
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An atom-efficient and convergent approach to the preparation of NS5A inhibitors by C–H activation†
Thomas O.
Moore
*,
Michael
Paradowski
and
Simon E.
Ward
Sussex Drug Discovery Centre, University of Sussex, Brighton, England BN1 9QJ, UK. E-mail: T.Moore@sussex.ac.uk
First published on 25th February 2016
Abstract
A novel approach of the convergent functionalisation of aryl dibromides to form NS5A type inhibitors using C–H activation is reported. The focus of investigation was to reduce the formation of homodimeric side product, as well as to investigate the scope of different aryl dibromides that were tolerated under the reaction conditions. The C–H activation methodology was found to give a viable synthetic route to NS5A inhibitors, with late stage functionalisation of the core portion of the molecule, albeit with some chemical functionalities not tolerated.
Introduction
The hepatitis C virus (HCV) is estimated to affect 170 million people worldwide and, without treatment, may lead to cirrhosis, hepatocellular carcinoma, liver failure and ultimately death.1–4 Until recently, the standard of care for HCV patients was a combination of lengthy regimens of pegylated interferon α (pegIFN-α) and ribavirin.5 Those poorly tolerated and moderately efficacious therapies contributed to the urgent need to identify improved treatments by oral administration. One major focus to achieve this was targeted at the nonstructural protein 5A (NS5A) which is enabled in the HCV genome and plays an essential role for the viral RNA replication.6–8 Those efforts led to the discovery of daclatasvir (BMS) (Fig. 1), a non ‘rule of 5’ compliant NS5a inhibitor exhibiting picomolar activity against HCV genotypes 1a and 1b.9 Subsequently, there have been over 70 patents and publications describing novel NS5A inhibitors related to Daclatasvir.9–14 Of these, many have maintained its symmetrical biphenylimidazole core with differentiation arising from key structural aryl alterations.15–18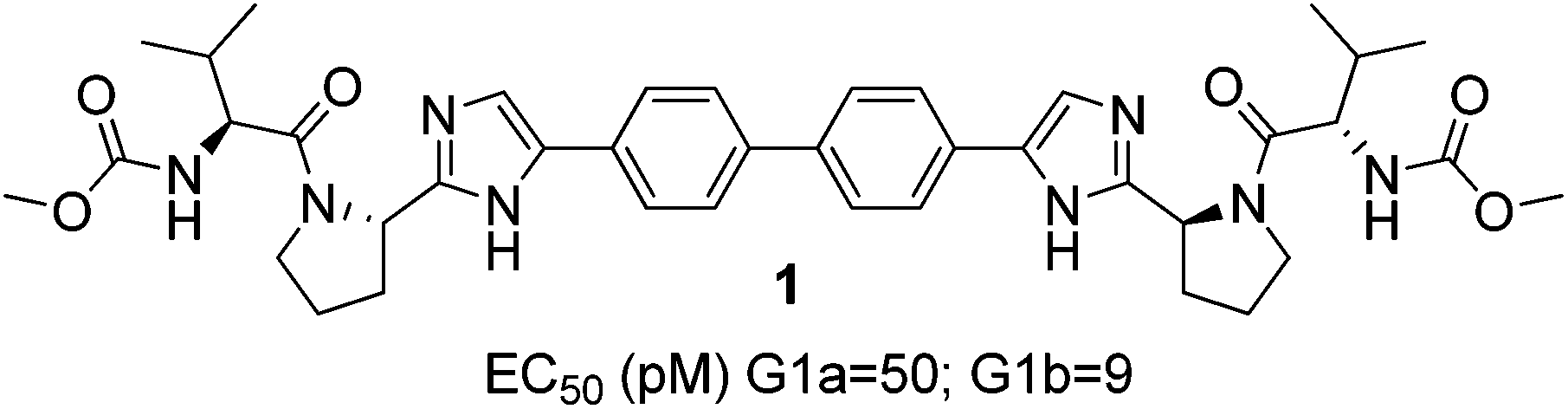 | ||
| Fig. 1 Structure of daclatasvir. | ||
The existing strategy for the preparation of dimeric NS5A inhibitors is by chain elongation from an aryl bis acetate core. However, this methodology is linear and does not allow rapid exploration of the central core unit. A number of reports and patents have also described the preparation of symmetrical aryl-imidazoles by a late stage cross coupling of bis-metalated aryl core to the required halogenated imidazole fragments.17–20 This methodology offers the advantage of a late diversification of the central core units but suffers from the lengthy, expensive and poor atom-efficient processes to install the pre-requisite organometallic reagents. However, to the best of our knowledge, none of the late stage cross coupling methodologies reported had the key pharmacophores comprising of a pair of (S)-methoxycarbonylvaline capped pyrrolidines already in place and required further synthetic steps to prepare the reported NS5A inhibitors.
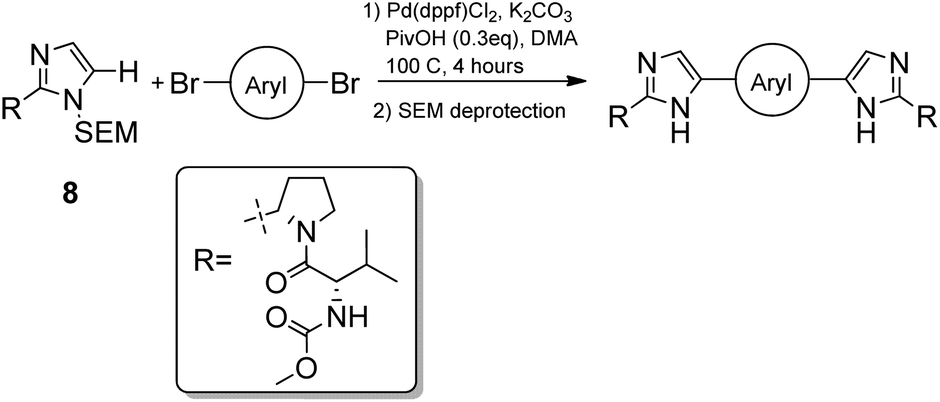
Our search for methodology that allowed us to form this bond as late as possible highlighted the opportunity to consider a double C–H activation strategy. The C–H activation of imidazoles in the desired C-5 position is one that has undergone much development in recent years.21–25 Arguably the key development was the establishment of broadly applicable conditions for the C–H activation of heterocycles by Fagnou,24 where the use of pivalic acid as an additive was found to greatly increase the rate and overall yield of reaction due to transition state stabilisation.26 Given that the SEM group has been found to be well tolerated under C–H activation conditions,27 a synthetic route based around the reaction between SEM-protected imidazoles and aryl dibromides was developed.
Results and discussion
N-Boc-L-prolinal 3 was obtained from the corresponding prolinol 2 under mild copper/TEMPO mediated aerobic oxidation conditions (Scheme 1). Although the Swern oxidation conditions gave 3 in quantitative yields, we had concerns about the potential racemisation of the carbonyl α-carbon due to the presence of triethylamine, and thus opted for milder conditions on scale up. Cyclisation of the prolinal 3 under glyoxal and ammonia conditions followed by removal of the BOC protecting group gave pyrrolidine 5 in 85% yield over three steps. Methyl carbamate-L-valine 6, obtained via reported Schotten–Bauman conditions from L-valine,28 was then coupled to imidazole pyrrolidine 5 by peptide bond formation, although this proved challenging due to high aqueous solubility of the product formed and its instability when purified by silica chromatography. Mild coupling reagents such as guanidinium salts and carbodiimides were found to be unsuitable mainly due to the difficulty separating the product and byproducts formed. The respective acid chloride was found to react cleanly with pyrrolidine 5, however we were concerned with the possibility of ketene formation from HCl elimination, leading to racemisation of the chiral valyl carbon as reported by Sauer.29 Pyridine was used instead of triethylamine to reduce the likelihood of racemisation and gave 7 in 97% yield. D-Valine-carbamate was also coupled with 5 and both NMR and HPLC experiments confirmed the lack of racemisation under the reaction conditions. Subsequent protection of imidazole 7 with 2-(trimethylsilyl)ethoxymethyl (SEM) chloride in THF with sodium hydride as a base gave the key C–H activation precursor 8 in 47% yield.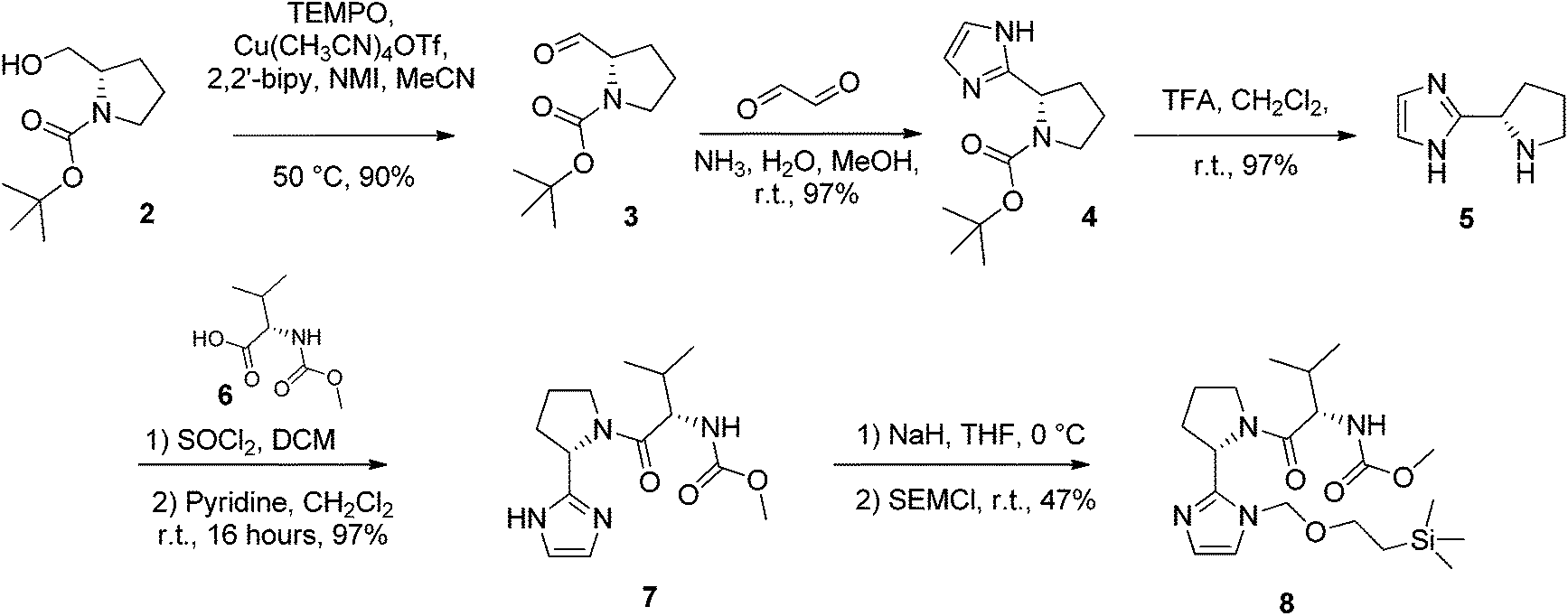 | ||
| Scheme 1 Preparation of the key (S)-methoxycarbonylvaline capped pyrrolidine 7. | ||
Pleasingly, 10 was obtained in moderate yields on first attempt by double C–H activation of 8 with 4-4′-dibromobiphenyl under the conditions reported by Fagnou (Pd(dppf)Cl2, PivOH, DMA, 100 °C, 4 h, Table 1).24 Unfortunately however a homo-coupled by-product 11 was produced, which proved difficult to separate from the product by standard purification techniques. In proof of concept studies on a simplified imidazole reagent carried out by our group, it was shown that optimisation of conditions focusing on the base, solvent and reaction temperature had the greatest effect on formation of this homo-coupled by-product. We therefore set about modifying these conditions to test whether it was possible to prevent any by-product formation. It was found that the previously established conditions30 were not only the highest yielding, but also exhibited very low amounts of homo-coupled product by LCMS analysis. When the reaction was carried out in dioxane at 100 °C, or in DMA at 60 °C (Table 1, entries 2 and 6), no homo-coupled product was observed by LCMS, but the drop in overall yield and rate of reaction in both of these cases made them impractical. As such, the Fagnou conditions were deemed the best suited for use in the double C–H activation. The 78% yield observed by LCMS for condition 5 equated to an 84% yield upon scale up and purification by column chromatography.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temperature | Yield 9a |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9a :9a |
|
a Yield of 9 and ratio of 10:9 determined by LCMS peak integration measured by adsorption at 254 nm. Conditions: (i) Pd(dppf)Cl2, PivOH, base, 4 hours. |
|||||
| 1 | K2CO3 | DMF | 100 °C | 65% | 0.13 |
| 2 | K2CO3 | Dioxane | 100 °C | 10% | 0 |
| 3 | K2CO3 | NMP | 100 °C | 38% | 0.05 |
| 4 | K2CO3 | THF | 100 °C | 25% | 0.11 |
| 5 | K2CO3 | DMA | 100 °C | 78% | 0.03 |
| 6 | K2CO3 | DMA | 60 °C | 1% | 0 |
| 7 | K2CO3 | DMA | 80 °C | 5% | 0.15 |
| 8 | K2CO3 | DMA | 120 °C | 69% | 0.07 |
| 9 | Cs2CO3 | DMA | 100 °C | 21% | 0.10 |
| 10 | KOAc | DMA | 100 °C | 3% | 0.23 |
| 11 | K3PO4 | DMA | 100 °C | 72% | 0.06 |
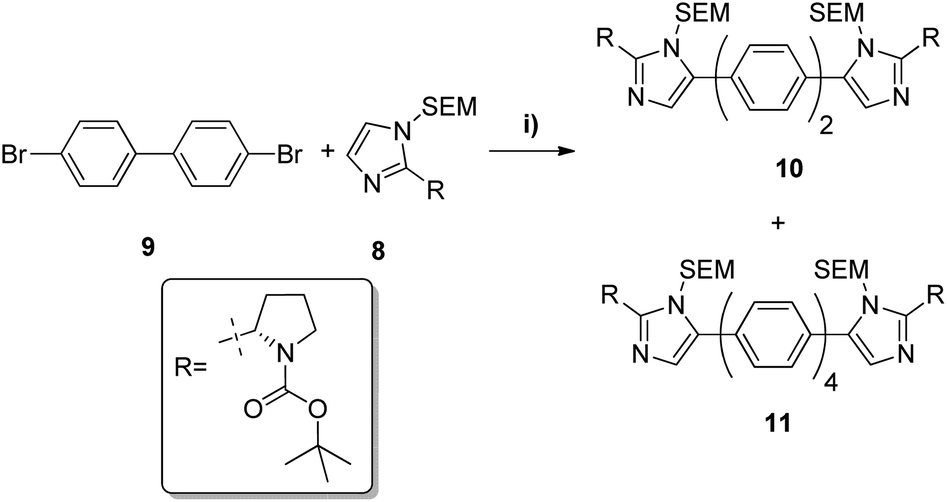
Removal of the SEM protecting group post C–H activation using organic solubilised fluoride ions proved unsuccessful even at elevated temperatures and extended reaction times. However, conditions involving trifluoroacetic acid at room temperature cleaved the SEM group slowly but in quantitative yield (Scheme 2). Despite some initial stability concerns, the methyl carbamate functionality proved to be stable under the TFA deprotection conditions.
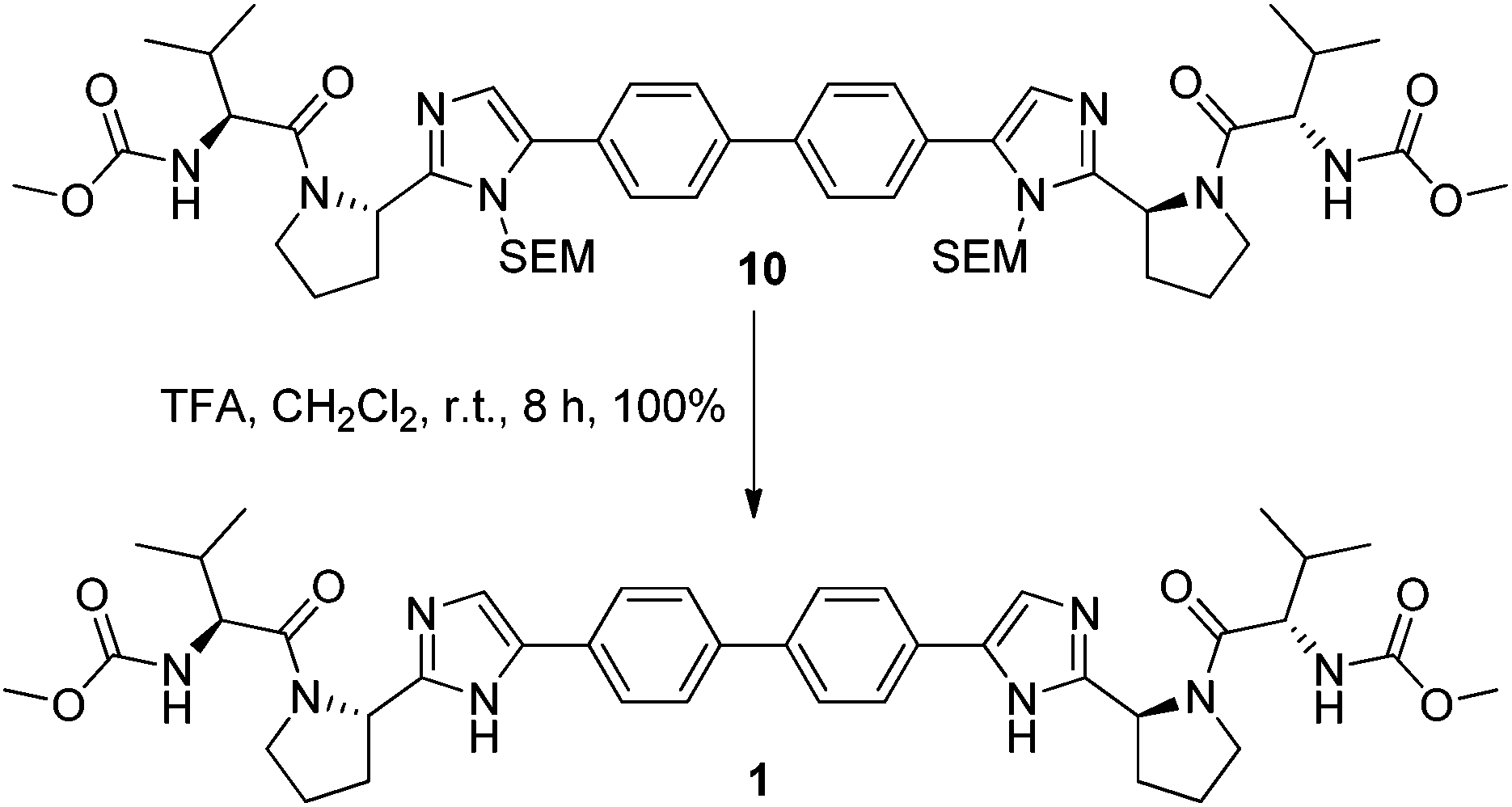 | ||
| Scheme 2 Deprotection step under protolytic conditions. | ||
With the synthesis for 1 optimised, we turned our attention to the scope of this methodology for the rapid synthesis of potential symmetrical NS5A inhibitors (Table 2). Under the reaction conditions developed above, bis-CH activated products were obtained in moderate to good yields over 2 steps. Purification of deprotected product was carried out by prep-scale LCMS mass directed auto purification, giving better separation of the product and homo-dimer by-product. Overall, of 16 different aryl dibromides tested, 9 successfully underwent double C–H activation. The yields of successful couplings consistently ranges from moderate to good on a 0.1 mmol scale. The proportion of homo-dimer to product, however, was found to vary greatly, with a tendency to be highest in electron rich aryl di-bromides, such as entries 4 and 7.
|
|
|||
|---|---|---|---|
| Entry | Aryl dibromide | Yielda (%) | HD:Pb |
| a Yield of product determined by LCMS peak integration measured by adsorption at 254 nm. Characterisation was carried out on the fully deprotected product (see Experimental section). b Ratio of homo-dimer to product. c Product decomposed under deprotection and characterisation was carried out on the SEM-protected product. | |||
| 1 |
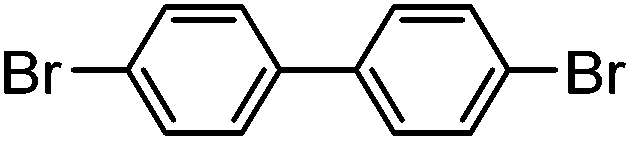
|
78% | 0.13 |
| 2 |
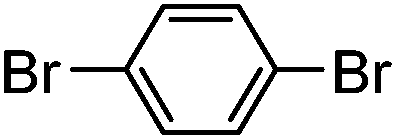
|
80% | 0 |
| 3 |
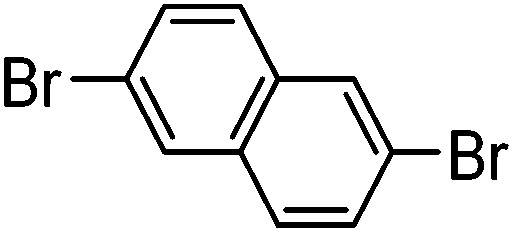
|
64% | 0.37 |
| 4 |
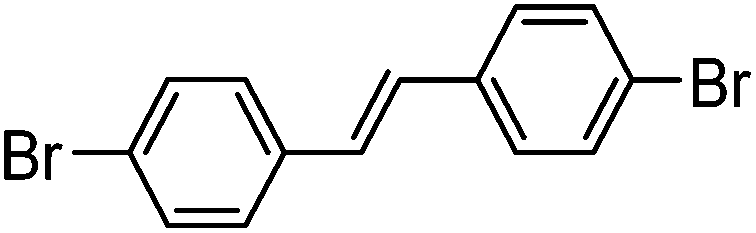
|
37% | 0.72 |
| 5 |
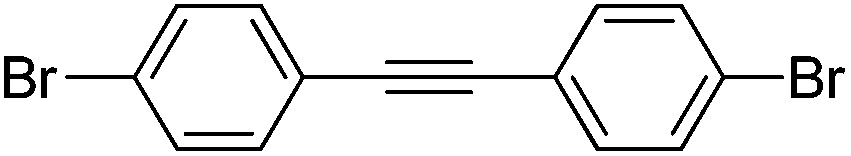
|
0% | — |
| 6 |
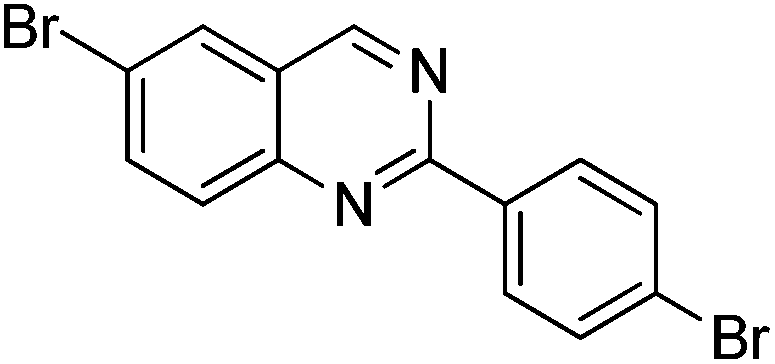
|
0% | — |
| 7 |
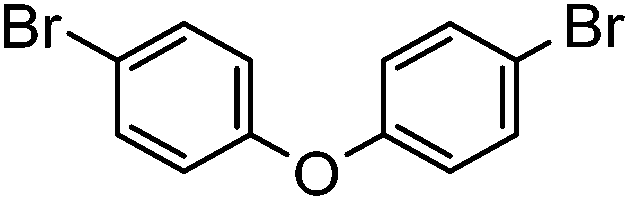
|
42% | 0.68 |
| 8 |
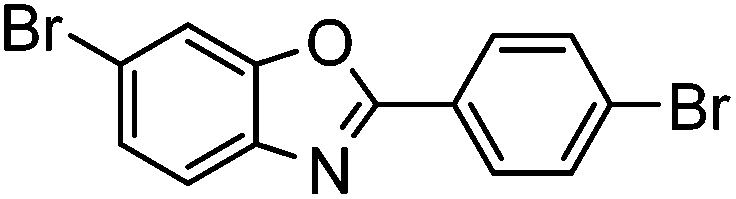
|
67% | 0.12 |
| 9c |
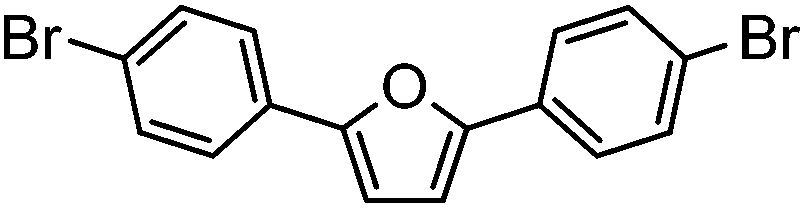
|
45% | 0.26 |
| 10c |
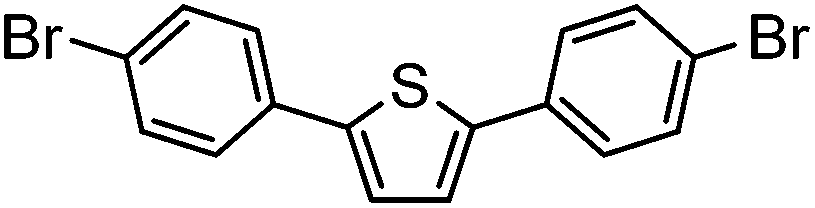
|
58% | 0.23 |
| 11 |
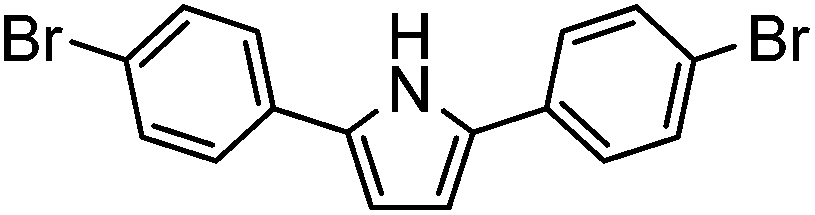
|
0% | — |
| 12 |
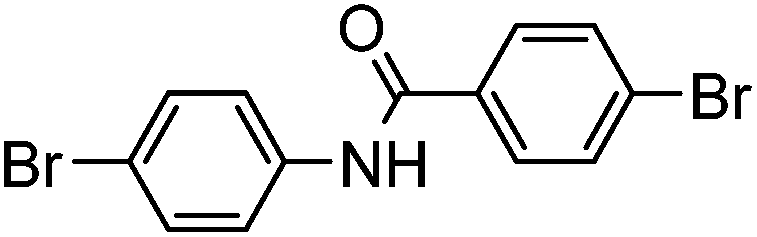
|
0% | — |
| 13 |
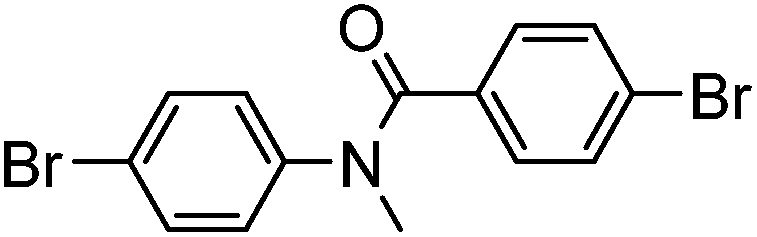
|
67% | 0 |
| 14 |
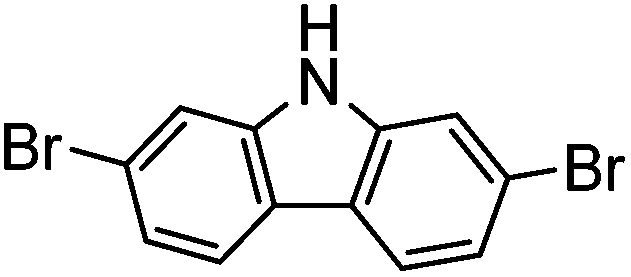
|
0% | — |
| 15 |
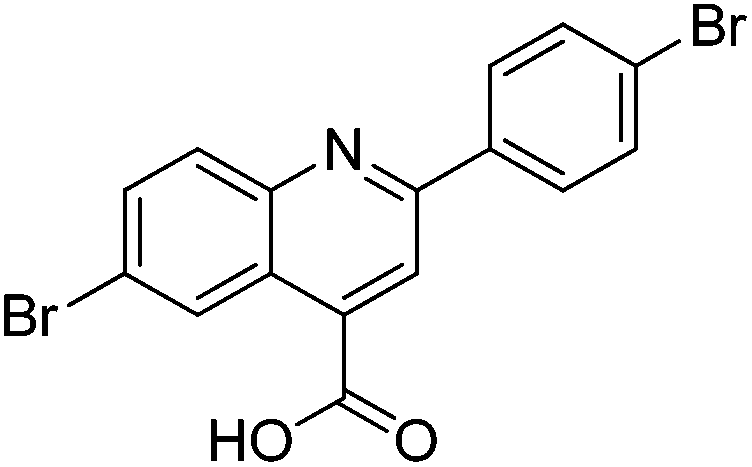
|
0% | — |
| 16 |
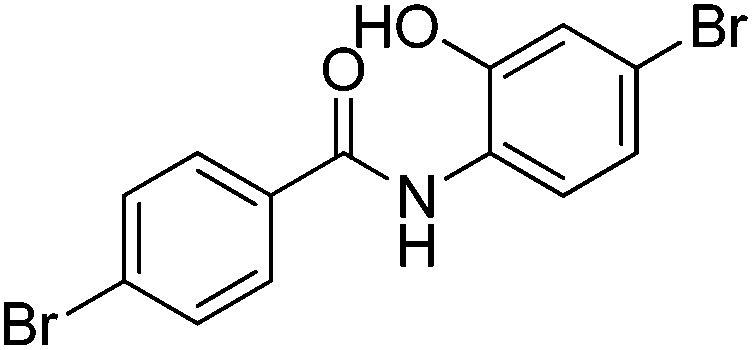
|
0% | — |
Several functional groups were found to not be compatible with the reaction conditions used. The presence of an alkyne (entry 5) seemed to prevent formation of the anticipated product, and instead produced many unidentifiable side-products. Coordination of alkynes to palladium centres through an η-2 binding mode is well precedented to undergo a wide variety of reactions, and as such this seems likely to be responsible for the lack of desired reactivity observed.31 Aryl dibromides containing a 2-phenylpyridine (entries 6 and 15) functionality also did not allow the reaction to proceed as hoped, with the reaction mixture containing starting materials and a mixture of side products after prolonged heating. There is much precedent for the C–H activation directing capability of pyridyl nitrogen's onto 2-phenyl groups,32 possibly accounting for the loss of desired reactivity, however there was no identifiable side product proving this hypothesis. Furthermore, no reaction was observed in the presence of aryl dibromides containing an N–H functionality (entries 11, 12, 14 and 16). However, when the amide group in entry 12 was masked with an N-methyl functionality the reaction proceeded well (entry 13). This is particularly surprising considering the presence of a carbamate N–H bond within the imidazole starting material, which is tolerated in the reaction conditions.
Subsequent deprotection under TFA conditions led to the preparation of the NS5A-like analogues. To our surprise, the furan and thiophene (entries 9 and 10) were found to decompose upon exposure to TFA during the deprotection. As they were also found to be unreactive towards milder TBAF conditions, they were purified and characterised as the SEM-protected analogue instead.
Conclusions
In summary, we have described a novel, green and efficient approach to the synthesis of symmetrical NS5A-like compounds such as daclatasvir. The overall synthesis was accomplished using a novel double C–H activation as a key step that allows for a rapid synthesis of analogues and late stage structural diversification, which is to our knowledge the first time that palladium catalysed C–H activation has been used to construct symmetrical molecules in such a way. However, we found that intolerance of some co-ordinating functionalities in the reaction conditions gave some limitation to the methodology, and this must be considered when hoping to attempt this strategy.Experimental section
General procedure for C–H activated cross coupling for compound without deprotection
The appropriate aryl dibromide (0.10 mmol), (S)-tert-butyl 2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pyrrolidine-1-carboxylate (7, 100 mg, 0.240 mmol), pivalic acid (6.1 mg, 0.060 mmol), K2CO3 (41 mg, 0.30 mmol) and Pd(ddpf)Cl2 (7.3 mg, 0.010 mmol) were placed in a sealed reaction vial. The reaction vial was placed under reduced pressure then purged with nitrogen, repeating the process 5 times. The contents of the sealed vial were dissolved in dimethylacetamide (1 mL), and the solvent was deoxygenated by bubbling with nitrogen for 20 minutes. The solution was then irradiated in a microwave at 100 °C for 4 hours. The resulting solution was quenched with water (15 mL), and extracted with EtOAc (2 × 10 mL). The organic phase was washed with concentrated sodium bicarbonate solution (2 × 15 mL) water (2 × 15 mL), and brine (2 × 15 mL), before being concentrated to a solid under reduced pressure. This solid was then purified to the desired product by mass-directed auto purification.General procedure for C–H activated cross coupling and subsequent SEM-removal
The appropriate aryl dibromide (0.10 mmol), (S)-tert-butyl 2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pyrrolidine-1-carboxylate (7, 100 mg, 0.240 mmol), pivalic acid (6.1 mg, 0.060 mmol), K2CO3 (41 mg, 0.30 mmol) and Pd(ddpf)Cl2 (7.3 mg, 0.010 mmol) were placed in a sealed reaction vial. The reaction vial was placed under reduced pressure then purged with nitrogen, repeating the process 5 times. The contents of the sealed vial were dissolved in dimethylacetamide (1 mL), and the solvent was deoxygenated by bubbling with nitrogen for 20 minutes. The solution was then irradiated in a microwave at 100 °C for 4 hours. The resulting solution was quenched with water (15 mL), and extracted with EtOAc (2 × 10 mL). The organic phase was washed with concentrated sodium bicarbonate solution (2 × 15 mL) water (2 × 15 mL), and brine (2 × 15 mL), before being concentrated to a solid under reduced pressure. This solid material was then redissolved in dichloromethane (0.5 mL) and trifluoroacetic acid (0.50 mL, 6.5 mmol) was added. The solution was then stirred in inert atmosphere for 4 hours, before being diluted with dichloromethane (15 mL). The solution was then washed with concentrated sodium bicarbonate solution (2 × 30 mL), taking care due to the production of gas as the solution is neutralised. The organic phase was then concentrated to a solid under reduced pressure. This solid was then purified to the desired product by mass-directed auto purification.Acknowledgements
Thanks to Dr Iain Day for his help with NMR spectroscopy, and Dr Alaa Abdul-Sada for his help with mass spectroscopy. Thanks to all members of the SDDC who have helped support this project.Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions and Calculator Plugins were used for structure property prediction and calculation, Marvin 15.8.10.0, 2015, ChemAxon (http://www.chemaxon.com).
References
- R. S. Brown, Nature, 2005, 436, 973–978 CrossRef CAS PubMed.
- D. Lavanchy, Liver Int., 2009, 29, 74–81 CrossRef PubMed.
- T. K. H. Scheel and C. M. Rice, Nat. Med., 2013, 19, 837–849 CrossRef CAS PubMed.
- D. L. Thomas, Nat. Med., 2013, 19, 850–858 CrossRef CAS PubMed.
- F. J. Torriani, M. Rodriguez-Torres, J. K. Rockstroh, E. Lissen, J. Gonzalez-García, A. Lazzarin, G. Carosi, J. Sasadeusz, C. Katlama, J. Montaner, H. Sette, S. Passe, J. De Pamphilis, F. Duff, U. M. Schrenk and D. T. Dieterich, N. Engl. J. Med., 2004, 351, 438–450 CrossRef CAS PubMed.
- R. Bartenschlager and V. Lohmann, J. Gen. Virol., 2000, 81, 1631–1648 CrossRef CAS PubMed.
- K. L. LeMay, J. Treadaway, I. Angulo and T. L. Tellinghuisen, J. Virol., 2013, 87, 1255–1260 CrossRef CAS PubMed.
- M. Hughes, S. Griffin and M. Harris, J. Gen. Virol., 2009, 90, 1329–1334 CrossRef CAS PubMed.
- M. Belema and N. A. Meanwell, J. Med. Chem., 2014, 57, 5057–5071 CrossRef CAS PubMed.
- M. Belema, O. D. Lopez, J. A. Bender, J. L. Romine, D. R. St. Laurent, D. R. Langley, J. A. Lemm, D. R. O'Boyle, J. H. Sun, C. Wang, R. A. Fridell and N. A. Meanwell, J. Med. Chem., 2014, 57, 1643–1672 CrossRef CAS PubMed.
- M. Belema, V. N. Nguyen, C. Bachand, D. H. Deon, J. T. Goodrich, C. A. James, R. Lavoie, O. D. Lopez, A. Martel, J. L. Romine, E. H. Ruediger, L. B. Snyder, D. R. S. Laurent, F. Yang, J. Zhu, H. S. Wong, D. R. Langley, S. P. Adams, G. H. Cantor, A. Chimalakonda, A. Fura, B. M. Johnson, J. O. Knipe, D. D. Parker, K. S. Santone, R. A. Fridell, J. A. Lemm, D. R. O'Boyle, R. J. Colonno, M. Gao, N. A. Meanwell and L. G. Hamann, J. Med. Chem., 2014, 57, 2013–2032 CrossRef CAS PubMed.
- D. R. St. Laurent, M. H. Serrano-Wu, M. Belema, M. Ding, H. Fang, M. Gao, J. T. Goodrich, R. G. Krause, J. A. Lemm, M. Liu, O. D. Lopez, V. N. Nguyen, P. T. Nower, D. R. O'Boyle, B. C. Pearce, J. L. Romine, L. Valera, J. H. Sun, Y. K. Wang, F. Yang, X. Yang, N. A. Meanwell and L. B. Snyder, J. Med. Chem., 2014, 57, 1976–1994 CrossRef CAS PubMed.
- M. Belema, V. N. Nguyen, D. R. St. Laurent, O. D. Lopez, Y. Qiu, A. C. Good, P. T. Nower, L. Valera, D. R. O'Boyle, J. H. Sun, M. Liu, R. A. Fridell, J. A. Lemm, M. Gao, J. O. Knipe, N. A. Meanwell and L. B. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 4428–4435 CrossRef CAS PubMed.
- M. Belema, V. N. Nguyen, J. L. Romine, D. R. St. Laurent, O. D. Lopez, J. T. Goodrich, P. T. Nower, D. R. O'Boyle, J. A. Lemm, R. A. Fridell, M. Gao, H. Fang, R. G. Krause, Y. K. Wang, A. J. Oliver, A. C. Good, J. O. Knipe, N. A. Meanwell and L. B. Snyder, J. Med. Chem., 2014, 57, 1995–2012 CrossRef CAS PubMed.
- D. A. DeGoey, J. T. Randolph, D. Liu, J. Pratt, C. Hutchins, P. Donner, A. C. Krueger, M. Matulenko, S. Patel, C. E. Motter, L. Nelson, R. Keddy, M. Tufano, D. D. Caspi, P. Krishnan, N. Mistry, G. Koev, T. J. Reisch, R. Mondal, T. Pilot-Matias, Y. Gao, D. W. A. Beno, C. J. Maring, A. Molla, E. Dumas, A. Campbell, L. Williams, C. Collins, R. Wagner and W. M. Kati, J. Med. Chem., 2014, 57, 2047–2057 CrossRef CAS PubMed.
- O. D. Lopez, V. N. Nguyen, D. R. St. Laurent, M. Belema, M. H. Serrano-Wu, J. T. Goodrich, F. Yang, Y. Qiu, A. S. Ripka, P. T. Nower, L. Valera, M. Liu, D. R. O'Boyle, J.-H. Sun, R. A. Fridell, J. A. Lemm, M. Gao, A. C. Good, N. A. Meanwell and L. B. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 779–784 CrossRef CAS PubMed.
- I. K. Mangion, C. Chen, H. Li, P. Maligres, Y. Chen, M. Christensen, R. Cohen, I. Jeon, A. Klapars, S. Krska, H. Nguyen, R. A. Reamer, B. D. Sherry and I. Zavialov, Org. Lett., 2014, 16, 2310–2313 CrossRef CAS PubMed.
- F. Wakenhut, T. D. Tran, C. Pickford, S. Shaw, M. Westby, C. Smith-Burchnell, L. Watson, M. Paradowski, J. Milbank, D. Stonehouse, K. Cheung, R. Wybrow, F. Daverio, S. Crook, K. Statham, D. Leese, D. Stead, F. Adam, D. Hay, L. R. Roberts, J.-Y. Chiva, C. Nichols, D. C. Blakemore, G. H. Goetz, Y. Che, I. Gardner, S. Dayal, A. Pike, R. Webster and D. C. Pryde, ChemMedChem, 2014, 9, 1387–1396 CrossRef CAS PubMed.
- M. Zhong, E. Peng, N. Huang, Q. Huang, A. Huq, M. Lau, R. Colonno and L. Li, Bioorg. Med. Chem. Lett., 2014, 24, 5738–5742 CrossRef CAS PubMed.
- T. D. Tran, F. Wakenhut, C. Pickford, S. Shaw, M. Westby, C. Smith-Burchnell, L. Watson, M. Paradowski, J. Milbank, R. A. Brimage, R. Halstead, R. Glen, C. P. Wilson, F. Adam, D. Hay, J.-Y. Chiva, C. Nichols, D. C. Blakemore, I. Gardner, S. Dayal, A. Pike, R. Webster and D. C. Pryde, ChemMedChem, 2014, 9, 1378–1386 CrossRef CAS PubMed.
- F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, J. Org. Chem., 2005, 70, 3997–4005 CrossRef CAS PubMed.
- F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, Eur. J. Org. Chem., 2006, 693–703 CrossRef CAS.
- F. Bellina, S. Cauteruccio, A. Di Fiore and R. Rossi, Eur. J. Org. Chem., 2008, 5436–5445 CrossRef CAS.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed.
- R. Goikhman, T. L. Jacques and D. Sames, J. Am. Chem. Soc., 2009, 131, 3042–3048 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- J. M. Joo, B. B. Touré and D. Sames, J. Org. Chem., 2010, 75, 4911–4920 CrossRef CAS PubMed.
- I. H. Bae, J. K. Choi, C. Chough, S. J. Keum, H. Kim, S. K. Jang and B. M. Kim, ACS Med. Chem. Lett., 2014, 5, 255–258 CrossRef CAS PubMed.
- J. C. Sauer, J. Am. Chem. Soc., 1947, 69, 2444–2448 CrossRef CAS.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00340k |
| This journal is © The Royal Society of Chemistry 2016 |