Design, synthesis and anticancer activities of novel otobain derivatives†
Received
22nd October 2015
, Accepted 17th November 2015
First published on 17th November 2015
Abstract
A series of novel racemic otobain derivatives was designed and synthesised using 2-piperonyl-1,3-dithianes in the conjugate addition–alkylation to 5H-furan-2-one, followed by cationic cyclisation. All the synthesised compounds were consequently evaluated for their anticancer activity against several human cancers in vitro. The efficacy of the most active compound 27g was comparable with etoposide, with IC50 values ranging from 1.06 μM to 4.16 μM in different cancer cell lines. Notably, compound 27g strongly induced cell cycle arrest and increased the expression of mitosis-specific markers MPM-2 and phosphorylated histone H3, but it did not trigger cell apoptosis. Further a colony formation assay showed that compound 27g effectively inhibited the anchor growth of lung cancer cells in a dose-dependent manner. More importantly, compound 27g at 40 mg kg−1 significantly suppressed tumour volume (P < 0.01) and tumour weight (P < 0.05) in a human lung cancer cell xenograft mouse model without causing systematic toxicity in mice. Our findings indicated that compound 27g has significant potential for further drug development.
Introduction
Cancer is a serious threat to human health. Approximately 14.1 million new cases of cancer were diagnosed worldwide in 2012. The number of new cases could further rise to 19.3 million by 2025.1 Although current therapies are effective in treating early stages of cancer, the survival rates of cancer remain limited.2 Therefore, the search for promising new approaches to treat this deadly disease is an urgent need. Among these approaches, natural compounds have been considered as a good source of novel anticancer drugs.
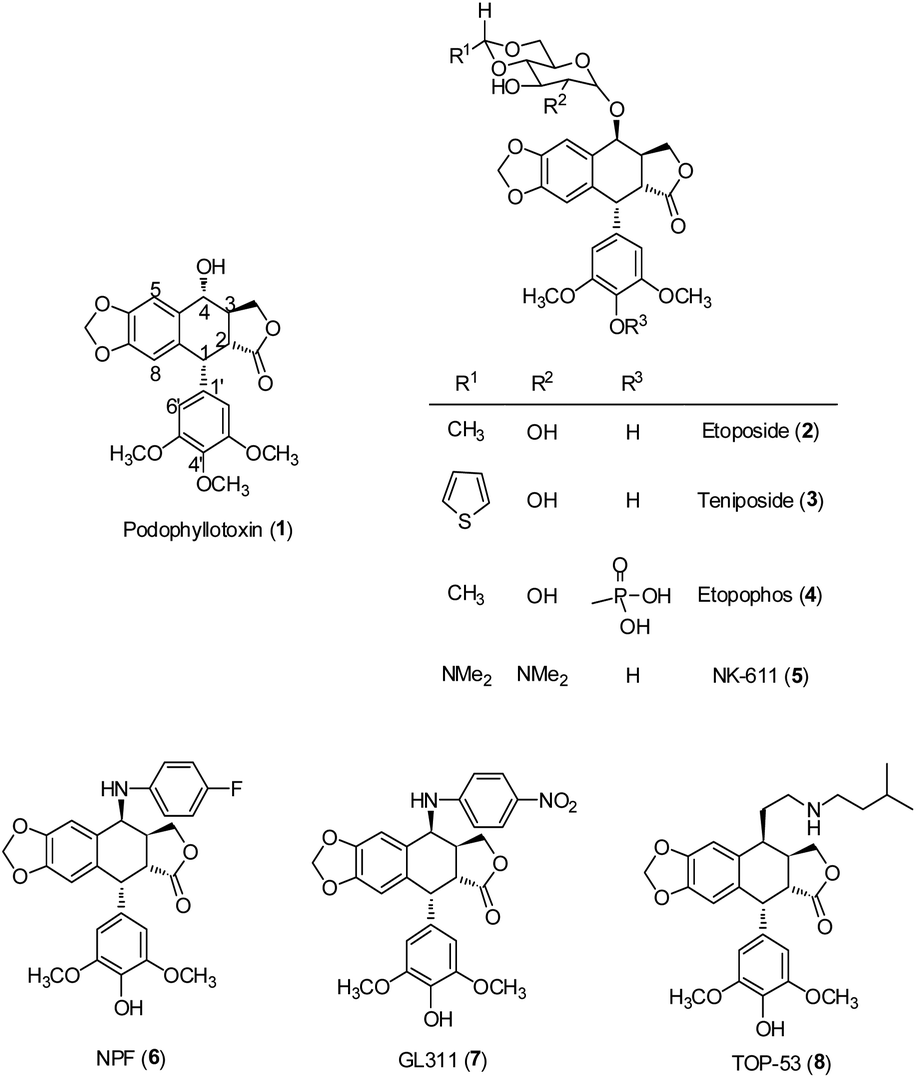
Lignans, a class of phenylpropanoid compounds widely distributed in the plant kingdom, have generated particular interest because of their broad range of biological activities, including antitumor,3–6 anti-inflammatory,6,7 cardiovascular,8 antioxidant,9–11 neuroprotective effects,11 antiviral actions12–14 and anti-liver fibrotic activities.15 The best known antitumor lignan is podophyllotoxin (PPT, 1), a bioactive component of Podophyllum peltatum and Podophyllum hexandrum.16 Its semisynthetic derivatives, including etoposide (VP-16, ETO, 2), teniposide (3) and etopophos (4) (Fig. 1), have been developed as clinically useful medicines against various cancers.17–19 However, their high toxicity, low water solubility and drug resistance have limited their application in cancer chemotherapy.20 To obtain better therapeutic agents, a great number of podophyllotoxin analogues have been synthesized. Among them, NK611 (5), NPF (6), GL-311 (7) and TOP53 (8) (Fig. 1) are presently under clinical trials. The clinical success of podophyllotoxin derivatives has stimulated great interest in the exploration of new lignans with improved antitumor activity.
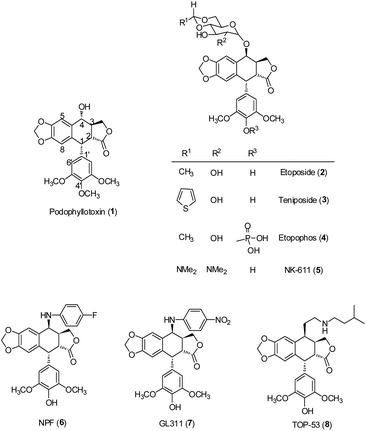 |
| | Fig. 1 Structures of PPT and related compounds. | |
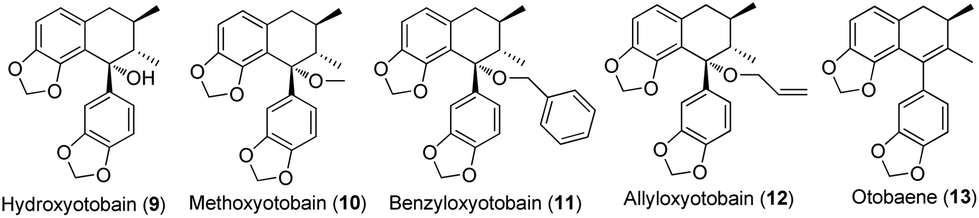
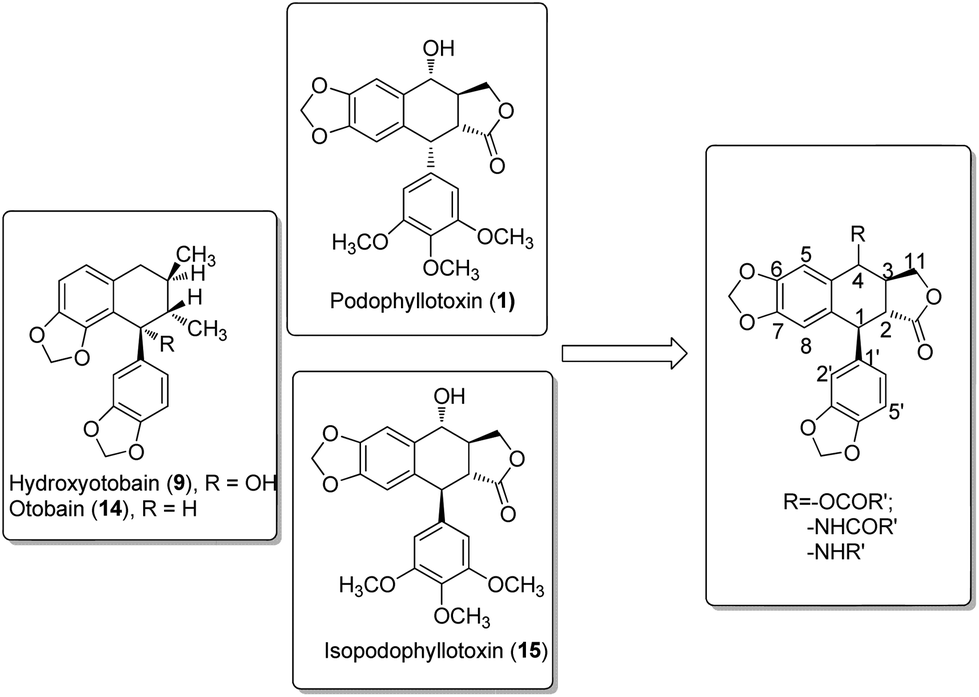
In our investigation on the bioactive components from the stem barks of Myristica fragrans Houtt, we isolated one lignan, hydroxyotobain (9) (Fig. 2) in 0.50% yield from ethanolic extracts.21 Preliminary structural modification of hydroxyotobain obtained methoxyotobain (10), benzyloxyotobain (11), allyloxyotobain (12) and otobaene (13) (Fig. 2).22 These compounds showed some anticancer activity, but their levels were still not ideal. On the one hand, considering that the hydroxy of hydroxyotobain was prone to an elimination reaction, otobain (14) was then designed. In addition, studies have shown that trans-fused γ-lactone is essential for the anticancer activity of podophyllotoxin derivatives.23,24 Therefore, the trans-fused γ-lactone was introduced to otobain. On the other hand, a large number of previous studies on podophyllotoxin derivatives focused on C-4 position modification,25 but very little work has been directed to the biological activity of isopodophyllotoxin (15) with the relative stereochemistry at positions 1, 2 and 3 in (1,2–2,3)-trans–trans. Recently, Thanh et al. reported that a novel aryl-tetralin lignan, cleistantoxin, which was isolated from the fruits of Cleistanthus indochinensis, shows potent activities against several human cancers in vitro.3,26 The structure of cleistantoxin is interesting as the relative stereochemistry of cleistantoxin at positions 1, 2 and 3 is similar to that of podophyllotoxin, whereas the benzodioxole structure of cleistantoxin at position 1 is similar to that of otobain. According to some literature reports on podophyllotoxin derivatives, we find that some podophyllotoxin ester, amide and amine derivatives show good anticancer activity.27–30 From these studies, we recently synthesised 20 novel racemic otobain ester, amide and amine derivatives (Fig. 3), which were stereochemically preferential compounds with a known synthetic route (Scheme 1). These compounds were evaluated for their cytotoxicities against a panel of human cancer cell lines and human immortal keratinocyte cells.
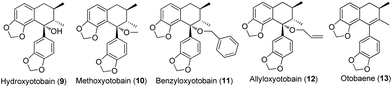 |
| | Fig. 2 Structures of hydroxyotobain and related compounds. | |
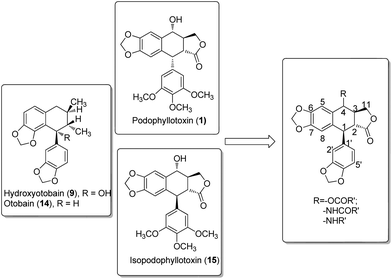 |
| | Fig. 3 Structures of synthesized otobain derivatives. | |
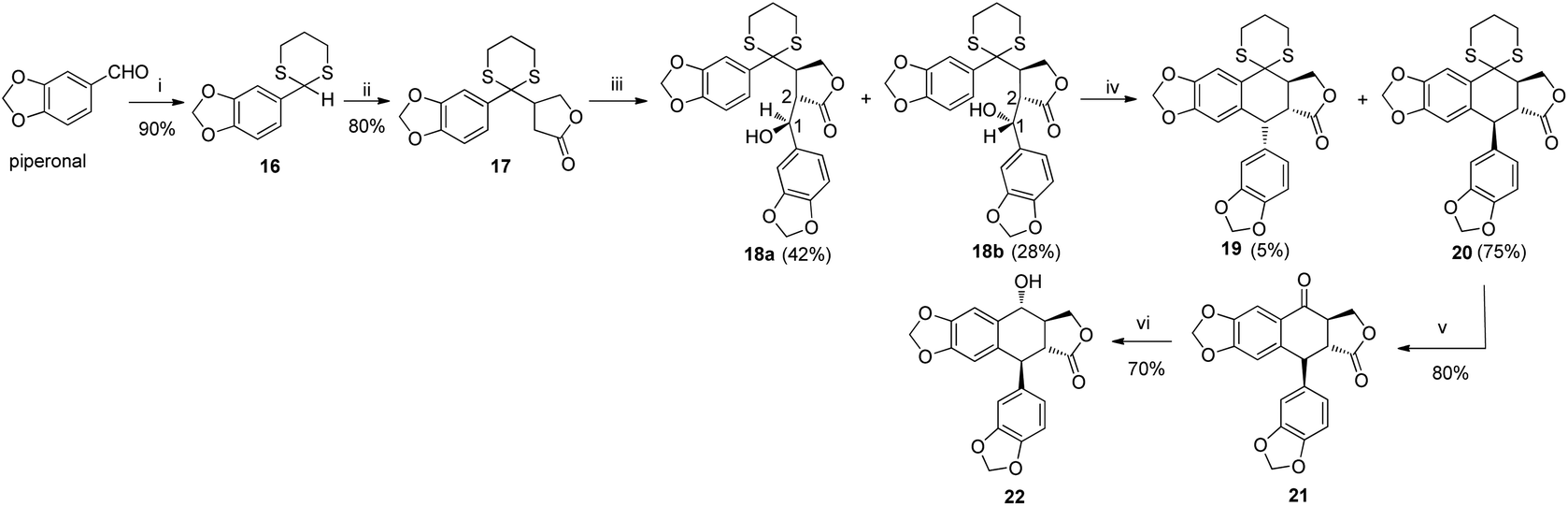
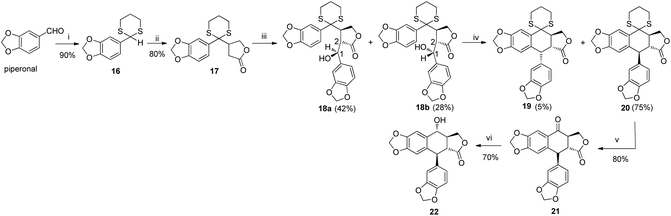 |
| | Scheme 1 Synthesis of 22. Reagents and conditions: (i) SH (CH2)3SH, p-TsOH, DCM, rt; (ii) n-BuLi, furan-2(5H)-one, THF, −78 °C; (iii) LDA, piperonal, THF, −78 °C; (iv) TFA, DCM, rt; (v) HgO, BF3·Et2O, THF/H2O (85/15), 0 °C–rt; (vi) NaBH4, THF, 0 °C. | |
Results and discussion
Chemistry
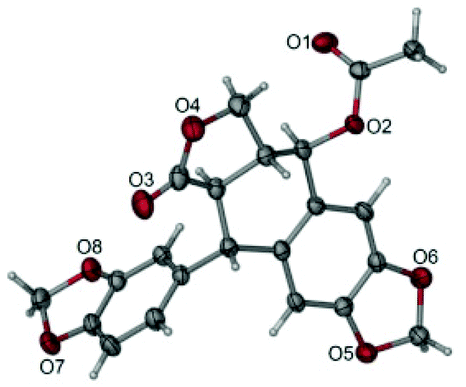
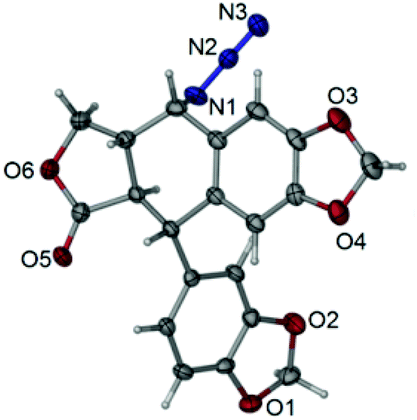
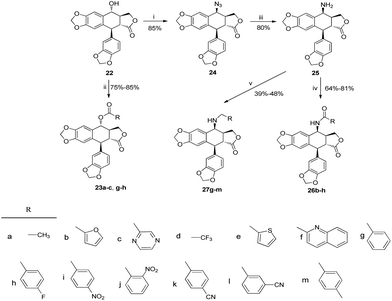
The total synthesis of otobain 22 was carried out by the synthetic sequence illustrated in Scheme 1. Compound 16 was prepared from piperonal by employing p-TsOH and 1,3-propanedithiol in excellent yield. Treatment of compound 16 with n-butyllithium by the addition of 2(5H)-furanone gave rise to compound 17 with good yield. The enolate derived from compound 17 by treating 17 with lithium diisopropylamide was captured by piperonal, which led to a mixture of diastereomers of compound 18 in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. Erythro18a and threo18b are distinguished based on the 1H NMR chemical shift of H-1 and JH1–H2 coupling constants (erythro: δH1 > 5.12 ppm, JH1–H2 (2–4 Hz); threo: δH1 ≤ 4.95 ppm, JH1–H2 (6–9 Hz)).31–33 Cyclisation of compound 18 under TFA in DCM yielded a mixture of compound 19 (cis–trans, podophyllotoxin-type; minor) and compound 20 (trans–trans, isopodophyllotoxin-type; major; JH1–H2 and JH2–H3 >10 Hz) in a 1:15 ratio. These results were consistent with those usually described for such a cyclisation reaction.34 The stereochemically preferential compound 20 was selected for use in subsequent syntheses. Deprotection of compound 20 with HgO and BF3·Et2O gave the ketoderivative 21 with no change in the relative stereochemistry at positions 1, 2 and 3 (JH1–H2 and JH2–H3 >10 Hz).34 Reduction of ketone 21 with NaBH4 afforded alcohol 22 in 70% yield. The assignment of relative stereochemistry for C-4 on compound 22 was based on JH3–H4 coupling constants: JH3–H4 ≥10 Hz indicates that H-3 and H-4 are in a trans relationship,35 which was further confirmed by the X-ray crystal of ester 23a (Fig. 4).36 The synthesis of otobain analogues 23a–c, g–h, 26b–h and 27g–m is described in Scheme 2. Compounds 23a–c and g–h were obtained in good yields by the reaction of compound 22 with the corresponding carboxylic acids in the presence of N,N′-dicyclohexylcarbodiimide and 4-(N,N-dimethylamino)pyridine. Intermediate 24 was prepared from compound 22 by employing the TFA/NaN3 reagent system. The assignment of relative stereochemistry for C-4 on compound 24 was based on JH3–H4 coupling constants: JH3–H4 ≈ 4 Hz indicates that H-3 and H-4 are in a cis relationship.35 The precise 3D structural information on compound 24 was determined by single-crystal X-ray diffraction as illustrated in Fig. 5.36 Compound 24 was then reduced under a hydrogen atmosphere with a Pd/C catalyst to yield the key intermediate 25. Compound 25 was coupled with various chlorides or anhydrides in dichloromethane to obtain compounds 26b–h in good yields. Compounds 27g–m were obtained in 39%–48% yields by the coupling compound 25 with the corresponding benzyl iodide. The structures of the target compounds were characterised by 1H NMR and 13C NMR spectral analyses.
:2 ratio. Erythro18a and threo18b are distinguished based on the 1H NMR chemical shift of H-1 and JH1–H2 coupling constants (erythro: δH1 > 5.12 ppm, JH1–H2 (2–4 Hz); threo: δH1 ≤ 4.95 ppm, JH1–H2 (6–9 Hz)).31–33 Cyclisation of compound 18 under TFA in DCM yielded a mixture of compound 19 (cis–trans, podophyllotoxin-type; minor) and compound 20 (trans–trans, isopodophyllotoxin-type; major; JH1–H2 and JH2–H3 >10 Hz) in a 1:15 ratio. These results were consistent with those usually described for such a cyclisation reaction.34 The stereochemically preferential compound 20 was selected for use in subsequent syntheses. Deprotection of compound 20 with HgO and BF3·Et2O gave the ketoderivative 21 with no change in the relative stereochemistry at positions 1, 2 and 3 (JH1–H2 and JH2–H3 >10 Hz).34 Reduction of ketone 21 with NaBH4 afforded alcohol 22 in 70% yield. The assignment of relative stereochemistry for C-4 on compound 22 was based on JH3–H4 coupling constants: JH3–H4 ≥10 Hz indicates that H-3 and H-4 are in a trans relationship,35 which was further confirmed by the X-ray crystal of ester 23a (Fig. 4).36 The synthesis of otobain analogues 23a–c, g–h, 26b–h and 27g–m is described in Scheme 2. Compounds 23a–c and g–h were obtained in good yields by the reaction of compound 22 with the corresponding carboxylic acids in the presence of N,N′-dicyclohexylcarbodiimide and 4-(N,N-dimethylamino)pyridine. Intermediate 24 was prepared from compound 22 by employing the TFA/NaN3 reagent system. The assignment of relative stereochemistry for C-4 on compound 24 was based on JH3–H4 coupling constants: JH3–H4 ≈ 4 Hz indicates that H-3 and H-4 are in a cis relationship.35 The precise 3D structural information on compound 24 was determined by single-crystal X-ray diffraction as illustrated in Fig. 5.36 Compound 24 was then reduced under a hydrogen atmosphere with a Pd/C catalyst to yield the key intermediate 25. Compound 25 was coupled with various chlorides or anhydrides in dichloromethane to obtain compounds 26b–h in good yields. Compounds 27g–m were obtained in 39%–48% yields by the coupling compound 25 with the corresponding benzyl iodide. The structures of the target compounds were characterised by 1H NMR and 13C NMR spectral analyses.
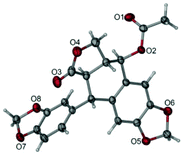 |
| | Fig. 4 X-ray crystal structure of compound 23a. | |
 |
| | Scheme 2 Synthesis of 23a–c, g–h, 26b–h and 27g–m. Reagents and conditions: (i) DCC, DMAP, RCOOH, DCM, 28 °C; (ii) TFA, NaN3, CH3Cl, rt; (iii) H2/Pd–C, ethyl acetate, rt; (iv) RCOCl or (RO)2CO, DMAP, DCM, rt; (v) RCH2I (from RCH2Br + NaI), 1,2-dichloroethane, 75–80 °C. | |
 |
| | Fig. 5 X-ray crystal structure of compound 24. | |
In vitro cytotoxicity assay
Target compounds 23a–c, g–h, 26b–h and 27g–m were evaluated for in vitro cytotoxicity against five human tumour cell lines and one human normal cell line using the SRB assay. Etoposide was used as a reference. The results showed that most of the compounds possessed considerable anticancer activity (Table 1). Lead compound 22 and ester derivatives (23a–c and g–h) showed poor activities. Aromatic heterocyclic esters (23b and 23c) possessed more potent antiproliferative activity than acetates (23a) and aromatic esters (23g and 23h), which implied that aromatic heterocyclic esters were better substituents in the C-4 position of otobain by the ester bond. Except for compounds 26g and 26h, most of the amide derivatives (26b–f) showed promising activity compared with ester derivatives (23a–c and g–h) and lead compound 22. The amine derivatives (27g–m) showed good activity against the HL-60 cancer cell line with IC50 values ranging from 1.06 μM to 15.47 μM, but only moderate activity against other cell lines. Notably, 27g showed potent actions against HL-60 (IC50 = 1.06 μM), SGC-901 (IC50 = 4.16 μM), MCF-7 (IC50 = 2.25 μM), HCT-116 (IC50 = 1.65 μM) and A549 (IC50 = 2.32 μM) cancer cell lines, whereas etoposide showed IC50 values of 0.38, 3.00, 1.68, 2.64 and 2.90 μM in the respective cell lines. Moreover, the cytotoxicity of 27g against HaCat (i.e., IC50 = 1.16 μM) was two-fold higher than that of etoposide (i.e., IC50 = 0.58 μM). These results suggested that the target compounds showed promising anticancer effects and structure–activity relationships.
Table 1 Cytotoxicity of compounds 23a–c, g–h, 26b–h and 27g–m
| Compounds |
IC50 (μmol L−1) |
| HL60a |
SGC-7901b |
MCF-7c |
HCT116d |
A549e |
HaCatf |
|
Human promyelocytic leukemia cells.
Human gastric cancer cells.
Human breast cancer cells.
Human colon carcinoma cell.
Human non-small cell lung cancer cells.
Human skin cell line/keratinocyte. IC50 is shown as mean ± standard deviation. The assays were performed in three independent experiments in triplicate. |
|
23a
|
>100 |
80.91 ± 2.3 |
61.11 ± 0.7 |
>100 |
>100 |
87.75 ± 1.9 |
|
23b
|
53.52 ± 2.7 |
28.95 ± 1.6 |
14.53 ± 0.4 |
41.22 ± 1.5 |
71.00 ± 1.3 |
66.55 ± 1.8 |
|
23c
|
35.57 ± 2.9 |
21.35 ± 1.7 |
17.08 ± 0.2 |
29.41 ± 0.5 |
24.50 ± 4.3 |
16.33 ± 0.4 |
|
23g
|
>100 |
— |
— |
— |
>100 |
— |
|
23h
|
>100 |
— |
— |
— |
>100 |
— |
|
26b
|
3.83 ± 0.3 |
15.22 ± 1.4 |
14.03 ± 1.5 |
11.52 ± 0.8 |
9.80 ± 0.5 |
6.81 ± 0.1 |
|
26c
|
6.71 ± 1.6 |
12.72 ± 0.3 |
16.06 ± 0.5 |
16.82 ± 0.4 |
11.15 ± 0.4 |
7.72 ± 0.2 |
|
26d
|
10.71 ± 0.9 |
36.13 ± 0.3 |
32.13 ± 2.8 |
15.24 ± 0.2 |
21.23 ± 0.6 |
12.39 ± 0.3 |
|
26e
|
7.22 ± 1.4 |
>100 |
47.15 ± 3.5 |
31.86 ± 1.2 |
22.48 ± 0.9 |
7.84 ± 0.2 |
|
26f
|
8.09 ± 0.1 |
73.40 ± 9.2 |
19.66 ± 1.4 |
14.79 ± 0.7 |
17.61 ± 0.7 |
11.49 ± 0.5 |
|
26g
|
>100 |
— |
— |
— |
>100 |
— |
|
26h
|
>100 |
— |
— |
— |
>100 |
— |
|
27g
|
1.06 ± 0.1 |
4.16 ± 0.4 |
2.25 ± 0.2 |
1.65 ± 0.1 |
2.32 ± 0.2 |
1.16 ± 0.1 |
|
27h
|
8.08 ± 0.2 |
26.19 ± 2.1 |
27.89 ± 2.0 |
12.64 ± 0.6 |
17.94 ± 2.0 |
6.42 ± 0.3 |
|
27i
|
4.65 ± 0.1 |
19.81 ± 0.5 |
7.91 ± 0.3 |
10.02 ± 0.3 |
10.21 ± 0.7 |
4.40 ± 0.2 |
|
27j
|
4.51 ± 0.4 |
18.48 ± 1.0 |
5.67 ± 0.1 |
7.73 ± 0.2 |
7.53 ± 0.5 |
3.94 ± 0.2 |
|
27k
|
4.90 ± 0.01 |
43.92 ± 3.4 |
61.44 ± 1.1 |
49.08 ± 5.7 |
20.52 ± 4.3 |
7.47 ± 0.3 |
|
27l
|
15.47 ± 0.6 |
>100 |
>100 |
>100 |
98.60 ± 11.0 |
26.38 ± 0.1 |
|
27m
|
7.16 ± 0.8 |
47.16 ± 4.2 |
41.55 ± 5.6 |
32.68 ± 2.2 |
67.99 ± 2.6 |
8.80 ± 0.2 |
|
22
|
>100 |
— |
— |
— |
>100 |
— |
| Etoposide |
0.38 ± 0.01 |
3.00 ± 0.3 |
1.68 ± 0.1 |
2.64 ± 0.1 |
2.90 ± 0.4 |
0.58 ± 0.02 |
Compound 27g arrests cell cycle
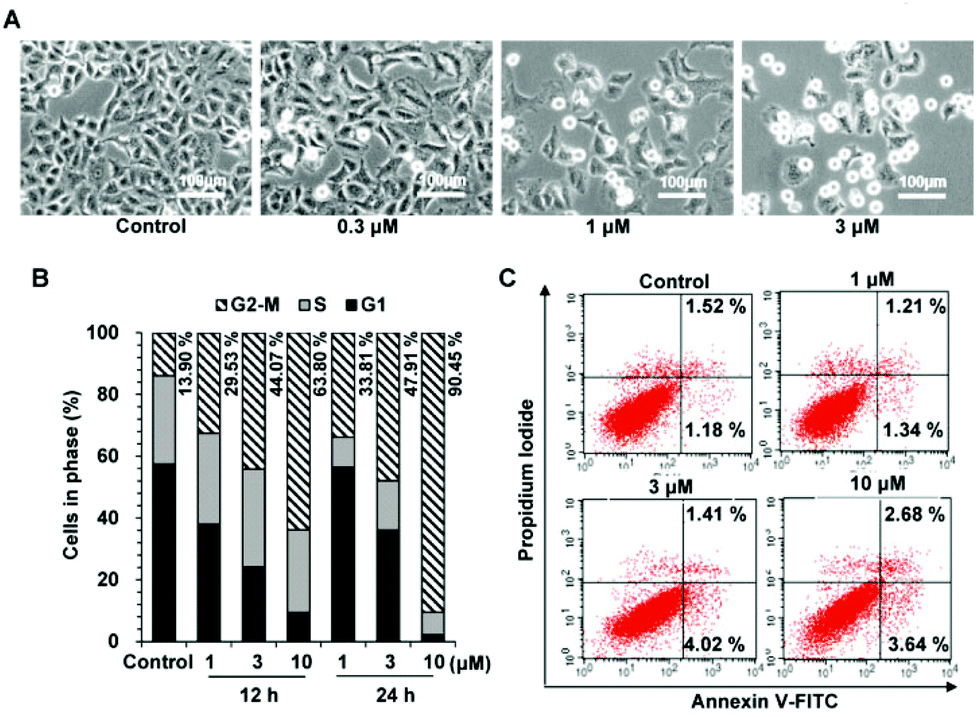
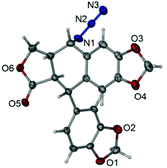
We found that cells treated with 27g decreased in size and became detached and round, a phenomenon that usually occurred during mitosis (Fig. 6A). Cell cycle analysis was further applied to examine how 27g inhibited cell growth. As shown in Fig. 6B, compound 27g dose-dependently arrested the cell cycle at the G2/M phase with a concomitant decrease in the G1 phase as compared with the DMSO control. Treatment of 27g with the indicated concentrations (1, 3 and 10 μM) for 12 h led to a marked increase in cell population in the G2/M phase from 13.09% to 63.80%, and a further increase to 90.45% after 24 h of treatment in A549 cancer cells.
 |
| | Fig. 6
27g dramatically blocked cell cycle, but had little effects on cell apoptosis. (A) A549 cells were incubated with different dilutions of 27g for 24 h. Morphological observation was visualized under a microscope at a magnification of 100. (B) Cells were incubated with the indicated concentrations of 27g for 12 h and 24 h, followed by being stained with propidium iodide (PI). The DNA contents were analyzed by using a flow cytometer. (C) Annexin V-FITC apoptosis was examined by using a flow cytometer in treated cells. | |
Apoptosis is a kind of programmed cell death, and an annexin V-FITC/propidium iodide binding assay was further carried out to test whether compound 27g induces cell apoptosis. Our results in Fig. 6C showed that compound 27g did not apparently induce cell apoptosis at the drug concentrations of 1–10 μM. Furthermore, there was no population of sub-G1 phase cells in the cell cycle assay, which was consistent with the results of apoptosis detection. Therefore, these results indicated that 27g inhibited cell proliferation by arresting the cell cycle in the G2/M phase without inducing apoptosis.
Compound 27g increases the expression of mitotic markers
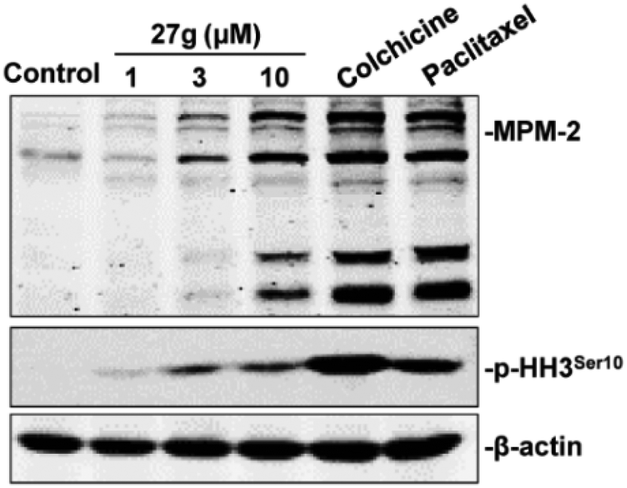
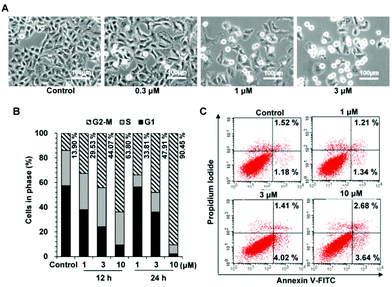
To further validate the inhibitory function of 27g on cell mitosis, we carried out a western blot assay to examine the expression levels of MPM-2 and phosphorylated histone H3 (p-HH3), which are two mitosis-specific markers. MPM-2 reflected the phosphorylation level of mitosis-specific proteins,37 and the expression of phosphorylation of histone H3 at Ser 10 peaked in the M phase.38 As shown in Fig. 7, MPM-2 expression was slightly increased when the cells were treated with 3 μM 27g, but it significantly increased under 10 μM 27g. Consistent with this result, p-HH3 remarkably increased in 27g-treated cells, which was consistent with the cell cycle arrest in mitosis mediated by 27g (Fig. 6B).
 |
| | Fig. 7 Compound 27g increased the expression of MPM-2 and phosphorylated histone H3. | |
Compound 27g inhibits colony formation of cancer cells
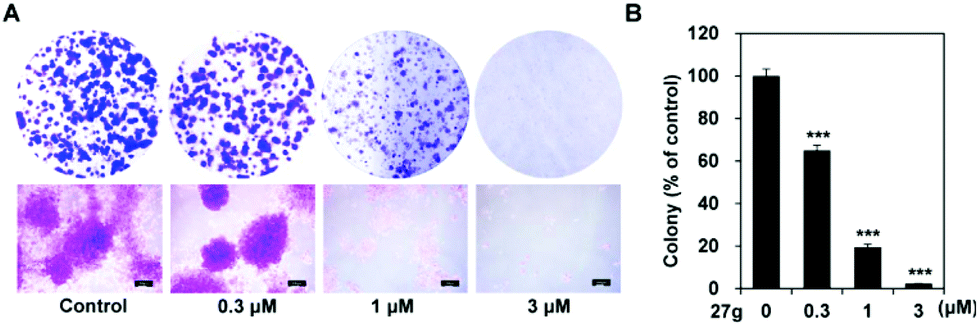
Colony formation assay is an in vitro cell survival assay based on the ability of a single cell to grow into a colony. This assay was carried out to test the inhibitory effect of compound 27g on the “unlimited” division of cancer cells. The results showed that compound 27g could block the colony formation of A-549 cells in a dose-dependent manner (Fig. 8). The number of colonies was significantly reduced by 27g even at 0.3 μM compared with the DMSO control group (P < 0.001). 27g at 1 μM apparently suppressed both the number and size of colonies (Fig. 8A and B) and almost blocked the anchor growth of lung cancer cells at 3 μM.
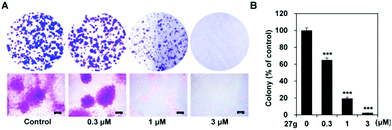 |
| | Fig. 8
27g inhibited colony formation of lung cancer cells. (A) A-549 cells were incubated with the indicated concentrations of 27g for 10 days and stained with 0.2% crystal violet. The colonies of the local region were visualized under a microscope (magnification; 40×). (B) The relative colony number is shown as mean ± SD. Statistical differences are noted; ***P < 0.001. | |
In vivo antitumor efficacy of compound 27g
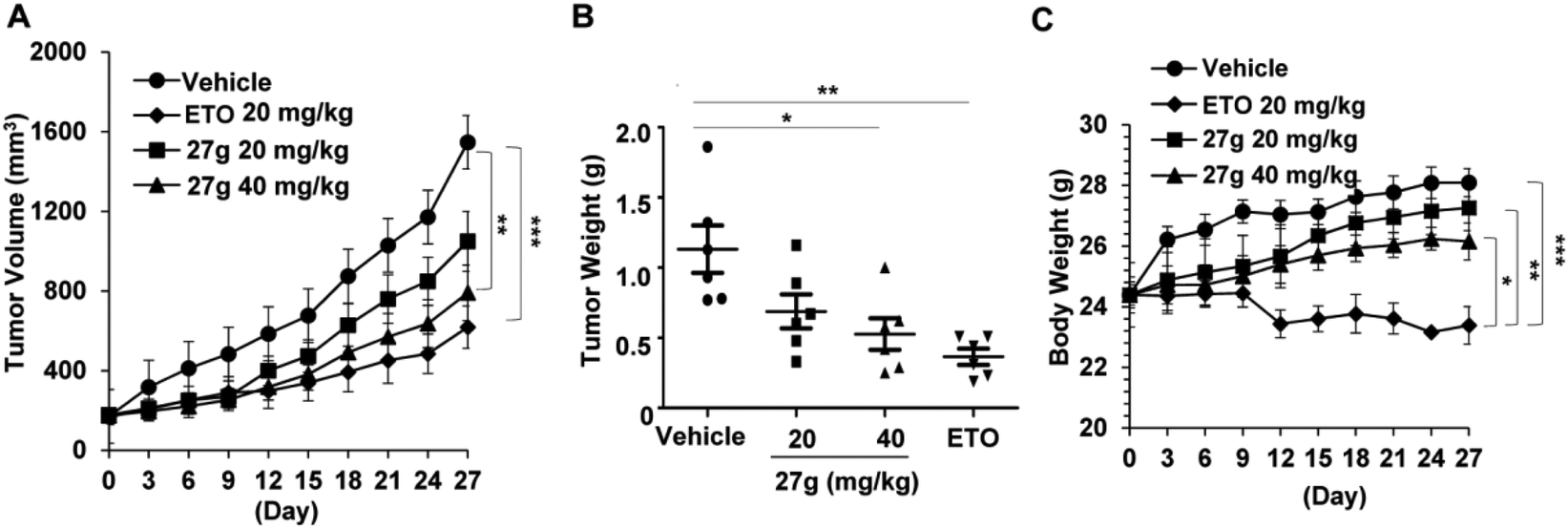
Based on the above in vitro cytotoxic activities, compound 27g was then chosen to investigate its anticancer properties in vivo. We established a xenograft mouse model of human lung cancer using A549 lung cancer cells. Compound 27g (20 and 40 mg per kg body weight) and etoposide (20 mg kg−1) were administered intraperitoneally for four weeks. The therapeutic efficacy of 27g is shown in Fig. 9. Compound 27g at 20 mg kg−1 marginally delayed tumour growth, but 27g at 40 mg kg−1 significantly suppressed tumour volume (P < 0.01) and tumour weight (P < 0.05). The inhibitory action of 27g was durable. Although etoposide blocked solid tumour growth more potently as compared with 27g, it was much more toxic than 27g because the body weights in the etoposide-treated group were reduced by 16.75% at the end of the experiments (P < 0.001). However, tumour-bearing mice were much more tolerant to 27g as the body weight of mice was not obviously affected after treatment with 27g. These data suggested that compound 27g might be an effective antitumor candidate with low toxicity.
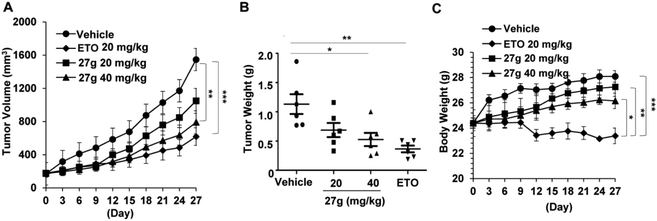 |
| | Fig. 9
27g suppressed tumor growth of xenograft lung tumors in vivo (n = 6 per group). (A) Tumor size was calculated by using the formula (major circumference × minor circumference2 × 0.5). Data are shown as mean ± SEM. (B) Tumor weight was measured when the nude mice were sacrificed. Each dot indicates the tumor weight of an individual mouse. (C) Body weight was measured every three days. Statistical differences among different groups are noted; *P < 0.05, **P < 0.01, ***P < 0.001. | |
Conclusions
In this study, a series of novel racemic otobain derivatives with ester, amide or amine linkages at the C-4 position was synthesised. These derivatives were examined for anti-proliferative activity against several cancer cell lines. Most of the compounds showed improved activity compared with lead compound 22. Compound 27g showed the most promising anticancer action with IC50 values ranging from 1.06 μM to 4.16 μM in various human cancer cells. Compound 27g was found to cause G2/M phase arrest in A-549 cells, but it did not induce apoptosis. Colony formation results showed that compound 27g effectively blocked the anchor growth of lung cancer cells. Compound 27g was a promising agent that significantly inhibited the growth of human lung cancer (P < 0.01). Additionally, 27g was relatively safe and tolerant compared with etoposide because it did not cause gross toxicity in mice at the tested dosages in vivo. Additional work on the enantioselective synthesis of compound 27g is currently underway in our laboratory.
Experimental section
Chemistry
Materials and methods.
All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. 1H (500 MHz) and 13C (125 MHz) NMR spectra were recorded on a Varian INOVA spectrometer with CDCl3 or DMSO-d6 and tetramethylsilane (TMS) as the internal standard. All chemical shift values were reported in units of δ (ppm). The following abbreviations were used to indicate the peak multiplicity: s = singlet; d = doublet; t = triplet; m = multiplet; br = broad. Analytical thin-layer chromatography (TLC) was carried out on precoated plates (silica gel 60 F254), and the spots were visualized with ultraviolet (UV) light. Flash column chromatography was carried out with silica gel (300–350 mesh). Melting points were determined on a Yanano MP 500. High-resolution mass data were obtained on a Bruker microOTOFQ II spectrometer.
The preparation and characterization details of intermediate compounds 16–22, 24, 25 are included within the ESI.†
General procedure for the synthesis of compounds 23a–c, g–h
A mixture of 23 (0.40 mmol), the corresponding acids RCO2H (0.48 mmol), N,N′-dicyclohexylcarbodiimide (DCC, 0.48 mmol) and 4-(N,N-dimethylamino)pyridine (DMAP, 0.04 mmol) in dry CH2Cl2 (25 mL) was stirred at 28 °C. Upon completion, 0.1 N HCl (25 mL) was added. The organic layer was washed with saturated brine (3 × 15 mL), dried over anhydrous Na2SO4, filtered, concentrated in vacuo, and recrystallized from ethyl acetate to give the products. The characterization and spectral data of the final compounds 24a–e are given below.
9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl acetate (23a).
White solid, yield: 85%, m.p. 237–239 °C; 1H NMR (500 MHz, CDCl3): δ 2.21(s, 3H), 2.66–2.74(m, 2H), 4.07(d, J = 10.5 Hz, 1H), 4.20–4.24(m, 1H), 4.42(dd, J1 = 9.3 Hz, J2 = 6.1 Hz, 1H), 5.92(s, 2H), 5.94(dd, J1 = 3.4 Hz, J2 = 1.4 Hz, 2H), 6.07(d, J = 9.6 Hz, 1H), 6.31(s, 1H), 6.55(s, 1H), 6.70(s, 1H), 6.72(d, J = 7.9 Hz, 1H), 6.78(d, J = 7.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 21.07, 45.42, 46.05, 46.25, 70.01, 72.84, 101.11, 101.51, 105.67, 108.27, 109.01, 109.59, 123.09, 127.95, 133.97, 135.89, 146.80, 147.03, 147.91, 147.96, 171.39, 174.04; HRMS (ESI): calc. for C22H18O8Na (M + Na)+ 433.0894, found: 433.0899.
9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl furan-2-carboxylate (23b).
White solid, yield: 81%, m.p. 236–239 °C; 1H NMR (500 MHz, CDCl3): δ 2.75–2.82(m, 2H), 4.10–4.12(m, 1H), 4.31(t, J = 9.6 Hz, 1H), 4.47–4.50(m, 1H), 5.92(d, J = 2.2 Hz, 2H), 5.95(s, 2H), 6.29(d, J = 8.0 Hz, 1H), 6.34(s, 1H), 6.58(s, 2H), 6.74(d, J = 7.7 Hz, 1H), 6.79–6.80(m, 2H), 7.29(d, J = 3.2 Hz, 1H), 7.66(s, 1H); 13C NMR (125 MHz, CDCl3): δ 45.44, 46.08, 46.30, 70.01, 73.33, 101.12, 101.52, 105.79, 108.30, 109.05, 109.63, 112.25, 119.31, 123.11, 127.76, 134.11, 135.85, 143.66, 146.83, 147.15, 147.28, 147.99, 148.05, 158.88, 173.99; HRMS (ESI): calc. for C25H18O9Na (M + Na)+ 485.0843, found: 485.0850.
9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl pyrazine-2-carboxylate (23c).
White solid, yield: 77%, m.p. 235–236 °C; 1H NMR (500 MHz, CDCl3): δ 2.82–2.96(m, 2H), 4.14(d, J = 10.6 Hz, 1H), 4.38(t, J = 9.7 Hz, 1H), 4.49–4.52(m, 1H), 5.94(s, 2H), 5.97(s, 2H), 6.37(s, 1H), 6.46(d, J = 9.4 Hz, 1H), 6.60(s, 1H), 6.77(d, J = 7.2 Hz, 1H), 6.82(d, J = 7.5 Hz, 2H), 8.80(s, 1H), 8.85(s, 1H), 9.40(s, 1H); 13C NMR (125 MHz, CDCl3): δ 45.32, 46.07, 46.24, 69.90, 74.86, 101.14, 101.59, 105.75, 108.31, 109.02, 109.70, 123.14, 127.17, 134.33, 135.71, 142.64, 144.80, 146.51, 146.87, 147.29, 148.01, 148.24, 148.32, 164.42, 173.75; HRMS (ESI): calc. for C25H18O8N2Na (M + Na)+ 497.0955, found: 497.0961.
9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl benzoate (23g).
White solid, yield: 80%, m.p. 247–249 °C; 1H NMR (500 MHz, CDCl3): δ 2.76–2.84(m, 2H), 4.13(d, 1H, J = 9.3 Hz), 4.32–4.36(m, 1H), 4.46(dd, J1 = 9.3 Hz, J2 = 5.8 Hz, 1H), 5.92–5.95(m, 4H), 6.32(d, J = 8.1 Hz, 1H), 6.36(s, 1H), 6.59(s, 1H), 6.76(d, J = 7.8 Hz, 1H), 6.80(d, J = 9.0 Hz, 2H), 7.49(t, J = 7.8 Hz, 2H), 7.63(t, J = 7.4 Hz, 1H), 8.10(d, J = 7.4 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 45.62, 46.13, 46.34, 70.15, 73.30, 101.12, 101.51, 105.85, 108.30, 109.06, 109.65, 123.12, 128.18, 128.71, 129.15, 129.89, 133.78, 134.06, 135.93, 146.82, 147.12, 147.96, 147.99, 166.84, 174.08; HRMS (ESI): calc. for C27H20O8Na (M + Na)+ 495.1050, found: 490.1052.
9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl 4-fluorobenzoate (23h).
White solid, yield: 75%, m.p. 262–264 °C; 1H NMR (500 MHz, CDCl3): δ 2.78–2.83(m, 2H), 4.13(d, J = 9.3 Hz, 1H), 4.31–4.35(m, 1H), 4.44–4.47(m, 1H), 5.92(s, 1H), 5.94(s, 1H), 5.95(d, J = 1.9 Hz, 2H), 6.31(d, J = 8.1 Hz, 1H), 6.36(s, 1H), 6.59(s, 1H), 6.75–6.81(m, 3H), 7.17(t, J = 8.6 Hz, 2H), 8.12(dd, J1 = 8.6 Hz, J2 = 5.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 45.58, 46.11, 46.30, 70.09, 73.47, 101.13, 101.54, 105.76, 108.30, 109.05, 109.68, 115.86(2JC–F = 21.3 Hz), 123.13, 125.41, 128.02, 132.48(d, 3JC–F = 8.7 Hz), 134.13, 135.87, 146.84, 147.14, 148.00, 165.24(d, 1JC–F = 253.7 Hz), 165.87, 174.01; HRMS (ESI): calc. for C27H19O8FNa (M + Na)+ 513.0956, found: 513.0961.
General procedure for the synthesis of compounds 26b–h
A mixture of 25 (0.70 mmol), chloride or anhydride (0.85 mmol), and 4-(N,N-dimethylamino)pyridine (DMAP, 1.12 mmol) in dry CH2Cl2 (25 mL) was stirred at room temperature under argon. Upon completion, the organic layer was washed with saturated brine (3 × 15 mL), dried over anhydrous Na2SO4, filtered, concentrated in vacuo, and purified by silica column chromatography (ethyl acetate/dichloromethane, 1:30–1:10) to give the products. The characterization and spectral data of the final compounds 26b–f are given below.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)furan-2-carboxamide (26b).
White solid, yield: 74%, m.p. 262–265 °C; 1H NMR (500 MHz, CDCl3) δ 2.76(dd, J1 = 14.2, J2 = 11.4 Hz, 1H), 2.82–2.89(m, 1H), 3.97(d, J = 9.7 Hz, 1H), 4.01(d, J = 11.9 Hz, 1H), 4.58(dd, J1 = 9.2, J2 = 6.9 Hz, 1H), 5.33(dd, J1 = 6.7, J2 = 3.8 Hz, 1H), 5.94–5.99(m, 4H), 6.39(s, 1H), 6.53(d, J = 6.7 Hz, 1H), 6.58(dd, J1 = 3.3, J2 = 1.6 Hz, 1H), 6.65(s, 1H), 6.75(s, 1H), 6.80–6.85(m, 2H), 7.23(d, J = 3.4 Hz, 1H), 7.50(s, 1H); 13C NMR (125 MHz, CDCl3): 42.24, 44.25, 46.17, 47.95, 68.60, 101.14, 101.58, 108.37, 108.97, 109.15, 109.79, 112.59, 115.58, 123.19, 127.85, 134.30, 135.82, 144.53, 146.80, 146.82, 147.18, 147.98, 148.54, 158.35, 174.83; HRMS (ESI): calc. for C25H19NO8Na (M + Na)+ 484.1003, found: 484.1045.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)pyrazine-2-carboxamide (26c).
White solid, yield: 66%, m.p. 263–265 °C; 1H NMR (500 MHz, CDCl3) δ 2.72–2.77(m, 1H), 2.85–2.92(m, 1H), 3.93(t, J = 10.1 Hz, 1H), 4.02(d, J = 11.3 Hz, 1H), 4.55–4.58(m, 1H), 5.37(dd, J1 = 7.1 Hz, J2 = 3.7 Hz, 1H), 5.91(d, J = 4.2 Hz, 2H), 5.96(d, J = 3.3 Hz, 2H), 6.39(s, 1H), 6.66(s, 1H), 6.72(s, 1H), 6.79–6.83(m, 2H), 7.98(d, J = 7.1 Hz, 1H), 8.54(s, 1H), 8.80(s, 1H), 9.44(s, 1H); 13C NMR (125 MHz, CDCl3): 42.41, 44.20, 46.24, 48.27, 68.31, 101.10, 101.54, 108.35, 109.02, 109.09, 109.82, 123.17, 127.91, 134.22, 135.91, 142.67, 143.52, 144.61, 146.84, 147.23, 147.90, 148.02, 148.61, 163.18, 174.51; HRMS (ESI): calc. for C25H19N3O7Na (M + Na)+ 496.1115, found: 496.1229.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)-2,2,2-trifluoroacetamide (26d).
White solid, yield: 81%, m.p. 228–230 °C; 1H NMR (500 MHz, CDCl3) δ 2.64(dd, J1 = 14.3 Hz, J2 = 11.5 Hz, 1H), 2.77–2.85(m, 1H), 3.78(dd, J1 = 11.0 Hz, J2 = 9.3 Hz, 1H), 3.93(d, J = 11.4 Hz, 1H), 4.48(dd, J1 = 9.2 Hz, J2 = 7.0 Hz, 1H), 5.17(dd, J = 7.2 Hz, J2 = 4.1 Hz, 1H), 5.90–5.93(m, 4H), 6.34(s, 1H), 6.54(d, J = 1.2 Hz, 1H), 6.66(s, 1H), 6.72(dd, J1 = 8.0, J2 = 1.5 Hz, 1H), 6.77(d, J = 7.9 Hz, 1H), 6.89(d, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): 41.98, 43.41, 46.17, 48.85, 67.80, 101.17, 101.75, 108.27, 108.76, 108.87, 109.85, 116.72(q, 1JC–F = 286.2 Hz), 123.30, 125.95, 134.53, 135.08, 146.90, 147.36, 148.00, 148.91, 157.41(q, 2JC–F = 37.5 Hz), 174.37; HRMS (ESI): calc. for C22H16NO7F3Na (M + Na)+ 486.0771, found: 486.0893.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)thiophene-2-carboxamide (26e).
White solid, yield: 68%, m.p. 261–263 °C; 1H NMR (500 MHz, CDCl3) δ 2.73(dd, J1 = 14.2 Hz, J2 = 11.3 Hz, 1H), 2.79–2.86(m, 1H), 3.97(t, J = 10.4 Hz, 2H), 4.56(dd, J1 = 9.2 Hz, J2 = 6.8 Hz, 1H), 5.30(dd, J1 = 6.4, J2 = 3.8 Hz, 1H), 5.92(d, J = 4.3 Hz, 2H), 5.96(d, J = 4.6 Hz, 2H), 6.20(d, J = 6.4 Hz, 1H), 6.37(s, 1H), 6.61(s, 1H), 6.73(s, 1H), 6.77(d, J = 7.9 Hz, 1H), 6.81(d, J = 7.9 Hz, 1H), 7.11(t, J = 4.3 Hz, 1H), 7.55(d, J = 4.4 Hz, 2H); 13C NMR (125 MHz, CDCl3): 42.25, 44.28, 46.19, 48.73, 68.73, 101.15, 101.60, 108.37, 108.91, 109.12, 109.80, 123.14, 127.84, 127.90, 128.89, 131.07, 134.39, 135.76, 137.43, 146.81, 147.22, 147.99, 148.55, 162.09, 174.87; HRMS (ESI): calc. for C25H19NO7SNa (M + Na)+ 500.0774, found: 500.0870.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)quinoline-2-carboxamide (26f).
White solid, yield: 64%, m.p. 179–181 °C; 1H NMR (500 MHz, CDCl3) δ 2.85–2.94(m, 2H), 3.98(dd, J1 = 10.8, J2 = 9.3 Hz, 1H), 4.03–4.05(m, 1H), 4.57–4.60(m, 1H), 5.49–5.41(m, 1H), 5.91(d, J = 3.9 Hz, 2H), 5.98(d, J = 2.3 Hz, 2H), 6.42(s, 1H), 6.75(s, 1H), 6.77(s, 1H), 6.86(s, 2H), 7.63(t, J = 7.5 Hz, 1H), 7.77(t, J = 7.2 Hz, 1H), 7.89(d, J = 8.1 Hz, 1H), 8.10(d, J = 8.5 Hz, 1H), 8.31(d, J = 8.5 Hz, 1H), 8.36(d, J = 8.5 Hz, 1H), 8.50(d, J = 7.6 Hz, 1H); 13C NMR(125 MHz, CDCl3): 42.51, 44.40, 46.14, 48.26, 68.57, 101.16, 101.56, 108.40, 109.11, 109.33, 109.76, 118.77, 123.08, 127.81, 128.35, 129.57, 129.83, 130.41, 134.12, 136.25, 137.89, 146.45, 146.78, 147.13, 148.05, 148.49, 148.60, 164.62, 174.99; HRMS (ESI): calc. for C30H22N2O7Na (M + Na)+ 545.1319, found: 545.1456.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)benzamide (26g).
White solid, yield: 78%, white solid, m.p. 280–281 °C; 1H NMR (500 MHz, CDCl3) δ 2.72(dd, J1 = 14.2 Hz, J2 = 11.4 Hz, 1H), 2.80–2.88(m, 1H), 3.92(dd, J1 = 10.7 Hz, J2 = 9.5 Hz, 1H), 3.97(d, J = 11.3 Hz, 1H), 4.57(dd, J1 = 9.3 Hz, J2 = 6.9 Hz, 1H), 5.33(dd, J1 = 6.5 Hz, J2 = 3.9 Hz, 1H), 5.92(dd, J1 = 8.3 Hz, J2 = 1.1 Hz, 1H), 5.95(dd, J1 = 6.6 Hz, J2 = 1.4 Hz, 1H), 6.36(d, J = 6.7 Hz, 1H), 6.37(s, 1H), 6.60(s, 1H), 6.73(s, 1H), 6.77(dd, J1 = 8.0 Hz, J2 = 1.3 Hz, 1H), 6.80(d, J = 7.9 Hz, 1H), 7.45–7.48(m, 2H), 7.58(t, J = 7.4 Hz, 1H), 7.79–7.81(m, 2H); 13C NMR (125 MHz, CDCl3): 42.30, 44.29, 46.20, 48.72, 68.75, 101.14, 101.57, 108.35, 108.91, 109.09, 109.81, 123.19, 127.06, 128.03, 128.88, 132.30, 133.28, 134.37, 135.79, 146.80, 147.21, 147.98, 148.50, 167.73, 174.85; HRMS (ESI): calc. for C27H21NO7Na (M + Na)+ 494.1210, found: 494.1342.
N-(9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)-4-fluorobenzamide (26h).
White solid, yield: 70%, m.p. 256–259 °C; 1H NMR (500 MHz, CDCl3) δ 2.70(dd, J1 = 14.1 Hz, J2 = 11.5 Hz, 1H), 2.80–2.87(m, 1H), 3.90(t, J = 10.1 Hz, 1H), 3.98(d, J = 11.3 Hz, 1H), 4.55(dd, J1 = 9.0 Hz, J2 = 7.1 Hz, 1H), 5.33(dd, J1 = 6.1 Hz, J2 = 4.2 Hz, 1H), 5.91(d, J = 6.0 Hz, 2H), 5.94(d, J = 5.1 Hz, 2H), 6.27(d, J = 6.4 Hz, 1H), 6.38(s, 1H), 6.61(s, 1H), 6.73(s, 1H), 6.77(d, J = 7.9 Hz, 1H), 6.80(d, J = 7.9 Hz, 1H), 7.13(t, J = 8.4 Hz, 2H), 7.80(dd, J1 = 8.4 Hz, J2 = 5.3 Hz, 2H); 13C NMR (125 MHz, CDCl3): 42.34, 44.28, 46.25, 48.83, 68.63, 101.10, 101.54, 108.32, 108.89, 109.02, 109.82, 115.83(d, 2JC–F = 21.3 Hz), 123.17, 128.05, 129.43(d, 3JC–F = 8.7 Hz), 129.54, 134.39, 135.79, 146.85, 147.27, 148.01, 148.57, 164.17(d, 1JC–F = 251.3 Hz), 166.60, 174.65; HRMS (ESI): calc. for C27H20NO7FNa (M + Na)+ 512.1116, found: 512.1188.
General procedure for the synthesis of compounds 27g–m
To a solution of substituted benzyl bromide (0.79 mmol) in acetone (3 mL) was added sodium iodide (128 mg, 0.85 mmol). The reaction mixture was stirred for 20 min and then filtered. The filtrate was evaporated to give the corresponding benzyl iodide. To 25 (0.66 mmol) in 1,2-dichloroethane (15 mL) were added substituted benzyl iodide (0.79 mmol) and anhydrous barium carbonate (0.79 mmol) under nitrogen. After stirring for 40 h at 75–80 °C, the mixture was filtered. The filtrate was evaporated to give a crude product which was purified by silica column chromatography (ethyl acetate/petroleum ether, 3:1). The characterization and spectral data of the final compounds 27g–m are given below.
5-(Benzo[d][1,3]dioxol-5-yl)-9-(benzylamino)-5,5a,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(8H)-one (27g).
White solid, yield: 48%, m.p. 196–199 °C; 1H NMR (500 MHz, CDCl3) δ 2.58–2.65(m, 1H), 3.23(dd, J1 = 13.9 Hz, J2 = 11.4 Hz, 1H), 3.89(d, J = 3.1 Hz, 1H), 3.90–3.92(m, 3H), 4.31(dd, J1 = 10.9 Hz, J2 = 8.2 Hz, 1H), 4.35–4.38(m, 1H), 5.87(dd, J1 = 4.5 Hz, J2 = 1.1 Hz, 2H), 5.93(d, J = 0.7 Hz, 2H), 6.32(s, 1H), 6.45(s, 1H), 6.60(s, 1H), 6.77(s, 2H), 7.30–7.33(m, 1H), 7.36–7.40(m, 4H); 13C NMR (125 MHz, CDCl3): δ 42.08, 45.20, 45.92, 54.52, 55.44, 67.99, 101.01, 101.25, 108.18, 108.45, 109.12, 109.91, 123.01, 127.58, 128.36, 128.73, 131.77, 133.20, 136.99, 139.83, 146.53, 146.55, 147.64, 147.83, 176.28; HRMS (ESI): calc. for C27H23O6NNa (M + Na)+ 480.1418, found: 480.1463.
5-(Benzo[d][1,3]dioxol-5-yl)-9-((4-fluorobenzyl)amino)-5,5a,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(8H)-one (27h).
White solid, yield: 45%, m.p. 187–189 °C; 1H NMR (500 MHz, CDCl3) δ 2.59–2.66(m, 1H), 3.21(dd, J1 = 13.9 Hz, J2 = 11.4 Hz, 1H), 3.84–3.87(m, 2H), 3.90–3.95(m, 2H), 4.28(dd, J1 = 11.0, J2 = 8.2 Hz, 1H), 4.35–4.38(m, 1H), 5.87–5.92(m, 4H), 6.33(s, 1H), 6.50(s, 1H), 6.59(s, 1H), 6.77(s, 2H), 7.06(t, J = 8.6 Hz, 2H), 7.34(dd, J1 = 8.4 Hz, J2 = 5.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 42.09, 45.13, 45.90, 53.67, 55.57, 67.91, 101.02, 101.30, 108.19, 108.41, 109.07, 109.98, 115.44(d, 2JC–F = 21.3 Hz), 123.01, 129.82(d, 3JC–F = 7.5 Hz), 131.64, 133.19, 135.55, 135.57, 136.94, 146.55, 146.58, 147.71, 147.85, 161.21(d, 1JC–F = 243.0 Hz), 176.17; HRMS (ESI): calc. for C27H22O6NFNa (M + Na)+ 498.1323, found: 498.1373.
5-(Benzo[d][1,3]dioxol-5-yl)-9-((4-nitrobenzyl)amino)-5,5a,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(8H)-one (27i).
Light yellow solid, yield: 43%, m.p. 210–212 °C; 1H NMR (500 MHz, CDCl3) δ 2.64–2.71(m, 1H), 3.19(dd, J1 = 13.8 Hz, J2 = 11.5 Hz, 1H), 3.90(d, J = 2.9 Hz, 1H), 3.94(d, J = 2.9 Hz, 1H), 3.97(d, J = 6.1 Hz, 1H), 4.16(d, J = 14.1 Hz, 1H), 4.28(dd, J1 = 10.9 Hz, J2 = 8.3 Hz, 1H), 4.40(t, J = 7.4 Hz, 1H), 5.89–5.93(m, 4H), 6.36(s, 1H), 6.58(d, J = 7.9 Hz, 2H), 6.78(s, 2H), 7.56(d, J = 8.4 Hz, 2H), 8.23(d, J = 8.4 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 42.13, 45.01, 45.38, 53.38, 56.06, 67.73, 101.07, 101.39, 108.23, 108.40, 108.98, 110.20, 123.02, 123.92, 128.80, 131.09, 133.25, 136.78, 146.62, 146.65, 147.19, 147.39, 147.89, 147.92, 175.90; HRMS (ESI): calc. for C27H22O8N2Na (M + Na)+ 525.1268, found: 525.1271.
5-(Benzo[d][1,3]dioxol-5-yl)-9-((2-nitrobenzyl)amino)-5,5a,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(8H)-one (27j).
Light yellow solid, yield: 39%, m.p. 163–165 °C; 1H NMR (500 MHz, CDCl3) δ 2.65–2.72(m, 1H), 3.18(dd, J1 = 14.0 Hz, J2 = 11.4 Hz, 1H), 3.93–3.96(m, 2H), 4.11(d, J = 13.7 Hz, 1H), 4.26–4.30(m, 2H), 4.45(dd, J1 = 8.0 Hz, J2 = 6.9 Hz, 1H), 5.89–5.95(m, 4H), 6.35(s, 1H), 6.56(s, 1H), 6.63(s, 1H), 6.79(s, 2H), 7.48–7.52(m, 1H), 7.62–7.67(m, 2H), 8.01(d, J1 = 8.1 Hz, J2 = 0.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 42.08, 45.11, 45.95, 51.38, 56.28, 67.90, 101.01, 101.32, 108.18, 108.50, 109.13, 109.99, 123.03, 125.05, 128.66, 131.25, 131.55, 133.20, 133.49, 134.82, 136.89, 146.54, 146.63, 147.80, 147.83, 149.19, 176.10; HRMS (ESI): calc. for C27H22O8N2Na (M + Na)+ 525.1268, found: 525.1291.
4-(((9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)amino)methyl)benzonitrile (27k).
White solid, yield: 47%, m.p. 213–215 °C; 1H NMR (500 MHz, CDCl3) δ 2.63–2.70(m, 1H), 3.180(dd, J1 = 13.9 Hz, J2 = 11.4 Hz, 1H), 3.88(d, J = 3.1 Hz, 1H), 3.91–3.95(m, 2H), 4.10(d, J = 13.9 Hz, 1H), 4.26(dd, J1 = 11.0 Hz, J2 = 8.2 Hz, 1H), 4.37–4.40(m, 1H), 5.89–5.93(m, 4H), 6.35(s, 1H), 6.54(s, 1H), 6.58(s, 1H), 6.77(s, 2H), 7.50(d, J = 8.1 Hz, 2H), 7.66(d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 42.11, 45.02, 45.88, 53.72, 55.99, 67.74, 101.06, 101.38, 108.22, 108.38, 108.98, 110.16, 111.43, 118.71, 123.03, 128.77, 131.16, 132.50, 133.24, 136.78, 145.16, 146.61, 146.64, 147.89, 175.91; HRMS (ESI): calc. for C28H22O6N2Na (M + Na)+ 505.1370, found: 505.1404.
3-(((9-(Benzo[d][1,3]dioxol-5-yl)-8-oxo-5,5a,6,8,8a,9-hexahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl)amino)methyl)benzonitrile (27l).
White solid, yield: 40%, m.p. 201–203 °C; 1H NMR (500 MHz, CDCl3) δ 2.62–2.69(m, 1H), 3.17(dd, J1 = 14.0 Hz, J2 = 11.4 Hz, 1H), 3.88–3.90(m, 2H), 3.94(d, J = 11.3 Hz, 1H), 4.06(d, J = 13.5 Hz, 1H), 4.27(dd, J1 = 11.0 Hz, J2 = 8.2 Hz, 1H), 4.37–4.40(m, 1H), 5.88–5.92(m, 4H), 6.34(s, 1H), 6.54(s, 1H), 6.59(s, 1H), 6.77(s, 2H), 7.48(t, J = 7.7 Hz, 1H), 7.60(d, J = 7.7 Hz, 1H), 7.64(d, J = 7.9 Hz, 1H), 7.66(s, 1H); 13C NMR (125 MHz, CDCl3): δ 42.12, 45.00, 45.89, 53.37, 55.94, 67.76, 101.05, 101.38, 108.22, 108.41, 109.06, 110.14, 112.72, 118.68, 122.99, 129.53, 131.18, 131.27, 131.66, 132.64, 133.22, 136.81, 141.28, 146.59, 146.63, 147.87, 175.95; HRMS (ESI): calc. for C28H22O6N2Na (M + Na)+ 505.1370, found: 505.1411.
5-(Benzo[d][1,3]dioxol-5-yl)-9-((4-methylbenzyl)amino)-5,5a,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(8H)-one (27m).
White solid, yield: 43%, m.p. 184–186 °C; 1H NMR (500 MHz, CDCl3) δ 2.40(s, 3H), 2.59–2.66(m, 1H), 3.26(dd, J1 = 13.8 Hz, J2 = 11.5 Hz, 1H), 3.87–3.94(m, 4H), 4.33(dd, J1 = 10.9 Hz, J2 = 8.2 Hz, 1H), 4.37–4.40(m, 1H), 5.90(d, J = 3.8 Hz, 2H)), 5.95(s, 2H), 6.35(s, 1H), 6.50(s, 1H), 6.63(s, 1H), 6.79(s, 2H), 7.22(d, J = 7.8 Hz, 2H), 7.28(d, J = 7.8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 21.17, 42.09, 45.21, 45.92, 54.24, 55.38, 68.04, 101.01, 101.25, 108.17, 108.51, 109.15, 109.88, 123.00, 128.33, 129.38, 131.87, 133.19, 136.82, 137.04, 137.24, 146.51, 146.53, 147.61, 147.83, 176.35; HRMS (ESI): calc. for C28H25O6NNa (M + Na)+ 494.1574, found: 494.1629.
Biology
Cell culture.
The human cancer cell lines were incubated at 37 °C in a humidified incubator with 5% CO2, including HL-60 (human promyelocytic leukemia cell line), SGC-7901 (gastric cancer), MCF-7 (breast cancer), HCT-116 (colon cancer), A549 (non-small cell lung cancer) and HaCat (human skin cell line/keratinocyte). All the cells were brought from American Type Culture Collection (ATCC; Manassas, VA, USA). The cells were cultured in RPMI-1640 (HL-60, SGC-7901, HaCat) or Dulbecco's modified Eagle's medium (DMEM; MCF-7, HCT-116, A549) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Logan, UT) and 1% penicillin–streptomycin antibiotics (HyClone Laboratories, Logan, UT).
Cell viability assay.
Cell viability was measured by the sulforhodamine B (SRB) assay as previously described.39 Briefly, cells in their log growth phase were harvested, counted and seeded in 96-well microtitre plates. After 24 h of incubation to allow cell attachment, the cells were incubated in fresh medium containing various concentrations (0.01–300 μM) of the tested compounds for an additional 72 h. Five replicate wells were set up for each experimental condition. The attached cells were then fixed with chilled trichloroacetic acid for 1 h at 4 °C, washed with phosphate-buffered saline (PBS) and dried, followed by staining with 0.04% sulforhodamine B dye for 0.5 h. The non-adsorbed dye was washed away by 1% acetic acid solution and the adsorbed dye was solubilized in 10 mM Tris-buffer. Before the optical density was measured at 515 nm on a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA, USA) with a GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA),40 the plates were gently shaken for 20 min on a mechanical shaker. The results are reported in terms of the half maximal inhibitory concentration (IC50) values. Etoposide served as the positive control. DMSO (0.1%) was used as a negative control unless special instructions are given. Three or more independent experiments were carried out.
Cell cycle analysis.
A549 cells were seeded in 6-well plates and incubated with compound 27g at concentrations of 1 μM, 3 μM, 10 μM for 12 h and 24 h. Then, both floating and adherent cells were collected, washed with ice-cold PBS and fixed in 70% ethanol at 4 °C overnight. The cells were then washed with PBS, incubated with 1 mg mL−1 DNase-free RNase A (OMEGA, Norcross, GA, USA) at 37 °C for 30 min, and stained with 50 mg mL−1 propidium iodide (Sigma, St Louis, MO, USA) in 0.1% Triton X-100 in the dark at room temperature for 30 min. The stained cells were analyzed with a Becton Dickinson flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).41
Cell apoptosis detection.
Annexin V-FITC Apoptosis Detection Kit (eBioscience/Bender MedSystems GmbH, Vienna, Austria) was used to detect apoptosis induced by compound 27g. A549 cells were seeded into 6-well plates (105 cells per well). After attachment, cells were treated with 1 μM, 3 μM, 10 μM of 27g for 24 h. All the cells were harvested and washed with ice-cold PBS. Annexin V-FITC and propidium iodide (PI) were added. The cells were incubated at room temperature for 30 min. Annexin V-FITC and PI fluorescence were monitored by using a Becton Dickinson FAC scan flow cytometer and the data were analyzed using Cell Quest software. The experiments were performed in three replicates.42
Colony formation assay.
A549 cells were seeded in 6-well plates at a density of 103 cells per well. After 24 h, the culture medium was removed and fresh medium with various concentrations of 27g was added. The medium was changed every three days. Colonies were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet solution. The images of the colonies were captured and the number of crystal violet stained colonies was counted randomly. GraphPad Prism 5 was used to do all statistical calculations. We used One-way ANOVA (and nonparametric) with post hoc Tukey's tests (compare all pairs of columns) for more than two groups. The data represented mean values ± SD. Statistical differences were compared between the groups and noted as ***P < 0.001.43
Western blotting assay.
After being treated with the indicated concentrations of compound 27g, the cells were harvested and lysed in ice-cold radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% SDS; 0.5% sodium deoxycholate; 0.5% Triton X-100) supplemented with 50 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor and phosphorylation protease inhibitor. Colchicine (about 4 times IC50) and paclitaxel (about 8 times IC50) were taken as the positive control. The lysates were centrifuged at 12000 rpm for 20 min and the protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). The lysates were mixed with loading buffer (10% SDS, 25% 2-mercaptoethanol, 50% glycerol, 0.05% bromophenol blue and 0.35 M Tris-HCl, pH 6.8), and then boiled for 10 min immediately. Fifty mg of total protein were loaded onto 12% SDS-polyacrylamide gels, transferred to nitrocellulose membrane, incubated with BSA, and probed with primary antibodies [phospho-histone H3 (Ser10) (Abcam, Burlingame, CA, USA), MPM-2 (Millipore, Billerica, MA, USA) and β-actin (Sigma, St Louis, MO, USA)] overnight at 4 °C. Either a goat anti-mouse or goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Pierce) was incubated for 1 h at room temperature. Protein bands were visualized by enhanced chemiluminescence detection using the Odyssey Infrared Imager (LI-COR® Biosciences, Lincoln, NE, USA).42
Xenograft mouse model.
BALB/cA nude male mice used in the study were purchased from National Rodent Laboratory Animal Resources (Shanghai, China). All treatments were administered according to the NIH standards established in the Guidelines for the Care and Use of Experimental Animals, and all the protocols were approved by East China Normal University.
A xenograft model was set up to study the inhibitory effects of 27gin vivo. Human lung cancer cells A549 (5 × 106 cells per mouse) were implanted subcutaneously into the right hind flanks of 4- to 5-week-old male nude mice (National Rodent Laboratory Animal Resources, Shanghai Branch, China). When the tumor volume reached a mean volume of approximately 150 mm3, the mice were randomized into treatment groups and were treated intraperitoneally with different compounds dissolved in the solvent vehicle (sterile PBS containing 1% DMSO, 20% PEG400 and 25% ethyl alcohol) for 28 days. The vehicle control group received 100 μL of the solvent vehicle every two days. The reference group received 20 mg per kg body weight of etoposide every 4 days and the tested compound groups received 20 mg per kg and 40 mg per kg body weight of 27g every other day. The major and minor circumferences of tumors were measured every 3 days with a vernier caliper until animals were sacrificed. The tumor volume was calculated by using the formula (major circumference × minor circumference2 × 0.5). On day 29, the mice were sacrificed and the tumors were removed and weighed.44
GraphPad Prism 5 was used to perform all statistical calculations. We used One-way ANOVA (and nonparametric) with post hoc Tukey's tests (compare all pairs of columns) for more than two groups. The data represented mean values ± SEM. Statistical differences were compared between the groups and noted as ***P < 0.001, **P < 0.01, *P < 0.05.
References
- F. Hamam, Food Nutr. Sci., 2014, 5, 2257–2264 CrossRef CAS.
- P. Kandra and H. P. Kalangi, Appl. Microbiol. Biotechnol., 2015, 99, 2055–2064 CrossRef CAS PubMed.
- V. T. T. Thanh, V. C. Pham, H. D. T. Mai, M. Litaudon, F. Gueritte, P. Retailleau, V. H. Nguyen and V. M. Chau, J. Nat. Prod., 2012, 75, 1578–1583 CrossRef CAS PubMed.
- W. Li, Y. N. Sun, X. T. Yan, S. Y. Yang, E. J. Kim, H. K. Kang and Y. H. Kim, J. Agric. Food Chem., 2013, 61, 10730–10740 CrossRef CAS PubMed.
- L. P. Liu, K. Han, W. Chen, Y. Y. Zhang, L. J. Tong, T. Peng, H. Xie, J. Ding and H. B. Wang, Bioorg. Med. Chem., 2014, 22, 4198–4203 CrossRef CAS PubMed.
- K. H. Kim, E. Moon, S. K. Ha, W. S. Suh, H. K. Kim, S. Y. Kim, S. U. Choi and K. R. Lee, Chem. Pharm. Bull., 2014, 62, 1136–1140 CrossRef CAS PubMed.
- S. S. Li, C. H. Chao, D. Y. Shen, H. H. Chan, C. H. Chen, Y. R. Liao, S. J. Wu, Y. L. Leu, Y. C. Shen, Y. H. Kuo, E. J. Lee, K. Qian, T. S. Wu and K. H. Lee, Bioorg. Med. Chem., 2011, 19, 677–683 CrossRef PubMed.
- J. Peterson, J. Dwyer, H. Adlercreutz, A. Scalbert, P. Jacques and M. L. McCullough, Nutr. Rev., 2010, 68, 571–603 CrossRef PubMed.
- J. Sun, J. Yu, P. C. Zhang, F. Tang, Y. D. Yue, Y. N. Yang, Z. M. Feng and X. F. Guo, J. Agric. Food Chem., 2013, 61, 4556–4562 CrossRef CAS PubMed.
- L. M. Kuo, L. J. Zhang, H. T. Huang, Z. H. Lin, C. C. Liaw, H. L. Cheng, K. H. Lee, S. L. Morris-Natschke, Y. H. Kuo and H. O. Ho, J. Nat. Prod., 2013, 76, 580–587 CrossRef CAS PubMed.
- H. Y. Yu, Z. Y. Chen, B. Sun, J. J. Liu, F. Y. Meng, Y. Liu, T. Tian, A. Jin and H. L. Ruan, J. Nat. Prod., 2014, 77, 1311–1320 CrossRef CAS PubMed.
- G. Y. Yang, R. R. Wang, H. X. Mu, Y. K. Li, X. N. Li, L. M. Yang, Y. T. Zheng, W. L. Xiao and H. D. Sun, J. Nat. Prod., 2013, 76, 250–255 CrossRef CAS PubMed.
- J. Li, A. P. Meng, X. L. Guan, J. Li, Q. Wu, S. P. Deng, X. J. Su and R. Y. Yang, Bioorg. Med. Chem. Lett., 2013, 23, 2238–2244 CrossRef CAS PubMed.
- H. Cui, B. Xu, T. Z. Wu, J. Xu, Y. Yuan and Q. Gu, J. Nat. Prod., 2014, 77, 100–110 CrossRef CAS PubMed.
- Y. C. Chen, C. C. Liaw, Y. B. Cheng, Y. C. Lin, C. H. Chen, Y. T. Huang, S. S. Liou, S. Y. Chen, C. T. Chien, G. C. Lee and Y. C. Shen, Bioorg. Med. Chem. Lett., 2013, 23, 880–885 CrossRef CAS PubMed.
- J. F. Liu, C. Y. Sang, X. H. Xu, L. L. Zhang, X. Yang, L. Hui, J. B. Zhang and S. W. Chen, Eur. J. Med. Chem., 2013, 64, 621–628 CrossRef CAS PubMed.
- H. Wang, L. J. Tang, Y. J. Tang and Z. P. Yuan, Bioorg. Med. Chem., 2014, 22, 6183–6192 CrossRef CAS PubMed.
- A. Kamal, T. S. Reddy, S. Polepalli, N. Shalini, V. G. Reddy, A. V. S. Rao, N. Jain and N. Shankaraiah, Bioorg. Med. Chem., 2014, 22, 5466–5475 CrossRef CAS PubMed.
- K. H. Lee and Z. Y. Xiao, Phytochem. Rev., 2003, 2, 341–362 CrossRef CAS.
- K. Kobayashi and M. J. Ratain, Cancer Chemother. Pharmacol., 1994, 34, 64–68 CrossRef.
- Z. J. Xu, S. X. Gao, M. Zhang and X. Z. Zou, Chin. J. Org. Chem., 2007, 27, 1254–1257 CAS.
-
Z. J. Xu, M.S. Thesis, East China Normal University, Shanghai, 2006 (in Chinese).
- J. D. Sellars and P. G. Steel, Eur. J. Org. Chem., 2007, 3815–3828 CrossRef CAS.
- Y. Q. Liu, D. F. Wei, Y. L. Zhao, W. D. Cheng, Y. Lu, Y. Q. Ma, X. Li, C. Han, Y. X. Wei, H. M. Cao and C. Y. Zhao, Bioorg. Med. Chem., 2012, 20, 6285–6295 CrossRef CAS PubMed.
- L. Kou, M. J. Wang, L. T. Wang, X. B. Zhao, X. Nan, L. Yang, Y. Q. Liu, S. L. Morris and K. H. Lee, Eur. J. Med. Chem., 2014, 75, 282–288 CrossRef CAS PubMed.
- V. T. T. Thanh, V. C. Pham, H. D. T. Mai, M. Litaudon, F. Gueritte, P. Retailleau, V. H. Nguyen and V. M. Chau, Planta Med., 2014, 80, 695–702 CrossRef PubMed.
- X. Li, W. Zhao, H. M. Li, D. J. Wan, D. S. Li, T. Chen and Y. J. Tang, Eur. J. Med. Chem., 2014, 80, 267–277 CrossRef PubMed.
- M. A. Shareef, D. Duscharla, G. Ramasatyaveni, N. R. Dhoke, A. Das, R. Ummanni and A. K. Srivastava, Eur. J. Med. Chem., 2015, 89, 128–137 CrossRef CAS PubMed.
- W. H. Cheng, B. Cao, H. Shang, C. Niu, L. M. Zhang, Z. H. Zhang, D. L. Tian, S. Zhang, H. Chen and Z. M. Zou, Eur. J. Med. Chem., 2014, 85, 498–507 CrossRef CAS PubMed.
- W. Zhao, L. Chen, H. M. Li, D. J. Wang, D. S. Li, T. Chen, Z. P. Yuan and Y. J. Tang, Bioorg. Med. Chem., 2014, 22, 2998–3007 CrossRef CAS PubMed.
- A. C. Ramos, R. Pelaez, J. L. Lopez, E. Caballero, M. Medarde and A. S. Feliciano, Tetrahedron, 2001, 57, 3963–3977 CrossRef CAS.
- A. G. Gonzalez, R. D. Rosa and J. M. Trujillo, Tetrahedron, 1986, 42, 3899–3904 CrossRef CAS.
- F. E. Ziegler and J. A. Schwartz, J. Org. Chem., 1978, 43, 985–991 CrossRef CAS.
- B. Madrigal, P. Puebla, R. Pelaez, E. Caballero and M. Medarde, J. Org. Chem., 2003, 68, 854–864 CrossRef CAS PubMed.
- Z. Z. Che, X. Yu, X. Y. Zhi, L. L. Fan, X. J. Yao and H. Xu, J. Agric. Food Chem., 2013, 61, 8148–8155 CrossRef CAS PubMed.
- X-ray data and ORTEP depiction for compounds 23a and 24, please see the ESI.† CCDC 1430247 (23a) and CCDC 1430248 (24) contain the supplementary crystallographic data for this paper.
- Y. M. Ma, Y. B. Zhou, C. M. Xie, D. M. Chen and J. Li, Acta Pharmacol. Sin., 2012, 33, 407–417 CrossRef CAS PubMed.
- S. Sense, Y. C. Lo, D. Huang, T. A. Zangle, A. A. Gholkar, L. Robert, B. Homet, A. Ribas, M. K. Summers, M. A. Teitell, R. Damoiseaux and J. Z. Torres, Cell Death Dis., 2014, 5, e1462 CrossRef PubMed.
- J. Q. Wang, Y. L. Liu, J. J. Zhao, W. Zhang and X. F. Pang, J. Sci. Food Agric., 2013, 93, 1492–1498 CrossRef CAS PubMed.
- D. K. Sharma, B. Rah, M. R. Lambu, A. Hussain, S. K. Yousuf, A. K. Tripathi, B. Singh, G. Jamwal, Z. Ahmed, N. Chanauria, A. Nargotra, A. Goswami and D. Mukherjee, Med. Chem. Commun., 2012, 3, 1082–1091 RSC.
- F. J. Dai, Y. H. Chen, Y. J. Song, L. Huang, D. Zhai, Y. M. Dong, L. Lai, T. Zhang, D. L. Li, X. F. Pang, M. Y. Liu and Z. F. Yi, PLoS One, 2012, 7, e52162 CAS.
- J. Q. Wang, L. Zhang, G. L. Chen, J. Zhang, Z. X. Li, W. Q. Lu, M. Y. Liu and X. F. Pang, Breast Cancer Res. Treat., 2014, 148, 279–289 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- B. B. Wang, W. W. Yu, J. W. Guo, X. W. Jiang, W. Q. Lu, M. Y. Liu and X. F. Pang, J. Pharmacol. Exp. Ther., 2015, 352, 129–138 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of all new compounds. CCDC 1430247 and 1430248. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02176f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.