
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of triazole-linked morpholino oligonucleotides via CuI catalysed cycloaddition†
Matthew J.
Palframan‡
,
Rima D.
Alharthy§‡
,
Paulina K.
Powalowska
and
Christopher J.
Hayes
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: chris.hayes@nottingham.ac.uk; Tel: +44 (0)115 951 3045
First published on 16th February 2016
Abstract
Triazole-linked morpholino (TLMO) oligonucleic acids were synthesised using the CuI catalysed (3 + 2) azide–alkyne cycloaddition (CuAAC) reaction. The modified DNA analogues were incorporated into 13-mer sequences via solid phase synthesis. UV melting experiments showed that the TLMO modification gives higher Tm values than the corresponding TLDNA modification.
Introduction
Click chemistry has recently emerged as a powerful tool in the field of nucleic acid research.1 In particular, the CuI catalysed (3 + 2) azide–alkyne cycloaddition (CuAAC)2 has been used to construct modified internucleotide linkages,3 to prepare nucleic acid conjugates,4 and as a strand ligation tool.5 Zerrouki et al., designed a novel triazole-linked DNA analogue (TLDNA) 1 using the CuAAC for oligomer elongation,6 and this preliminary work has been significantly extended by Brown et al. The artificial TLDNA retains good aqueous solubility, is stable towards enzymatic degradation,3a and can be read by polymerases,7 thus making it capable of in vitro transcription8 and rolling circle amplification.7 Furthermore, and perhaps most impressively, Brown has demonstrated that genes containing TLDNA 1 are functional in vivo in Escherichia coli and in human cells.9 Given the biocompatibility of the TLDNA 1 with DNA processing enzymes, it is curious that the thermal stability of complementary duplexes is reduced. A recent study on the structural basis of this phenomenon reported that the TLDNA modification leads to less optimal stacking interactions and distortion in the backbone at, and adjacent to, the site of the triazole.10 Whilst high melting temperatures are not required for all uses of modified nucleic acids, the formation of stable duplexes is a requirement for therapeutic applications of oligonucleotides, and as such TLDNAs 1 do not represent good drug candidates.As part of our own research aimed at developing therapeutic nucleic acids, we wondered if the thermal stability of triazole-containing duplexes could be improved by the addition of further modifications to the backbone. Thus we decided to examine triazole-linked morpholino (TLMO) hybrid structures 2 (Fig. 1) as they could combine the ease of synthesis of the TLDNAs 1 with the increased melting temperatures associated with morpholino drug candidates.11,12 The TLMO hybrid 2 can be disconnected to reveal the azide 4 and the alkyne-substituted morpholine 3 as potential precursors for the proposed CuAAC reaction (Fig. 1).
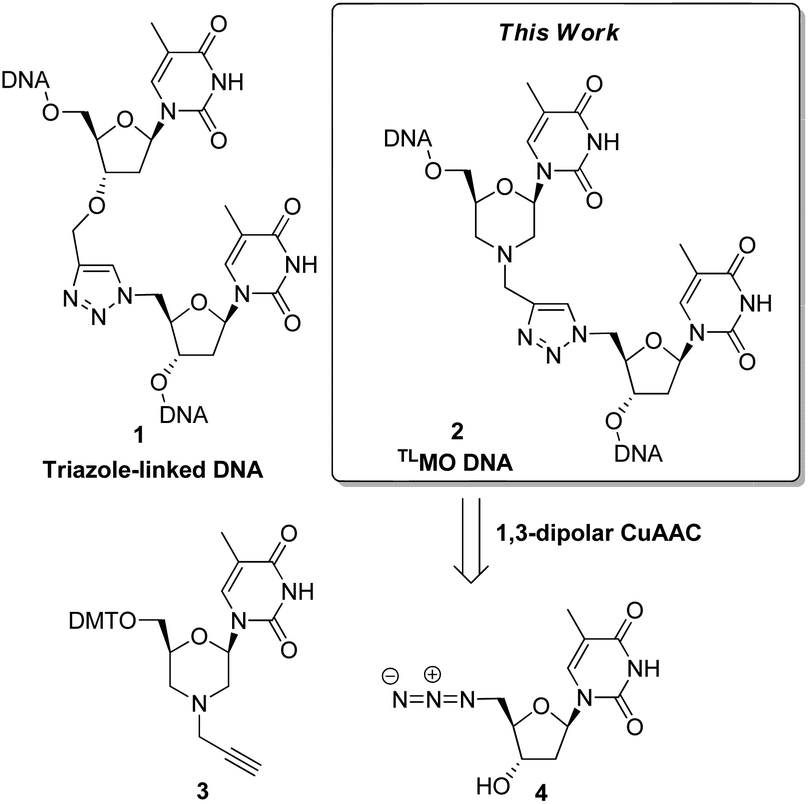 | ||
| Fig. 1 Approach to triazole-linked morpholino (TLMO) hybrid structures 2. | ||
Our initial route to 3 proceeded via the morpholine 6, which was readily prepared from 5-methyl uridine 5 in good yield by oxidative cleavage and subsequent reductive amination13 (Scheme 1). Although the N-alkylation of 6 did produce the desired N-propargyl morpholine 3, only a low yield (36%) of the desired alkyne was obtained. The main side reaction was over alkylation of the thymine base in addition to N-alkylation of the morpholine, and an alternative route was explored. Thus, oxidative cleavage of 5 in the presence of propargylamine first gave 7, which upon treatment with sodium cyanoborohydride/AcOH gave the desired product 3 in good overall yield (71%) (Scheme 1).
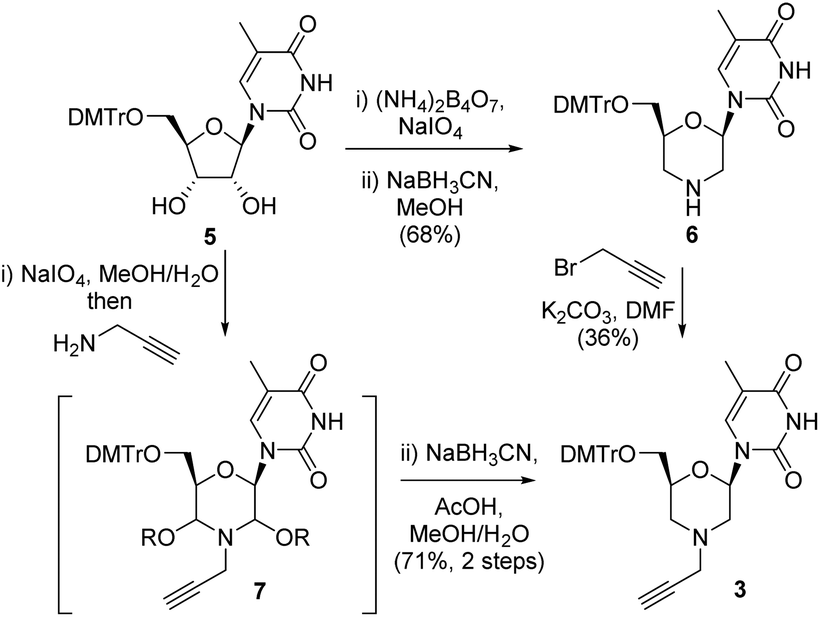 | ||
| Scheme 1 Synthesis of the alkyne morpholine nucleoside 3. | ||
Following formation of the alkyne-containing morpholine nucleoside 3, our attention then turned to the synthesis of the azide cycloaddition partner. Pleasingly, the 3′-TBSO-protected-5′-deoxy-5′-azido thymidine 10 was readily prepared from the alcohol 8via a two-step sequence involving mesylate formation and displacement with sodium azide. The 3′-OH-5′-deoxy-5′-azido thymidine 4 was then synthesised from 10via TBAF deprotection (Scheme 2). Alternatively, 4 could be accessed directly from thymidine via selective 5′-tosylation and subsequent azide displacement using the known procedure (38%, 2 steps).14
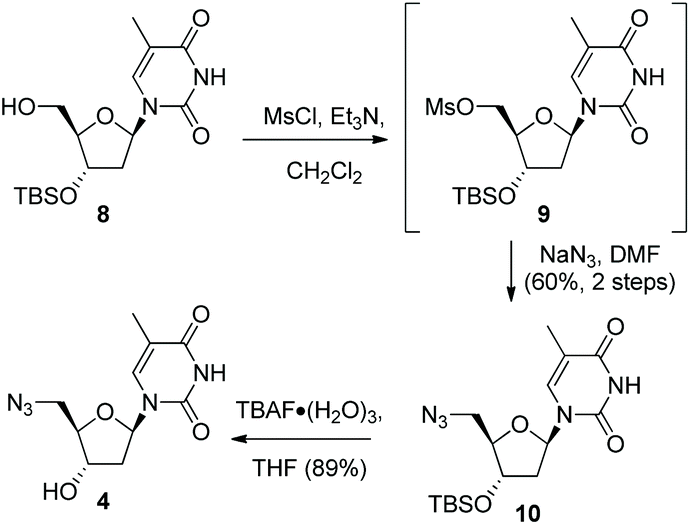 | ||
| Scheme 2 Synthesis of 5′-deoxy-5′-azido thymidine 4. | ||
With the required coupling partners in hand, we next examined the key Cu-catalysed cycloaddition step. As we needed access the 3′-alcohol 12 to prepare phosphoramidite 13, we explored two routes for its synthesis. The first was cycloaddition of 3 and the protected azide 10 (i.e.3 + 10 → 11), followed by TBS deprotection (11 → 12), and the second was initial deprotection (10 → 4, Scheme 2) followed by cycloaddition with 3. A range of catalysts and solvents were initially screened, and it was quickly found that the use of copper(I) iodide in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tBuOH:H2O (3:2:1) with microwave heating (80 °C) was optimal (Scheme 3).3a Under these conditions, cycloaddition of the acetylene 3 with the TBS-protected azide 10 gave the triazole-linked morpholino (TLMO) dimer 11 in good yield, and TBAF deprotection of 11 gave the desired alcohol 12 in good yield (Scheme 3). We were pleased to find that the alternative cycloaddition of 4 with 3 also proceeded in good yield to give the alcohol 12 directly, and this was adopted as our favoured route due to an improved overall yield and easier of purification of 12 by column chromatography. Finally, the TLMO 12 was converted to the 3′-cyanoethyl phosphoramidite 13 (74%) under standard conditions (Scheme 3).
:tBuOH:H2O (3:2:1) with microwave heating (80 °C) was optimal (Scheme 3).3a Under these conditions, cycloaddition of the acetylene 3 with the TBS-protected azide 10 gave the triazole-linked morpholino (TLMO) dimer 11 in good yield, and TBAF deprotection of 11 gave the desired alcohol 12 in good yield (Scheme 3). We were pleased to find that the alternative cycloaddition of 4 with 3 also proceeded in good yield to give the alcohol 12 directly, and this was adopted as our favoured route due to an improved overall yield and easier of purification of 12 by column chromatography. Finally, the TLMO 12 was converted to the 3′-cyanoethyl phosphoramidite 13 (74%) under standard conditions (Scheme 3).
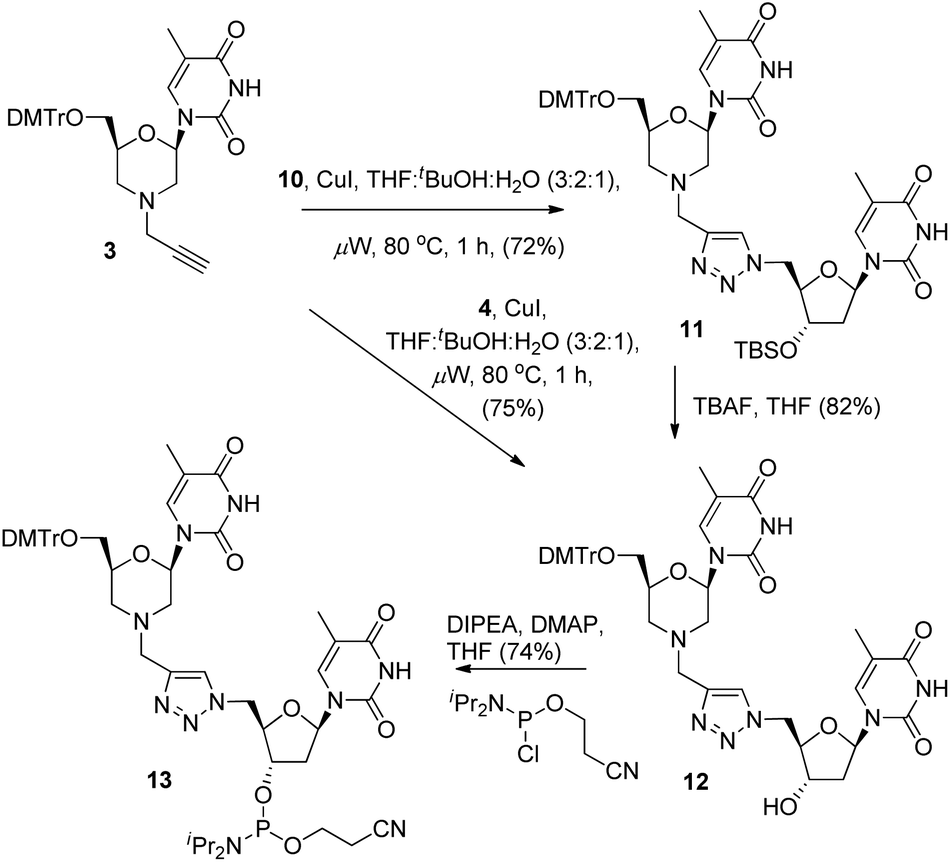 | ||
| Scheme 3 Synthesis of 3′-cyanoethyl phosphoramidite 13. | ||
To provide a direct comparison of the new TLMO hybrid 2 to the triazole-linked DNA analogue (TLDNA) 1, we next prepared the phosphoramidite 17. This reagent facilitates incorporation of the triazole modification 1 into oligonucleotide sequences via solid-phase synthesis as opposed to the fragment ligation method used previously by Brown et al.10 The phosphoramidite 17 was readily prepared from 14via 3′-O-alkylation to give the alkyne 15, Cu-catalysed cycloaddition with 4 to provide the triazole-containing dimer 16 and then conversion to 17 in the usual manner (Scheme 4).
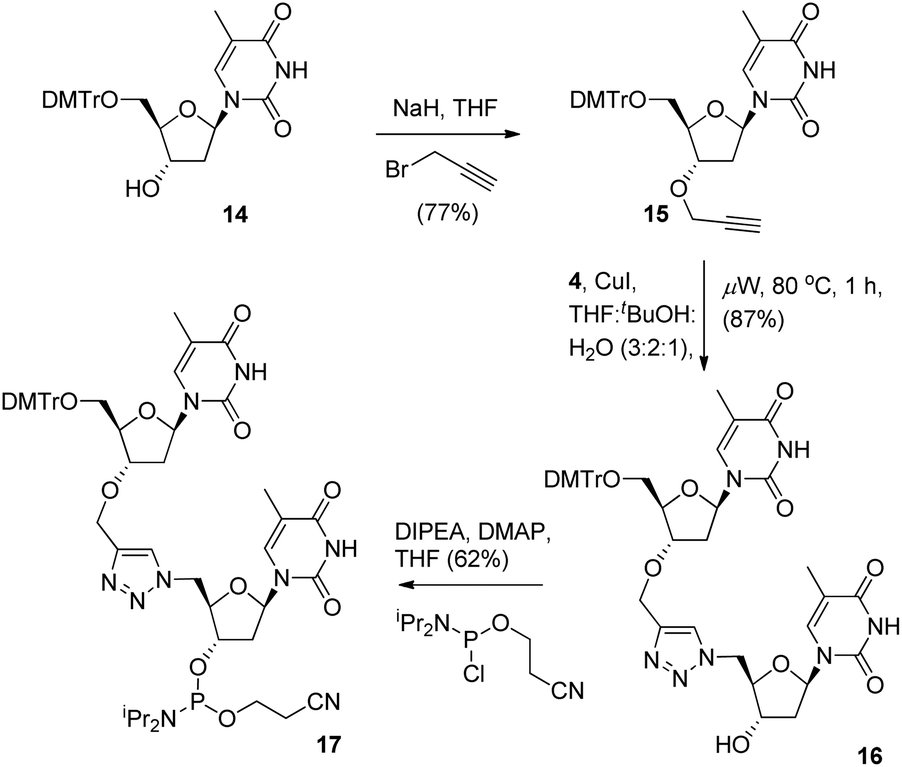 | ||
| Scheme 4 Synthesis of 3′-cyanoethyl phosphoramidite 17. | ||
Pleasingly, the modified phosphoramidites 13 and 17 were fully compatible with solid-phase oligonucleotide synthesis and we prepared the TLMO-containing oligomer 21 and the known TLDNA oligomer 2210 in good yield (Table 1). Stock aqueous solutions (pH 7) of the oligomers 21 and 22 were readily prepared, and no adverse solubility issues were observed. As Brown et al. have already reported UV-melting data of 22 duplexed with its complimentary DNA strand 18,10 we also prepared 18 so that we could directly compare the Tm values of 18 + 21, 18 + 22 and the unmodified duplex (18 + 20) under the same conditions. In order to assess the potential use of the TLMO-modification in therapeutically useful oligomers, we also synthesised the complimentary RNA oligonucleotide 19, as this simulates an intracellular mRNA target. The integrity of the oligomers 18–22 was confirmed by ESI mass spectrometry (Table 1) and HPLC (see ESI†).
| Identifier | Sequence 5′ → 3′ | m/z | |
|---|---|---|---|
| Calculated | Found | ||
a
 indicates the position of the morpholine-triazole modification 13.
b indicates the position of the morpholine-triazole modification 13.
b
 indicates the position of the triazole modification 17. indicates the position of the triazole modification 17.
|
|||
| 18 (DNA) | d(GCTGCAAACGTCG) | 3953.55 | 3953.73 |
| 19 (RNA) | GCUGCAAACGUCG | 4133.49 | 4133.63 |
| 20 | d(CGACGTTTGCAGC) | 3944.53 | 3944.72 |
| 21 | d(CGACG TGCAGC) TGCAGC) |
3944.64 | 3944.80 |
| 22 | d(CGACG TGCAGC) TGCAGC) |
3945.63 | 3945.78 |
Thermal stabilities of the TLMO 21, TLDNA 22, and unmodified DNA 20 duplexed with complimentary DNA 18 (Fig. 2) and RNA 19 (Fig. 3) were then determined by UV melting experiments15 (Table 2). Pleasingly, the Tm values of the control DNA 18:DNA 20 (62.4 °C) (entry 1, Table 2), and the DNA 18:TLDNA 22 (55.1 °C) (entry 3, Table 2) duplexes were in close agreement with those reported previously by Brown (62.89 °C and 55.30 °C respectively).10 The TLMO-containing oligomer 21 duplexed to DNA 18 gave a Tm value of 56.1 °C (entry 2, Table 2), which represents a small increase (ΔTm 1.0 °C) over that determined for 22, but still represents a significant decrease from the unmodified DNA (ΔTm −6.3 °C). As mentioned above, duplexes with RNA provide a more meaningful comparison for future therapeutic applications and the Tm value of RNA 19 duplexed with unmodified DNA 20 was determined (58.5 °C) as a control (entry 4, Table 2). In contrast to the duplexes with DNA, the Tm of TLMO 21 (56.6 °C) was much closer to that of the unmodified DNA:RNA than was TLDNA 22 (54.1 °C) with RNA 19 (ΔTm −1.9 °C for 21vs. −4.4 °C for 22) (entries 5 and 6, Table 2), thus demonstrating that the addition of the morpholine modification can regain half of the Tm lost by incorporating the triazole internucleotide linkage.
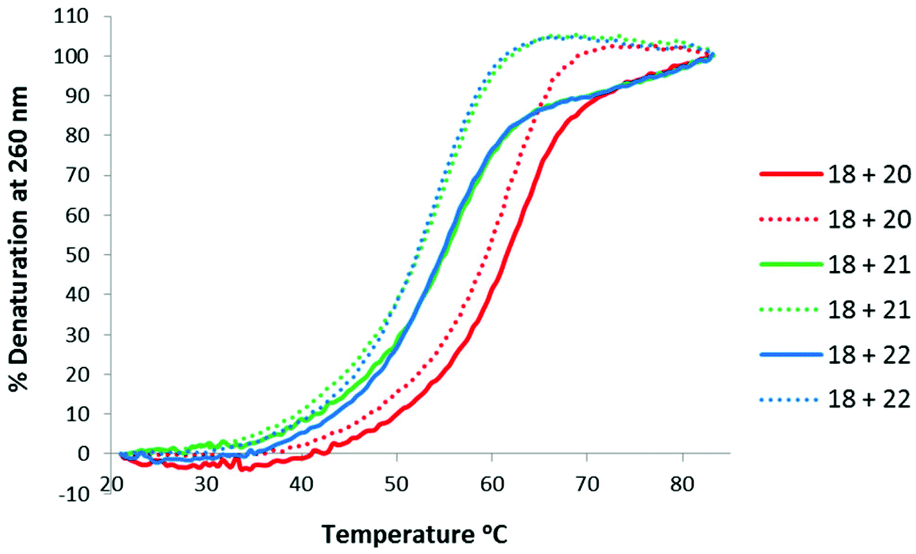 | ||
| Fig. 2 UV-melting curves for oligomers (3 μM) 20, 21 and 22 duplexed with DNA 18. Dotted lines represent cooling curves. | ||
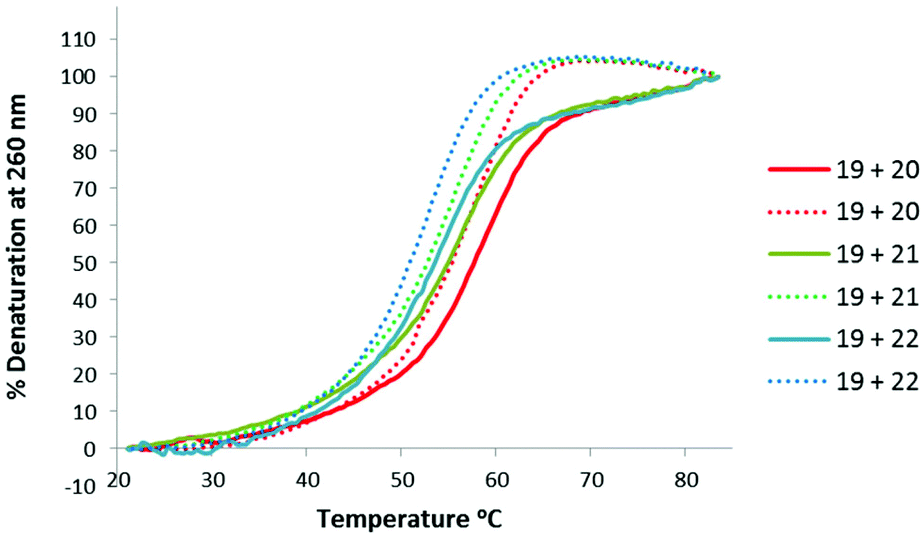 | ||
| Fig. 3 UV-melting curves for oligomers (3 μM) 20, 21 and 22 duplexed with RNA 19. Dotted lines represent cooling curves. | ||
| Entry | Oligomers |
T
ma °C |
T
mb |
|---|---|---|---|
| a T m values for 3 μM oligo samples. Values in parentheses refer to cooling curves. b ΔTm per modification relative to the control DNA 20. | |||
| 1 | 18 + 20 | 62.4 (61.9) | N/A |
| 2 | 18 + 21 | 56.1 (54.7) | −6.3 (−7.2) |
| 3 | 18 + 22 | 55.1 (54.1) | −7.3 (−7.8) |
| 4 | 19 + 20 | 58.5 (58.2) | N/A |
| 5 | 19 + 21 | 56.6 (55.3) | −1.9 (−2.9) |
| 6 | 19 + 22 | 54.1 (52.8) | −4.4 (−5.4) |
Further structural studies are underway in order to fully assess the duplexes formed by TLMO-modified oligomers, before selecting the best candidates for biological evaluation.
Experimental¶
5′-O-DMTr-morpholino thymidine (6)
5′-O-DMTr-5-methyluridine 5 (700 mg, 1.25 mmol) was dissolved in dry MeOH (10 mL) under an argon atmosphere. Ammonium biborate (328 mg, 2.50 mmol), sodium periodate (535 mg, 2.50 mmol) were added to the reaction mixture. After stirring for 3 h at room temperature, the mixture was filtered and sodium cyanoborohydride (155 mg, 2.50 mmol) was added to the filtrate in one portion with stirring. Stirring continued for 6 h followed by evaporation to afford a residue, which was dissolved in EtOAc (10 mL), washed with brine (3 × 10 mL). The organic phase was dried over MgSO4, filtered and evaporated and purified by column chromatography eluting with (CHCl3:MeOH, 25:1) to afford 6 as a colourless foam (460 mg, 68% over three steps); Rf 0.28 (CHCl3:MeOH, 25:1); [α]25D +47 (c 0.61, CHCl3); νmax/cm−1 (CHCl3) 3389, 2933, 2838, 2103, 1684, 1609, 1487 and 1455; 1H NMR (400 MHz, CDCl3) 7.47–7.42 (2H, m, Ar), 7.36–7.27 (6H, m, Ar), 7.32 (1H, s, C6H), 7.24–7.19 (1H, m, Ar), 6.84 (4H, d, J 8.9, Ar), 5.77 (1H, dd, J 10.0, 2.7, C1′H), 4.01(1H, dtd, J 10.7, 4.9, 2.2, C4′H), 3.79 (6H, s, OCH3), 3.27 (1H, dd, J 9.7, 5.1, C5′HH), 3.15 (1H, dd, J, 12.5, 2.7, C2′HH), 3.11–3.02 (2H, m, C5′HH and C3′HH), 2.68–2.58 (2H, m, C3′HH, C2′HH), 1.95 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) 164.1 (C), 158.5 (C), 150.4 (C), 144.8 (C), 135.9 (C), 135.8 (C), 135.4 (CH), 130.1 (CH), 130.0 (CH), 128.1 (CH), 127.8 (CH), 126.9 (CH), 113.1 (CH), 110.9 (C), 86.1 (C), 80.5 (CH), 78.0 (CH), 64.5 (CH2), 55.2 (CH3), 49.0 (CH2), 46.9 (CH2), 12.9 (CH3); HRMS m/z (ES+) Found 566.2245 (M + Na, C31H33N3NaO6 requires 566.2245).
N′-propargyl-5′-O-DMTr-morpholino thymidine (3)
To a stirred solution of 5′-O-DMTr 5-methyluridine (5) (725 mg, 1.29 mmol) in MeOH (12 mL) under an argon atmosphere, was added a solution of sodium periodate (304 mg, 1.42 mmol) in water (2 mL) dropwise over 5 min, followed by propargyl amine (103 μL, 1.62 mmol) in one portion. The resulting solution was stirred at room temperature for 3 hours, during which time a white precipitate formed, the mixture was filtered. To the stirred solution of the filtrate was added sodium cyanoborohydride (162 mg, 2.58 mmol) followed by the dropwise addition of acetic acid (110 μL, 1.93 mmol). The reaction was stirred for 12 h at room temperature. The volatile organic were removed by evaporation. The residue was partitioned between sat. NaHCO3 (50 mL) and EtOAc (50 mL), the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were washed with brine (3 × 50 mL), dried over MgSO4, and evaporated. The residue was purified by silica gel chromatography, eluting with DCM:MeOH (40:1) to afford the title compound (529 mg, 71%) as a colourless foam; Rf 0.17 (DCM:MeOH 25:1); [α]25D +30 (c 0.93, CHCl3); νmax/cm−1 (CHCl3) 3390, 3196, 2955, 1933, 1838, 1694, 1633, 1609, 1583, 1491, 1456; 1H NMR (400 MHz, chloroform-d) δ 9.95 (1H, br s, NH), 7.48–7.44 (2H, m, Ar), 7.37–7.27 (6H, m, Ar), 7.32 (1H, s, C6H), 7.24–7.20 (1H, m, Ar), 6.84 (4H, d, J 8.9, Ar), 5.93 (1H, dd, J 9.8, 2.7, C1′H), 4.12 (1H, m, C4′H), 3.79 (6H, s, OCH3), 3.45–3.44 (2H, m, NCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 3.34 (1H, dd, J 9.6, 5.3, C5′HH), 3.11 (1H, dd, J 9.6, 5.4, C5′HH), 2.97 (1H, br d, J 10.5, C3′HAHB), 2.84 (1H, br d, J 11.4, C2′HH), 2.32 (1H, t, J 2.3, CCH), 2.34–2.23 (2H, m, C3′HH, C2′HH), 1.96 (3H, s, CH3); 13C NMR (101 MHz, chloroform-d) 164.1 (C), 158.6 (C), 150.3 (C), 144.8 (C), 136.0 (C), 135.8 (C), 135.6 (CH), 130.13 (CH), 130.09 (CH), 128.2 (CH), 127.9 (CH), 126.9 (CH), 113.2 (CH), 110.9 (C), 86.2 (C), 79.6 (CH), 77.4 (C), 75.6 (CH), 74.5 (CH), 64.6 (CH2), 55.3 (CH3), 54.6 (CH2), 52.8 (CH2), 46.4 (CH2), 12.7 (CH3); 1H NMR (400 MHz, Benzene-d6) δ 9.88 (1H, s, NH), 7.68 − 7.59 (2H, m, Ph), 7.52–7.40 (4H, m, Ar), 7.23–7.15 (2H, m, Ph), 7.11 − 7.01 (1H, m, Ph), 6.94 (1H, q, J 1.2, C6H), 6.81–6.71 (4H, m, Ar), 5.95 (1H, dd, J 9.7, 2.7, C1′H), 3.96–3.90 (1H, m, C4′H), 3.38 (1H, dd, J 9.6, 5.4, C5′HH), 3.30 (3H, s, OCH3), 3.29 (3H, s, OCH3) 3.17 (1H, dd, J 9.6, 5.1, C5′HH), 3.01 (1H, dd, J 17.5, 2.4, NCHHCCH), 2.92 (1H, dd, J 17.5, 2.4, NCHHCCH), 2.63 (1H, d, J 10.4, C2′HH), 2.47 (1H, d, J 10.9, C3′HH), 2.16 (1H, t, J 11.0, C3′HH), 2.16 (1H, t, J 10.4, C3′HH), 1.90 (1H, t, J 2.4, CCH), 1.66 (3H, d, J 1.2, CH3); 13C NMR (101 MHz, Benzene-d6) δ 163. 9 (C4), 159.3 (2 × ArC), 150.3 (C2), 145.7 (PhC), 136.4 (ArC), 136.3 (ArC), 135.1 (C6H), 130.6 (4 × ArCH), 128.7 (2 × PhCH), 127.9 (2 × PhCH), 127.2 (PhCH), 113.6 (4 × ArCH), 110.8 (C5H), 86.7 (Ar3CO), 80.2 (C1′H), 78.0 (C), 75.9 (C4′H), 74.3 (CH), 65.1 (C5′H2), 54.8 (2 × OCH3 and C2′H2), 52.6 (C3′H2), 46.3 (NCH2C), 12.6 (CH3). HRMS (ESI) C34H36N3O6 (M + H+) requires 582.2599, found 582.2569.
CH), 3.34 (1H, dd, J 9.6, 5.3, C5′HH), 3.11 (1H, dd, J 9.6, 5.4, C5′HH), 2.97 (1H, br d, J 10.5, C3′HAHB), 2.84 (1H, br d, J 11.4, C2′HH), 2.32 (1H, t, J 2.3, CCH), 2.34–2.23 (2H, m, C3′HH, C2′HH), 1.96 (3H, s, CH3); 13C NMR (101 MHz, chloroform-d) 164.1 (C), 158.6 (C), 150.3 (C), 144.8 (C), 136.0 (C), 135.8 (C), 135.6 (CH), 130.13 (CH), 130.09 (CH), 128.2 (CH), 127.9 (CH), 126.9 (CH), 113.2 (CH), 110.9 (C), 86.2 (C), 79.6 (CH), 77.4 (C), 75.6 (CH), 74.5 (CH), 64.6 (CH2), 55.3 (CH3), 54.6 (CH2), 52.8 (CH2), 46.4 (CH2), 12.7 (CH3); 1H NMR (400 MHz, Benzene-d6) δ 9.88 (1H, s, NH), 7.68 − 7.59 (2H, m, Ph), 7.52–7.40 (4H, m, Ar), 7.23–7.15 (2H, m, Ph), 7.11 − 7.01 (1H, m, Ph), 6.94 (1H, q, J 1.2, C6H), 6.81–6.71 (4H, m, Ar), 5.95 (1H, dd, J 9.7, 2.7, C1′H), 3.96–3.90 (1H, m, C4′H), 3.38 (1H, dd, J 9.6, 5.4, C5′HH), 3.30 (3H, s, OCH3), 3.29 (3H, s, OCH3) 3.17 (1H, dd, J 9.6, 5.1, C5′HH), 3.01 (1H, dd, J 17.5, 2.4, NCHHCCH), 2.92 (1H, dd, J 17.5, 2.4, NCHHCCH), 2.63 (1H, d, J 10.4, C2′HH), 2.47 (1H, d, J 10.9, C3′HH), 2.16 (1H, t, J 11.0, C3′HH), 2.16 (1H, t, J 10.4, C3′HH), 1.90 (1H, t, J 2.4, CCH), 1.66 (3H, d, J 1.2, CH3); 13C NMR (101 MHz, Benzene-d6) δ 163. 9 (C4), 159.3 (2 × ArC), 150.3 (C2), 145.7 (PhC), 136.4 (ArC), 136.3 (ArC), 135.1 (C6H), 130.6 (4 × ArCH), 128.7 (2 × PhCH), 127.9 (2 × PhCH), 127.2 (PhCH), 113.6 (4 × ArCH), 110.8 (C5H), 86.7 (Ar3CO), 80.2 (C1′H), 78.0 (C), 75.9 (C4′H), 74.3 (CH), 65.1 (C5′H2), 54.8 (2 × OCH3 and C2′H2), 52.6 (C3′H2), 46.3 (NCH2C), 12.6 (CH3). HRMS (ESI) C34H36N3O6 (M + H+) requires 582.2599, found 582.2569.
5′-O-Mesyl-3′-O-tert-butyldimethylsilyl deoxythymidine (9)
To a stirred solution of 5′-OH-3′-O-tert-butyldimethylsilyl deoxythymidine (1.06 g, 3.0 mmol) in dichloromethane (15 mL) at 0 °C was added triethylamine (0.84 mL, 6.0 mmol) followed by the dropwise addition of mesyl chloride (277 μL, 3.6 mmol). The resulting solution was stirred at 0 °C for 1 h, then warmed to room temperature, and stirred for a further 3 hours. The reaction was quenched by the addition of water (50 mL), the layers were separated, and the aqueous layer was extracted with DCM (2 × 50 mL). The combined organic layers were washed with sat NH4Cl (50 mL), sat. NaHCO3 (50 mL), brine (50 mL), dried over MgSO4, and evaporated to afford the title compound (1.29 g, Quant.) as a yellow foam, which was used without further purification; νmax/cm−1 (CHCl3) 3393, 3006, 2955, 2930, 2885, 1690, 1471, 1362, 1320, 1257, 1176, 1132, 1085 and 1062; 1H NMR (400 MHz, chloroform-d) δ 9.20 (1H, s, NH), 7.31 (1H, q, J 1.2, C6H), 6.28 (1H, t, J 6.7, C1′H), 4.450 (1H, dd, J 11.2, 3.0, C5′HH) 4.45–4.38 (1H, m, C4′H), 4.36 (1H, dd, J 11.2, 3.6, C5′HH), 4.05 (1H, app. q, J 3.6, C3′H), 3.06 (3H, s, SO2CH3), 2.28 (1H, ddd, J 13.6, 6.4, 3.9, C2′HH), 2.17 (1H, dt, J 13.6, 6.8, C2′HH), 1.93 (3H, d, J 1.3, CH3), 0.88 (9H, s, SiC(CH3)3), 0.09 (6H, s, Si(CH3)2); 13C NMR (101 MHz, chloroform-d) δ 163.9 (C4), 150.4 (C2), 135.6 (C6H), 111.7 (C5), 85.4 (C4′H, 84.3 (C1′H), 71.5 (C3′H), 68.5 (C6′H2), 40.6 (C2′H2), 37.8 (SO2CH3), 25.7 (SiC(CH3)3) 17.9, (SiC(CH3)3) 12.6 (CH3), −4.6 (SiCH3), −4.8 (SiCH3); HRMS (ESI) C17H31N2O7SSi (M + H) requires 435.1616; found 436.1624 and C17H30N2NaO7SSi (M + Na) requires 457.1435; found 457.1444.5′-Azide-3′-O-tert-butyldimethylsilyl deoxythymidine (10)
A solution of 5′-O-mesyl-3′-O-tert-butyldimethylsilyl deoxythymidine (9) (1.29 g, 3.0 mmol) and sodium azide (580 mg, 9.0 mmol) in dry DMF (12 mL) under argon was heated to 100 °C for 14 h. The reaction was cooled to room temperature, diluted with water (100 mL) and extracted with diethyl ether (3 × 100 mL). The combined organic layers were washed with brine (3 × 75 mL), over MgSO4, and evaporated. The residue was purified by silica gel chromatography, eluting with petrol:diethyl ether (1:1 to 0:1) to afford the title compound (686 mg, 61%) as a white foam; Rf 0.18 (petrol:diethyl ether 1:1); [α]22D +80 (c 0.78, CHCl3); νmax/cm−1 (CHCl3) 3087, 3062, 3010, 2928, 2855, 2104, 1959, 1701, 1670, 1624, 1554, 1509, 1480, 1448, 1381, 1349, 1309, 1249, 1178, 1153; 1H NMR (500 MHz, chloroform-d) δ 8.95 (1H, s, NH), 7.31 (1H, q, J 1.2, C6H), 6.24 (1H, t, J 6.6, C1′H), 4.34 (1H, dt, J 7.0, 4.3, C3′H), 3.93 (1H, dt, J 4.4, 3.5, C4′H), 3.70 (1H, dd, J 13.3, 3.4, C5′HH), 3.49 (1H, dd, J 13.3, 3.6, C5′HH), 2.28 (1H, ddd, J 13.6, 6.6, 4.3, C2′HH), 2.16 (1H, dt, J 13.7, 6.9, C2′HH), 1.94 (3H, d, J 1.3, CH3), 0.88 (9H, s, SiC(CH3)3), 0.08 (6H, s, Si(CH3)2); 13C NMR (126 MHz, chloroform-d) δ 163.8 (C4), 150.4 (C2), 135.5 (C6H), 111.5 (C5), 84.9 (C4′H, and C1′H), 71.8 (C3′H), 51.9 (C6′H2), 40.8 (C2′H2), 25.8 (SiC(CH3)3), 18.04 (SiC(CH3)3), 12.8 (CH3), −4.54 (SiCH3), −4.77 (SiCH3); HRMS (ESI) C16H28N5O4Si (M + H) requires 382.1905; found 382.1910 and C16H27N5NaO4Si (M + Na) requires 404.1725; found 404.1737.
5′-Azide-3′-OH deoxythymidine (4)
To a stirred solution of 5′-azido-3′-O-tert-butyldimethylsilyl deoxythymidine (10) (151 mg, 400 μmol) in THF (2.0 mL) under an argon atmosphere, was added tetrabutylammonium fluoride trihydrate (189 mg, 600 μmol). The resulting solution was stirred at room temperature for 12 hours, and then the volatile organics were evaporated. The residue was purified by silica gel chromatography, eluting with EtOAc:MeOH (19:1) to afford the title compound (95 mg, 89%) as a white foam; Rf 0.20 (EtOAc); [α]22D +114 (c 1.0, CHCl3); νmax/cm−1 (CHCl3); 3390, 3009, 2956, 2105, 1690, 1471, 1438, 1262; 1H NMR (500 MHz, Methanol-d4) δ 7.54 (1H, q, J 1.2, C6H), 6.26 (1H, t, J 6.8, C1′H)), 4.34 (1H, dt, J 6.5, 4.1, C3′H), 3.96 1H, (dt, J 5.0, 3.8, C5′H), 3.63 (1H, dd, J 13.2, 3.7, C5′HH), 3.57 (1H, dd, J 13.2, 5.1, C5′HH), 2.31 (1H, dd, J 13.7, 6.6, C2′HH), 2.25 (1H, ddd, J 13.7, 6.6, 3.9, C2′HH), 1.89 (3H, d, J 1.3, CH3).13C NMR (126 MHz, Methanol-d4) δ 166.3 (C4), 152.3 (C2), 137.7 (C6H), 111.9 (C5), 86.4 (C4′H or C1′H) 86.3 (C4′H or C1′H), 72.5 (C3′H), 53.4 (C6′H2), 40.2 (C2′H2), 12.5 (CH3); HRMS (ESI) C10H14N5O4 (M + H) requires 268.1040; found 268.1044 and C10H14N5NaO4 (M + Na) requires 290.0859; found 290.0858.
TLMorpholino-3′-O-tert-butyl silyl dimer T-T (11)
To a microwave vial containing the morpholino thymidine (3) (290 mg, 500 μmol) and the azide thymidine (10) (190 mg, 500 μmol) in THF:tBuOH:H2O (3:2:1 ratio, total volume 2 mL) was added copper iodide (47.0 mg, 250 μmol). The vial was sealed, stirred and irradiated in a Biotage microwave at 80 °C (approximately power of irritation 16 W) for 3½ h. After cooling to room temperature the vial was removed, and the solvents were removed in vacuo to afford a residue, which was purified by silica gel chromatography, eluting with DCM:MeOH (40:1 to 30:1) to afford the title compound (339 mg, 72%) as a colourless foam; Rf 0.21 (DCM:MeOH 25:1); [α]24D +66 (c 0.74, CHCl3); νmax/cm−1 (CHCl3) 3603, 3390, 3305, 3200, 2934, 2838, 2552, 1905, 1713, 1681, 1633, 1611, 1584, 1490, 1456; 1H NMR (400 MHz, chloroform-d) δ 9.49 (1H, s, NH), 9.38 (1H, s, NH), 7.61 (1H, s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN), 7.46–7.37 (2H, m, Ph), 7.35–7.15 (8H, m, 4 × Ar, C6H, 3 × Ph), 6.86–6.76 (4H, m, Ar), 6.68 (1H, d, J 1.4, C6H), 6.07 (1H, t, J 6.6, C1′H), 5.79 (1H, dd, J 9.7, 2.6, C1′H),4.72–4.56 (2H, m, NCH2C), 4.45 (1H, dt, J 7.0, 5.2, C3′H), 4.15–4.00 (2H, m, C4′H and C4′H), 3.80 (1H, d, J 13.9, OC5′HH) 3.78 (6H. s, OCH3), 3.68 (1H, d, J 13.9, OC5′HH), 3.26 (1H, dd, J 9.7, 5.3, NC5′HH), 3.09–2.97 (2H, m, NC5′HH and NCHH), 2.91 (1H, d, J 10.9, NCHH), 2.36–2.18 (2H, m, C2′H2), 2.10 (1H, t, 10.7, NCHH), 2.04 (1H, t, 10.7, NCHH), 1.92 (3H, d, J 1.2, CH3), 1.92 (3H, d, J 1.2, CH3), 0.89 (9H, s, SiC(CH3)3), 0.11 (3H, s, SiCH3), 0.08 (3H, s, SiCH3); 13C NMR (101 MHz, chloroform-d) δ 163.8 (2 × C4), 158.6 (2 × ArC), 150.3 (C2), 150.2 (C2), 144.8 (ArC), 143.86 (C) 136.4 (C6H), 135.9 (ArC), 135.8 (ArC), 135.62 (C6H), 130.2 (2 × ArCH), 130.14 (2 × ArCH), 128.23 (2 × PhCH), 127.9 (2 × PhCH), 126.9 (PhCH), 124.7 (CHN), 113.2 (4 × ArCH), 111.6 (C5), 110.9 (C5), 86.6 (Ar3CO), 86.2 (C1′H), 84.2 (C4′H), 79.8 (C1′H), 75.8 (C4′H), 72.0 (C3′H), 64.6 (NC5′H2), 55.9 (NCH2), 55.3 (2 × OCH3), 54.4 (NCH2), 52.8 (OC5′H2), 50.9 (NCH2C), 39.4 (C2′H2), 25.8 (SiC(CH3)3), 17.9 (SiC(CH3)3), 12.70 (2 × CH3), −4.5 (SiCH3), −4.6 (SiCH3); HRMS (ESI +ve) C50H63N8O10Si (M + H+) requires 963.4431, found 963.4436, and C50H62N8NaO10Si (M + Na+) requires 985.4250, found 985.4231.
CHN), 7.46–7.37 (2H, m, Ph), 7.35–7.15 (8H, m, 4 × Ar, C6H, 3 × Ph), 6.86–6.76 (4H, m, Ar), 6.68 (1H, d, J 1.4, C6H), 6.07 (1H, t, J 6.6, C1′H), 5.79 (1H, dd, J 9.7, 2.6, C1′H),4.72–4.56 (2H, m, NCH2C), 4.45 (1H, dt, J 7.0, 5.2, C3′H), 4.15–4.00 (2H, m, C4′H and C4′H), 3.80 (1H, d, J 13.9, OC5′HH) 3.78 (6H. s, OCH3), 3.68 (1H, d, J 13.9, OC5′HH), 3.26 (1H, dd, J 9.7, 5.3, NC5′HH), 3.09–2.97 (2H, m, NC5′HH and NCHH), 2.91 (1H, d, J 10.9, NCHH), 2.36–2.18 (2H, m, C2′H2), 2.10 (1H, t, 10.7, NCHH), 2.04 (1H, t, 10.7, NCHH), 1.92 (3H, d, J 1.2, CH3), 1.92 (3H, d, J 1.2, CH3), 0.89 (9H, s, SiC(CH3)3), 0.11 (3H, s, SiCH3), 0.08 (3H, s, SiCH3); 13C NMR (101 MHz, chloroform-d) δ 163.8 (2 × C4), 158.6 (2 × ArC), 150.3 (C2), 150.2 (C2), 144.8 (ArC), 143.86 (C) 136.4 (C6H), 135.9 (ArC), 135.8 (ArC), 135.62 (C6H), 130.2 (2 × ArCH), 130.14 (2 × ArCH), 128.23 (2 × PhCH), 127.9 (2 × PhCH), 126.9 (PhCH), 124.7 (CHN), 113.2 (4 × ArCH), 111.6 (C5), 110.9 (C5), 86.6 (Ar3CO), 86.2 (C1′H), 84.2 (C4′H), 79.8 (C1′H), 75.8 (C4′H), 72.0 (C3′H), 64.6 (NC5′H2), 55.9 (NCH2), 55.3 (2 × OCH3), 54.4 (NCH2), 52.8 (OC5′H2), 50.9 (NCH2C), 39.4 (C2′H2), 25.8 (SiC(CH3)3), 17.9 (SiC(CH3)3), 12.70 (2 × CH3), −4.5 (SiCH3), −4.6 (SiCH3); HRMS (ESI +ve) C50H63N8O10Si (M + H+) requires 963.4431, found 963.4436, and C50H62N8NaO10Si (M + Na+) requires 985.4250, found 985.4231.
TLMorpholino-3′-OH dimer T-T (12)
:tBuOH:H2O (3:2:1 ratio, total volume 1.75 mL) was added copper iodide (33.0 mg, 175 μmol). The vial was sealed, stirred and irradiated in a Biotage microwave at 80 °C (approximately power of irritation 16 W) for 3½ h. After cooling to room temperature the vial was removed, and the solvents were removed in vacuo to afford a residue, which was purified by silica gel chromatography, eluting with DCM:MeOH (12:1 to 10:1) to afford the title compound (223 mg, 75%) as a white foam; Rf 0.12 (DCM:MeOH 10:1); [α]24D −7.13 (c 1.0, CHCl3); νmax/cm−1 (ATR) 3390, 3008, 2961, 1690, 1608, 1509, 1490, 1456; 1H NMR (500 MHz, chloroform-d) δ 9.93 (1H, s, NH), 9.86 (1H, s, NH), 7.72 (1H, s, CCHN), 7.48–7.41 (2H, m, Ph), 7.37–7.25 (7H, m, 4 × Ar, C6H, 2 × Ph), 7.26–7.18 (1H, m, Ph), 6.95 (1H, s, C6H), 6.89–6.80 (4H, m, Ar), 6.07 (1H, t, J 6.6, C1′H), 5.75 (1H, dd, J 8.8, C1′H), 4.74–4.68 (2H, m, NCH2C), 4.51 (1H, bs, C3′H), 4.39 (1H, bs, OH), 4.24 (1H, m, C4′H), 4.14–4.05 (1H, m, C4′H), 3.89 (1H, d, J 13.6, OC5′HH), 3.80 (6H, s, 2 × OCH3), 3.63 (1H, d, J 13.6, OC5′HH), 3.30 (1H, dd, J 9.7, 5.1, NC5′HH), 3.10 (1H, dd, J 9.7, 5.0, NC5′HH), 3.07–2.97 (2H, m, NCHH and NCHH), 2.33–2.26 (2H, m, C2′H2), 2.18 (1H, t, J 11.0, NCHH), 1.97 (1H, t, J 10.4, NCHH), 1.92 (3H, s CH3), 1.89 (3H, s, CH3); 13C NMR (126 MHz, chloroform-d) δ 164.0 (C4), 163.9 (C4), 158.5 (2 × ArC), 150.5 (C2), 150.4 (C2), 144.6 (C), 143.8 (ArC), 136.6 (C6H), 135.8 (ArC), 135.7 (ArC), 135.5 (C6H), 130.1 (2 × ArCH), 130.0 (2 × ArCH), 128.1 (2 × ArCH), 127.8 (2 × ArCH), 126.9 (PhCH), 124.6 (CHN), 113.1 (4 × ArCH), 111.3 (C5), 111.0 (C5), 86.52 (C1′H), 86.1 (Ar3CO), 83.9 (C4′H), 79.9 (C1′H), 75.7 (C4′H), 71.6 (C3′H), 64.3 (NC5′H2), 55.4 (NCH2), 55.2 (2 × OCH3), 54.9 (NCH2), 52.7 (OC5′H2), 51.4 (NCH2C), 38.9 (C2′H2), 12.6 (CH3), 12.5 (CH3); HRMS (ESI +ve) C44H49N8O10 (M + H+) requires 849.3566, found 849.3644.
:MeOH (12:1 to 10:1) to afford the title compound (251 mg, 82%) as a white foam; Rf 0.12 (DCM:MeOH 10:1).
TLMorpholino phosphoramidite T-T (13)
To a stirred solution of the TLMorpholino-3-OH dimer T-T (12) in DCM under an argon atmosphere at room temperature was added N,N-diisopropylethylamine (98 μL, 561 μmol) followed by 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (59.0 mg, 266 μmol) dropwise over 1 minute, then stirred at room temperature for 24 h. The solvent was blown off with a stream of nitrogen gas, and the residue was purified by silica column chromatography, eluting with DCM:MeOH (20:1 to 15:1) to afford an analytical pure sample of the title compound (38 mg, 14%) as a white foam along with the bulk material (167 mg ca. 60%) containing small amounts of 2-cyanoethyl N,N-dipropylphosphonamidate as an off white foam; Rf 0.15 (DCM:MeOH 15:1); νmax/cm−1 (CHCl3) 3698, 3665, 3391, 3212, 2857, 2552, 2300, 2105, 2047, 1908, 1731, 1681, 1633, 1592, 1490 and 1455; 1H NMR (500 MHz, chloroform-d) δ 8.66 (1H, s, NH), 8.66 (1H, s, NH), 7.63 and 7.59 (1H, 2 × s, CCHN), 7.48–7.41 (2H, m, Ph), 7.35–7.23 (7H, m, 4 × Ar, C6H, 2 × Ph), 7.23–7.17 (1H, m, Ph), 6.87–6.77 (5H, m, Ar and C6H), 6.12 (1H, m, C1′H), 5.77 (1H, m, C1′H), 4.82–4.54 (3H, m, NCH2C and C3′H), 4.34–4.22 (1H, m, C4′H), 4.07–4.02 (1H, m, C4′H), 3.90–3.85 (1H, m, OCHH), 3.84–3.79 (1H, m, OC5′HH), 3.79 (6H, s, 2 × OCH3), 3.84–3.68 (1H, m, OCHH), 3.69–3.54 (3H, m, OC5′HH and 2 × NCH), 3.26 (1H, dd, J 9.7, 5.1, NC5′HH), 3.08–2.96 ((2H, m, NC5′HH and NCHH), 2.92 (1H, m, NCHH), 2.75–2.68 (1H, m, CHHCN), 2.71–2.59 (1H, m, CHHCN), 2.51–2.22 (2H, m, C2′H2), 2.09 (1H, t, J 11.0, NCHH), 2.07–1.97 (1H, m, NCHH), 1.92 (3H, s CH3), 1.90 (3H, s, CH3), 1.20 (12H, m, NCH(CH3)2); 13C NMR (500 MHz, chloroform-d) due to the presence of a diastereomeric mixture at the phosphorus(III) centre and coupling from phosphorus the 13C NMR could not be unambiguously assigned but the spectra are included in this ESI;†31P NMR (162 MHz, CDCl3) δ 149.2; HRMS (ESI +ve) C53H66N10O11P (M + H+) requires 1049.4645, found 1049.4653 and C53H65N10NaO11P (M + Na+) requires 1071.4464, found 1071.4489.
3′-O-Propargyl thymidine (15)
To a stirred solution of 5′-O-DMT thymidine (1.40 g, 2.55 mmol) in THF (25 mL) at 0 °C was added sodium hydride (257 mg, 6.4 mmol) in small portions over 5 min. The resulting solution was stirred at 0 °C for 30 min, then at room temp for 1 h. The solution was cooled to 0 °C and propargyl bromide (285 μL, 3.18 mmol) was added. The solution was stirred at 0 °C for 30 min, then at room temp for 5 h. The reaction was quenched by the addition of water (1 mL), and the volatile organic were removed by evaporation. The residue was partitioned between water (25 mL) and DCM (25 mL), the aquous layer was extracted with DCM (3 × 25 mL). The combined organic layers were washed with brine (2 × 25 mL), dried over MgSO4, and evaporated in vacuo to afford a residue, which was purified by silica gel chromatography, eluting with DCM:MeOH (38:1 to 19:1) to afford the title compound (1.15 g, 77%) as a white foam; Rf 0.4 (DCM:MeOH 19:1); [α]24D 33.0 (c 1.0, CHCl3); νmax/cm−1 (ATR); 3385, 3190, 2950, 1930, 1695, 1631, 1611, 1491; 1H NMR (400 MHz, chloroform-d) δ 8.75 (1H, br s, NH), 7.63 (1H, s, C6H), 7.46–7.41 (2H, m, Ar), 7.36–7.25 (7H, m, Ar), 6.89–6.85 (4H, m, Ar), 6.36 (1H, d, J 8.0 and 5.7, C1′H), 4.53 (1H, dt, J 5.3 and 2.2, C4′H), 4.21 (1H, dd, J 15.9 and 2.3, OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) HCCH), 4.21–4.16 (1H, m, C5′H), 4.16 (1H, dd, J 15.9 and 2.3, OCHCCH), 3.82 (6H, s, 2 × OCH3), 3.50 (1H, dd, J 10.6 and 3.0, C6′HH), 3.38 (1H, dd, J 10.6 and 2.7, C6′HH), 2.54 (1H, ddd, J 13.9, 5.7 and 2.2, C2′HH), 2.44 (1H, t, J 2.3, CCH), 2.26 (1H, ddd, J 13.9, 8.0 and 6.3, C2′HH), 1.53 (3H, s, CH3); 13C NMR (101 MHz, chloroform-d) δ 163.8 (C4), 158.8 (2 × C), 150.4 (C2), 144.5 (C), 135.6 (C6H), 135.5 (2 × C), 130.2 (4 × CH), 128.2 (2 × CH), 128.1 (2 × CH), 127.3 (CH), 113.4 (4 × ArCH), 111.3 (C5H), 87.1 (C), 84.91 (C1′H), 84.0 (C5′H), 79.2 (C), 78.6 (C4′H), 75.2 (CH), 63.6 (C5′H2), 56.7(CH2), 55.4 (2 × OCH3), 37.9 (C2′H2), 12.0 (CH3); HRMS (ESI +ve) C34H34N2NaO7 (M + Na+) requires 605.2258, found 605.2247.
HCCH), 4.21–4.16 (1H, m, C5′H), 4.16 (1H, dd, J 15.9 and 2.3, OCHCCH), 3.82 (6H, s, 2 × OCH3), 3.50 (1H, dd, J 10.6 and 3.0, C6′HH), 3.38 (1H, dd, J 10.6 and 2.7, C6′HH), 2.54 (1H, ddd, J 13.9, 5.7 and 2.2, C2′HH), 2.44 (1H, t, J 2.3, CCH), 2.26 (1H, ddd, J 13.9, 8.0 and 6.3, C2′HH), 1.53 (3H, s, CH3); 13C NMR (101 MHz, chloroform-d) δ 163.8 (C4), 158.8 (2 × C), 150.4 (C2), 144.5 (C), 135.6 (C6H), 135.5 (2 × C), 130.2 (4 × CH), 128.2 (2 × CH), 128.1 (2 × CH), 127.3 (CH), 113.4 (4 × ArCH), 111.3 (C5H), 87.1 (C), 84.91 (C1′H), 84.0 (C5′H), 79.2 (C), 78.6 (C4′H), 75.2 (CH), 63.6 (C5′H2), 56.7(CH2), 55.4 (2 × OCH3), 37.9 (C2′H2), 12.0 (CH3); HRMS (ESI +ve) C34H34N2NaO7 (M + Na+) requires 605.2258, found 605.2247.
Triazole-T-T dimer (16)
To a microwave vial containing the 3′-O-propargyl thymidine (15) (1.164 g, 2.0 mmol) and the azide thymidine (4) (534 mg, 2.0 mmol) in THF:tBuOH:H2O (3:2:1 ratio, total volume 12 mL) was added copper iodide (188 mg, 1.0 mol). The vial was sealed, stirred and irradiated in a Biotage microwave at 80 °C (approximately power of irritation 16 W) for 3½ h. After cooling to room temperature the vial was removed, and the solvents were removed in vacuo to afford a residue, which was purified by silica gel chromatography, eluting with DCM:MeOH (20:1 to 10:1) to afford the title compound (1.47 g, 87%) as a white foam; Rf 0.35 (DCM:MeOH 10:1); [α]24D 5.2 (c 1.0, CHCl3); νmax/cm−1 (ATR) 3392, 3010, 2963, 1691, 1604, 1493, 1460; 1H NMR (400 MHz, DMSO-d6) δ 11.35 (1H, br s, NH), 11.31 (1H, br s, NH), 8.08 (1H, s, triazole-CH), 7.51 (1H, s, C6H), 7.40–7.29 (4H, m, Ar), 7.33 (1H, s, C6H), 7.28–7.22 (5H, m, Ar), 6.90 (4H, d, J 8.7 Hz, Ar), 6.16 (1H, app t, J 6.4 Hz C1H), 6.14 (1H, app t, J 6.4 Hz C1H), 5.50 (1H, d, J 4.4, OH), 4.70 (1H, dd, J 14.3 and 4.4, NC5′HH), 4.59 (1H, dd, J 14.3 and 7.7, NC5′HH), 4.60–4.52 (2H, m, OCH2C), 4.45–4.37 (1H, m, C3′H), 4.31–4.24 (1H, m, C3′H), 4.10–4.02 (2H, m, C4′H and C4′H), 3.74 (6H, s, 2 × OCH3), 3.26 (1H, dd, J 10.5, 3.8 Hz, OC5′HH), 3.17 (1H, dd, J 10.5, 3.2 Hz, OC5′HH), 2.43–2.25 (2H, m, C2′HH), 2.25–2.04 (2H, m, C2′HH), 1.78 (3H, s, CH3), 1.43 (3H, s, CH3); 13C NMR (101 MHz, DMSO) δ 163.6 (2 × C4), 158.17 (2 × C), 150.37 (2 × C2), 144.6 (C), 143.7 (C), 136.0 (C6), 135.5 (C6), 135.4 (C), 135.1 (C), 129.7 (4 × CH), 127.9 (2 × CH), 127.6 (2 × CH), 126.8 (CH), 124.7 (triazole-CH), 113.3 (4 × ArCH), 109.8 (C5H), 109.7 (C5H), 86.04 (C), 84.02 (CH), 83.93 (CH), 83.78 (CH), 82.87 (CH), 78.75 (CH), 70.72 (CH), 63.77 (CH2), 61.77 (CH2), 55.05 (2 × OCH3), 51.17 (CH2), 37.9 (C2′H2), 36.5 (C2′H2), 12.1 (CH3), 11.9 (CH3); HRMS (ESI +ve) C44H47N7NaO11 (M + Na+) requires 872.3226, found 872.3215.
Triazole-T-T dimer phosphoramidite (17)
To a stirred solution of the triazole-T-T dimer (16) (493 mg, 0.58 mmol) in DCM (1.2 mL) under an argon atmosphere at room temperature was added N,N-diisopropylethylamine (182 μL, 1.05 mmol) followed by 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (125 mg, 0.58 mmol) dropwise over 1 minute, then stirred at room temperature for 24 h. The solvent was blown off with a stream of nitrogen gas, and the residue was purified by silica column chromatography, eluting with DCM:MeOH (20:1 to 15:1) to afford the title compound (391 mg, ca. 64%) containing 2-cyanoethyl N,N-diisopropylphosphonamidate as an off white foam; Rf 0.21 (DCM:MeOH 15:1); νmax/cm−1 (ATR) 3700, 3669, 3384, 3219, 2860, 2550, 2310, 2109, 1911, 1733, 1686, 1495 and 1459; 1H NMR (500 MHz, chloroform-d) δ 9.20 (2H, br s, NH), 7.69 and 7.65 (1H, 2 × s, triazole-CH), 7.60 (1H, s, C6H), 7.42–7.37 (2H, m, Ar), 7.37–7.21 (7H, m, Ar), 6.90–6.83 (4H, m, Ar), 6.78–6.73 (1H, 2 × s, C6H), 6.38–6.32 (1H, m, C1H), 6.21–6.13 (1H, m, C1H), 4.78–4.57 (5H, m, 2 × CH2 and OCH), 4.40–4.11 (3H, m, 3 × CH), 3.99–3.70 (2H, m, CH2), 3.81 (6H, s, 2 × OCH3), 3.68–3.45 (3H, m, 2 × CH and CHH), 3.38–3.32 (1H, m, CHH), 2.80–2.75 (1H, m, CHHCN) 2.70–2.65 (1H, m, CHHCN), 2.57–2.20 (4H, m, C2′HH and C2′HH), 1.90–1.87 (3H, m, CH3), 1.47 (3H, s, CH3); 1.26–1.18 (12H, m, NCH(CH3)2); δ13C NMR (500 MHz, chloroform-d) due to the presence of a diastereomeric mixture at the phosphorus(III) centre and coupling from phosphorus the 13C NMR could not be unambiguously assigned but the spectra are included in this ESI;†31P NMR (162 MHz, CDCl3) δ 149.1; HRMS (ESI +ve) C53H65N9O12P (M + H+) requires 1050.4485, found 1050.4464.
Conclusions
We have shown that the CuAAC reaction can be used to synthesise a new DNA mimic containing a triazole-linked morpholino (TLMO) internucleotide modification. Phosphoramidite reagents 13 and 17 were synthesised and their compatibility with automated solid phase synthesis was demonstrated. UV melting studies showed that incorporation of the TLMO modification provided an improved Tm value for binding to RNA when compared to the previously reported triazole-containing oligomers. Structural characterisation, and biological evaluation of the TLMO-modified oligomers is underway and the results of this work will be reported in due course.Acknowledgements
We thank King Abdulaziz University, Saudi Arabia (Scholarship to R. D. A.) and the BBSRC (DTP Scholarship for P. K. P.) for providing financial support for this work.Notes and references
- (a) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 29, 1388–1405 RSC; (b) A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258–1267 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed; (b) A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2009, 131, 3958 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- (a) R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859–6864 CrossRef CAS PubMed; (b) J. Qiu, A. H. El-Sagheer and T. Brown, Chem. Commun., 2013, 49, 6959–6961 RSC.
- (a) R. Lucas, V. Neto, A. Hadj Bouazza, R. Zerrouki, R. Granet, P. Krausz and Y. Champavier, Tetrahedron Lett., 2008, 49, 1004–1007 CrossRef CAS; (b) R. Lucas, R. Zerrouki, R. Granet, P. Krausz and Y. Champavier, Tetrahedron, 2008, 64, 5467–5471 CrossRef CAS.
- A. H. El-Sagheer, A. P. Sanzone, R. Gao, A. Tavassoli and T. Brown, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11338–11343 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Commun., 2011, 47, 12057–12058 RSC.
- (a) A. P. Sanzone, A. H. El-Sagheer, T. Brown and A. Tavassoli, Nucleic Acids Res., 2012, 40, 10567–10575 CrossRef CAS PubMed; (b) C. N. Birts, A. P. Sanzone, A. H. El-Sagheer, J. P. Blaydes, T. Brown and A. Tavassoli, Angew. Chem., Int. Ed., 2014, 53, 2362–2365 CrossRef CAS PubMed.
- A. Dallmann, A. H. El-Sagheer, C. Griesinger, L. Dehmel, N. P. Ernsting, C. Mügge and T. Brown, Chem. – Eur. J., 2011, 17, 14714–14717 CrossRef CAS PubMed.
- (a) J. Summerton and D. Weller, Antisense Nucleic Acid Drug Dev., 1997, 7, 187–195 CrossRef CAS PubMed; (b) J. Summerton, Biochim. Biophys. Acta, 1999, 1489, 141–158 CrossRef CAS PubMed; (c) J. E. Summerton, Lett. Pept. Sci., 2003, 10, 215–236 CrossRef CAS.
- T. K. Warren, A. C. Shurtleff and S. Bavari, Antiviral Res., 2012, 94, 80–88 CrossRef CAS PubMed.
- (a) N. Zhang, C. Tan, P. Cai, P. Zhang, Y. Zhao and Y. Jiang, Bioorg. Med. Chem., 2009, 17, 2441–2446 CrossRef CAS PubMed; (b) N. Zhang, C. Tan, P. Cai, Y. Jiang, P. Zhang and Y. Zhao, Tetrahedron Lett., 2008, 49, 3570–3573 CrossRef CAS.
- D. Pan, J. Sun, H. Jin, Y. Li, L. Li, Y. Wu, L. Zhanga and Z. Yang, Chem. Commun., 2015, 51, 469–472 RSC.
- (a) B. Froehler, P. Ng and M. Matteucci, Nucleic Acids Res., 1988, 16, 4831–4839 CrossRef CAS PubMed; (b) E. P. Stirchak, J. E. Summerton and D. D. Weller, Nucleic Acids Res., 1989, 17, 6129–6141 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and copies of 1H, 13C, 31P NMR spectra for all compounds. See DOI: 10.1039/c6ob00007j |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia. Tel: +96 6(12)2577246; E-mail: iaaalharte@kau.edu.sa |
| ¶ For general experimental details please see the ESI.† |
| This journal is © The Royal Society of Chemistry 2016 |