D. Mohanty, Nucleic Acids Res., 2010, 38, W487 CrossRef CAS PubMed.
- N. Khaldi, F. Seifuddin, G. Turner, D. Haft, W. Nierman, K. Wolfe and N. Fedorova, Fungal Genet. Biol., 2010, 47, 736 CrossRef CAS PubMed.
- M. Umemura, H. Koike, N. Nagano, T. Ishii, J. Kawano, N. Yamane, I. Kozone, K. Horimoto, K. Shin-ya, K. Asai, J. Yu, J. Bennett and M. Machida, PLoS One, 2013, 8, e84028 Search PubMed.
- T. Weber, C. Rausch, P. Lopez, I. Hoof, V. Gaykova and D. H. Huson, J. Biotechnol., 2009, 140, 13 CrossRef CAS PubMed.
- H. Tae, E. Kong and K. Park, BMC Bioinf., 2007, 8, 327 CrossRef PubMed.
- M. Medema, Y. Paalvast, D. Nguyen, A. Melnik, P. Dorrestein, E. Takano and R. Breitling, PLoS Comput. Biol., 2014, 10, e1003282 Search PubMed.
- P. Arnison, et al. , Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- J. Velsquez and W. van der Donk, Curr. Opin. Chem. Biol., 2011, 15, 11–21 CrossRef PubMed.
- A. de Jong, A. J. van Heel, J. Kok and O. Kuipers, Nucleic Acids Res., 2010, 38, W647651 CrossRef PubMed.
- A. van Heel, A. de Jong, M. Montalban-Lopez, J. Kok and O. Kuipers, Nucleic Acids Res., 2013, 41, W448–W453 CrossRef PubMed.
- J. Li, X. Qu, X. He, L. Duan, G. Wu, D. Bi, Z. Deng, W. Liu and H. Ou, PLoS One, 2012, 7, e45878 CAS.
- M. Maksimova, I. Pelczerb and J. Link, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15223–15228 CrossRef PubMed.
- N. Leikoski, D. Fewer, J. Jokela, M. Wahlsten, L. Rouhiainen and K. Sivonen, Appl. Environ. Microbiol., 2010, 76, 701–709 CrossRef CAS PubMed.
- R. Hammami, A. Zouhir, J. Ben Hamida and I. Fliss, BMC Microbiol., 2007, 7, 89–94 CrossRef PubMed.
- Y. Xu, R. Kersten, S. Nam, L. Lu, A. Al-Suwailem, H. Zheng, W. Fenical, P. Dorrestein, B. Moore and P. Qian, J. Am. Chem. Soc., 2012, 134, 8625–8632 CrossRef CAS PubMed.
- A. Hill and R. Mortishire-Smith, Rapid Commun. Mass Spectrom., 2005, 19, 3111–3118 CrossRef CAS PubMed.
- S. Wolf, S. Schmidt, M. Mller-Hannemann and S. Neumann, BMC Bioinf., 2010, 11, 148 CrossRef PubMed.
- M. Krauss, H. Singer and J. Hollender, Anal. Bioanal. Chem., 2010, 397, 943–951 CrossRef CAS PubMed.
- L. Ridder, J. J. J. van der Hooft, S. Verhoeven, R. C. H. de Vos, R. J. Bino and J. Vervoort, Anal. Chem., 2013, 85, 6033–6040 CrossRef CAS PubMed.
- Y. Wang, G. Kora, B. Bowen and C. Pan, Anal. Chem., 2014, 86, 9496–9503 CrossRef CAS PubMed.
- R. Mistrik, J. Lutisan, Y. Huang, M. Suchy, J. Wang and M. Raab, 9th International Conference of the Metabolomics Society, Glasgow, Scotland, 2013 Search PubMed.
- A. Klitgaard, A. Iversen, M. Andersen, T. Larsen, J. Frisvad and K. Nielsen, Anal. Bioanal. Chem., 2014, 406, 1933–1943 CrossRef CAS PubMed.
- H. Shen, N. Zamboni, M. Heinonen and J. Rousu, Metabolites, 2013, 3, 484–505 CrossRef CAS PubMed.
- F. Allen, A. Pon, M. Wilson, R. Greiner and D. Wishart, Nucleic Acids Res., 2014, 42, W94–W99 CrossRef CAS PubMed.
- F. Allen, R. Greiner and D. Wishart, Metabolomics, 2015, 11, 98–110 CrossRef CAS.
- M. Gerlich and S. Neumann, J. Mass Spectrom., 2013, 48, 291–298 CrossRef CAS PubMed.
- J. Ng, N. Bandeira, W. Liu, M. Ghassemian, T. Simmons, W. Gerwick, R. Linington, P. Dorrestein and P. Pevzner, Nat. Methods, 2009, 6, 596–599 CrossRef CAS PubMed.
- A. Ibrahim, L. Yang, C. Johnston, X. Liu, B. Ma and N. Magarveya, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19196–19201 CrossRef CAS PubMed.
- S. Kim, N. Gupta and P. Pevzner, J. Proteome Res., 2008, 7, 3354–3363 CrossRef CAS PubMed.
- H. Mohimani, S. Kim and P. Pevzner, J. Proteome Res., 2013, 12, 1560–1568 CrossRef CAS PubMed.
- X. Tang, P. Thibault and R. Boyd, Int. J. Mass Spectrom. Ion Processes, 1992, 122, 153–179 CrossRef CAS.
- H. Mohimani, W. Liu, R. Kersten, B. Moore, P. Dorrestein and P. Pevzner, J. Nat. Prod., 2014, 77, 1902–1909 CrossRef CAS PubMed.
- P. Pevzner, V. Dancik and C. Tang, J. Comput. Biol., 2000, 7, 777–787 CrossRef CAS PubMed.
- H. Mohimani, W. Liu, Y. Liang, S. Gaudenico, W. Fenical, P. Dorrestein and P. Pevzner, J. Comput. Biol., 2011, 18, 1371–1381 CrossRef CAS PubMed.
- E. Bolton, Y. Wang, P. Thiessen and S. Bryant, Annu. Rep. Comput. Chem., 2008, 4, 217–241 CAS.
- H. Pence and A. Williams, J. Chem. Educ., 2010, 87, 1123–1124 CrossRef CAS.
- M. Kanehisa, S. Goto, Y. Sato, M. Furumichi and M. Tanabe, Nucleic Acids Res., 2012, 40, 109–114 CrossRef PubMed.
- R. Caspi, T. Altman, R. Billington, K. Dreher, H. Foerster, C. A. Fulcher, T. A. Holland, I. M. Keseler, A. Kothari, A. Kubo, M. Krummenacker, M. Latendresse, L. A. Mueller, Q. Ong, S. Paley, P. Subhraveti, D. S. Weaver, D. Weerasinghe, P. Zhang and P. D. Karp, Nucleic Acids Res., 2014, 42, D459–D471 CrossRef CAS PubMed.
- S. Caboche, M. Pupin, V. Leclre, A. Fontaine, P. Jacques and G. Kucherov, Nucleic Acids Res., 2008, 36, D326–D331 CrossRef CAS PubMed.
- M. H. Medema, et al. , Nat. Chem. Biol., 2015, 11, 625–631 CrossRef CAS PubMed.
- J. Blunt, M. Munro and H. Laatsch, Antimarin database, University of Canterbury, University of Gottingen, Christchurch, New Zealand, Gottingen, Germany, 2007 Search PubMed.
- H. Lam, E. Deutsch, J. Eddes, J. Eng, N. King, S. Stein and R. Aebersold, Proteomics, 2007, 7, 655–667 CrossRef CAS PubMed.
- S. Heller, Todays Chemist at Work, 1999, 8, 45–46 Search PubMed.
- D. Wishart, C. Knox, A. Guo, R. Eisner, N. Young, B. Gautam, D. Hau, N. Psychogios, E. Dong, S. Bouatra, R. Mandal, I. Sinelnikov, J. Xia, L. Jia, J. Cruz, E. Lim, C. Sobsey, S. Shrivastava, P. Huang, P. Liu, L. Fang, J. Peng, R. Fradette, D. Cheng, D. Tzur, M. Clements, A. Lewis, A. de Souza, A. Zuniga, M. Dawe, Y. Xiong, D. Clive, R. Greiner, A. Nazyrova, R. Shaykhutdinov, L. Li, H. Vogel and I. Forsythe, Nucleic Acids Res., 2009, 37, D603–D610 CrossRef CAS PubMed.
- H. Horai, M. Arita, S. Kanaya, Y. Nihei, T. Ikeda, K. Suwa, Y. Ojima, K. Tanaka, S. Tanaka, K. Aoshima, Y. Oda, Y. Kakazu, M. Kusano, T. Tohge, F. Matsuda, Y. Sawada, M. Hirai, H. Nakanishi, K. Ikeda, N. Akimoto, T. Maoka, H. Takahashi, T. Ara, N. Sakurai, H. Suzuki, D. Shibata, S. Neumann, T. Iida, K. Tanaka, K. Funatsu, F. Matsuura, T. Soga, R. Taguchi, K. Saito and T. Nishioka, J. Mass Spectrom., 2010, 45, 703–714 CrossRef CAS PubMed.
- C. Smith, G. O'Maille, E. Want, C. Qin, S. Trauger, T. Brandon, D. Custodio, R. Abagyan and G. Siuzdak, Ther. Drug Monit., 2005, 6, 747–751 CrossRef.
- P. Ausloos, et al. , J. Am. Soc. Mass Spectrom., 1999, 10, 287–299 CrossRef CAS.
- S. Stein and D. Scott, J. Am. Soc. Mass Spectrom., 1994, 5, 859–866 CrossRef CAS.
- L. Domokos, D. Hennberg and B. Weimann, Anal. Chim. Acta, 1984, 165, 61–74 CrossRef CAS.
- J. Halket, D. Waterman, A. Przyborowska, R. Patel, P. Fraser and P. Bramley, J. Exp. Bot., 2005, 56, 219–243 CrossRef CAS PubMed.
- R. Craig, J. Cortens, D. Fenyo and R. Beavis, J. Proteome Res., 2006, 5, 1843–1849 CrossRef CAS PubMed.
- H. Lam and R. Aebersold, Methods Mol. Biol., 2010, 604, 95–103 CAS.
- H. Lam, E. Deutsch and R. Aebersold, J. Proteome Res., 2010, 9, 605–610 CrossRef CAS PubMed.
- B. Milman and I. Zhurkovich, Anal. Chem. Res., 2014, 1, 8–15 CrossRef PubMed.
- H. Mohimani, R. Kersten, W. Liu, M. Wang, S. Purvine, S. Wu, H. Brewer, L. Pasa-Tolic, B. Moore, P. Pevzner and P. Dorrestein, ACS Chem. Biol., 2014, 9, 1545–1551 CrossRef CAS PubMed.
- D. Nguyen, C. Wu, W. Moree, A. Lamsa, M. Medema, X. Zhao, R. Gavilan, M. Aparicio, L. Atencio, C. Jackson, J. Ballesteros, J. Sanchez, J. Watrous, V. Phelan, C. van de Wiel, R. Kersten, S. Mehnaz, R. de Mot, E. Shank, P. Charusanti, H. Nagarajan, B. Duggan, B. Moore, N. Bandeira, K. Palsson, B. Pogliano, M. Gutierrez and P. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2611–E2620 CrossRef CAS PubMed.
- W. Liu, A. Lamsa, W. Wong, P. Boudreau, R. Kersten, Y. Peng, W. Moree, B. Duggan, B. Moore, W. Gerwick, R. Linington, K. Pogliano and P. Dorrestein, J. Antibiot., 2014, 67, 99–104 CrossRef CAS PubMed.
- J. Yang, L. Sanchez, C. Rath, X. Liu, P. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. Wong, R. Linington, L. Zhang, H. Debonsi, W. Gerwick and P. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- W. J. Moree, V. V. Phelana, C. H. Wu, N. Bandeira, D. S. Cornett, B. M. Duggan and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13811–13816 CrossRef CAS PubMed.
- K. Duncan, M. Crsemann, A. Lechner, A. Sarkar, J. Li, N. Ziemert, M. Wang, N. Bandeira, B. Moore, P. Dorrestein and P. Jensen, Chem. Biol., 2015, 22, 460–471 CrossRef CAS PubMed.
- M. Traxler, J. Watrous, T. Alexandrov, P. Dorrestein and R. Kolter, mBio, 2013, 4, e00459-13 CrossRef PubMed.
- J. Winnikoff, E. Glukhov, J. Watrous, P. Dorrestein and W. Gerwick, J. Antibiot., 2014, 67, 105–112 CrossRef CAS PubMed.
- M. Wilson, T. Mori, C. Rckert, A. Uria, M. Helf, K. Takada, C. Gernert, U. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. Helfrich, A. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- A. Bouslimani, C. Rath, M. Wang, Y. Guoc, A. Gonzalez, D. Berg-Lyon, G. Gail Ackermann, G. Christensen, T. Nakatsujig, L. Zhang, A. W. Borkowskig, M. J. Meehan, K. Dorrestein, R. Gallog, N. Bandeira, R. Knight, T. Alexandrov and P. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2120–E2129 CrossRef CAS PubMed.
- A. Edlund, Y. Yang, S. Yooseph, A. Hall, D. Nguyen, P. Dorrestein, K. Nelson, X. He, R. Lux, W. Shi and J. McLean, ISME J., 2015 DOI:10.1038/ismej.2015.72.
- M. I. Vizcaino and J. M. Crawford, Nat. Chem., 2015, 7, 411–417 CrossRef CAS PubMed.
- H. Mohimani, Y. Liang, W. Liu, P. Hsieh, P. Dorrestein and P. Pevzner, J. Proteomics, 2011, 11, 3642–3650 CrossRef CAS PubMed.
- D. Kavan, M. Kuzma, K. Lemr, K. Schug and V. Havlicek, J. Am. Soc. Mass Spectrom., 2013, 24, 1177–1184 CrossRef CAS PubMed.
- J. Allmer, Expert Rev. Proteomics, 2011, 8, 645–657 CrossRef PubMed.
- V. Dancik, T. Addona, K. Clauser, J. Vath and P. Pevzner, J. Comput. Biol., 1999, 6, 327–342 CrossRef CAS PubMed.
- B. Ma, K. Zhang, C. Hendrie, C. Liang, M. Li, A. Doherty-Kirby and G. Lajoie, Rapid Commun. Mass Spectrom., 2003, 17, 2337–2342 CrossRef CAS PubMed.
- A. Frank and P. Pevzner, Anal. Chem., 2005, 77, 964–983 CrossRef CAS.
- J. A. Taylor and R. S. Johnson, Anal. Chem., 2000, 73, 2594–2604 CrossRef.
- J. Novak, K. Lemr, K. A. Schug and V. Havlicek, J. Am. Soc. Mass Spectrom., 2015, 26, 1780–1786 CrossRef CAS PubMed.
- N. Bandeira, J. Olsen, M. Mann and P. Pevzner, Bioinformatics, 2008, 24, 416–423 CrossRef PubMed.
- A. Guthals, C. Boucher and N. Bandeira, J. Comput. Biol., 2014, 22, 353–366 CrossRef PubMed.
- H. Mohimani, W. Liu, J. Mylne, A. Poth, M. Colgrave, D. Tran, M. Selsted, P. Dorrestein and P. Pevzner, J. Proteome Res., 2011, 10, 4505–4512 CrossRef CAS PubMed.
- Q. Zhang, M. Ortega, Y. Shi, H. Wang, J. Melby, W. Tang, D. Mitchell and W. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12031–12036 CrossRef CAS PubMed.
- K. Blin, D. Kazempour, W. Wohlleben and T. Weber, PLoS One, 2014, 9, 489420 Search PubMed.
- N. Bandeira, V. Pham, P. Pevzner, D. Arnott and J. Lill, Nat. Biotechnol., 2008, 26, 1336–1338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |