
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated glycan assembly of xyloglucan oligosaccharides†
Pietro
Dallabernardina
ab,
Frank
Schuhmacher
ab,
Peter H.
Seeberger
ab and
Fabian
Pfrengle
*ab
aDepartment of Biomolecular Systems, Max-Planck-Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany
bFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany. E-mail: Fabian.Pfrengle@mpikg.mpg.de
First published on 3rd November 2015
Abstract
We report the automated glycan assembly of oligosaccharide fragments related to the hemicellulose xyloglucan (XG). Iterative addition of monosaccharide and disaccharide building blocks to a solid support provided seven cellulose and xyloglucan fragments including XXGG- and XXXG-type oligosaccharides.
Introduction
Xyloglucans (XGs) are the most prevalent class of hemicelluloses in the cell wall of higher plants.1 They are constructed from a cellulosic backbone consisting of β-(1→4)-linked D-glucopyranoses highly substituted with α-D-xylopyranose residues in the C6 position of the glucose units.2 Further monosaccharides such as galactose and fucose may be attached to the xylose substituents. Generally, xyloglucans occur in two major forms. While the XXGG-type consists of two xylose-substituted glucose moieties separated by two unsubstituted glucose residues, in the XXXG-type three xylose-substituted residues are separated by only one unsubstituted glucose (Fig. 1b).1,3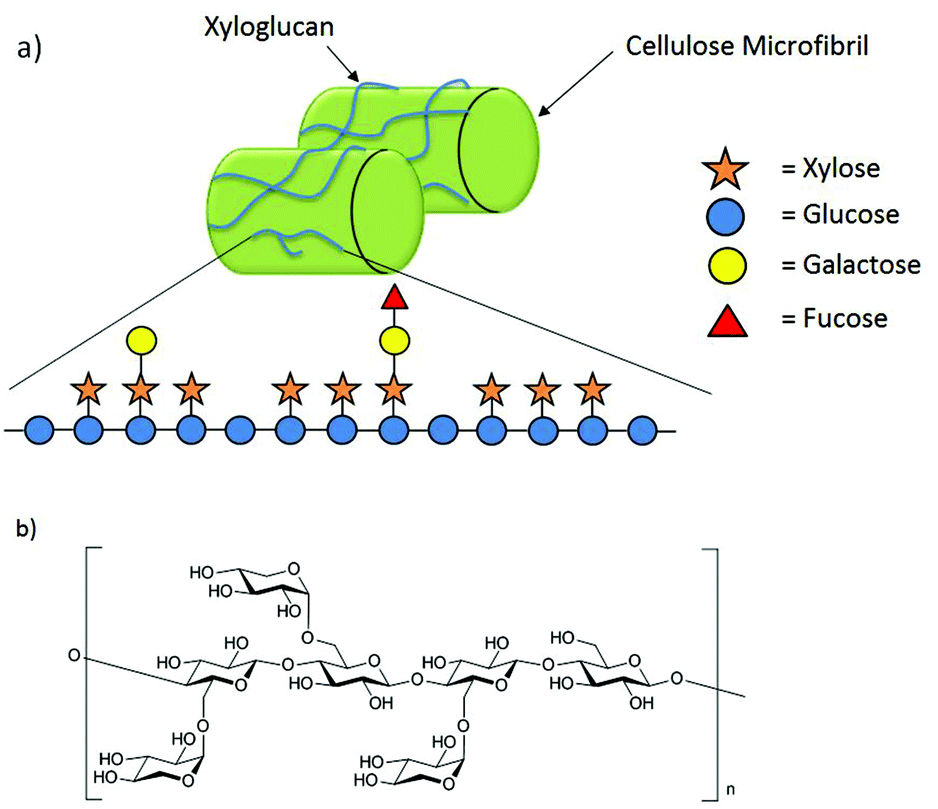 | ||
| Fig. 1 (a) The hemicellulose xyloglucan interconnects cellulose microfibrils through hydrogen bonding; (b) XXXG-type of XG oligosaccharides. | ||
Despite the structural diversity found in different species, the functions of xyloglucan in plant growth and development are conserved among all species of flowering plants.4 Xyloglucans are strongly bonded through non-covalent interactions to the cellulose microfibrils in the cell wall (Fig. 1a).2 These interactions are not only essential for the structural capacity of the cell wall but also its flexibility during synthesis.2,4–7 Xyloglucans store energy6 and are signaling molecules.4
Homogeneous, well-defined oligosaccharide samples obtained by chemical synthesis are essential molecular tools for investigating the structure and function of xyloglucans. Few chemical syntheses of XG oligosaccharides have been reported.8–11 The multi-step total synthesis of an XG-derived nonasaccharide9 relied on a late-stage introduction of a Fuc-Gal-Xyl side chain. Large defined xyloglucan fragments have also been prepared by enzymatic coupling of XG oligosaccharides obtained from enzymatic degradation of xyloglucan polymers.12 This approach is limited only in the diversity of the products due to restricted accessibility of the oligosaccharide building blocks. Recently, automated glycan assembly13–15 has emerged as a powerful tool for the facile and expedient generation of plant carbohydrate libraries.16,17 Here, we report the automated synthesis of seven cellulose and XG oligosaccharides based on the linear assembly of the glycans on solid support using monosaccharide and disaccharide building blocks (BB).
Results and discussion
Initially we investigated the possibility that XG oligosaccharides could be assembled exclusively from monosaccharide BBs. Two glucose BBs (1 and 2) and one xylose BB (3) were designed for automated synthesis. Glucose BBs 1 and 2 were equipped in the C2 position with benzoyl esters (Bz) as participating protecting groups to favour the formation of β-linkages. Fluorenylmethoxycarbonyl (Fmoc) and levulinoyl (Lev) were used as temporary protecting groups to enable either elongation of the β-1,4-glucan backbone or substitution of the 6-position with xylose. All remaining positions were permanently protected as benzyl ethers. Both building blocks contain phosphate leaving groups to enable smooth chain elongation.18,19 Xylose BB 3 was protected in all positions with benzyl groups and used directly as the thioglycoside.With BBs 1–3 in hand,20–23 we explored their potential for the automated glycan assembly of trisaccharide 5 (Scheme 1a). TMSOTf-promoted glycosylation of linker-functionalized resin 4 with two times 3.7 equivalents BB 1 followed by Fmoc-deprotection and another glycosylation with BB 2 provided the disaccharide for installation of the α-xyloside. After removal of the Lev-group we investigated the NIS/TfOH-promoted glycosylation of the second glucose unit with BB 3. At temperatures as low as −35 °C, full conversion was achieved and the desired trisaccharide obtained in 29% yield after cleavage of the photo-labile linker in a commercial flow photoreactor.17 Unfortunately the reaction resulted in a mixture of α/β-isomers with a relative ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 1b). The two stereoisomers were unambiguously assigned by MALDI mass spectrometry and NMR-spectroscopy (Scheme 1c/d). With less of the desired α-isomer being obtained than in similar solution–phase reactions,12 and since multiple α-xylosides have to be installed in the synthesis of larger XG oligosaccharides, BB 3 was deemed not applicable for our purposes.
:1 (Scheme 1b). The two stereoisomers were unambiguously assigned by MALDI mass spectrometry and NMR-spectroscopy (Scheme 1c/d). With less of the desired α-isomer being obtained than in similar solution–phase reactions,12 and since multiple α-xylosides have to be installed in the synthesis of larger XG oligosaccharides, BB 3 was deemed not applicable for our purposes.
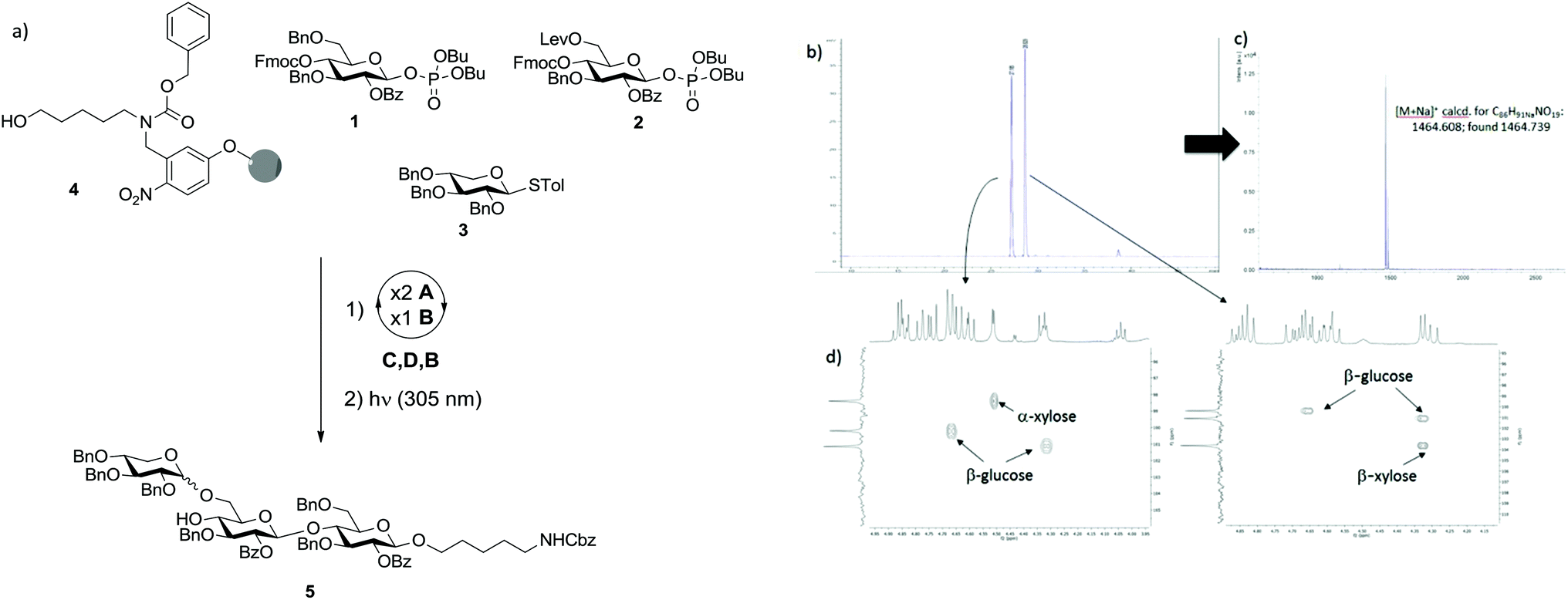 | ||
| Scheme 1 (a) Automated glycan assembly of trisaccharide 5. Reagents and conditions: 2 × 3.7 equiv. BB 1 or BB 2, TMSOTf, CH2Cl2, −30 °C (5 min) or −35 °C (5 min) → −15 °C (30 min) (Module A); 3 cycles of 20% Et3N in DMF, 25 °C (5 min) (Module B); N2H4·OAc (155 mM) in pyridine/AcOH/H2O 4:1:0.25, 25 °C (30 min) (Module C); 2 × 3.7 equiv. BB 3 NIS/TfOH, CH2Cl2/dioxane 2:1, −55 °C (5 min) → −35 °C (40 min) (Module D); (b) HPLC-chromatogram of the crude fully protected anomeric mixture; (c) MALDI-TOF analysis after separation of the diastereomers by preparative HPLC; (d) HSQC of the anomeric region of the two isolated stereoisomers. | ||
Based on this result we decided to employ a disaccharide BB containing a preinstalled α-xylosidic linkage. We envisioned that the synthesis of disaccharide BB 10 would not require more steps than the synthesis of BB 2 (Scheme 2). Glucose derivative 7 was selectively glycosylated with xylopyranose imidate 6 to provide disaccharide 8 as the major product. The transformation proceeded in rather low yield due to the formation of the β isomer and the bis-glycosylated compounds as side products.12 Subsequently, the C4-hydroxyl of glucose was protected as Lev-ester and resulting thioglycoside 9 converted into corresponding phosphate 10. Installation of an Fmoc-group at C4 failed due to steric hinderance.
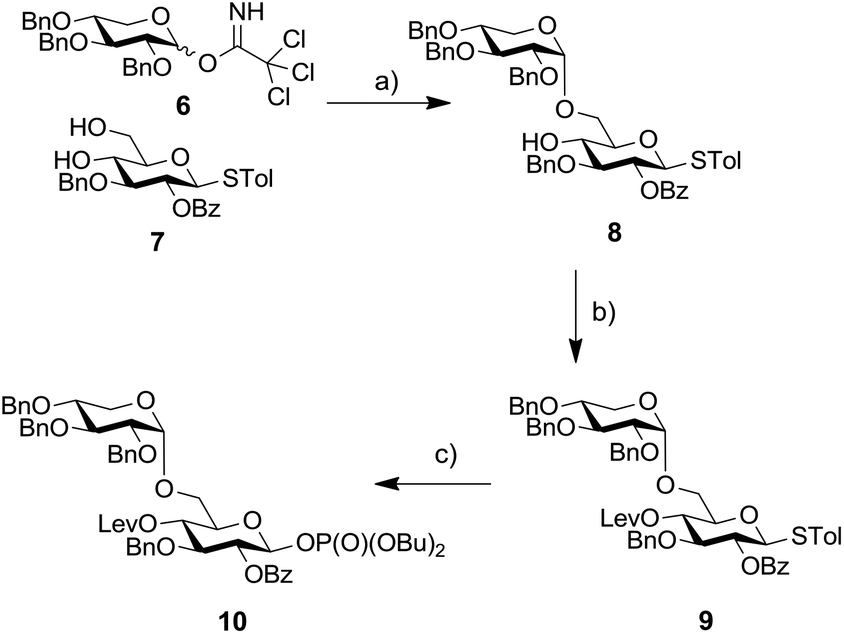 | ||
| Scheme 2 Synthesis of disaccharide BB 10. Reagents and conditions: (a) TMSOTf, CH2Cl2, −78 °C → −10 °C, 39%; (b) LevOH, DIC, DMAP, CH2Cl2, 84%; (c) HOP(O)(OBu)2, NIS, TfOH, CH2Cl2, 0 °C, 57%. | ||
With disaccharide BB 10 in hand, xyloglucan fragments were produced using an automated oligosaccharide synthesizer (Scheme 3). Disaccharides had been successfully used during automated glycan assembly previously and iterative addition of BBs 1 and 10 to linker-functionalized resin 4 provided the protected form of 13 with full conversion after cleavage of the oligosaccharide from the solid support.24 However, oligosaccharides containing two α-xyloside substituents were formed with reduced efficiency and the desired products were accompanied by deletion sequences missing one of the disaccharide units. Still, fully deprotected compounds 14, 16 and 17 were isolated in overall yields of 9–16% after cleavage from the solid support, treatment with sodium methoxide and a final hydrogenation reaction. Octasaccharide 15 containing three disaccharide units was assembled in 2% overall yield when using three instead of two times 3.7 equivalents BB 10 for the two key glycosylations. Due to steric hindrance the assembly of chains containing more than three disaccharide units is not practical. To complete the set of molecular tools for xyloglucan biology, two cellulose fragments (11, 12) were prepared. Couplings using 3.7 equivalents of BB 1 just once were sufficient to ensure complete glycosylations.
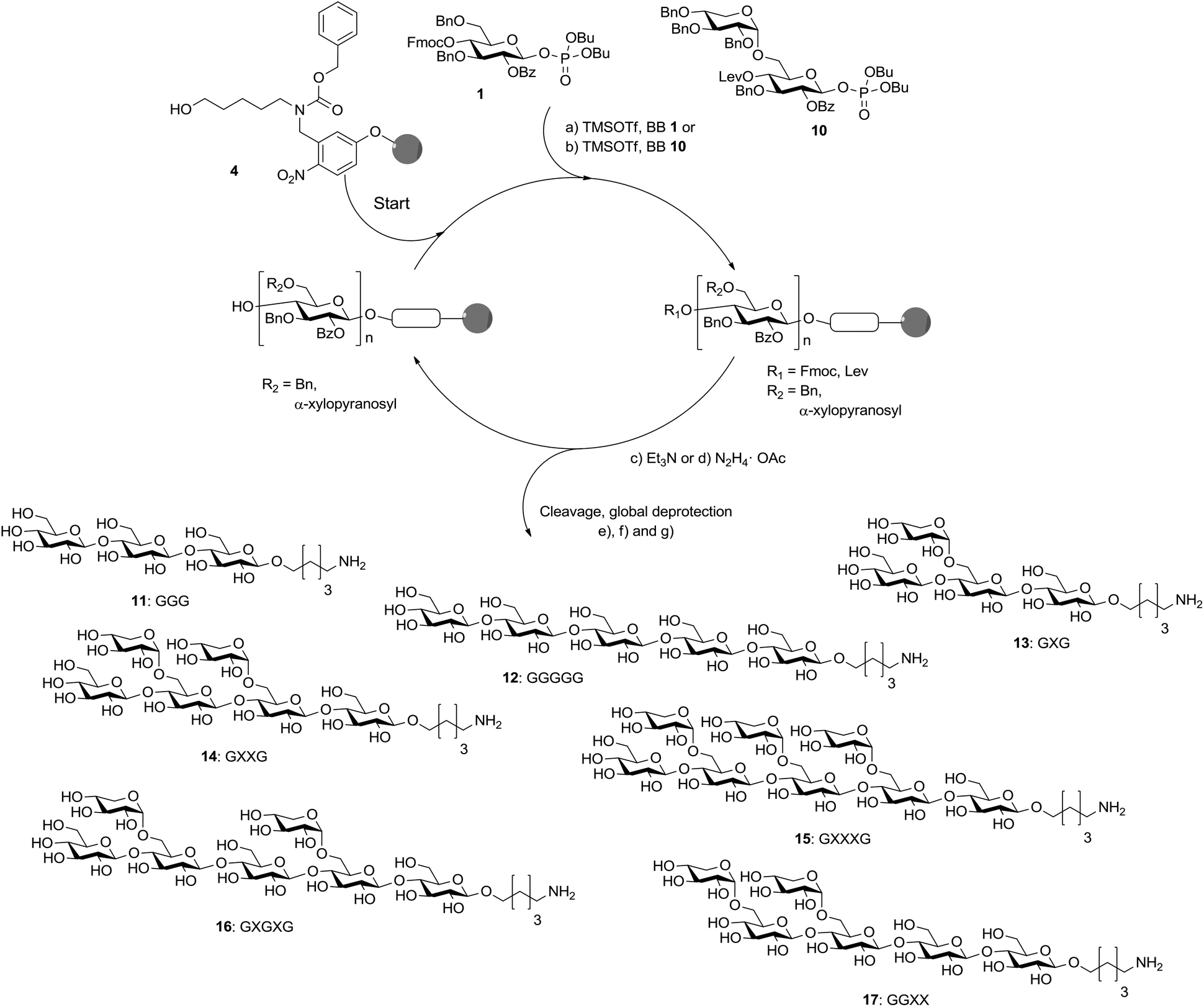 | ||
| Scheme 3 Automated glycan assembly of XG oligosaccharides. Reagents and conditions: (a) 1 or 2 × 3.7 equiv. BB 1, TMSOTf, CH2Cl2, −30 °C (5 min) → −15 °C (30 min) (Module A); (b) 2 or 3 × 3.7 equiv. BB 10, TMSOTf, CH2Cl2, −35 °C (5 min) → −15 °C (35 min) (Module A); (c) 3 cycles of 20% NEt3 in DMF, 25 °C (5 min) (Module B); (d) N2H4·OAc (155 mM) in pyridine/AcOH/H2O 4:1:0.25, 25 °C (30 min) (Module C); (e) CH2Cl2, hν (305 nm); (f) NaOMe, THF/MeOH, 12 h; (g) H2, Pd/C, EtOAc/MeOH/H2O/HOAc, 12 h. 11: 28%; 12: 23%; 13: 15%; 14: 16%; 15: 2%; 16: 10%; 17: 9%. The letter code below the structures refers to a common nomenclature of XG oligosaccharides.25 | ||
Conclusions
In conclusion, seven oligosaccharides related to cellulose and xyloglucan were prepared by automated glycan assembly. The use of a disaccharide BB enabled the synthesis of oligosaccharides containing up to three α-xylosidic linkages. These XG glycans will be used as molecular probes to investigate the specificities of xyloglucan-modifying enzymes and cell wall glycan-directed antibodies.Experimental section
Synthesizer modules and conditions
Linker-functionalized resin 4 (16.9 μmol of hydroxyl groups) was placed in the reaction vessel of the automated oligosaccharide synthesizer and swollen for at least 30 min in DCM. Before every synthesis the resin was washed with DMF, THF and DCM. Subsequently the glycosylation (Module A and D) and deprotection (Module B and C) steps were performed. Mixing of the components was accomplished by bubbling Argon through the reaction mixture.Module A: glycosylation with glycosyl phosphates
The resin (16.9 μmol of hydroxyl groups) was swollen in DCM (2 mL) and the temperature of the reaction vessel was adjusted to −30 °C. Prior to the glycosylation reaction the resin was washed with TMSOTf in DCM and then DCM only. For the glycosylation reaction the DCM was drained and a solution of phosphate BB (3.7 equiv. in 1 mL DCM) was delivered to the reaction vessel. After the set temperature was reached, the reaction was started by the addition of TMSOTf in DCM (3.7 equiv. in 1 mL DCM). The glycosylation was performed for 5 min at −30 °C or −35 °C and then at −15 °C for 30 or 35 minutes. Subsequently the solution was drained and the resin was washed three times with DCM. The whole procedure was performed once, twice or three times depending on the conversion of the acceptor sites. Afterwards the resin was washed three times with DCM at 25 °C.Module B: Fmoc deprotection
The resin was washed with DMF, swollen in 2 mL DMF and the temperature of the reaction vessel was adjusted to 25 °C. Prior to the deprotection step the DMF was drained and the resin was washed with DMF three times. For Fmoc deprotection 2 mL of a solution of 20% Et3N in DMF was delivered to the reaction vessel. After 5 min the solution was drained and the whole procedure was repeated another two times. After Fmoc deprotection was complete the resin was washed with DMF, THF and DCM.Module C: Lev deprotection
Prior to the deprotection step the resin was washed with DCM three times, swollen in 1.3 mL DCM and the temperature of the reaction vessel was adjusted to 25 °C. For Lev deprotection 0.8 mL of a solution of 150 mM N2H4·AcOH in Pyridine/AcOH/H2O 4:1:0.25 was delivered to the reaction vessel. After 30 min the solution was drained and the deprotection step was repeated two times. After Lev deprotection was complete the resin was washed with DCM, DMF, THF and again DCM three times each.
Module D: glycosylation with thioglycosides
The resin (16.9 μmol of hydroxyl groups) was swollen in DCM (2 mL) and the temperature of the reaction vessel was adjusted to −30 °C. Prior to the glycosylation reaction the resin was washed with TMSOTf in DCM and DCM. For the glycosylation reaction the DCM was drained and a solution of the thioglycoside BB (3.7 equiv. in 1 mL DCM) was delivered to the reaction vessel. After the set temperature was reached, the reaction was started by the addition of NIS (4.44 equiv.) and TfOH (0.44 equiv.) in 1 mL DCM/dioxane (2:1). The glycosylation was performed for 5 min at −55 °C and then for 40 min at −30 °C. Subsequently the solution was drained and the resin was washed with DCM. The whole procedure was repeated once to ensure full conversion of all acceptor sites. Afterwards the resin was washed three times with DCM at 25 °C.
Cleavage from the solid support
After assembly of the oligosaccharides cleavage from the solid support was accomplished by modification of a previously published protocol,18 using the Vapourtec E-Series UV-150 photoreactor Flow Chemistry System. The medium pressure metal halide lamp is filtered using the commercially available red filter. The resin, suspended in DCM, was loaded into a plastic syringe. The suspension was then pumped using a syringe pump (PHD2000, Harvard Aparatus) at 1 mL min−1 through a 10 mL reactor, constructed of 1/8 inch o.d. FEP tubing. The total volume within the photoreactor was 9 mL. The temperature of the photoreactor was maintained at 20 °C and the lamp power was 80%. The exiting flow was deposited in a 10 mL syringe containing a filter, with a collection flask beneath the syringe.Global deprotection of the assembled oligosaccharides
The protected oligosaccharide was dissolved in THF (3 mL) and NaOMe (0.5 M in MeOH, 0.5 mL) was added. The reaction mixture was stirred overnight and subsequently neutralized by addition of prewashed Amberlite IR-120 resin. The resin was filtered off and the solvents were removed in vacuo. The crude product was purified by preparative HPLC, dissolved in a mixture of EtOAc/MeOH/AcOH/H2O (4:2:2:1, 3 mL) and the resulting solution was added to a round-bottom flask containing Pd/C (10% Pd, 10–20 mg). The suspension was saturated with H2 for 30 min and stirred under an H2-atmosphere overnight. After filtration of the reaction mixture through a syringe filter the solvents were evaporated to provide the fully deprotected oligosaccharide.
Acknowledgements
We gratefully acknowledge financial support from the Max Planck Society, the German Research Foundation (DFG, Emmy Noether program PF850/1-1 to FP), the Fonds der chemischen Industrie (Liebig-fellowship to FP) and an ERC Advanced Grant (AUTOHEPARIN to PHS). We thank Dr K. Gilmore for critically proofreading this manuscript.Notes and references
- A. Ebringerova, Z. Hromadkova and T. Heinze, Adv. Polym. Sci., 2005, 186, 1–67 CrossRef CAS.
- M. Pauly, P. Albersheim, A. Darvill and W. S. York, Plant J., 1999, 20, 629–639 CrossRef CAS PubMed.
- W. D. Bauer, K. W. Talmadge, K. Keegstra and P. Albersheim, Plant Physiol., 1973, 51, 174–187 CrossRef CAS PubMed.
- K. H. Caffall and D. Mohnen, Carbohydr. Res., 2009, 344, 1879–1900 CrossRef CAS PubMed.
- T. Hayashi, Annu. Rev. Plant Biol., 1989, 40, 139–168 CrossRef CAS.
- M. S. Buckeridge, H. P. dos Santos and M. A. S. Tine, Plant Physiol. Biochem., 2000, 38, 141–156 CrossRef CAS.
- N. C. Carpita and D. M. Gibesut, Plant J., 1993, 3, 1–30 CrossRef CAS PubMed.
- A. Wotowic and J.-C. Jacquinet, Carbohydr. Res., 1990, 205, 235–245 CrossRef.
- K. Sakai, Y. Nakahara and T. Ogawa, Tetrahedron Lett., 1990, 31, 3035–3038 CrossRef CAS.
- D. K. Watt, D. J. Brasch, D. S. Larsen, L. D. Melton and J. Simpson, Carbohydr. Res., 2000, 325, 300–312 CrossRef CAS PubMed.
- D. K. Watt, D. J. Brasch, D. S. Larsen, L. D. Melton and J. Simpson, Carbohydr. Res., 1996, 286, 1–15 CrossRef.
- R. Faurè, M. Saura-Valls, H. Brumer, A. Planas, S. Cottaz and H. Driguez, J. Org. Chem., 2006, 71, 5151–5161 CrossRef PubMed.
- L. Kröck, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschädler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS PubMed.
- D. Schmidt, F. Schumacher, A. Geissner, P. H. Seeberger and F. Pfrengle, Chem. – Eur. J., 2015, 21, 5709–5713 CrossRef CAS PubMed.
- M. P. Bartetzko, F. Schumacher, H. S. Hahm, P. H. Seeberger and F. Pfrengle, Org. Lett., 2015, 17, 4344–4347 CrossRef CAS PubMed.
- S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5858–5861 CrossRef CAS PubMed.
- O. J. Plante, R. B. Andrade and P. H. Seeberger, Org. Lett., 1999, 1, 211–214 CrossRef CAS PubMed.
- S. David, A. Malleron and C. Dini, Carbohydr. Res., 1989, 188, 193–200 CrossRef CAS.
- T. Polat and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 12795–12800 CrossRef CAS PubMed.
- Y.-H. Wanga, H.-W. Yeha, H.-W. Wanga, C.-C. Yua, J.-H. Guha, D.-Z. Liub and P.-H. Lianga, Carbohydr. Res., 2013, 375, 118–135 CrossRef PubMed.
- B. Lüning, T. Norberg and J. Tejbrant, J. Carbohydr. Chem., 1992, 11, 933–943 CrossRef.
- For the HPLC-chromatogramm see ESI.†.
- S. C. Fry, W. S. York, P. Albersheim, A. Darvill, T. Hayashi, J.-P. Joseleau, Y. Kato, E. P. Lorences, G. A. Maclachlan, M. McNeil, A. J. Mort, J. S. G. Reid, H. U. Seitz, R. R. Selvendran, A. G. J. Voragen and A. R. White, Physiol. Plant., 1993, 89, 1–3 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for solution- and automated solid-phase reactions and characterization data for all compounds. See DOI: 10.1039/c5ob02226f |
| This journal is © The Royal Society of Chemistry 2016 |