
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cyclic 1,3-diols as scaffolds for spatially directed libraries†
Gurpreet
Singh
and
Jeffrey
Aubé‡
*
The University of Kansas Chemical Methodologies and Library Development Center, 2034 Becker Drive, Del Shankel Structural Biology Center, Lawrence, Kansas 66047, United States. E-mail: jaube@unc.edu
First published on 12th April 2016
Abstract
A useful design element in small molecule libraries is spatial diversity, in which binding moieties are systematically directed toward different regions of three-dimensional space. One way of achieving this is through the use of conformationally diverse scaffolds onto which various binding moieties can be placed. Such scaffolds can represent synthetic challenges of their own. In this paper, we describe a new route to chiral, cyclic 1,3-diol building blocks that features silylated dithianes as relay linchpins. These diols are subsequently used for the construction of a 24-compound pilot library bearing two amino acid residues.
Introduction
It is well appreciated that bioactivity of drug or probe molecules can strongly depend on the conformational state of the compound of interest. Accordingly, careful consideration of stereochemical and conformational issues can enhance the utility of small-molecule libraries for broad screening. Aside from the obvious possibility that only some isomers of a given structure might be sufficiently potent to register as a hit in a high-throughput screen, some authors have suggested that high sp3 content in target compounds is positively correlated with progression through the drug discovery pipeline.1Although early combinatorial libraries were based on peptides and therefore intrinsically stereochemically rich, the development of the next generation of libraries depended strongly on available chemistry suitable for parallel synthesis, which often led to an over-emphasis on achiral compounds.2 Subsequently, various approaches to creating libraries with a strong emphasis on stereochemical diversity have appeared. One is to use chiral natural product templates, which more often than not are generated from naturally occurring sources.3 For fully synthetic libraries constructed under the rubric of diversity-oriented synthesis, the use of chiral building blocks is a hallmark of strategies such as “build/couple/pair”,4 “click/click/cyclize”,5 and by systematically varying the configuration and hybridization of a key stereocenter.6
Recently, we published an approach toward small-molecule libraries based on the stereochemical and conformational diversification of a piperidine template (Fig. 1).7 In this work, 1,3-allylic strain was used both to control substitution patterns in the central ring and to impose a degree of conformational diversity on the final products. In this paper, we disclose a simpler approach in which 1,3-diols attached to 5- and 6-membered rings serve as attachment points for chemical diversity (here, amino acid-based substituents) and the standard conformational tendencies of the rings are used to provide a modest degree of conformational flexibility (Fig. 1b). For example, substituents are displayed via dramatically different vectors when attached to cis vs. trans 1,3-disubstituted cyclopentenes; a further degree of diversity is obtained from the conformational mobility of rings having particular substitution patterns.
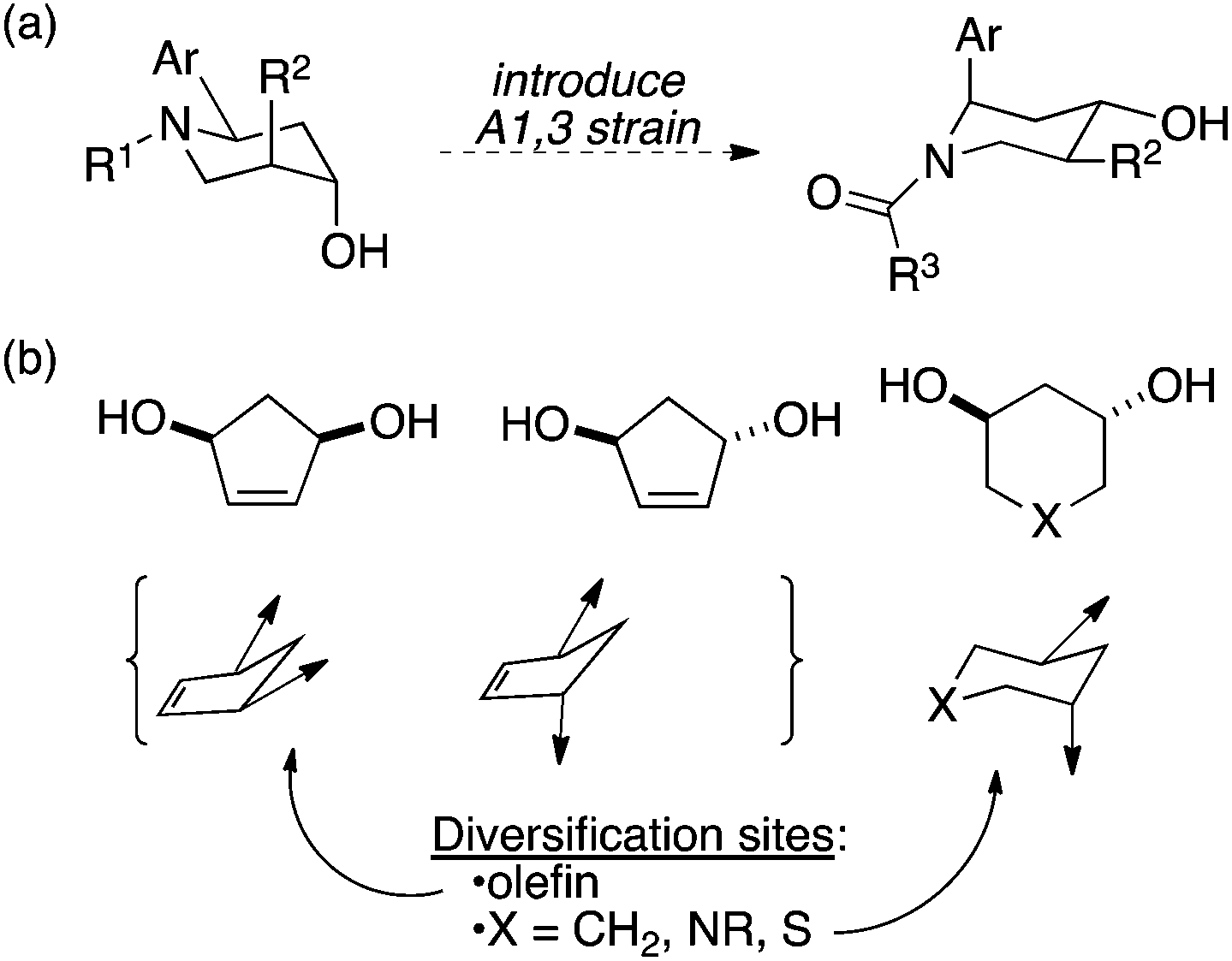 | ||
| Fig. 1 (a) Use of 1,3-allylic strain to establish conformational diversity in a small molecule library.7 (b) This work: conformational diversity encouraged by placing groups onto cyclic 1,3-diols. | ||
These rings also permit the incorporation of additional diversity elements. For example, the presence of a double bond in the cyclopentene-based scaffold serves as a potential site for additional hydroxyl groups or transformation into acyclic scaffolds using ring-opening metathesis, while the 6-membered rings can be substituted by carbon, sulfur, or nitrogen, the last of which is a natural site for substitution as well. In this paper, we describe (1) the synthesis of diols suitable for the display of binding moieties, including a new route to chiral, cyclic 1,3-diol building blocks of general utility in synthesis and (2) the construction of a pilot library that displays two amino acid residues through these linkers.
Results and discussion
Synthesis of chiral scaffolds
The necessary building blocks were prepared in enantiomerically pure form. The four stereoisomers of mono-TIPS protected cyclopentenediol 3 were prepared from 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 by the straightforward modification of our previously published route to 4-silyloxy-cyclopenten-2-ones (Scheme 1a).9 The diols were prepared and used as TIPS derivatives to ensure stepwise substitution of the cyclopentenediols by diversifying groups. The conversion of trans-(S,S)-3 to the corresponding cis isomer was accomplished by a Mitsunobu reaction, which generates an interesting molecule that is chiral solely by virtue of the placement of a protecting group on what would otherwise be a meso diol. This represents an unusually valuable use of a protecting group and molecules like (1R,4S)-3 may well find other applications in synthesis. Here, the protecting/desymmetrizing group helps to control stepwise substitution in the library synthesis (see below).
8 by the straightforward modification of our previously published route to 4-silyloxy-cyclopenten-2-ones (Scheme 1a).9 The diols were prepared and used as TIPS derivatives to ensure stepwise substitution of the cyclopentenediols by diversifying groups. The conversion of trans-(S,S)-3 to the corresponding cis isomer was accomplished by a Mitsunobu reaction, which generates an interesting molecule that is chiral solely by virtue of the placement of a protecting group on what would otherwise be a meso diol. This represents an unusually valuable use of a protecting group and molecules like (1R,4S)-3 may well find other applications in synthesis. Here, the protecting/desymmetrizing group helps to control stepwise substitution in the library synthesis (see below).
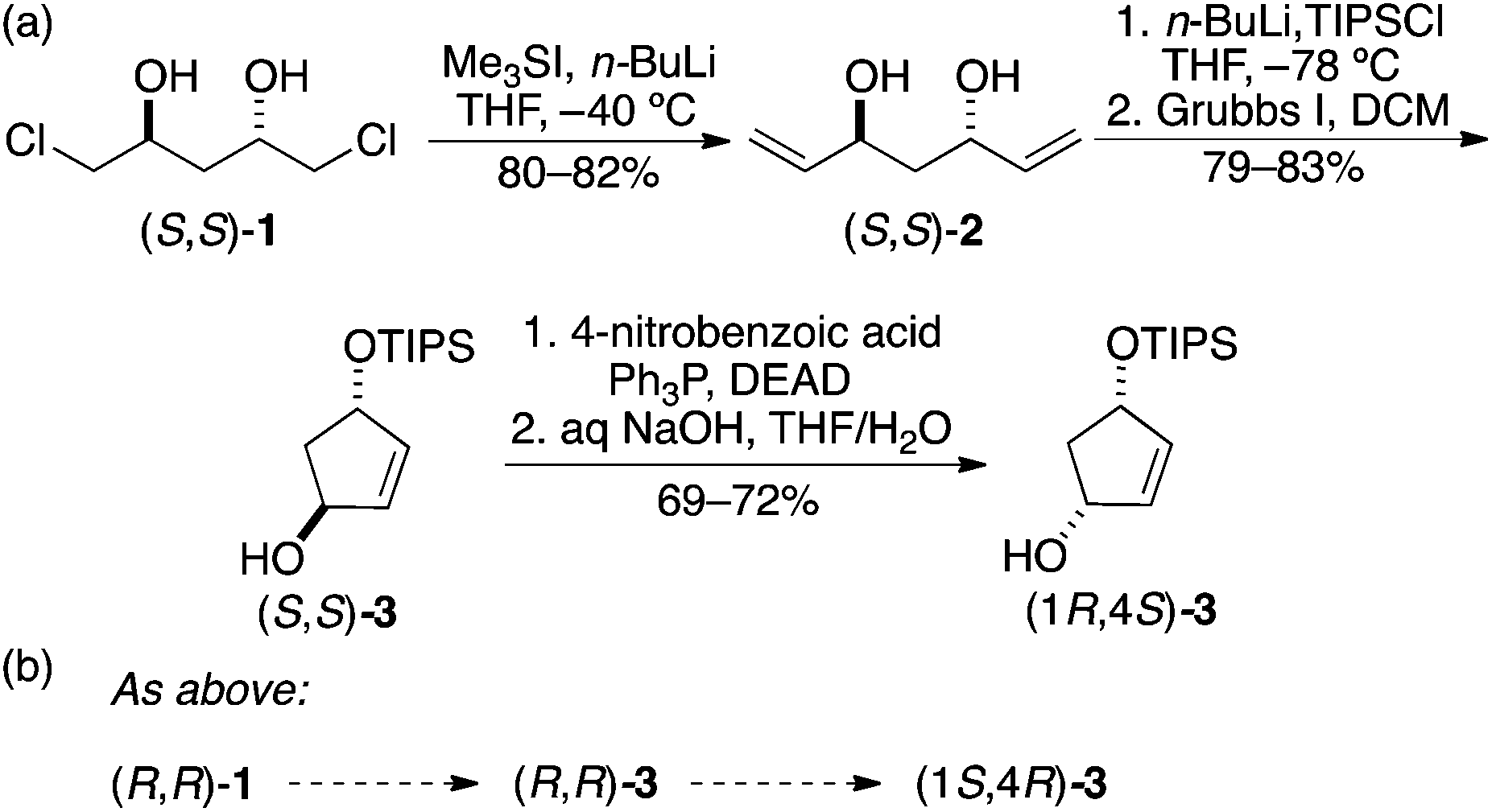 | ||
| Scheme 1 Formation of (a) (1R,4S)-3 and (b) the enantiomeric series. | ||
Although cyclohexane-1,-3-diols have been used as precursors in the syntheses of biologically important natural products and pharmaceutically important compounds,10 most of their reported syntheses result in moderate yield and selectivities.11 Enantioenriched synthesis of these diols has also been targeted by different groups however involve multi-step synthesis and one or more enzymatic desymmetrization steps.12 Similarly, there are only a few approaches to the synthesis of 3,5-dihydroxy piperidines. In 2006, Cossy reported a synthesis of enantioenriched 3,5-dihydroxy piperidine via ring-expansion of 4-hydroxyproline,13 and Bäckvall reported a combined enzymatic/metallocatalysis route to related materials.14
We considered that one convenient approach would be to subject compound 115 to dual displacement of the two chlorine atoms by a one-atom, doubly-activated, nucleophile (Fig. 2). A clue for accomplishing this was provided by the work of Linclau, who used a dithiane-based linchpin for the preparation of carbonucleosides.16 Such relay approaches have been largely popularized by Smith,17 (who used this chemistry for the preparation of piperidines in a diversity-oriented synthesis context18). The primary challenge in adapting this route to our present purpose was to subvert the dominant regiochemistry of the Linclau route, which proceeded according to the Baldwin paradigm to afford a five-membered ring with >10:1 selectivity.19 We felt that this would be possible were we able to prevent epoxide formation prior to the second nucleophilic (i.e., cyclization) attack, which we anticipated accomplishing through protection. Success also depended upon the initial attack occurring at the epoxide and not the chloride atom.
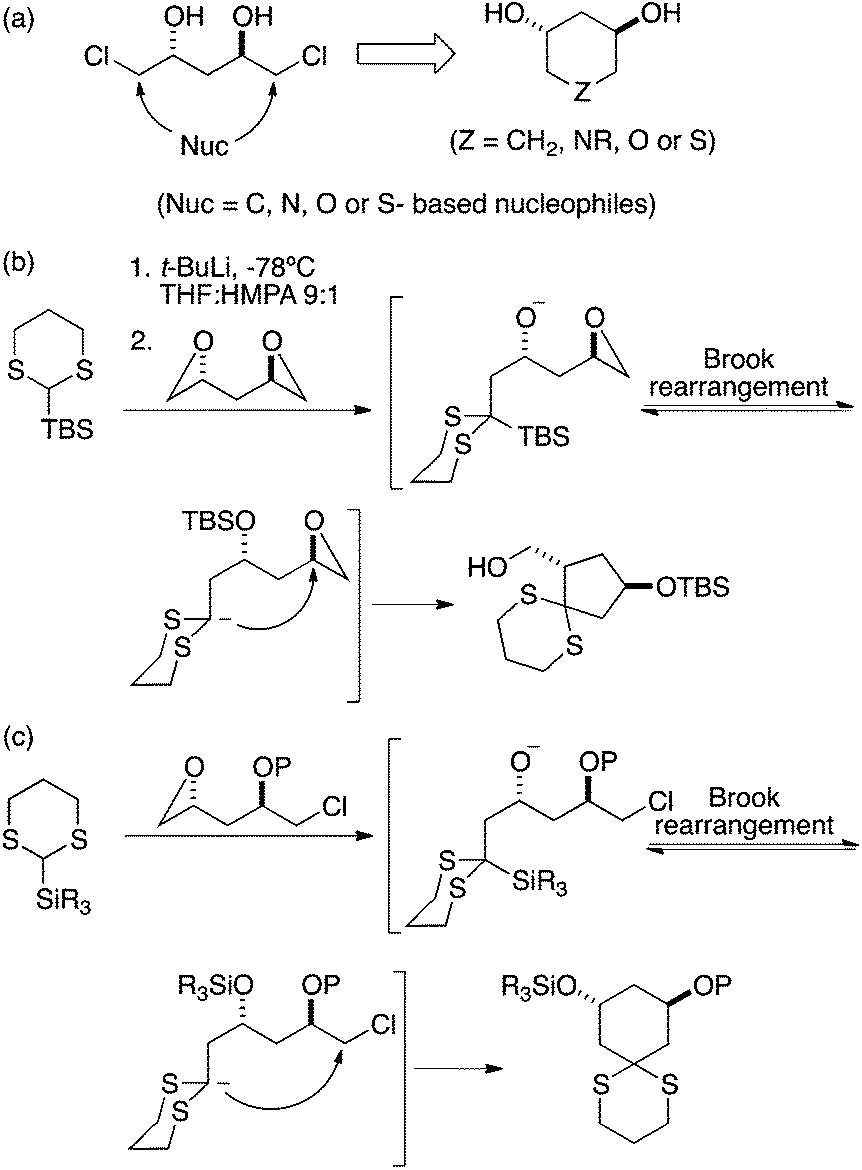 | ||
| Fig. 2 (a) Plan for making chiral six-membered ring trans diols. (b) Linclau's route to chiral cyclopentanes using a dithiane linchpin approach.16 (c) Proposed modification to direct the relay route toward six-membered ring formation. | ||
This plan worked as expected (Scheme 2). Addition of 1 equiv. of n-BuLi to a precooled solution of dichlorodiol (R,R)-1 in THF followed by the addition of 1 equiv. of TIPSCl yielded the mono silylated product 4; interestingly, no epoxide formation was observed under these conditions. The remaining chlorohydrin was converted to epoxide in excellent yield using powdered potassium hydroxide in anhydrous diethyl ether. Lithiation of 2-TBS-dithiane was carried out at room temperature in a 9:1 mixture of THF/HMPA using n-BuLi as base. The resulting yellow solution was added to a pre-cooled solution of the epoxide (1R,3R)-5 in 9:1 THF/HMPA at −78 °C. The reaction mixture was maintained at −78 °C for 0.5 h and then raised to −40 °C for 2 h. Consumption of the starting material was observed by TLC at −40 °C, indicating opening of the epoxide. Further allowing the reaction to stir at rt for 18 h formed the cyclized product 6 in 88% yield. The disilyl cyclohexanediol (8R,10R)-6 was treated with 1 M TBAF in THF to produce C2-symmetric 1,3-cyclohexanediol (R,R)-7 in 94% yield.
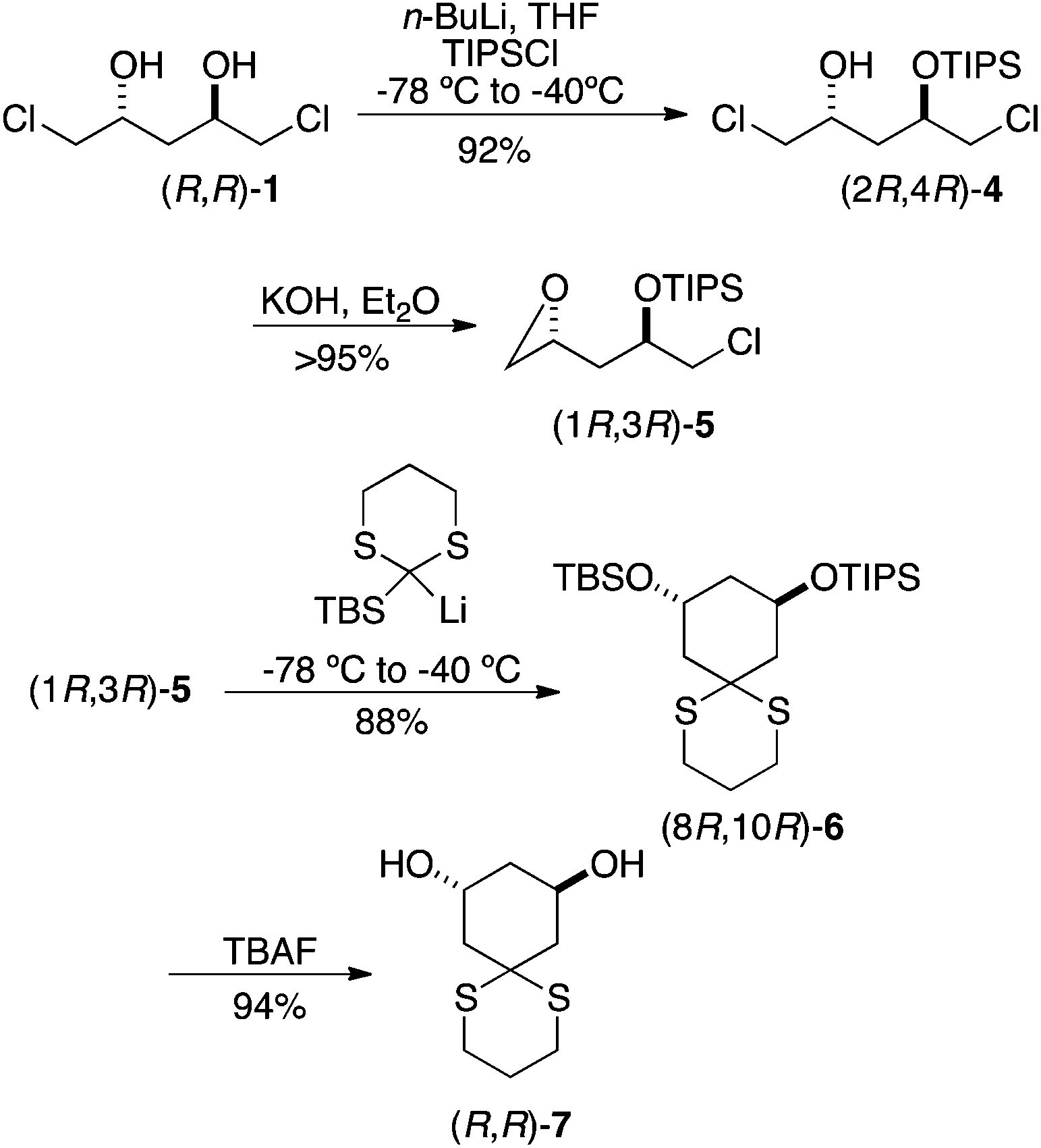 | ||
| Scheme 2 Formation of chiral cyclohexane-1,3-diols. | ||
A similar protocol was applied to afford mono-protected diols by replacing the linchpin reagent with TMS-dithiane and selective removal of the more labile TMS ether, which occurred spontaneously upon conversion of 8 to either alkane 9 or ketone 10 (Scheme 3). Having previously encountered unexpected difficulties in monosilylation of symmetrical diols,9 this was viewed as an attractive entry into these monoprotected building blocks for library synthesis. As demanded by parity conservation, application of these reaction sequence beginning with (S,S)-1 led to the enantiomers of these building blocks (not shown).
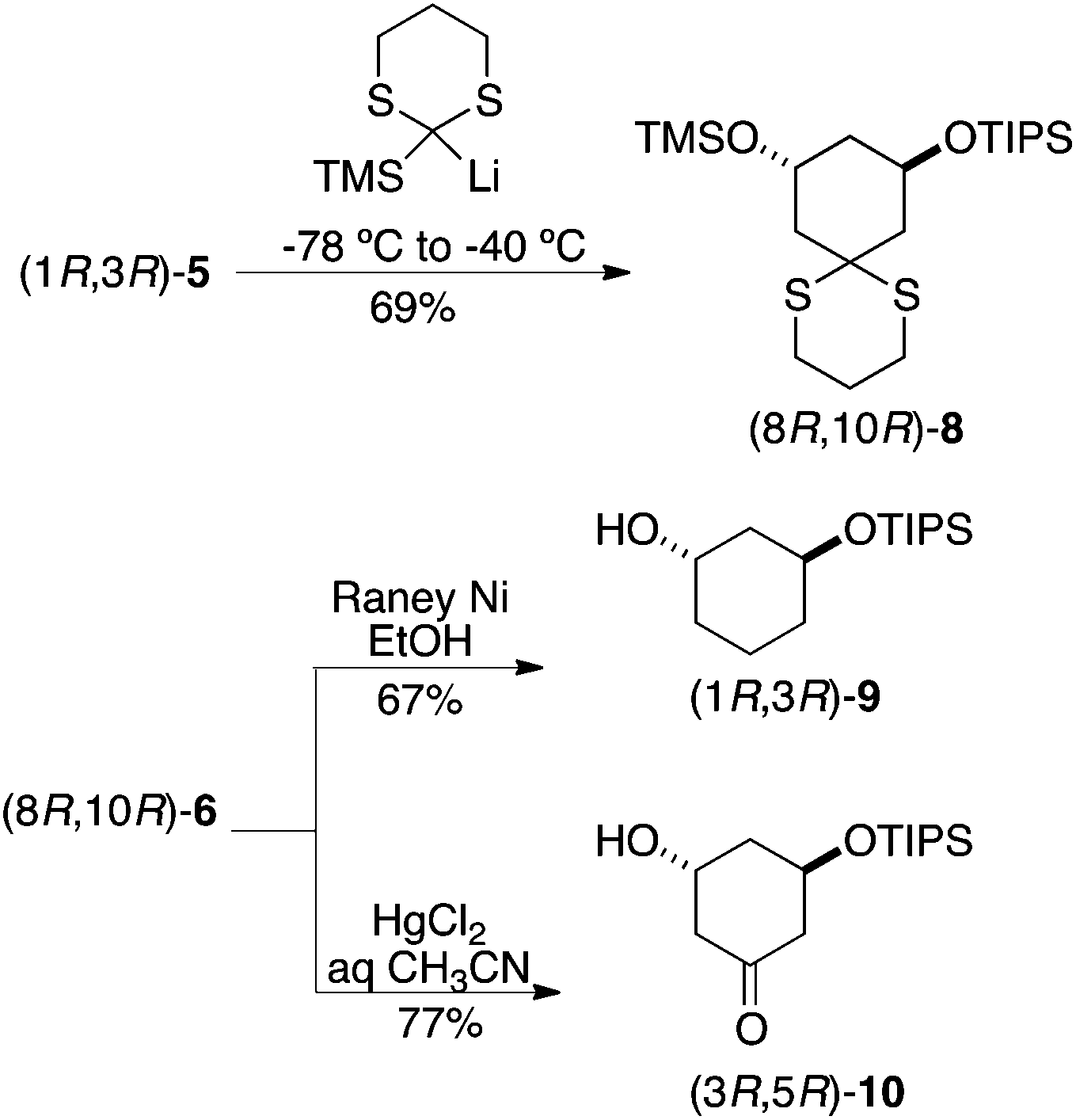 | ||
| Scheme 3 Application to unsymmetrically substituted cyclohexane-1,3-diols. | ||
Application of the above strategy to heterocycles synthesis did not require formation and isolation of a precursor epoxide (although it is very likely that one is formed in situ), but did benefit from conversion of the dichloride bis-electrophile to the corresponding diiodide and carrying out the reactions at ca. 100 °C in EtOH for 16 h. As shown in Scheme 4, enantiopure monoprotected or unprotected 3,5-dihydroxypiperidines and -thianes were obtained from common intermediates 4. As above, either enantiomeric series may be obtained in this way.
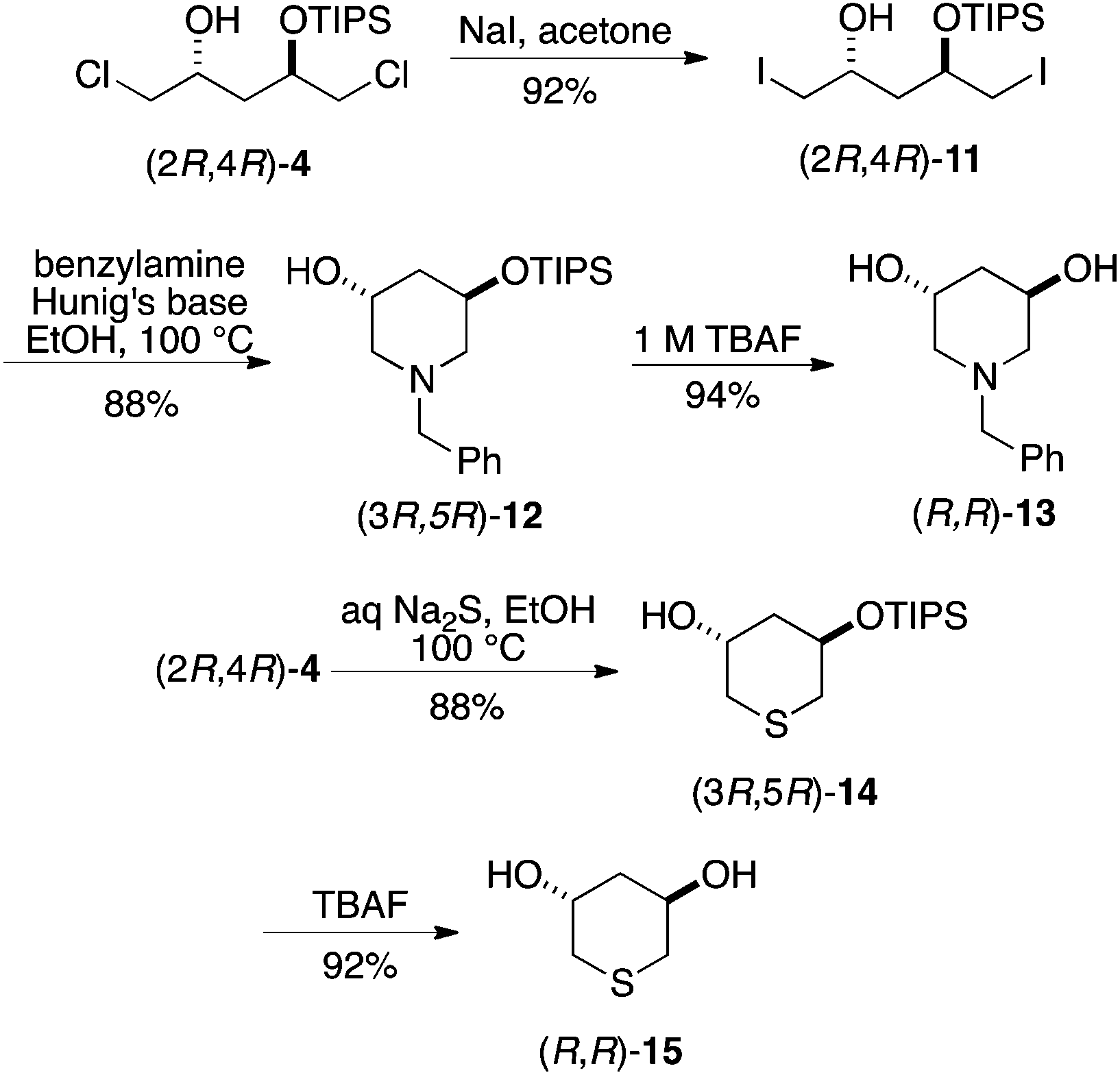 | ||
| Scheme 4 Application to piperidine and thiane synthesis. | ||
Pilot library construction
The utility of the above building blocks was explored in the context of a library in which a given diol would be substituted by a range of amino acids through a carbonate linker, chosen for a good blend of synthetic accessibility and stability under ordinary assay conditions. The virtues of using a monoprotected diol are that (1) it is possible to enhance yields and avoid doubly modified diols in the first reaction step and (2) one is able to avoid stereoisomeric mixtures that would be generated attachment of two different chiral amino acids to a meso centroid. We chose a selection of 4 amino acids for the initial coupling step (alanine, arginine, tryptophan, and proline), 6 for the second coupling (phenylalanine, tert-butyl serine, lysine, methionine, tert-butyl tyrosine, and tert-butyl glutamic acid), and (R,R)-3 for this initial library, targeting a total of 24 compounds.Following some experimentation, it was determined that the best route for conversion of the open alcohol to a carbamate was via a p-nitro carbonate, which was readily synthesized as shown in Scheme 5. Thereafter, 16 was treated with a set of amino acid esters in the presence of catalytic amount of DMAP in THF at 100 °C to afford 17a–d in 79–83% yield. These carbamates were isolated on 600–700 mg scale and fully characterized.
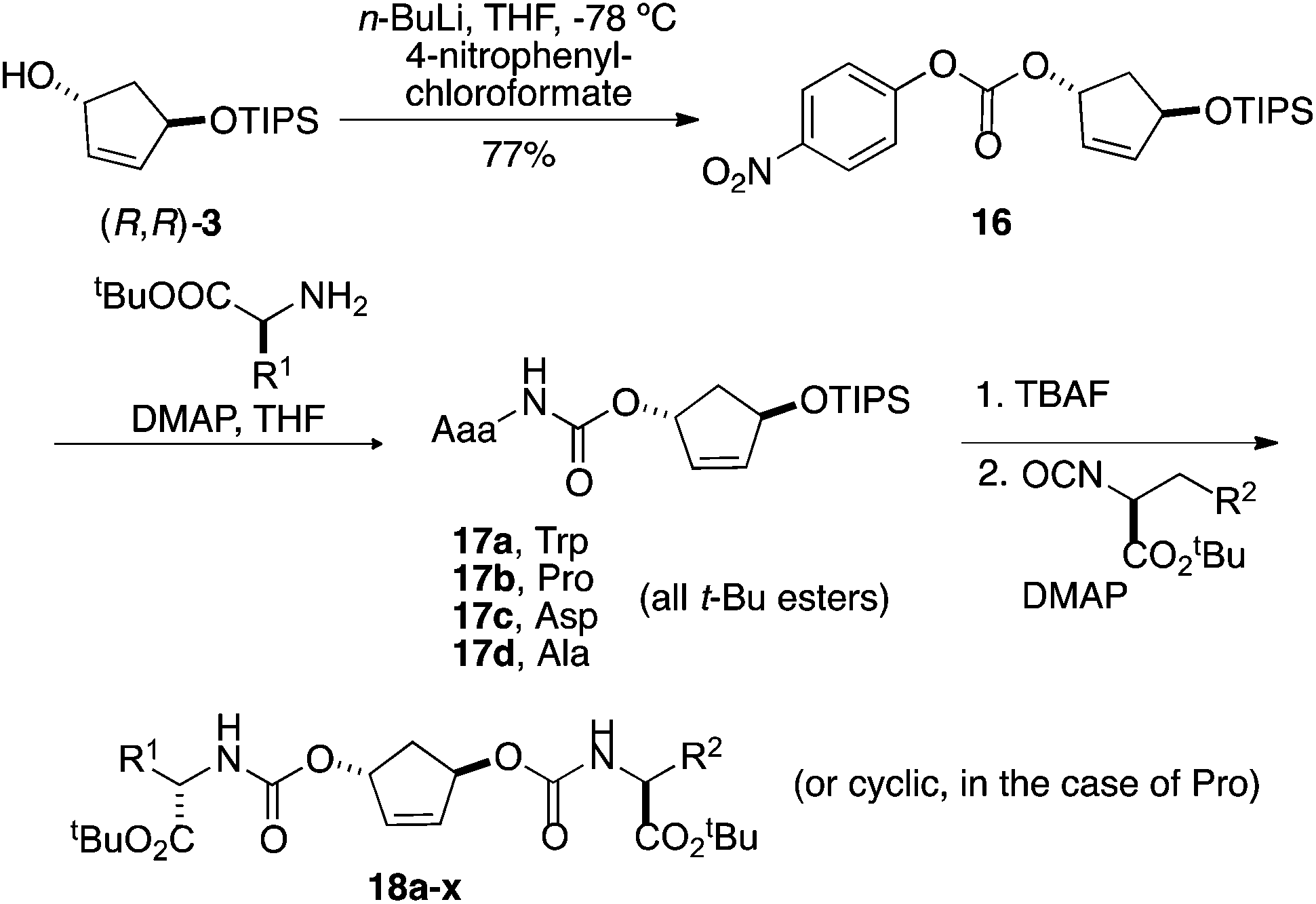 | ||
| Scheme 5 Preparation of pilot library. | ||
The silyl carbamates 17 were deprotected using TBAF and the resulting crude alcohols treated with amino acid isocyanate. Initial attempts, in which isocyanate was reacted with alcohol in refluxing toluene for 48 h, were very slow and left a significant amount of starting material. Ultimately, reacting alcohols with excess of neat isocyanates (3 equiv.) in the presence of catalytic amount of N,N-dimethylaminopyridine at 100 °C led to successful completion of the reactions. To complete the construction of this demonstration library, alcohols 17a–d were reacted with six different amino acid isocyanates in two-dram vials on a 24-well heating block. The reaction mixture was cooled to room temperature and was dissolved in dichloromethane. The crude reaction mixtures were subjected to solid-phase extraction to remove unreacted isocyanates followed by mass-directed automated purification, resulting in 18 biscarbamates in 5–71 mg quantities and purities of >90%. The purification results of the 24 compounds are listed in the Table 1. No obvious trends regarding success in this library format are evident from these results.
| Entry | Compound | R1 | R2 | Amount (mg) | Purity (%) |
|---|---|---|---|---|---|
| 1 | 18a | 3-Indolyl-ylmethyl | PhCH2 | 24 | 100 |
| 2 | 18b | 3-Indolyl-ylmethyl | t BuOCH2 | 62 | 98 |
| 3 | 18c | 3-Indolyl-ylmethyl | (CH3)2CHCH2 | 34 | 94 |
| 4 | 18d | 3-Indolyl-ylmethyl | CH3SCH2CH2 | 25 | 79 |
| 5 | 18e | 3-Indolyl-ylmethyl | t BuO2CCH2CH2 | 54 | 99 |
| 6 | 18f | 3-Indolyl-ylmethyl | p-tBuOPhCH2 | 25 | 92 |
| 7 | 18g | H2NCOCH2 | PhCH2 | 27 | 100 |
| 8 | 18h | H2NCOCH2 | t BuOCH2 | 44 | 98 |
| 9 | 18i | H2NCOCH2 | (CH3)2CHCH2 | 39 | 39 |
| 10 | 18j | H2NCOCH2 | CH3SCH2CH2 | 26 | 100 |
| 11 | 18k | H2NCOCH2 | t BuO2CCH2CH2 | 66 | 98 |
| 12 | 18l | H2NCOCH2 | p-tBuOPhCH2 | 22 | 39 |
| 13 | 18m | L-Proline | PhCH2 | 34 | 100 |
| 14 | 18n | L-Proline | t BuOCH2 | 10 | 98 |
| 15 | 18o | L-Proline | (CH3)2CHCH2 | 18 | 97 |
| 16 | 18p | L-Proline | CH3SCH2CH2 | 71 | 100 |
| 17 | 18q | L-Proline | t BuO2CCH2CH2 | 25 | 56 |
| 18 | 18r | L-Proline | p-tBuOPhCH2 | 34 | 98 |
| 19 | 18s | CH3 | PhCH2 | 5 | 78 |
| 20 | 18t | CH3 | t BuOCH2 | 27 | 96 |
| 21 | 18u | CH3 | (CH3)2CHCH2 | 29 | 41 |
| 22 | 18v | CH3 | CH3SCH2CH2 | 39 | 98 |
| 23 | 18w | CH3 | t BuO2CCH2CH2 | 33 | 92 |
| 24 | 18x | CH3 | p-tBuOPhCH2 | 38 | 65 |
Finally, a few examples of C2-symmetrical analogs were created directly from the corresponding fully unprotected diols (Scheme 6). Thus 19 was synthesized directly from (S,S)-2 using ring-closing metathesis and 20 by deprotecting (1S,3S)-9, and (S,S)-13 and (S,S)-15 as shown in Scheme 4. Individual diols were then directly reacted with 3 equiv. of isocyanate to afford the depicted derivatives in moderate yields following chromatography.
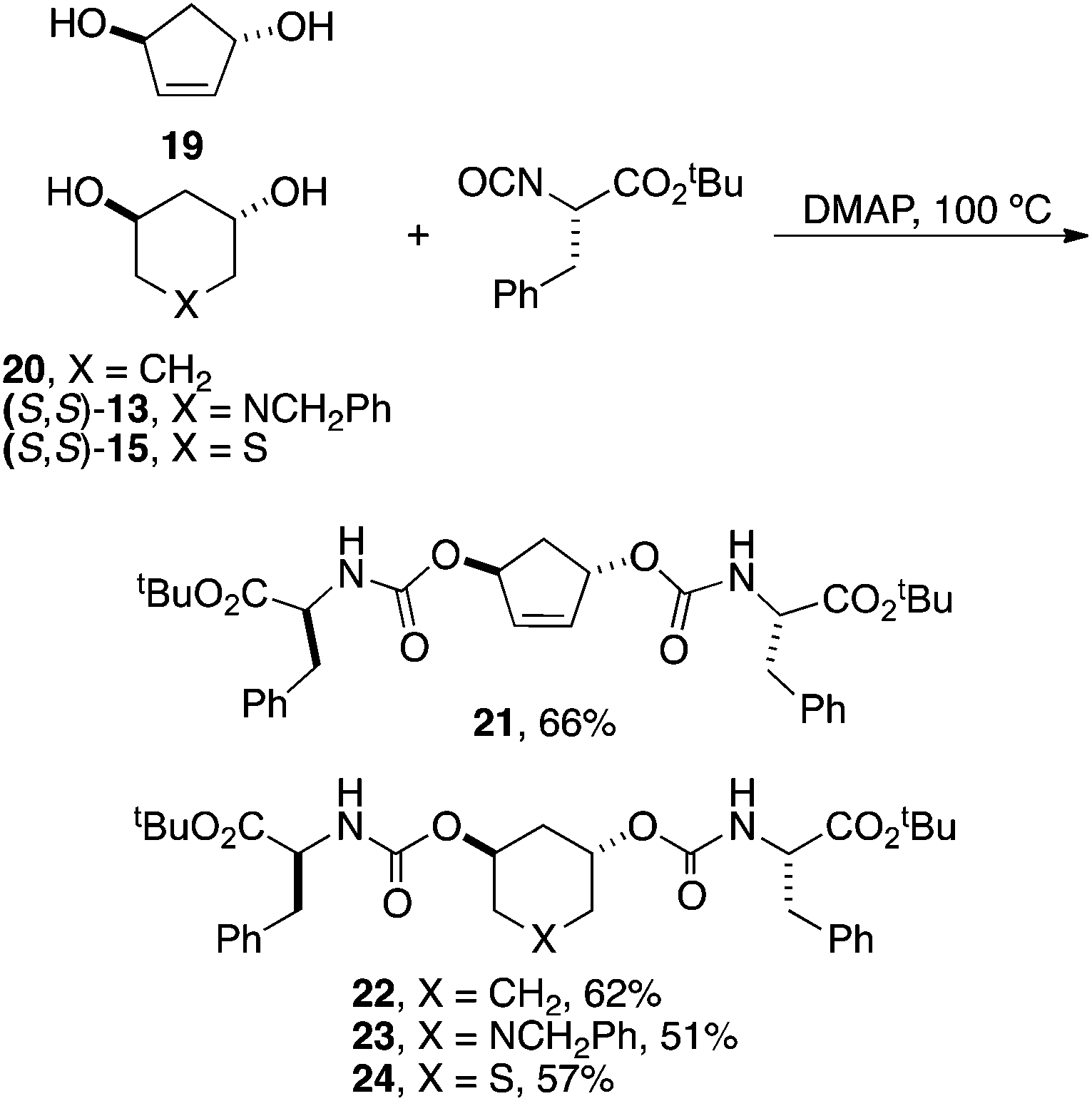 | ||
| Scheme 6 Synthesis of C2-symmetric derivatives. | ||
Conclusions
Desired elements of small molecule library construction include easy chemistry that may be scaled up without undue experimental cautions and easy access to building blocks. In the present case, we sought to address these needs by developing routes to straightforward-looking diols that nonetheless are non-trivial to make by other means. These routes to 5- and 6-membered ring diols all emanate from the common precursor 1, which is easily made in either enantiomeric form via Noyori reduction of the corresponding ketone on multigram scale. Moreover, the strategic use of alcohol protection enhances the suitability of these building blocks in library synthesis. These simple building blocks may also find other applications in organic chemistry. Finally, the potential utilization of these scaffolds in library synthesis is demonstrated through the preparation of 22 amino-acid-decorated versions of the scaffolds, which sets the stage for full library construction and downstream validation via biological screening.Acknowledgements
We thank Patrick Porubsky for carrying out mass-directed fractionation of pilot library compounds. This work was supported by the National Institute of General Medical Sciences through P41GM089154.Notes and references
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- J. Aubé, S. A. Rogers and C. Santini, in Comprehensive Organic Synthesis, ed. G. A. Molander and P. Knochel, Elsevier, Oxford, 2nd edn, 2014, vol. 9, pp. 1–27 Search PubMed.
- (a) U. Abel, C. Koch, M. Speitling and G. Hansske Friedrich, Curr. Opin. Chem. Biol., 2002, 6, 453–458 CrossRef CAS PubMed; (b) R. A. Bauer, J. M. Wurst and D. S. Tan, Curr. Opin. Chem. Biol., 2010, 14, 308–314 CrossRef CAS PubMed.
- (a) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS PubMed; (b) S. Dandapani and L. A. Marcaurelle, Curr. Opin. Chem. Biol., 2010, 14, 362–370 CrossRef CAS PubMed; (c) L. A. Marcaurelle, E. Comer, S. Dandapani, J. R. Duvall, B. Gerard, S. Kesavan, M. D. Lee IV, H. Liu, J. T. Lowe and J.-C. Marie, J. Am. Chem. Soc., 2010, 132, 16962–16976 CrossRef PubMed; (d) M. E. Fitzgerald, C. A. Mulrooney, J. R. Duvall, J. Wei, B.-C. Suh, L. B. Akella, A. Vrcic and L. A. Marcaurelle, ACS Comb. Sci., 2012, 14, 89–96 CrossRef PubMed.
- (a) A. Zhou, D. Rayabarapu and P. R. Hanson, Org. Lett., 2008, 11, 531–534 CrossRef PubMed; (b) A. Rolfe, G. H. Lushington and P. R. Hanson, Org. Biomol. Chem., 2010, 8, 2198–2203 RSC.
- (a) S. Kumar, P. D. Thornton and C. Santini, ACS Comb. Sci., 2013, 15, 564–571 CrossRef CAS PubMed; (b) T. O. Painter, J. R. Bunn, F. J. Schoenen, J. T. Douglas, V. W. Day and C. Santini, J. Org. Chem., 2013, 78, 3720–3730 CrossRef CAS PubMed.
- T. C. Coombs, G. H. Lushington, J. Douglas and J. Aubé, Angew. Chem., Int. Ed., 2011, 50, 2734–2737 CrossRef CAS PubMed.
- A. Whitehead, M. D. McReynolds, J. D. Moore and P. R. Hanson, Org. Lett., 2005, 7, 3375–3378 CrossRef CAS PubMed.
- G. Singh, A. Meyer and J. Aube, J. Org. Chem., 2014, 79, 452–458 CrossRef CAS PubMed.
- (a) K. Prasad and O. Repic, Tetrahedron Lett., 1984, 25, 2435–2438 CrossRef CAS; (b) R. R. Sicinski and H. F. DeLuca, Bioorg. Med. Chem. Lett., 1995, 5, 899–904 CrossRef CAS; (c) T. Honda, K. Endo and S. Ono, Chem. Pharm. Bull., 2000, 48, 1545–1548 CrossRef CAS PubMed; (d) R. Hayashi, S. Fernandez and W. H. Okamura, Org. Lett., 2002, 4, 851–854 CrossRef CAS PubMed; (e) K. C. Nicolaou, Y. H. Lim, J. L. Piper and C. D. Papageorgiou, J. Am. Chem. Soc., 2007, 129, 4001–4013 CrossRef CAS PubMed; (f) K. Sokolowska, D. Carballa, S. Seoane, R. Perez-Fernandez, A. Mourino and R. R. Sicinski, J. Org. Chem., 2015, 80, 165–173 CrossRef CAS PubMed.
- (a) K. Tamao, T. Nakajima, R. Sumiya, H. Arai, N. Higuchi and Y. Ito, J. Am. Chem. Soc., 1986, 108, 6090–6093 CrossRef CAS PubMed; (b) D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS; (c) A. Mattson, N. Oehrner, K. Hult and T. Norin, Tetrahedron: Asymmetry, 1993, 4, 925–930 CrossRef CAS; (d) A. Mattson, C. Orrenius, N. Oehrner, C. R. Unelius, K. Hult and T. Norin, Acta Chem. Scand., 1996, 50, 918–921 CrossRef CAS; (e) T. Hirata, S. Izumi, M. Aoki, S. Gotoh and R. Utsumi, Chirality, 1997, 9, 250–253 CrossRef CAS; (f) B. A. Persson, F. F. Huerta and J.-E. Bäckvall, J. Org. Chem., 1999, 64, 5237–5240 CrossRef CAS; (g) T. Kawabata, R. Stragies, T. Fukaya, Y. Nagaoka, H. Schedel and K. Fuji, Tetrahedron Lett., 2003, 44, 1545–1548 CrossRef CAS.
- (a) H. Suemune, M. Takahashi, S. Maeda, Z. F. Xie and K. Sakai, Tetrahedron: Asymmetry, 1990, 1, 425–428 CrossRef CAS; (b) K. Kato, H. Suemune and K. Sakai, Tetrahedron Lett., 1992, 33, 3481–3482 CrossRef CAS; (c) U. R. Kalkote, S. R. Ghorpade, S. P. Chavan and T. Ravindranathan, J. Org. Chem., 2001, 66, 8277–8281 CrossRef CAS PubMed.
- R. Roudeau, D. Gomez Pardo and J. Cossy, Tetrahedron, 2006, 62, 2388–2394 CrossRef CAS.
- B. Olofsson, K. Bogar, L. Fransson Ann-Britt and J. Bäckvall, J. Org. Chem., 2006, 71, 8256–8260 CrossRef CAS PubMed.
- (a) S. D. Rychnovsky, G. Griesgraber and J. P. Powers, Org. Synth., 2000, 77, 1–11 CrossRef CAS; (b) S. D. Rychnovsky, G. Griesgraber, S. Zeller and D. J. Skalitzky, J. Org. Chem., 1991, 56, 5161–5169 CrossRef CAS.
- (a) L. M. H. Leung, A. J. Boydell, V. Gibson, M. E. Light and B. Linclau, Org. Lett., 2005, 7, 5183–5186 CrossRef CAS PubMed; (b) L. M. H. Leung, V. Gibson and B. Linclau, J. Org. Chem., 2008, 73, 9197–9206 CrossRef CAS PubMed.
- A. B. Smith and C. M. Adams, Acc. Chem. Res., 2004, 37, 365–377 CrossRef CAS PubMed.
- A. B. Smith III, H. Han and W.-S. Kim, Org. Lett., 2011, 13, 3328–3331 CrossRef PubMed.
- (a) G. Stork and J. F. Cohen, J. Am. Chem. Soc., 1974, 96, 5270–5272 CrossRef CAS; (b) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, spectra, and chromatographs of selected compounds. See DOI: 10.1039/c6ob00598e |
| ‡ Current address: UNC Eshelman School of Pharmacy, 125 Mason Farm Road, CB 7363, University of North Carolina, NC, 27517 USA. |
| This journal is © The Royal Society of Chemistry 2016 |