
Cyclopent-2-enylaluminium as allylzinc precursor for the diastereoselective allylmetallation of non-racemic imines: applications to the synthesis of enantiomerically enriched heterocycles†
Michaël
Coffinet
,
Samantha
Lamy
,
Florian
Jaroschik
and
Jean-Luc
Vasse
*
Institut de Chimie Moléculaire de Reims, CNRS (UMR 7312) and Université de Reims, 51687 Reims Cedex 2, France. E-mail: jean-luc.vasse@univ-reims.fr
First published on 5th November 2015
Abstract
The generation of cyclopent-2-enylzinc from cyclopentadiene based on a titanium-catalyzed hydroalumination/transmetallation sequence is described. Applied to the allylmetallation of phenylglycinol-derived imines, this sequence leads to homoallylic amines with moderate to good stereoselectivities. The synthesis of disubstituted azetidines and piperidines illustrates the potential of the method.
Amenable to a large range of synthetic transformations, homoallylic amines are key intermediates for the preparation of synthetically and naturally occurring nitrogen-containing molecules of biological interest.1 Among the efficient methods which give access to homoallylic amines in diastereo- and enantiomerically pure forms, the allylmetalation of non-racemic imines is emerging. While a plethora of examples of imine allylmetalation using linear allymetals has been reported in the literature,2 only a few examples concern cyclic allylmetals.3 Notably, no example is reported to our knowledge in the specific case of cyclopenten-2-ylmetal.4 In this context, the development of a method allowing a diastereoselective cyclopenten-2-ylmetallation of imines is of particular interest, since the resulting homoallylic amines may constitute tunable intermediates for azetidines or piperidines synthesis (Fig. 1).
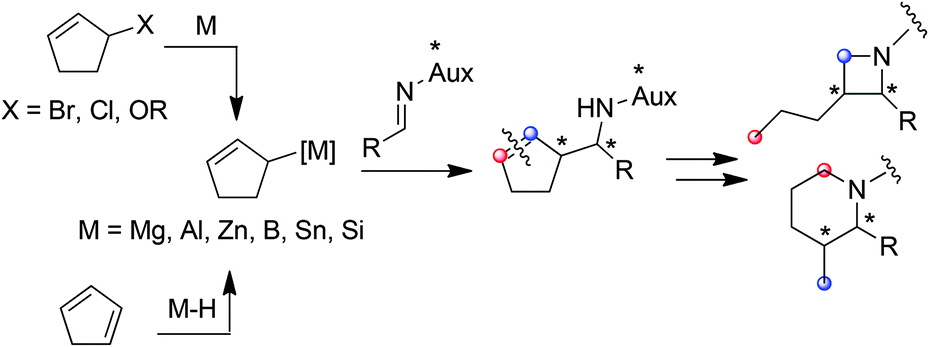 | ||
| Fig. 1 Generation of a cyclopent-2-enylmetal, a potential access to heterocycles through allymetallation of imine. | ||
Typically, allylmetals are prepared from the corresponding bromide,5 chloride,6 alcohol7 or acetate.8 Allylmetals can alternatively be prepared from conjugated dienes via a regioselective metal-catalyzed hydrometallation (Fig. 1).9
The latter approach is of particular interest since cyclic allyl halides are not readily available, some being unstable, thus limiting the synthetic applications.
Recently, we described the regioselective titanium-catalyzed hydroalumination of conjugated dienes applied to pentafulvenes.10 The resulting allylaluminium reacts with aldehydes and ketones to afford homoallylic alcohols in a highly diastereoselective manner (Scheme 1).
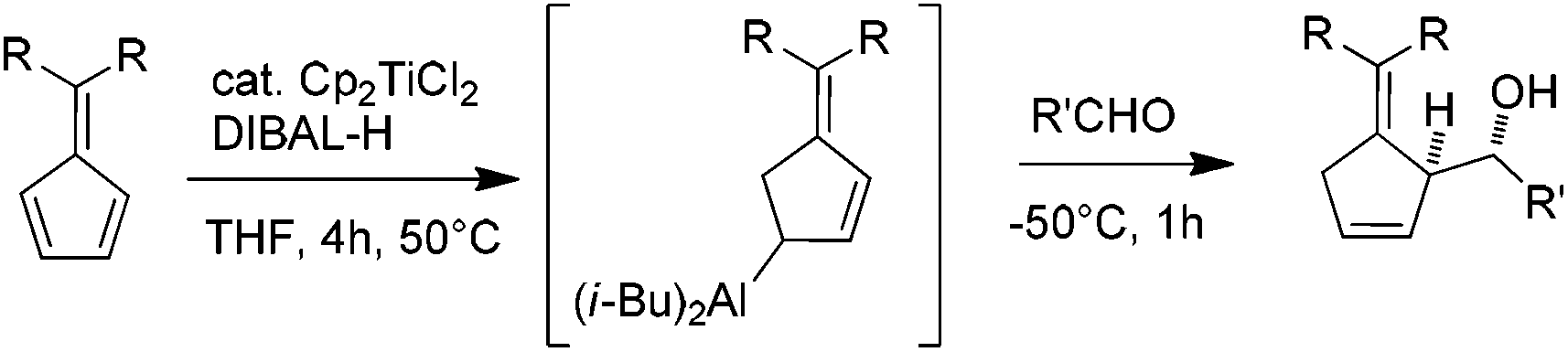 | ||
| Scheme 1 Titanium-catalyzed hydroalumination of pentafulvenes/addition onto aldehydes. | ||
The extension of this method to cyclopentadiene would provide a synthetically useful pathway for the generation of a 5-membered cyclic allylmetal, which coupled to imines, would give an access to original homoallylic amines.
In this communication, we disclose the titanium-mediated generation of cyclopent-2-enylaluminium and its use as allylzinc precursor in the diastereoselective synthesis of enantiomerically enriched homoallylamines, which can be further converted into azetidine or piperidine.
The study began by investigating the possibility of generating 5-membered cyclic allylaluminium by Cp2TiCl2-catalyzed hydroalumination of cyclopentadiene according to our recently reported method. Thus, to a solution of freshly cracked cyclopentadiene (3 equiv.) and Cp2TiCl2 (0.1 equiv.), was added Dibal-H (1.2 equiv.). The mixture was stirred for 4 h at 40 °C, then, benzaldehyde (1 equiv.) was added at 0 °C. The expected homoallylic alcohol 1 was isolated in 87% yield as a single diastereomer (Scheme 2).
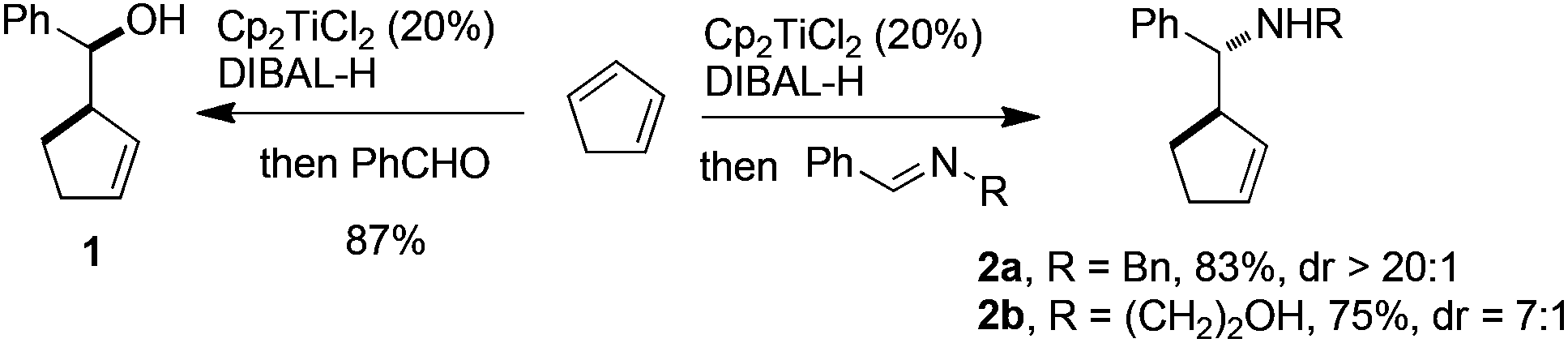 | ||
| Scheme 2 Initial results. | ||
The reactivity of the cyclic allylaluminium reagent was next tested towards two achiral imines. Nearly complete conversion was observed with both benzylamine- and ethanolamine-derived imines. In the latter case, two equiv. of allylaluminium reagent were used, one being presumably consumed by the hydroxyl group. The diastereoselectivity, determined by 1H NMR analysis of the crude reaction mixture, was satisfying in both cases in favor of the anti isomer (Scheme 2).
The encouraging reactivity of allylaluminium reagent towards imines prompted us to test a non-racemic phenylglycinol-derived imine.11,12 In that case, 2.4 equiv. of allylaluminium reagent were used, and the imine was added at low temperature. Unfortunately, despite the major formation of the anti over the syn isomers, a low diastereoselectivity within the couple of anti isomers was observed (Scheme 3).
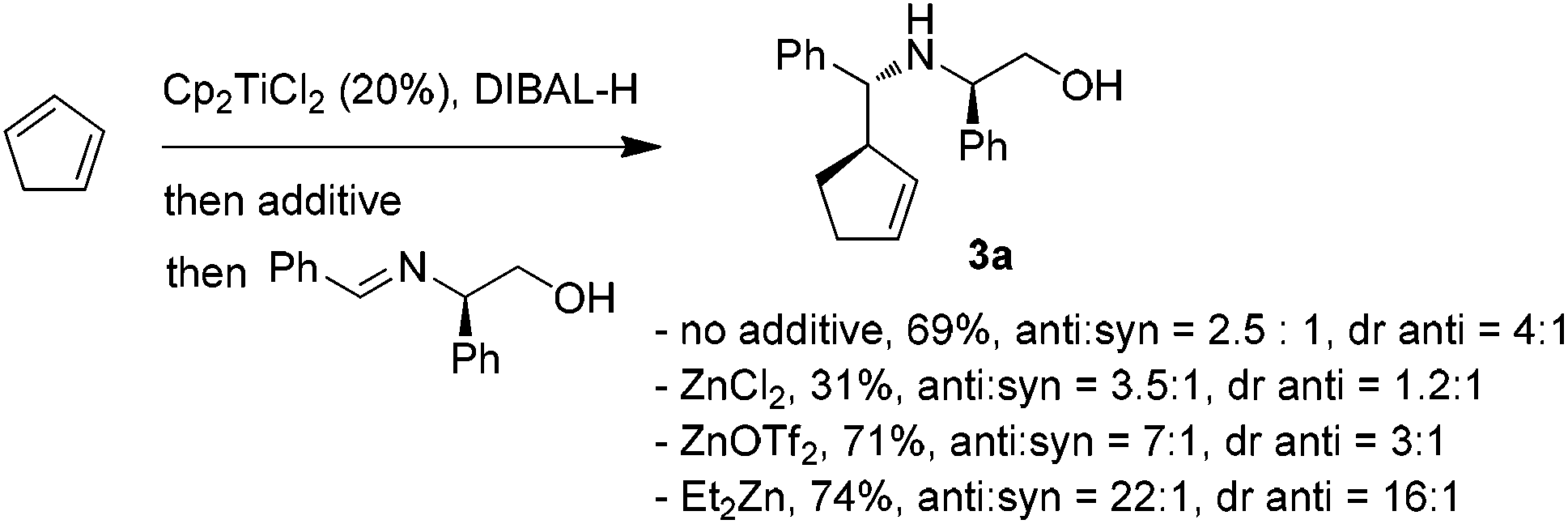 | ||
| Scheme 3 Addition of cyclopent-2-enylaluminium onto phenylgly-cinol-derived imine. Influence of zinc additives. | ||
To envision a diastereoselective process with cyclic allylmetals, it is essential that a kinetic or dynamic kinetic process operates within the organometallic reagent.13 Therefore, one may assume that the low stereoselectivity observed in our case could originate from (i) the absence of a beneficial resolution of the two enantiomeric forms of the organoaluminium or/and (ii) the coordination mode of aluminium to phenylglycinol-derived imine which does not allow an efficient diastereofacial discrimination.
In contrast, the addition of racemic cyclic allylzincs to non-racemic imines is known to proceed in a highly diastereoselective manner.3a Thus, the possibility of generating the analogous allylzinc was next examined by testing zinc-based additives (1 equiv. with respect to DIBAL-H) as Al→Zn transmetallation promotors.5a While the use of ZnCl2 decreased the yield and the stereoselectivity, an improvement of the anti/syn ratio was noticed with Zn(OTf)2. Finally, the best result was obtained by employing Et2Zn. In this case, a nearly complete anti selectivity was observed with a 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio in favor of the R,R,R isomer (Scheme 3).
:1 diastereomeric ratio in favor of the R,R,R isomer (Scheme 3).
With these new conditions in hand, a series of non-racemic imines were tested (Table 1). The reaction is quite general, providing diversely substituted homoallylic amines. Thus, homoallylic amines bearing aryl (entries 1–5), heteroaryl (entries 6–8), alkenyl (entries 9–11) and alkyl (entries 12 and 13) fragments could be obtained.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Aux | syn/antia | dr antia | 3 (%) |
| a Determined by 1H NMR analysis of the crude reaction mixture. b Yield of the major isomer Isolated in the pure form. c Yield of the isolated mixture of unseparable anti isomers. | |||||
| 1 | Ph | (R)-CHPh-CH2OH | 1:22 |
16:1 |
3a (74%)b |
| 2 | Ph | (S)-S(O)-tBu | 1:14 |
5:1 |
3′a (76%)b |
| 3 | 4-MeO-C6H4 | (R)-CHPh-CH2OH | 1:11 |
14.5:1 |
3b (63%)b |
| 4 | 4-Br-C6H4 | (R)-CHPh-CH2OH | 1:8 |
9:1 |
3c (62%)b |
| 5 | 4-CO2Me-C6H4 | (R)-CHPh-CH2OH | 1:5.8 |
7.3:1 |
3d (58%)b |
| 6 | 3-Pyridyl | (R)-CHPh-CH2OH | 1:5.7 |
12:1 |
3e (49%)b |
| 7 | 3-Pyridyl | (S)-S(O)-tBu | 1:10 |
5.8:1 |
3′e (57%)c |
| 8 | 3-Furyl | (R)-CHPh-CH2OH | 1:10 |
11:1 |
3f (78%)c |
| 9 | (E)-Ph-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
(R)-CHPh-CH2OH | 1:8 |
4.4:1 |
3g (56%)c |
| 10 | (E)-Ph-CHCH |
(S)-S(O)-tBu | 1 < 20 | 1:1 |
3′g (51%)c |
| 11 | (E)-Ph-CHCCH3 |
(R)-CHPh-CH2OH | 1:8 |
7.4:1 |
3h (76%)b |
| 12 | n-C4H9 | (R)-CHPh-CH2OH | 1:19 |
5:1 |
3i (29%)c |
| 13 | i-Pr | (R)-CHPh-CH2OH | 1:13 |
16:1 |
3j (60%)b |
Coincidently, the major isomers obtained from (S)-tert-butyl sulfinamide and (R)-Phenylglyninol-derived imines are identically (R,R) configurated, in regards to newly formed stereogenic centers. Imines derived from the Elman's auxiliary are known for inducing a high level of diastereoselectivity,14 however in the present reaction conditions, imines derived from phenylglycinol led to superior stereoselectivities (Table 1, entries 2, 7 and 10 vs. entries 1, 6 and 9). In the latter case, a general tendency indicates that higher stereoselectivities are obtained with imines bearing bulky substituents. While the diastereomeric ratios were typically good with an aryl or a large alkyl group, the selectivity dropped with linear alkyl and alkenyl ones. It could also be noticed that the presence of an extra coordinating group within the substrate affects the selectivity of the reaction. Finally, the major isomer could be isolated in the pure form in many cases. The absolute configuration of the major isomer was assigned by X-ray analysis of crystals grown from 3b and 3h (Fig. 2).
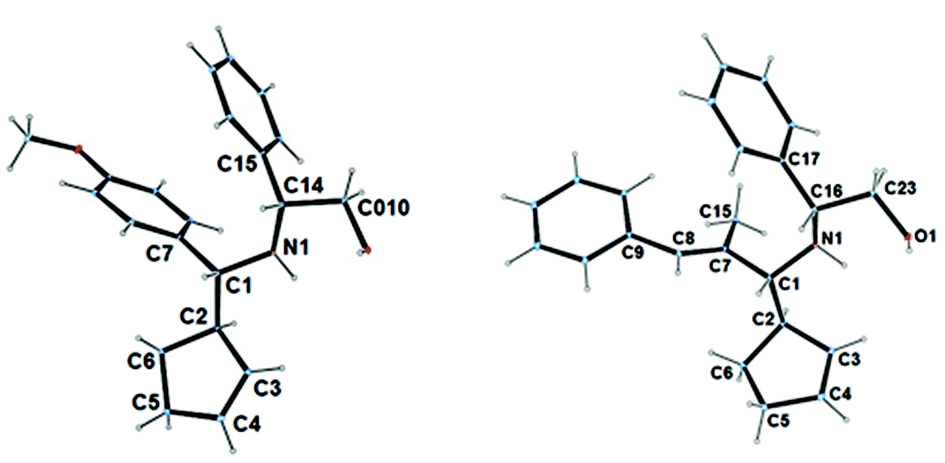 | ||
| Fig. 2 Crystal structure of 3b and 3h (CCDC 1431555–1431556). | ||
Protected homoallylic amines are useful synthetic intermediates which can be readily converted into valuable building blocks by selective transformation of the CC double bond.1a,15 To illustrate the synthetic potential of the method, we decided to convert the cyclopentenyl fragment into a bis electrophilic synthon.
Attempt to cleave the amino-alcohol fragment from diastereomerically pure 3a using NaIO4 gave disappointing results,16 nevertheless, the primary Boc-protected homoallylic amine 4 was obtained in two steps by standard Pb(OAc)4-mediated removal of the chiral auxiliary residue and condensation with Boc2O.17 Alternatively, methanolysis of diastereomerically pure 3′a18 and condensation with Boc2O also gave 4. Subsequent ozonolysis, followed by reduction with NaBH4,19 gave the corresponding diol which was immediately converted into piperidine 5 in 62% yield by mesylation and displacement (Scheme 4).
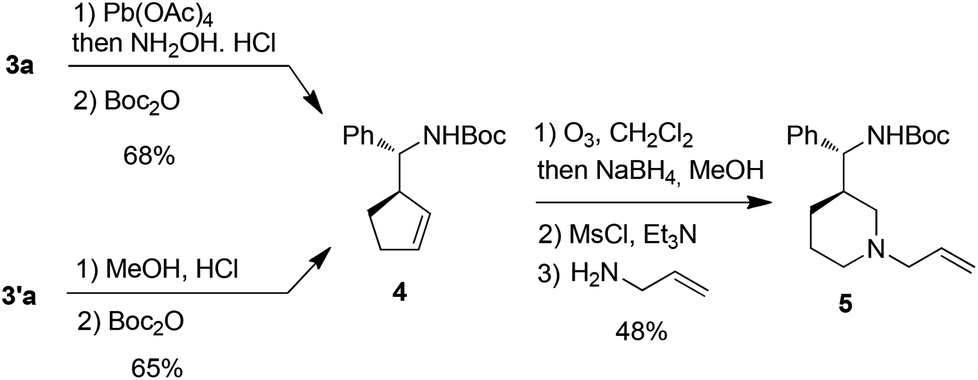 | ||
| Scheme 4 Synthesis of piperidine 5. | ||
To envision an access to heterocycles, a selective transformation of each of the two alcohol chains was next planned. Firstly, ozonolysis and reduction were applied to 620 and the diol intermediate underwent the selective protection of the less sterically hindered primary alcohol using TBDPSCl21 to afford 7. Secondly, the same sequence applied to 8, followed by treatment with mesyl chloride led to the oxazine 9 as the major product along with the corresponding benzoyl-azetidine22 (Scheme 5).
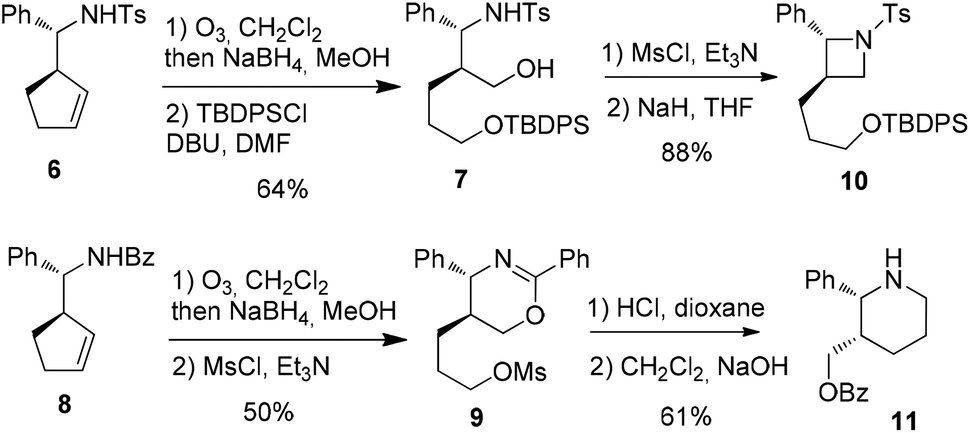 | ||
| Scheme 5 Selective lateral chains transformations and access to azetidine 10 and piperidine 11. | ||
Finally, the trans-2,3-disubstituted azetidine 10 was obtained from 7 by activation of the remaining alcohol chain with mesyl chloride and NaH-promoted cyclization. In parallel, the hydrolysis23 of 9 and subsequent basic treatment afforded the cis-2,3-disubstituted piperidine 11 (Scheme 5).
Conclusions
In summary, the diastereoselective cyclopent-2-en-1-ylmetallation of imines derived from phenylglycinol is described. The key allylzinc reagent was generated via a sequential titanium-catalyzed hydroalumination of cyclopentadiene followed by Al to Zn transmetallation. This method allows the preparation of diversely substituted homoallylic amines which constitute interesting building-blocks for the synthesis of nitrogen-containing heterocycles. This approach was illustrated by the synthesis of 2,3-disubstituted trans azetidine and cis piperidine in the enantiomerically enriched form. Further investigations regarding imines/metals interactions aimed at extending the method to other allylic fragments are currently underway.Acknowledgements
M. C. thanks the Ministère de la Recherche et de l'Enseigne-ment Supérieur for a PhD scholarship. The Université de Reims-Champagne-Ardenne and the CNRS for financial support, Sylviane Chevreux, Sylvie Lanthony and Dominique Harakat for technical assistance are gratefully acknowledged.Notes and references
- (a) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed; (b) K. L. Tan and E. N. Jacobsen, Angew. Chem., Int. Ed., 2007, 46, 1315–1317 CrossRef CAS PubMed; (c) R. Wada, T. Shibuguchi, S. Makino, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7687–7691 CrossRef CAS PubMed; (d) R. Kargbo, Y. Takahashi, S. Bhor, G. R. Cook, G. C. Lloyd-Jones and I. R. Shepperson, J. Am. Chem. Soc., 2007, 129, 3846–3847 CrossRef CAS PubMed; (e) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2007, 129, 15398–15404 CrossRef CAS PubMed; (f) M. Fujita, T. Nagano, U. Schneider, T. Hamada, C. Ogawa and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 2914–1915 CrossRef CAS PubMed; (g) J. A. Sirvent, F. Foubelo and M. Yus, Eur. J. Org. Chem., 2013, 2461–2247 CrossRef CAS; (h) W.-H. Chiou, Y.-W. Wang, C.-L. Kao, P. C. Chen and C.-C. Wu, Organometallics, 2014, 33, 4240–4244 CrossRef CAS.
- (a) X.-W. Sun, M.-H. Xu and G.-Q. Lin, Org. Lett., 2006, 8, 4979–4982 CrossRef CAS PubMed; (b) P. M. A. Rabbat, S. C. Valdez and J. L. Leighton, Org. Lett., 2006, 8, 6119–6121 CrossRef CAS PubMed; (c) J. D. Huber and J. L. Leighton, J. Am. Chem. Soc., 2007, 129, 14552–14553 CrossRef CAS PubMed; (d) D. Huber, N. R. Perl and J. L. Leighton, Angew. Chem., Int. Ed., 2008, 47, 3037–3039 CrossRef PubMed; (e) M. I. Feske, A. B. Santanilla and J. L. Leighton, Org. Lett., 2010, 12, 688–691 CrossRef PubMed; (f) M. Lui, A. Shen, X.-W. Sun, F. Deng, M.-H. Xu and G.-Q. Lin, Chem. Commun., 2010, 46, 8460–8462 RSC.
- (a) L. R. Reddy, B. Hu, M. Prashad and K. Prasad, Org. Lett., 2008, 10, 3109–3112 CrossRef CAS PubMed; (b) I. L. Lysenko, H. G. Lee and J. K. Cha, Org. Lett., 2009, 11, 3132–3134 CrossRef CAS PubMed.
- (a) C. Samann and P. Knochel, Synthesis, 2013, 1070–1076 Search PubMed; (b) L. R. Reddy and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 6230–6231 CrossRef CAS PubMed.
- (a) Z. Peng, T. D. Blümke, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 8516–8519 CrossRef CAS PubMed; (b) H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376–5377 CrossRef CAS PubMed; (c) P. H. Lee, K. Lee and S. Chang, Synth. Commun., 2001, 31, 3189–3196 CrossRef CAS; (d) F. A. Khan and B. Prabhudas, Tetrahedron, 2000, 56, 7595–7599 CrossRef CAS; (e) S. Harthong, T. Billard and B. R. Langlois, Synthesis, 2005, 2253–2263 CAS; (f) D. Oguro and H. Watanabe, Tetrahedron, 2011, 67, 777–781 CrossRef CAS; (g) N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2008, 6, 2782–2789 RSC; (h) H.-H. Tan, C.-Z. Tao, Y.-Q. Hou, L. Luo, L. Liu and Q.-X. Guo, Chin. J. Chem., 2005, 23, 237–241 CrossRef; (i) X.-X. Peng, Y.-J. Deng, X.-L. Yang, L. Zhang, W. Yu and B. Han, Org. Lett., 2014, 16, 4650–4653 CrossRef CAS PubMed.
- H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376–5377 CrossRef CAS PubMed.
- (a) T. Hirashita, S. Kambe, H. Tsuji, H. i. Omori and S. Araki, J. Org. Chem., 2004, 69, 5054–5059 CrossRef CAS PubMed; (b) N. Selander, S. Sebelius, C. Estay and K. Szabo, Eur. J. Org. Chem., 2006, 4085–4087 CrossRef CAS; (c) R. Alam, M. Raducan, L. Eriksson and K. J. Szabo, Org. Lett., 2013, 15, 2546–2549 CrossRef CAS PubMed; (d) G. Fontana, A. Lubineau and M.-C. Scherrmann, Org. Biomol. Chem., 2005, 3, 1375–1380 RSC.
- (a) G. P. Howell, A. J. Minnaard and B. L. Feringa, Org. Biomol. Chem., 2006, 4, 1278–1283 RSC; (b) S. Hikichi, Y. Gao and F. Sato, Tetrahedron Lett., 1997, 38, 2867–2870 CrossRef CAS; (c) T.-Z. Zhang, L.-X. Daia and X.-L. Hou, Tetrahedron: Asymmetry, 2007, 18, 251–259 CrossRef CAS.
- (a) Y. Sasaki, C. Zhong, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 1226–1227 CrossRef CAS PubMed; (b) S. Kobayashi and K. Nishio, J. Org. Chem., 1994, 59, 6620–6628 CrossRef CAS; (c) J. Y. Wu, B. Moreau and T. Ritter, J. Am. Chem. Soc., 2009, 131, 12915–12917 CrossRef CAS PubMed; (d) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 725–732 CrossRef PubMed.
- J. Joseph, F. Jaroschik, K. V. Radhakrishnan, D. Harakat, J.-L. Vasse and J. Szymoniak, Chem. – Eur. J., 2014, 20, 5433–5438 CrossRef CAS PubMed.
- For examples of diastereoselective allylation of phenylglycinol-derived imines, see: (a) M. Ahari, A. Joosten, J.-L. Vasse and J. Szymoniak, Synthesis, 2008, 61–68 CAS; (b) R. W. Bates, K. Sivarajan and B. F. Straub, J. Org. Chem., 2011, 76, 6844–6848 CrossRef CAS PubMed; (c) J.-M. Huang, X.-X. Wang and Y. Dong, Angew. Chem., Int. Ed., 2011, 50, 924–927 CrossRef CAS PubMed.
- (R)-Phenylglycinol was the only chiral aminoalcohol tested.
- (a) G. Alvaro and D. Savoia, Synlett, 2002, 651–673 CrossRef CAS; (b) H. Ding and G. K. Friedstad, Synthesis, 2005, 2815–2829 CAS.
- (a) D. Chen and M.-H. Xu, Chem. Commun., 2013, 49, 1327–1329 RSC; (b) G. Kolodney, G. Sklute, S. Perrone, P. Knochel and I. Marek, Angew. Chem., Int. Ed., 2007, 46, 9291–9294 CrossRef CAS PubMed; (c) A. Shen, M. Liu, Z.-S. Jia, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 5154–5157 CrossRef CAS PubMed; (d) M. S. Maji, R. Fröhlich and A. Studer, Org. Lett., 2008, 7, 1847–1850 CrossRef PubMed; (e) P. Wipf and J. G. Pierce, Org. Lett., 2005, 7, 3537–3540 CrossRef CAS PubMed.
- (a) Q. Chen, X.-L. Qiu and F.-L. Qing, J. Org. Chem., 2006, 71, 3762–3767 CrossRef CAS PubMed; (b) P.-O. Delaye, T. K. Pradhan, E. Lambert, J.-L. Vasse and J. Szymoniak, Eur. J. Org. Chem., 2010, 3395–3406 CrossRef CAS; (c) T. J. Senter, M. L. Schulte, L. C. Konkol, T. E. Wadzinski and C. W. Lindsley, Tetrahedron Lett., 2013, 54, 1645–1648 CrossRef CAS PubMed; (d) D. Enders, J. Schankat and M. Klatt, Synthesis, 1994, 795–800 CAS; (e) P. V. Ramachandran and T. E. Burghardt, Chem. – Eur. J., 2005, 11, 4387–4395 CrossRef CAS PubMed; (f) H. Huang, H. Wang, A. Kozekova, C. J. Rizzo and M. P. Stone, J. Am. Chem. Soc., 2011, 133, 16101–16110 CrossRef CAS PubMed; (g) A. A. Kudale, P. Anaspure, F. Goswami and M. Voss, Tetrahedron Lett., 2014, 55, 7219–7221 CrossRef CAS; (h) G. Lesma, B. Danieli, A. Sacchetti and A. Silvani, J. Org. Chem., 2006, 71, 3317–3320 CrossRef CAS PubMed; (i) S. Chandrasekhar, S. S. Sultana, N. Kiranmai and C. Narsihmulu, Tetrahedron Lett., 2007, 48, 2373–2375 CrossRef CAS.
- Similar unsuccessful results using NaIO4 were previously reported see: A. K. Awasthi, M. L. Boys, K. J. Cain-Janicki, P.-J. Colson, W. W. Doubleday, J. E. Duran and P. N. Farid, J. Org. Chem., 2005, 70, 5387–5397 CrossRef CAS PubMed.
- (a) T. Vilaivan, C. Winotapan, V. Banphavichit, T. Shinada and Y. Ohfune, J. Org. Chem., 2005, 70, 3464–3471 CrossRef CAS PubMed; (b) P.-O. Delaye, J.-L. Vasse and J. Szymoniak, Org. Lett., 2012, 14, 3004–3007 CrossRef CAS PubMed.
- (a) G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS; (b) M. B. Tait, S. Butterworth and J. Clayden, Org. Lett., 2015, 17, 1236–1239 CrossRef CAS PubMed.
- Q. Chen, X.-L. Qui and F.-L. Qing, J. Org. Chem., 2006, 71, 3762–3767 CrossRef CAS PubMed.
- A NTs protection was chosen to avoid the competitive cyclic carbamate formation frequently observed with Boc-protected amines. For a recent example see: J. K. Aaseng and O. R. Gautun, Tetrahedron, 2014, 70, 5057–5063 CrossRef CAS.
- S. Sumi, K. Matsumoto, H. Tokuyama and T. Fukuyama, Tetrahedron, 2003, 59, 8571–8587 CrossRef CAS.
- Interestingly, the benzoylazetidine could be converted into 9 in a clean three step sequence, see ESI.†.
- Y. M. Lee, D. J. Baek, S. Lee, D. Kim and S. Kim, J. Org. Chem., 2011, 76, 408–416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR data of all new compounds. CCDC 1431555 (3b) and 1431556 (3h). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02184g |
| This journal is © The Royal Society of Chemistry 2016 |