
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigations of ion transport through nanoscale polymer membranes by fluorescence quenching of CdSe/CdS quantum dot/quantum rods†
Jan-Philip
Merkl
*a,
Christopher
Wolter
a,
Sandra
Flessau
a,
Christian
Schmidtke
a,
Johannes
Ostermann
ab,
Artur
Feld
a,
Alf
Mews
a and
Horst
Weller
*abc
aInstitute of Physical Chemistry, Grindelallee 117, 20146 Hamburg, and the Hamburg Center for Ultrafast Imaging, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: weller@chemie.uni-hamburg.de; merkl@chemie.uni-hamburg.de
bCenter for Applied Nanotechnology (CAN) GmbH, Grindelallee 117, 20146 Hamburg, Germany
cDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O BOX 80203 Jeddah 21589, Saudi Arabia
First published on 11th March 2016
Abstract
Detailed steady-state and time-resolved fluorescence quenching measurements give deep insight into ion transport through nanometer thick diblock copolymer membranes, which were assembled as biocompatible shell material around CdSe/CdS quantum dot in quantum rods. We discuss the role of polymer chain length, intermolecular cross-linking and nanopore formation by analysing electron transfer processes from the photoexcited QDQRs to Cu(II) ions, which accumulate in the polymer membrane. Fluorescence investigations on single particle level additionally allow identifying ensemble inhomogeneities.
Diffusion of molecules and ions within polymer membranes is an emerging field for future products, such as organic solar cells, polymeric batteries, fuel cells as well as encapsulated nanomaterials.1,2 Due to progressive miniaturization in all technical fields, the synthesis of nanoscale membranes and the study of diffusion within these membranes is important for fundamental and product based research.3–5 Furthermore, the exponential rise of nanomaterial use in consumer products and the accompanied unavoidable release of nanoparticles (NPs) into the environment raises concerns of long-term stability.6–8
Encapsulation in amphiphilic polymer micelles is one of the main strategies to transfer water insoluble NPs into aqueous solution.9 A cross-linkage between the single polymer molecules leads to enhanced stability due to fixation of the micelle.9–12 The diffusion of molecules and ions through such membranes or shells may be used for accessibility assessment or for modulating the catalytic activity13 and the analytical properties14,15 of the NPs. Consequently, the surface coating of nanomaterials not only ensures water solubility but also determines the interaction with the surrounding media.10 It reveals on one hand a physical barrier controlling the mass transport between the NP and the environment and, on the other hand, determines the adsorption of proteins and peptides and therefore the biological fate of these particles.16–18 Furthermore, the particle accessibility is of upmost importance for Cd(II)-based NP use in biological environment because of the potential leakage of toxic Cd(II) ions from not perfectly encapsulated QDQRs.19
In the past, we studied the encapsulation of spherical CdSe/CdS/ZnS quantum dots (QDs) into micelles using amphiphilic poly(isoprene)-block-poly(ethylene oxide) (PI-b-PEO) polymers and showed that parameters such as the ratio of PI-b-PEO/QD,20 size of PI-b-PEO,21 chemical reactions with radical initiators11 and polymerizable monomers e.g. styrene and divinylbenzene22 influence the accessibility of various ions to the QDs. This encapsulation technique was also used to transfer elongated CdSe/CdS quantum dots in quantum rods (QDQRs) into water, preventing their extraordinary fluorescent properties.23,24 Thereby the PI-b-PEO forms spherical micelles surrounding the QDQRs.24,25 However, due to their elongated shape and curvature, the QDQR may hinder the formation of a perfectly dense micelle, which will be addressed here investigating the particle accessibility using a detailed study of the fluorescence quenching of encapsulated QDQRs on particle ensembles and on single particle level. The PL quenching can directly be attributed to the diffusion of PL quenchers in the NP shell and, consequently, allows to study the influences of chemical cross-linking reactions in the QDQRs ligand shell. QDQRs (length: 30 nm, aspect ratio 8.4, ESI Fig. 1†) were transferred to water using two PI-b-PEOs of different molecular weights (PI-b-PEO (1): Mn = 4.3 kDa, Mw = 4.6 kDa and (2): Mn = 13.6 kDa, Mw = 14.3 kDa, ratio PI/PEO = 1/2), which were chosen because they lead to pronounced differences in the quenching behaviour of encapsulated QDs.21 Prior to phase transfer the QDQRs were coated with PI-DETA (Mn = 1.3 kDa, Mw = 1.3 kDa). Then Polymer and AIBN were added and the solution was transferred to water and cross-linked for 4 hours at 80 °C. The ratio of PI-b-PEO/QDQRs was 1800/1, because lower excesses lead in formation of clustered QDQRs.25
As expected, the encapsulated QDQRs exhibit different hydrodynamic sizes in water. Fig. 1A shows the DLS volume distribution maximum of (1) and (2) at the size of 28 nm and 44 nm, respectively (intensity distribution and comments on the precision of determining the NPs size is given in the ESI†). We attribute the hydrophobic PI polymer region as the key of efficient shielding from the environment,11,20–22,26,27 while similar hydrophobic coatings are known to be of significant importance for shielding and biological fate of gold NPs.16,28 The hydrophobic part is also a unique feature with respect to other QD surface coatings which provides substantial PL of the QD, even under copper catalysed 1,3-dipolar Huisgen cycloaddition conditions.20–22,29–32 Based on the typical C–C bond length, we estimate the upper limit for the PI-block length in (1) and (2) to be 5.8 nm and 16.2 nm, respectively.
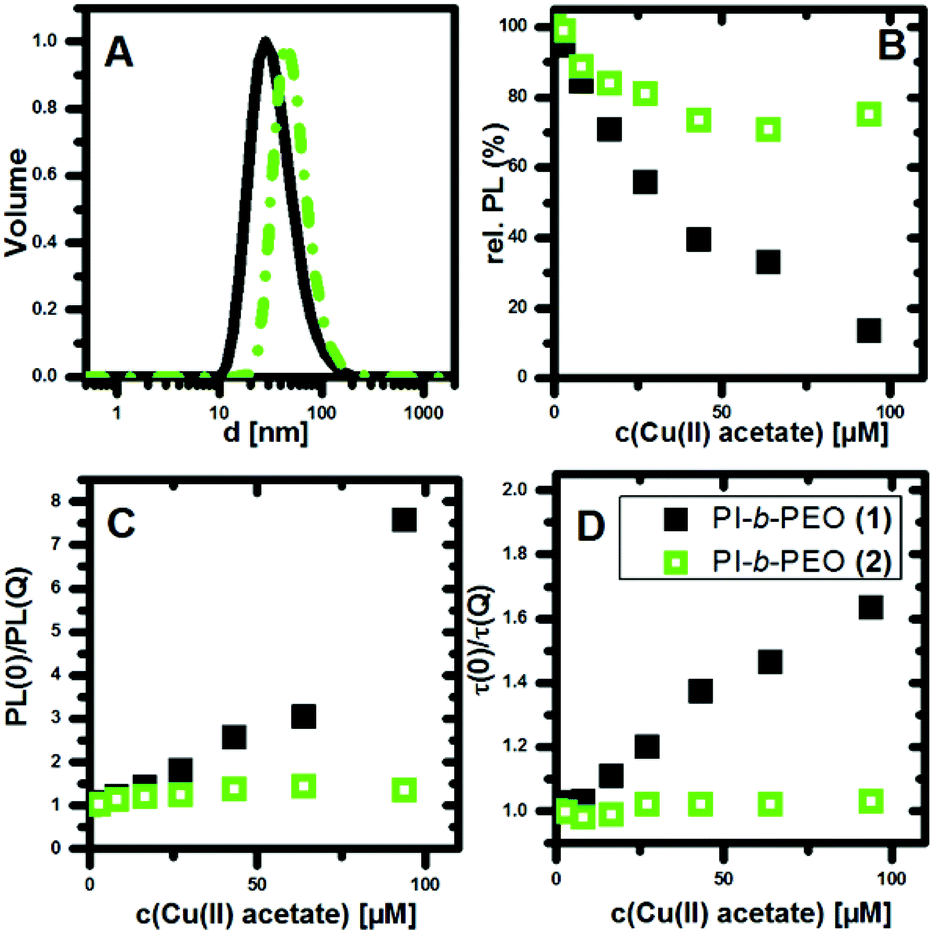 | ||
| Fig. 1 A: Normalized volume weighted size distribution of the encapsulated QDQRs with two different polymer coatings (thin coating: black solid line, thick coating: green dotted line). B: Relative fluorescence Intensity of encapsulated QDQRs (100 nM) with PI-b-PEO (1) (black filled squares) and PI-b-PEO (2) (green empty squares) diblock copolymer while incubation with copper(II) acetate in water. The addition of the copper(II) acetate was proceeded subsequently and between each measurement an equilibration time of 15 min was used. C: Stern–Volmer plot of the combined quenching, D: Stern–Volmer plot of the average fluorescence lifetime. | ||
We exposed the encapsulated QDQRs (100 nM) to different concentrations of copper(II) acetate in a cuvette and measured the relative PL-intensity and the respective PL decay curves. The PL of semiconductor NP is very sensitive to Cu(II) ions, which result in an efficient PL quenching upon contact of semiconductor NP and Cu(II) ion.29,30,32,33 The contact of one single Cu(II) ion can quench the PL of a semiconductor nanocrystal, as demonstrated for the case of CdS QDs.34 It is believed that the adsorbed Cu(II) is reduced to Cu(I), creating a sub band gap below the conduction band of the QD, leading to trap emission in the long wavelength region, thus quenching the PL.34 Here we observe that the PL of QDQRs with native ligands is also instantaneously quenched upon Cu(II) addition (ESI Fig. 8†), however without rise of surface defect trap emission.20,21
In general, fluorescence quenching is classified into two mechanisms. The static quenching is caused by the formation of a non-fluorescent complex with the quencher and the PL lifetime τ of the unaffected fluorophores is unchanged. In contrast, the dynamic quenching, resulting from dynamic encounters between the fluorophore and quencher, shortens the PL lifetime.35
Here, we use the distinction between these two processes to study the ion permeability of QDQR surrounding polymer membranes. Because Cu(II) ions in direct contact to the semiconductor NP result in complete PL quenching, we interpret the occurrence of static quenching by diffusion of Cu(II) through the polymer shell to the QDQRs surface.21 Contrary, the dynamic PL quenching process can be attributed to either electron or energy36 transfer from QDQRs to Cu(II) ions diffusing in the vicinity of the QDQRs. Albeit the exact mechanism of this dynamic quenching remains unclear, both energy- (Förster radii of approx. 2 nm)36 and electron transfer processes require proximity between QDQRs and the quenchers and thus penetration of Cu(II) into the hydrophobic PI-part of the micelle.
As shown in Fig. 1B, the PL intensity of the QDQRs encapsulated with the smaller polymer (1) is reduced significantly upon Cu(II) addition, whereas the PL of the QDQRs encapsulated with the larger polymer (2) is reduced only by 20% and then reaches a saturation plateau. The Stern–Volmer formalism is a useful tool to describe this fluorescence quenching process (eqn (1)), where the Stern–Volmer-constant KSV is a measure for the efficiency of the PL quenching and [Q] the concentration of the quencher (Fig. 1C).37,38
 | (1) |
The dynamic quenching may be expressed by eqn (2) where τ(0) and τ(Q) are the fluorescence lifetimes in absence and presence of the quencher, respectively. Kdyn is the Stern–Volmer constant for dynamic quenching.
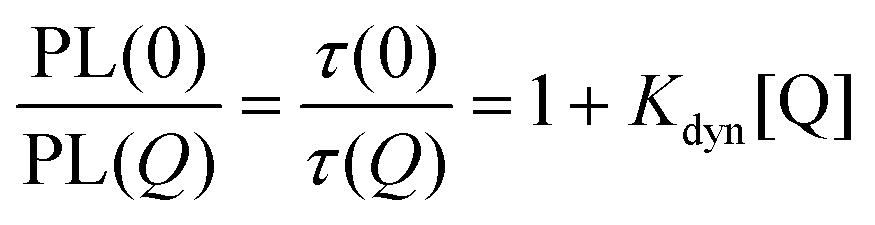 | (2) |
The Stern–Volmer plots of the PL intensity and the fluorescence lifetime are shown in the Fig. 1C and D. These highlight the fact, that substantial PL-quenching is only observed for QDQRs encapsulated with the small polymer (1). Especially no dynamic quenching occurs when the QDQRs are surrounded with PI-b-PEO (2), manifested by constant τ(0)/τ(Q) values, where PI-b-PEO (1) coated QDQRs exhibit subsequent reduction of the PL-lifetime when Cu(II) is added (Fig. 1D). At the highest quencher concentration (c(Cu(II) = 90 μM)) approx. 75% of the PI-b-PEO (1) encapsulated QDQRs are quenched statically, while those who remain luminescent exhibit shorter PL lifetimes. Moreover, 20% of the particles which are surrounded by PI-b-PEO (2) are quenched statically, while the rest is utmost perfectly shielded from the Cu(II) influence.
As a further proof, we conducted confocal fluorescence microscopy on QDQRs which were deposited on a glass cover slide mounted at the bottom of a flow cell. A detailed description of the procedure can be found in the ESI.† Due to the fact that water is not an optimal solvent for this experimental setup,39 we used PEO300 and ensured that the shielding trend reported above is also present in the ensemble quenching assay with PEO300 as the solvent. Then, we spin-coated a diluted nanoparticle solution to ensure sufficient interparticle distance for single particle detection. Signals from objects with more than one encapsulated QDQR were omitted based on the spectral width of the associated emission spectra (ESI). The excitation power was kept at 14 nW in the focal volume to avoid multiphoton processes.25,39 This investigation confirmed the ensemble PL-quenching assay, where some QDQRs vanished instantaneously, while others showed neither interaction with the quencher nor a reduced PL-lifetime, respectively. Representative fluorescence time traces of single encapsulated QDQRs before and after the addition of copper(II) acetate are shown in ESI Fig. 6,† which make reliable blinking analysis not possible.
The investigation of the PL lifetimes, however, gives insight into the diffusion of Cu(II) through the polymer membranes (1) and (2). Histograms of the average PL lifetime in the absence (Fig. 2A and B) and presence (Fig. 2C and D) of Cu(II) clearly revealed a reduction of the PL lifetime only for QDQRs coating with polymer shell (1) indicating substantial influence of the Cu(II) ions. The thick polymer shell (2) prevents this dynamic quenching mechanism.
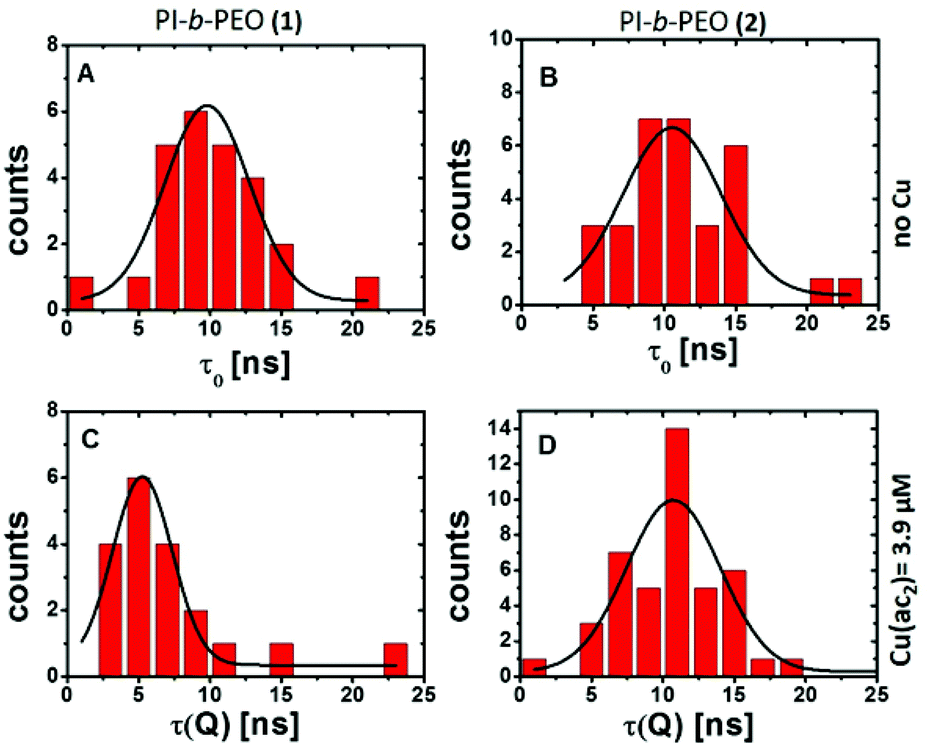 | ||
| Fig. 2 Histogram (binning 1 ns) of the average fluorescence lifetimes before (τ0, A, B) and after (τ(Q), C, D) the addition of copper(II) acetate to the QDQRs encapsulated with the PI-b-PEO (1) (left A, C) and the PI-b-PEO (2) (right B, D) and the associated fit (fitting parameters given in Table ESI 1†). | ||
This can only be explained by close proximity between QDQRs and Cu(II) (typically <1 nm for electron transfer), indicating Cu(II) penetration into the hydrophobic PI part of the micelle of PI-b-PEO (1). This may be due to imperfections in the ligand shell, such as nanopores or holes, which allow the accumulation of Cu(II) but prevent direct surface access.
This is in agreement with a recent report,21 where three different species of surface coatings of QDs were identified: dense coatings, which shield the particles well from the influence of Cu(II), permeable coatings which allow Cu(II) diffusion to the semiconductor surface, and partial permeable membranes, which prevent static, but allow dynamic quenching. The proportion of the latter two is highest, when small polymers (PI-b-PEO (1)) are used for encapsulation, while dense coatings are predominant, when QDs are encapsulated in PI-b-PEO (2).21 The here presented results underline these assumptions, which are also supported by the widefield microscopy investigation presented in the ESI.†
The PI block length of approximately 4.3 kDa (PI-b-PEO (2)) can be seen as a critical molar mass for effective shielding. We also notice that this block length, with addition of the PI-DETA block, is similar to the reported entanglement length of PI of 5.5 kDa.40 We, therefore, explain the different shielding by different coiling of the PI chains of PI-DETA and PI-b-PEO. Obviously, the PI chains interpenetrate as almost linear chains when PI-b-PEO (1) is used, which results in free space, where Cu(II) can accumulate (see Scheme 1). On the contrary, a more effective and flexible interaction occurs, when the PI moieties of PI-b-PEO (2) can entangle among each other and the PI-DETA (see Scheme 1), which may result in a coiled, perfectly dense PI-region surrounding the QDQRs. This prevents the accumulation of Cu(II) in the membrane and thus the dynamic quenching reaction. It is reasonable that in the latter case a more homogeneous shrinking of the PI shell may compensate contraction during cross-linking, whereas percolation with nanopore formation results in the more stiff arrangement of the PI chains.
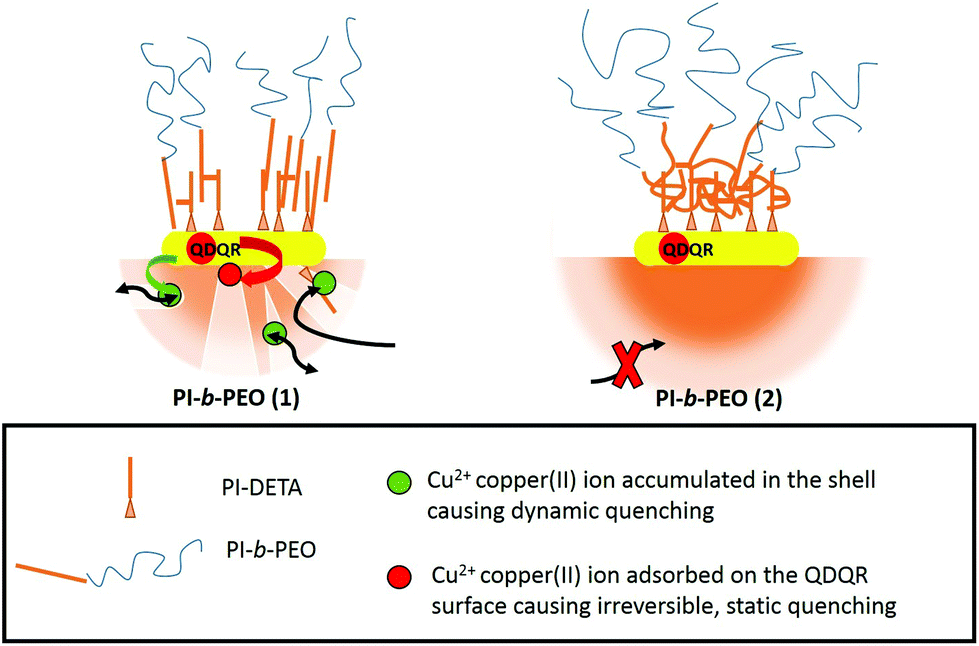 | ||
| Scheme 1 Schematic representation of the different polymer membranes surrounding the QDQRs encapsulated with PI-b-PEO (1) (left) and (2) (right). The upper part schematizes the different arrangement of the PI-DETA and PI-b-PEO, while the lower part shows the polymer membranes with or without nano-pores and holes. | ||
As a first and very brief estimation, we used the Stern–Volmer constant for dynamic quenching as a measure for the presence of Cu(II) in close distance to the QDQR and determined the diffusion coefficient of Cu(II) in the nanopores to be approx. 3 × 10−7 cm2 s−1, which is 40 times slower than ion diffusion in water (equation and assumptions given in the ESI†).41 The dynamic quenching reaction is similar to PI-b-PEO (1) encapsulated QDs, which indicates, that the membrane structure around QDs and QDQRs does not differ significantly.21
For a better understanding of the parameters determining the sample homogeneities and the intermolecular cross-linking, we investigated the influence of a variety of criteria, such as the polymer excess during encapsulation, the crosslinking time using AIBN, and a subsequent emulsion polymerization step with styrene and divinylbenzene on the shielding of the QDQRs.11,21,22 We added 800 equivalents of copper(II) acetate to a solution of PI-b-PEO (2) encapsulated QDQRs (c = 100 nM) to study only the particles of higher Cu(II) resistivity. We followed the temporal evolution of the quenching by acquisition of a variety of spectra during the first minutes and on longer time scales of up to one week. This allows to analyse the long term shielding of these particles, which may be of interest for in vivo application.42 Additionally protons and hydroxyl ions were used as PL quenchers to model different environments. Details on the experimental procedure and PL-intensity plots can be found in the ESI.†
Cross-linking of the micelles is essential to increase the stability against quenchers (ESI Fig. 8†). The temporal evolution of the relative PL intensity of QDQRs, which were cross-linked for various times during the encapsulation process, in the presence of 800 equivalents Cu(II) is shown in Fig. 3. Cross-linking reactions times of 4–10 h are necessary to form a dense PI-region and effectively shield the particles. For the EP similar shielding is achieved by reaction times of at least 1 hour (ESI Fig. 10 and 11†).
 | ||
| Fig. 3 Relative PL intensity upon the addition of 800 eq. copper(II) acetate of QDQRs encapsulated with PI-b-PEO (2) under different cross-linking times using AIBN as a radical initiator. The different cross-linking times are colour- and symbol-coded: 0 min (filled black squares), 30 min (empty red spheres), 2 h (filled blue stars), 3 h (empty olive triangles), 4 h (filled green diamonds), 5 h (empty dark-blue pentagons), 10 h (filled grey triangles). | ||
The main difference between the EP and the radically initiated AIBN cross-linkage is observed at short reaction times, where only the EP provides sufficient shielding against protons (ESI Fig. 9 and 11†). This can be attributed to the different nature of the polymerizable unit and the radical source, where reaction times can be considered comparable under the reaction conditions [respective 10 hour half times provided by the suppliers: AIBN 65 °C (Treaction = 80 °C), VA-044 44 °C (Treaction = 60 °C)]: AIBN initiates the cross-linkage of two neighbouring isoprene double bounds, while during the EP new material is brought into the QDQR shell. Imperfections in the ligand shell are likely to have a local lack of polymer ligands, which may hinder their cross-linkage. Contrarily, styrene and divinylbenzene swell the micelle during the initial step of the EP and their polymerization is initiated by oligomer radicals, which enter the seeds after being formed in solution.43 So, the better shielding after short reactions times of the seeded emulsion polymerization samples may be due to the diffusion of styrene and divinylbenzene to the imperfections in the shell. Their subsequent polymerization creates a rigid and hydrophobic polymer network, which closes the respective imperfection and hinders quencher diffusion towards the QDQR surface.
Conclusions
We report a Cu(II) ion and proton accessibility study of polymer encapsulated QDQRs based on fluorescence quenching analysis. This sensitive assay allows us to study the diffusion of Cu(II) in nanoscale membranes in ensemble and on single particles. It revealed that accessibility is favoured, when a small polymer surrounds the QDQR. The occurrence of two different PL quenching mechanisms allows distinguishing between QDQRs surrounded by perfect and imperfect shells, which exhibit holes or nano-pores. The accessibility of elongated QDQRs can be fine-tuned by chemical cross-link reactions in the surrounding polymer layer and the influence of these reactions can also be tracked using the same PL quenching assay.Acknowledgements
This work was supported by the Chemical Industry Fund, VCI: German Chemical Industry Association, and the German–American Fulbright Program. The authors thank Dr. Alexey Chernikov (Columbia University, New York City), Julian Miranda (Florida State University, Tallahassee) and Prof. Dr. Volker Abetz and Dr. Son C. Nguyen (University of Hamburg) for fruitful discussions. Further, we thank Daniel Meyer for the illustration of the TOC graphic.References
- T. C. Merkel, B. D. Freeman, R. J. Spontak, Z. He, I. Pinnau, P. Meakin and A. J. Hill, Science, 2002, 296, 519–522 CrossRef CAS PubMed.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- D. Anselmetti and A. Gölzhäuser, Angew. Chem., Int. Ed., 2014, 53, 12300–12302 CAS.
- D. Anselmetti and A. Gölzhäuser, Angew. Chem., 2014, 126, 12498–12500 CrossRef.
- R. Ishimatsu, J. Kim, P. Jing, C. C. Striemer, D. Z. Fang, P. M. Fauchet, J. L. McGrath and S. Amemiya, Anal. Chem., 2010, 82, 7127–7134 CrossRef CAS PubMed.
- B. Pelaz, G. Charron, C. Pfeiffer, Y. Zhao, J. M. de la Fuente, X.-J. Liang, W. J. Parak and P. del Pino, Small, 2013, 9, 1573–1584 CrossRef CAS PubMed.
- I. Corsi, G. N. Cherr, H. S. Lenihan, J. Labille, M. Hassellov, L. Canesi, F. Dondero, G. Frenzilli, D. Hristozov, V. Puntes, C. Della Torre, A. Pinsino, G. Libralato, A. Marcomini, E. Sabbioni and V. Matranga, ACS Nano, 2014, 8, 9694–9709 CrossRef CAS PubMed.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- G. Palui, F. Aldeek, W. T. Wang and H. Mattoussi, Chem. Soc. Rev., 2015, 44, 193–227 RSC.
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed.
- C. Schmidtke, H. Lange, H. Tran, J. Ostermann, H. Kloust, N. G. Bastús, J.-P. Merkl, C. Thomsen and H. Weller, J. Phys. Chem. C, 2013, 117, 8570–8578 CAS.
- W. Wang, A. Kapur, X. Ji, M. Safi, G. Palui, V. Palomo, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 5438–5451 CrossRef CAS PubMed.
- Y. Que, C. Feng, S. Zhang and X. Huang, J. Phys. Chem. C, 2015, 119, 1960–1970 CAS.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- S. Schlücker, Angew. Chem., 2014, 126, 4852–4894 CrossRef.
- T. A. Larson, P. R. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182–9190 CrossRef CAS PubMed.
- J. Ostermann, C. Schmidtke, C. Wolter, J.-P. Merkl, H. Kloust and H. Weller, Beilstein J. Nanotechnol., 2015, 6, 232–242 CrossRef PubMed.
- D. Maiolo, P. Del Pino, P. Metrangolo, W. J. Parak and F. Baldelli Bombelli, Nanomedicine, 2015, 10, 3231–3247 CrossRef CAS PubMed.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- J.-P. Merkl, J. Ostermann, C. Schmidtke, H. Kloust, R. Eggers, A. Feld, C. Wolter, A.-M. Kreuziger, S. Flessau, H. Mattoussi and H. Weller, in Proc. of SPIE 8955, ed. W. J. Parak, M. Osinski and K. I. Yamamoto, 2014, vol. 8955, p. 89551X Search PubMed.
- J. Ostermann, J.-P. Merkl, S. Flessau, C. Wolter, A. Kornowksi, C. Schmidtke, A. Pietsch, H. Kloust, A. Feld and H. Weller, ACS Nano, 2013, 7, 9156–9167 CrossRef CAS PubMed.
- H. Kloust, C. Schmidtke, J.-P. Merkl, A. Feld, T. Schotten, U. E. A. Fittschen, M. Gehring, J. Ostermann, E. Pöselt and H. Weller, J. Phys. Chem. C, 2013, 117, 23244–23250 CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- J. Dimitrijevic, L. Krapf, C. Wolter, C. Schmidtke, J.-P. Merkl, T. Jochum, A. Kornowski, A. Schüth, A. Gebert, G. Hüttmann, T. Vossmeyer and H. Weller, Nanoscale, 2014, 6, 10413–10422 RSC.
- M. Rafipoor, C. Schmidtke, C. Wolter, C. Strelow, H. Weller and H. Lange, Langmuir, 2015, 31, 9441–9447 CrossRef CAS PubMed.
- A. Feld, J.-P. Merkl, H. Kloust, S. Flessau, C. Schmidtke, C. Wolter, J. Ostermann, M. Kampferbeck, R. Eggers, A. Mews, T. Schotten and H. Weller, Angew. Chem., Int. Ed., 2015, 54, 12468–12471 CrossRef CAS PubMed.
- A. Feld, J.-P. Merkl, H. Kloust, S. Flessau, C. Schmidtke, C. Wolter, J. Ostermann, M. Kampferbeck, R. Eggers, A. Mews, T. Schotten and H. Weller, Angew. Chem., 2015, 127, 12645–12648 CrossRef.
- F. Schulz, T. Vossmeyer, N. G. Bastús and H. Weller, Langmuir, 2013, 29, 9897–9908 CrossRef CAS PubMed.
- C. Schieber, A. Bestetti, J. P. Lim, A. D. Ryan, T. Nguyen, R. Eldridge, A. R. White, P. A. Gleeson, P. S. Donnelly, S. J. Williams and P. Mulvaney, Angew. Chem., 2012, 124, 10675–10679 CrossRef.
- C. Schieber, A. Bestetti, J. P. Lim, A. D. Ryan, T.-L. Nguyen, R. Eldridge, A. R. White, P. A. Gleeson, P. S. Donnelly, S. J. Williams and P. Mulvaney, Angew. Chem., Int. Ed., 2012, 51, 10523–10527 CrossRef CAS PubMed.
- A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffé and I. Texier, Bioconjugate Chem., 2010, 21, 583–588 CrossRef CAS PubMed.
- G. G. Beaune, S. Tamang, A. Bernardin, P. Bayle-Guillemaud, D. Fenel, G. Schoehn, F. Vinet, P. Reiss and I. Texier, ChemPhysChem, 2011, 12, 2247–2254 CrossRef CAS PubMed.
- A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffé and I. Texier, Bioconjugate Chem., 2010, 21, 583–588 CrossRef CAS PubMed.
- A. V. Isarov and J. Chrysochoos, Langmuir, 1997, 13, 3142–3149 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag, Heidelberg, 4th edn, 2006 Search PubMed.
- J. U. Sutter, D. J. S. Birch and O. J. Rolinski, Meas. Sci. Technol., 2012, 23, 55103 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science+Buisness Media, New York, 3rd edn, 2006 Search PubMed.
- O. Stern and M. Volmer, Phys. Zeitschrift, 1919, 20, 183–188 CAS.
- S. Flessau, C. Wolter, E. Pöselt, E. Kroger, A. Mews and T. Kipp, Phys. Chem. Chem. Phys., 2014, 16, 10444–10455 RSC.
- L. J. Fetters, D. J. Lohse and R. H. Colby, Physical Properties of Polymers Handbook, Springer New York, New York, NY, 2007 Search PubMed.
- M. Smoluchowski, Ann. Phys., 1915, 48, 1103–1112 Search PubMed.
- W. G. Kreyling, A. M. Abdelmonem, Z. Ali, F. Alves, M. Geiser, N. Haberl, R. Hartmann, S. Hirn, D. J. de Aberasturi, K. Kantner, G. Khadem-Saba, J.-M. Montenegro, J. Rejman, T. Rojo, I. R. de Larramendi, R. Ufartes, A. Wenk and W. J. Parak, Nat. Nanotechnol., 2015, 10, 619–623 CrossRef CAS PubMed.
- J. W. Vanderhoff, J. F. Vitkuske, E. B. Bradford and T. Alfrey, J. Polym. Sci., 1956, 20, 225–234 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, particle characterization, data acquisition, widefield images, representative PL time traces, extracted stretched exponential factors, long term PL stability assessment at different pH values and in the presence of quenching Cu(II) ions. See DOI: 10.1039/c5nr08318d |
| This journal is © The Royal Society of Chemistry 2016 |