
Determination of rate constants for trifluoromethyl radical addition to various alkenes via a practical method†
M.
Hartmann
,
Y.
Li
and
A.
Studer
*
Organisch-Chemisches Institut, Westfäliscihe Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 5th November 2015
Abstract
A simple and practical method for the determination of rate constants for trifluoromethyl radical addition to various alkenes by applying competition kinetics is introduced. In the kinetic experiments the trifluoromethyl radicals are generated in situ from a commercially available hypervalent-iodine-CF3 reagent (Togni-reagent) by SET-reduction with TEMPONa in the presence of TEMPO and a π-acceptor. From the relative ratio of TEMPOCF3 and CF3-addition product formed, which is readily determined by 19F-NMR spectroscopy, rate constants for trifluoromethyl radical addition to the π-acceptor can be calculated. The practical method is also applicable to measure rate constants for the addition of other perfluoroalkyl radicals to alkenes as documented for CF3CF2-radical addition reactions.
Introduction
Fluorinated compounds are highly valuable in medicinal chemistry, in the agrochemical industry, and also in materials science.1–5 It is therefore not surprising that various synthetic methods for the preparation of fluorinated or perfluoroalkylated compounds have been developed. Several reviews covering the state-of-the-art in this active research area have recently appeared.6 The trifluoromethyl substituent, as judged from the high recent activities, has attracted particular attention along these lines, and ionic as well as transition-metal-based processes have been developed for the introduction of the trifluoromethyl group.6Radical chemistry has also been successfully applied to alkene and arene trifluoromethylation and many methods for the generation of the trifluoromethyl radical have been introduced.7 The trifluoromethyl and more generally perfluoroalkyl radicals exhibit high reactivity which largely derives from their high electrophilicity and their pyramidal geometry.8,9 Surprisingly, despite the great importance of the trifluoromethyl radical in organic synthesis, to our knowledge only very few absolute rate constants for the addition of the CF3-radical to alkenes have been disclosed to date. Ingold, Lusztyk and Dolbier Jr measured the rate constant for CF3-radical addition to styrene, to pentafluorostyrene (C6F5CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and to α-methylstyrene by laser flash photolysis studies.10 However, a larger list on rate constants of the CF3-radical addition to different π-acceptors is clearly missing. Kinetic data are highly important for reaction design in radical chemistry. Herein, we introduce a novel highly practical method to determine rate constants for CF3-radical addition to various acceptors by competition kinetics. Kinetic experiments are very easy to conduct and special equipment such as a laser flash photolysis apparatus is not necessary. The method relies on 19F-NMR spectroscopy which is nowadays available in most research laboratories. We will also show that the novel approach can be applied to measure rate constants for the addition of the CF3CF2-radical to π-acceptors.
CH2) and to α-methylstyrene by laser flash photolysis studies.10 However, a larger list on rate constants of the CF3-radical addition to different π-acceptors is clearly missing. Kinetic data are highly important for reaction design in radical chemistry. Herein, we introduce a novel highly practical method to determine rate constants for CF3-radical addition to various acceptors by competition kinetics. Kinetic experiments are very easy to conduct and special equipment such as a laser flash photolysis apparatus is not necessary. The method relies on 19F-NMR spectroscopy which is nowadays available in most research laboratories. We will also show that the novel approach can be applied to measure rate constants for the addition of the CF3CF2-radical to π-acceptors.
Results and discussion
We recently introduced the use of sodium aminoalkoxide 2 (TEMPONa), which is readily generated by the reduction of the commercially available 2,2,6,6-tetramethylpiperidine-N-oxyl radical (TEMPO) in tetrahydrofuran (THF), as a mild single electron transfer (SET) reagent for clean generation of the trifluoromethyl radical from the hypervalent I(III)–CF3 reagent 1 (Togni-reagent, Scheme 1).11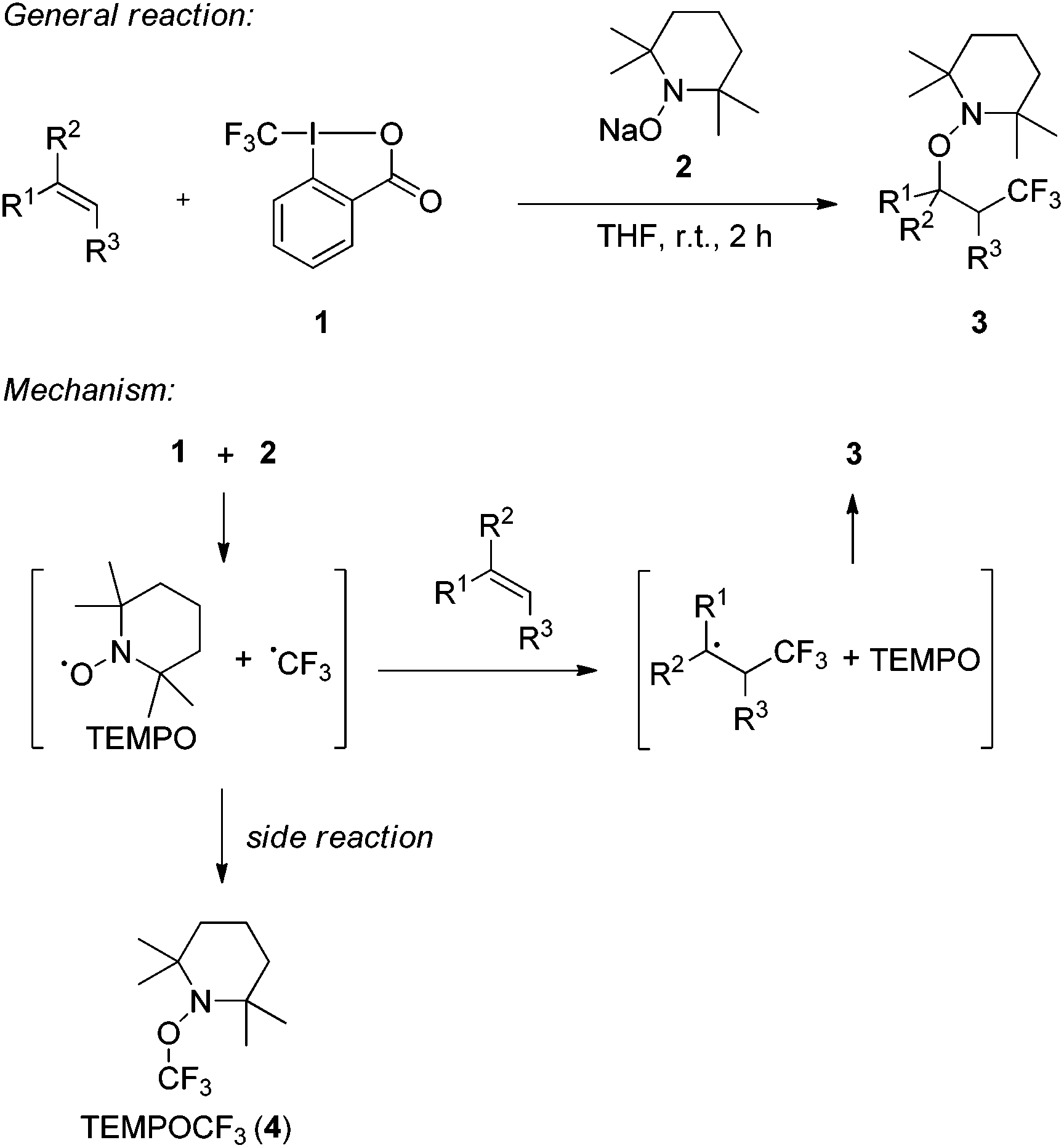 | ||
| Scheme 1 Radical trifluoromethylaminoxylation of alkenes mediated by TEMPONa. General reaction and mechanism. | ||
Reaction of the reagent 16b with TEMPONa 2 in the presence of an alkene provided the corresponding trifluoromethylaminoxylation product 3 in good to excellent yields.11 This sequence proceeds via the following mechanism. Reaction of TEMPONa with 1 first generates the CF3-radical along with the persistent TEMPO radical.12 Na-ortho-iodobenzoate is formed as a side product (not shown in the scheme). The CF3-radical then adds to the alkene to give the corresponding adduct radical which is eventually trapped by TEMPO to provide the trifluoromethylaminoxylated product 3.13 If the addition of the CF3-radical to the alkene is slow, competing direct trapping of the CF3 radical by TEMPO to give TEMPOCF3 (4) can occur as a side reaction. To suppress the formation of 4 in preparative experiments, the concentration of TEMPO was kept low by adding TEMPONa via a syringe pump and by generally using the alkene in excess.
Based on these findings, we decided to use the direct TEMPO-trapping of the CF3-radical (side reaction in the preparative experiments) as a radical clock14 for determining rate constants for CF3-radical addition to various alkenes. If the trifluoromethylaminoxylation depicted in Scheme 1 is conducted with a large excess of the alkene the concentration of the alkene can be considered to be constant during the whole reaction (pseudo-first order conditions). Moreover, if free TEMPO is added to the reaction mixture its concentration is held constant throughout the whole process because the amount of TEMPO generated in the reaction of 1 with 2 gets consumed in the formation of 3 or TEMPOCF3. Therefore, the concentration of added free TEMPO corresponds to the constant concentration of free TEMPO in the reaction mixture during the whole process. By knowing the rate constant for the reaction of TEMPO with the CF3-radical, one can determine the rate constant for CF3-radical addition to an alkene by simply determining the relative ratio of TEMPOCF3 (4) with respect to the trifluoromethylaminoxylation product 3 formed. This ratio can readily be determined by 19F-NMR spectroscopy without any prior work-up of the reaction mixture. The rate constant for addition kadd can then be calculated with eqn (2) which evolves from kinetic eqn (1) where the alkene and the TEMPO concentrations are constant and known:
 | (1) |
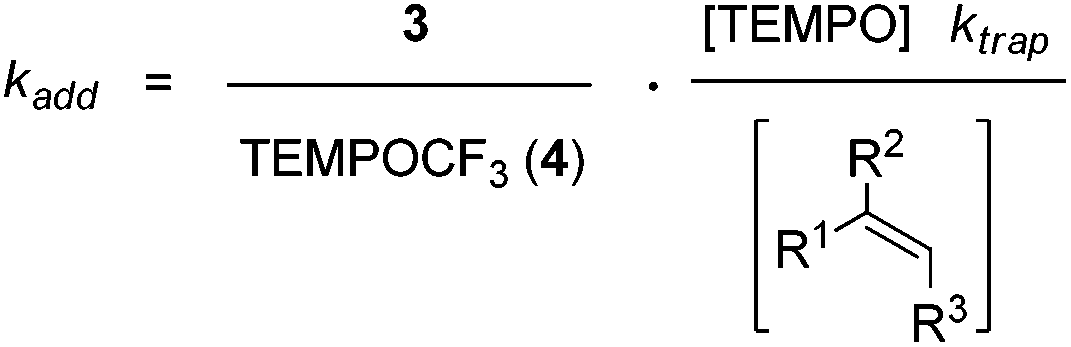 | (2) |
In order to apply eqn (2), the rate constant for CF3-radical trapping by TEMPO (ktrap) had to be determined first. To this end, we used the known absolute rate constant for CF3-radical addition to styrene (kadd = 5.3 × 107 M−1 s−1)10 as a radical clock and conducted a series of trifluoromethylaminoxylations with styrene (20 equiv.) and TEMPONa (1.2 equiv.) in the presence of TEMPO (3.3 equiv.). From the ratio of 4 (TEMPOCF3) and the trifluoromethylaminoxylated product 3a the rate constant of ktrap was calculated using eqn (3) evolved from kinetic eqn (1) (Scheme 2). Reactions were very clean, and in the 19F-NMR spectrum only the resonances belonging to 3a and 4 were observed. The kinetic experiment was repeated 5 times and the product ratio was averaged over all these runs. A value of 8.1 (±0.3) × 108 M−1 s−1 was obtained for TEMPO-trapping (ktrap) of the CF3-radical at room temperature.
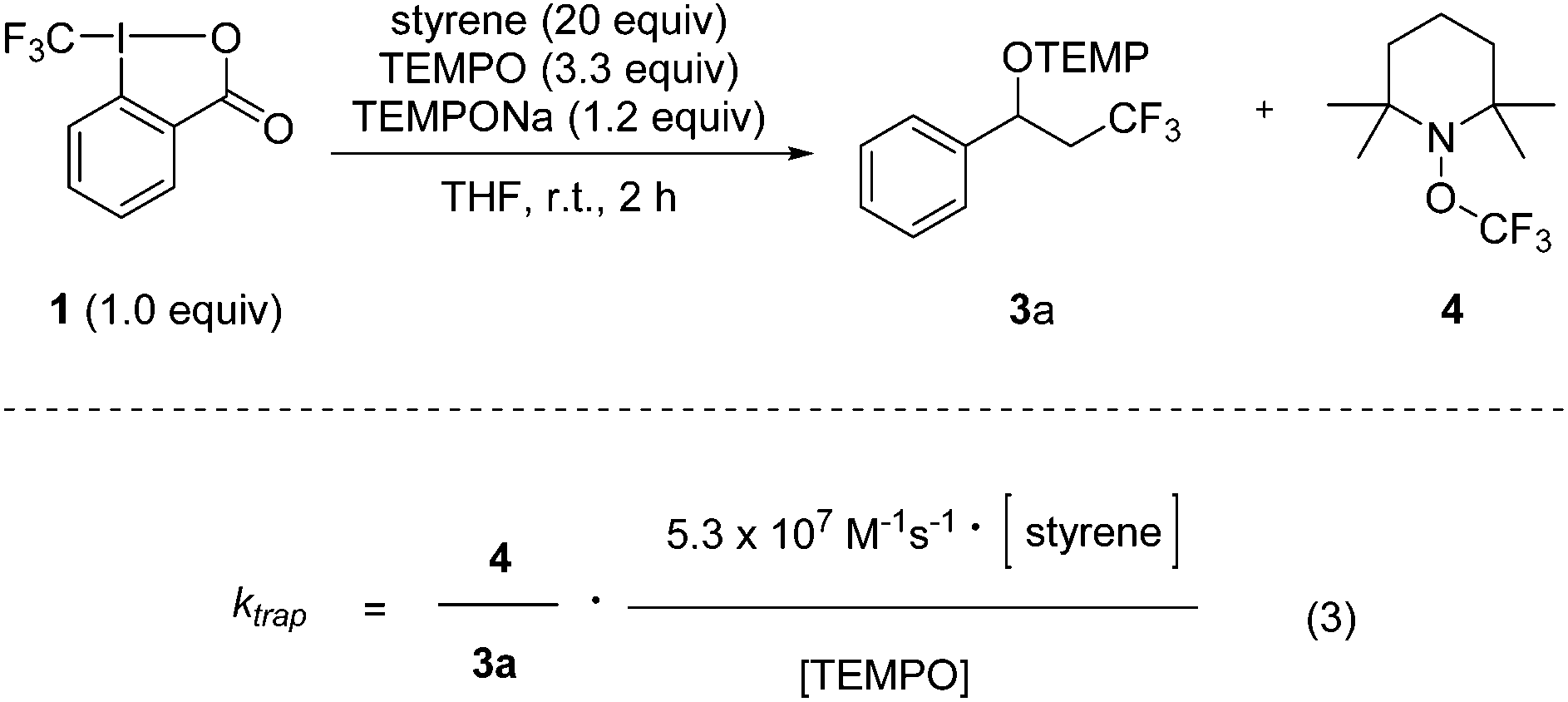 | ||
| Scheme 2 Determination of the rate constant for CF3-radical trapping with TEMPO. | ||
With this novel radical clock in hand we then determined rate constants for radical addition of the trifluoromethyl radical to various π-acceptors by competition kinetics using eqn (2) with ktrap = 8.1 × 108 M−1 s−1. The alkene was used in a 20 fold excess in these kinetic experiments (pseudo-first order). Reactions were clean and the product ratio was readily determined by 19F-NMR spectroscopy of the reaction mixture.15 Experiments were repeated at least 2 times and data obtained were averaged. The rate constants determined for various acceptors are summarized in Table 1.

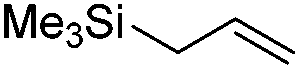


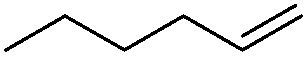

It is expected that the trifluoromethyl radical behaves in alkene addition reactions as the perfluoropropyl radical for which a larger set of data is available in the literature.9c Reactions which occur via early transition states will be fast and kinetics will be governed by polar effects (electron rich double bonds are more reactive as compared to electron deficient radical acceptors).
Comparing the three known rate constants for CF3-radical addition10 to styrene derivatives with the corresponding values for CF3CF2CF2-radical addition, we assume the CF3-radical due to its higher electrophilicity to be generally slightly more reactive than the perfluoropropyl radical. For the second order rate constant of the CF3-radical addition to α-methylstyrene we measured a value of 7.6 × 107 M−1 s−1 which is similar to the reported rate constant for the CF3CF2CF2-radical adding to α-methylstyrene (7.8 × 107 M−1 s−1).10 Our value compares well with the rate constant measured by using laser flash photolysis (8.7 × 107 M−1 s−1),10 validating our novel practical method. Small differences are expected due to the fact that solvent polarity should influence to some extent the rate constants.9e The laser flash-photolysis experiments were conducted in Freon 11310 whereas our kinetics were run in THF.
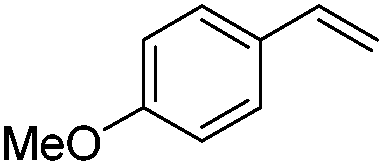
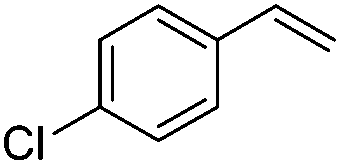
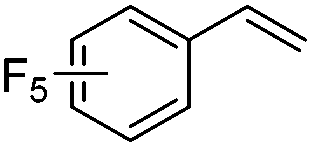
As expected based on polar effects, the rate constant increases for the para-methoxy-substituted styrene (11 × 107 M−1 s−1) as compared to the parent styrene and the electron-poorer pentafluorostyrene showed a significantly smaller rate constant (1.9 × 107 M−1 s−1). For 4-chlorostyrene (6.5 × 107 M−1 s−1) and styrene similar values were obtained. Notably, the rate constant measured for CF3-addition to C6F5CHCH2 is in good agreement with the literature value (2.6 × 107 M−1 s−1),10 further confirming the usefulness of the introduced practical and simple method. The reported rate constant for the corresponding CF3CF2CF2-radical addition is smaller (1.3 × 107 M−1 s−1).10
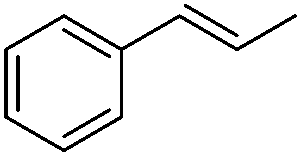
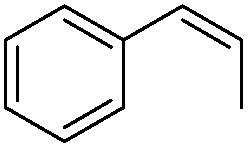
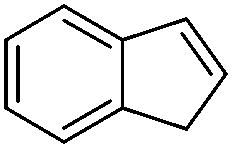
Steric factors affect CF3-radical addition rate constants and for trans-β-methylstyrene a value of 1.1 × 107 M−1 s−1 was measured. Interestingly, cis-β-methylstyrene reacts measurably more slowly (0.62 × 107 M−1 s−1). In the latter case due to the cis-methyl substituent the vinyl group and the activating phenyl substituent are not in perfect conjugation and this likely explains the lower reactivity of the cis-derivative as compared to its trans-congener. For indene a rate constant of 1.5 × 107 M−1 s−1 was measured. In line with other alkenes, CF3CF2CF2-radical addition to β-methyl styrene (configuration not specified in the paper) was reported to occur with a smaller rate constant (0.38 × 107 M−1 s−1).9c
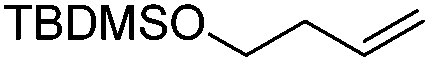
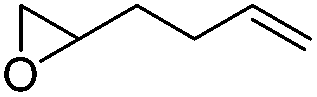
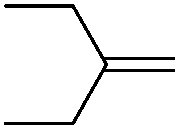
We also investigated rate constants for addition to non-activated double bonds and examined 1-hexene first. As expected, due to the lower nucleophilicity of this alkene as compared to styrene, a smaller rate constant was measured (0.63 × 107 M−1 s−1). 5-Hexen-1-ol reacted slightly faster with a rate constant of 0.88 × 107 M−1 s−1 and a slightly lower rate constant was obtained for the corresponding TBDMS-protected derivative (0.54 × 107 M−1 s−1). The same rate constant was also measured for the lower homologue TBDMSOCH2CH2CHCH2 (0.54 × 107 M−1 s−1). Allyl trimethylsilane is electronically activated by the silyl group and as compared to the unactivated 1-hexene a 2 fold larger rate constant was found (1.2 × 107 M−1 s−1). A slightly larger value (1.5 × 107 M−1 s−1) was measured for butylvinyl ether and to our surprise bis allyl ether reacts fast with the trifluoromethyl radical (2.0 × 107 M−1 s−1). Under the applied conditions, 5-exo cyclization was fully suppressed and we only observed the trifluoromethylaminoxylation product (see ESI†). As expected, a remote epoxide entity does not alter the addition rate constant to a large extent (0.72 × 107 M−1 s−1). Due to the higher nucleophilicity of the double bond in 2-ethyl-1-butene as compared to 1-hexene, the rate constant increases (2.0 × 107 M−1 s−1).
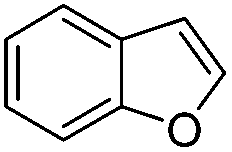
Whereas we failed to measure rate constants for CF3-radical addition to non-activated arenes, for a single example (benzofuran) our method was applicable and a value of 0.13 × 107 M−1 s−1 was obtained. As previously shown,11 the CF3-radical adds to the α-position of the O-atom in benzofuran and the benzylic adduct radical gets trapped by TEMPO with complete diastereocontrol (trans-product). Hence in the kinetic competition experiment only two resonances were identified in the 19F-NMR spectrum. For values <106 M−1 s−1 we see the lower limit for a measurable rate constant using our novel method. If the addition reaction with the trifluoromethyl radical is too slow, direct TEMPO trapping becomes the exclusive pathway.
To document the generality of our method we also applied the approach to measure rate constants for pentafluoroethyl radical addition to alkenes. The CF3CF2-Togni-reagent 5 was previously introduced by us.11 As for the CF3-case, the TEMPO-trapping rate constant had to be determined first. To this end, we chose the known rate constant for the addition of the CF3CF2-radical to α-methylstyrene (9.4 × 107 M−1 s−1)9c as a radical clock in competition experiments (Scheme 3). Reactions were conducted by using a large excess of α-methylstyrene (20 equiv.) in the presence of 2.9 equiv. of free TEMPO. TEMPONa (1.2 equiv.) was added. Along with the pentafluoroethylaminoxylation product 6a we also identified the perfluoroalkylated styrene 7a as a side product (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6, see ESI†). 7a either derives from TEMPOH elimination in product 6a or via direct H-abstraction of the benzylic adduct radical by TEMPO. Since 6a and 7a both derive from CF3CF2-radical addition to α-methylstyrene, the rate constant ktrap for the reaction of the CF3CF2-radical with TEMPO can be calculated from the ratio of TEMPOC2F5 (8) and the sum of products 6a and 7a according to eqn (4). The kinetic experiment was repeated 5 times and 8.6 (±0.3) × 108 M−1 s−1 was measured for the second order TEMPO-trapping rate constant ktrap at room temperature, similar to the rate constant for TEMPO-trapping of the CF3-radical.
:1.6, see ESI†). 7a either derives from TEMPOH elimination in product 6a or via direct H-abstraction of the benzylic adduct radical by TEMPO. Since 6a and 7a both derive from CF3CF2-radical addition to α-methylstyrene, the rate constant ktrap for the reaction of the CF3CF2-radical with TEMPO can be calculated from the ratio of TEMPOC2F5 (8) and the sum of products 6a and 7a according to eqn (4). The kinetic experiment was repeated 5 times and 8.6 (±0.3) × 108 M−1 s−1 was measured for the second order TEMPO-trapping rate constant ktrap at room temperature, similar to the rate constant for TEMPO-trapping of the CF3-radical.
 | ||
| Scheme 3 Determination of the rate constant for C2F5-radical trapping with TEMPO and addition rate constants to various styrene derivatives. | ||
Finally, the TEMPO/CF3CF2-radical trapping reaction was used as a radical clock to determine rate constants for the addition of the CF3CF2-radical to styrene, para-methoxy styrene and para-chloro styrene. Kinetic experiments were performed as described above for the reactions with the Togni-reagent 2. The measured rate constants for C2F5-radical addition compared to CF3-radical addition are slightly larger and governed by polar effects. This is in agreement with literature reports.9c,10 Since these three experiments were mainly conducted to show the potential of the method, additional rate constants for CF3CF2-radical additions were not determined.
Summary and conclusions
In conclusion, we have introduced a novel practical method for the determination of rate constants for the addition of the trifluoromethyl and the pentafluoroethyl radical to various alkenes (20 values measured). Kinetic competition experiments are very easy to conduct and special equipment is not necessary. As an analytical tool 19F-NMR spectroscopy is used. The 19F-NMR spectra are recorded directly on the reaction mixture and workup or product isolation is not necessary. This is important because some perfluoroalkylated compounds are volatile and product loss during isolation will lead to errors in the kinetic data. Internal standards are not required to run these kinetic competition experiments.We determined rate constants for the TEMPO-trapping of the trifluoromethyl and pentafluoroethyl radical. These reactions have then been used as radical clocks in kinetic competition experiments. As is known for perfluoroalkyl radicals, rate constants for radical additions to alkenes are large and show polar effects. The trifluoromethyl radical is generally slightly more reactive than the heptafluoropropyl radical but slightly less reactive than the perfluoroethyl radical.16 Importantly, our method should be generally applicable to measure kinetics for alkene perfluoroalkyl radical additions. A prerequisite is that the corresponding perfluoroalkyl-Togni-reagent can be prepared and that at least one absolute alkene addition rate constant for this particular perfluoroalkyl radical is known from the literature to clock the TEMPO trapping reaction.
Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for financial support.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2015, 115, 2432 Search PubMed.
- C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- (a) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (c) T. Besset, T. Poisson and X. Pannecouke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (d) C. Zhang, Adv. Synth. Catal., 2014, 356, 2895 CrossRef CAS; (e) S. Barata-Vallejo, B. Lantano and A. Postigo, Chem. – Eur. J., 2014, 20, 16806 CrossRef CAS PubMed; (f) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (g) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (h) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (i) P. Chen and G. Liu, Synthesis, 2013, 2919 CAS.
- (a) W. R. Dolbier Jr. and X. X. Rong, Chem. Rev., 1996, 96, 1557 CrossRef; (b) W. R. Dolbier Jr. in Topics in Current Chemistry, ed. R. D. Chambers, Springer, Berlin, 1997, vol. 192, p. 97 Search PubMed; (c) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- J. M. Tedder and J. C. Walton, Tetrahedron, 1980, 36, 701 CrossRef CAS.
- (a) W. R. Dolbier Jr. and X. X. Rong, J. Fluorine Chem., 1995, 72, 235 CrossRef; (b) B. Delest, A. B. Shtarev and W. R. Dolbier Jr., Tetrahedron, 1998, 54, 9273 CrossRef CAS; (c) D. V. Avila, K. U. Ingold, J. Lusztyk, W. R. Dolbier, H.-Q. Pan and M. Muir, J. Am. Chem. Soc., 1994, 116, 99 CrossRef CAS; (d) X. X. Rong, H.-Q. Pan, W. R. Dolbier and B. E. Smart, J. Am. Chem. Soc., 1994, 116, 4521 CrossRef CAS; (e) L. Zhang, W. R. Dolbier Jr., B. Sheeller and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 6362 CrossRef CAS.
- D. A. Avila, K. U. Ingold, J. Lusztyk, W. R. Dolbier Jr. and H.-Q. Pan, J. Org. Chem., 1996, 61, 2027 CrossRef CAS.
- Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 CrossRef CAS PubMed.
- (a) T. Vogler and A. Studer, Synthesis, 2008, 1979 CAS; (b) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed.
- The use of TEMPONa as a SET reagent is not limited to the generation of trifluoromethyl radicals. We also developed analogous procedures for aryl, azidyl or bissulfonamidyl radical generation, see: (a) M. Hartmann, Y. Li and A. Studer, J. Am. Chem. Soc., 2012, 134, 16516 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Org. Lett., 2013, 15, 4548 CrossRef CAS PubMed; (c) Y. Li, M. Hartmann and A. Studer, Chem. Commun., 2015, 51, 5706 RSC.
- M. Newcomb, Tetrahedron, 1993, 49, 1151 CrossRef CAS.
- In some experiments we identified in very small amounts of a doublet at around −60 ppm in the 19F-NMR spectrum. The corresponding product could not be isolated and assigned. However, since it is always the same side product for different alkenes it cannot be derived from CF3-radical addition to the alkene. Therefore, kinetics will not be disturbed. We also performed the reaction with styrene without any workup and directly subjected the crude reaction mixture to 19F NMR-analysis to determine whether CF3H is generated. However, only 3a and 4 were identified in the crude reaction mixture indicating that H-abstraction by the CF3 radical does not occur. Additional experiments on selected examples (4 systems) with an internal standard also revealed perfect mass balance showing that the Togni reagent is fully converted to either the perfluoroalkylation product or the direct trapping product (see ESI†). In agreement with this observation, we never identified TEMPO-trapping products derived from H-abstraction from the solvent THF or from an alkene.
- Pauling-electronegativity for F = 4.0 and CF3 = 3.4, see: E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, Universal Science Books, Sausalito, California, 2006, p. 12 Search PubMed. σmeta-Substituent constant for CF3 = 0.46 and F = 0.34 indicating that the CF3 group shows a stronger inductive effect, see: E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, Universal Science Books, Sausalito, California, 2006, p. 445 Search PubMed. Hence, the pentafluoroethyl radical is likely slightly more electronegative. In contrast, the CF3-radical is smaller than the CF3CF2-radical and obviously the electronic effect overrides the steric effect: the CF3CF2-radical shows higher reactivity than the CF3-radical.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02210j |
| This journal is © The Royal Society of Chemistry 2016 |