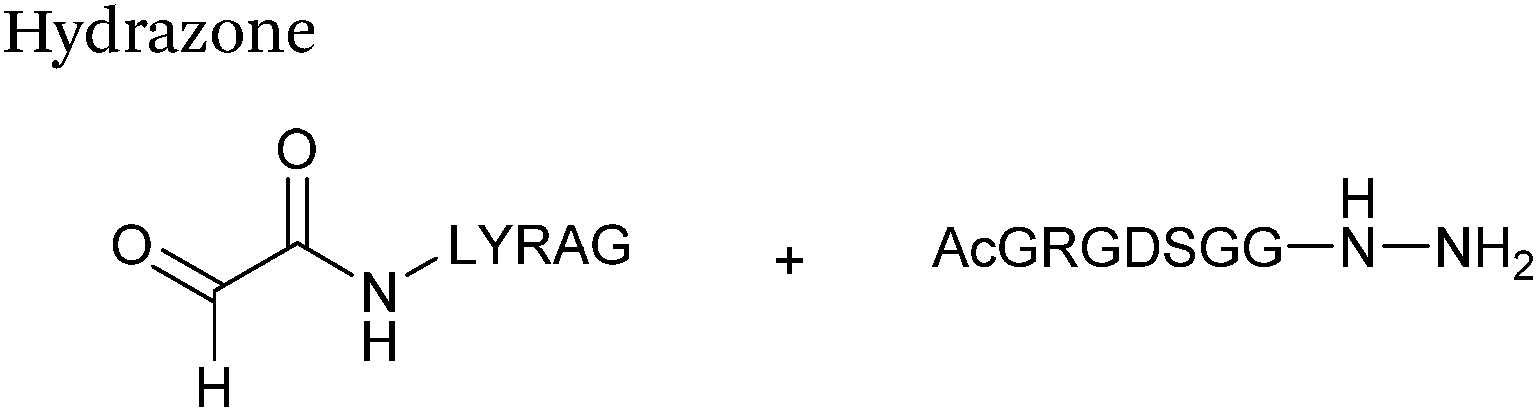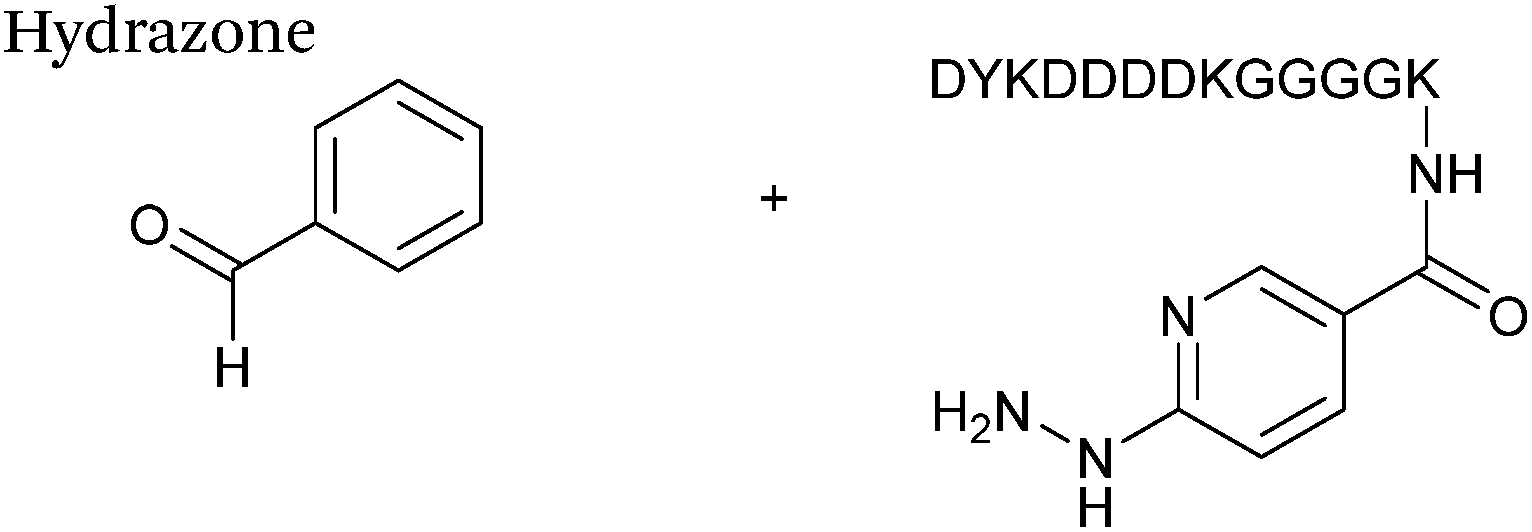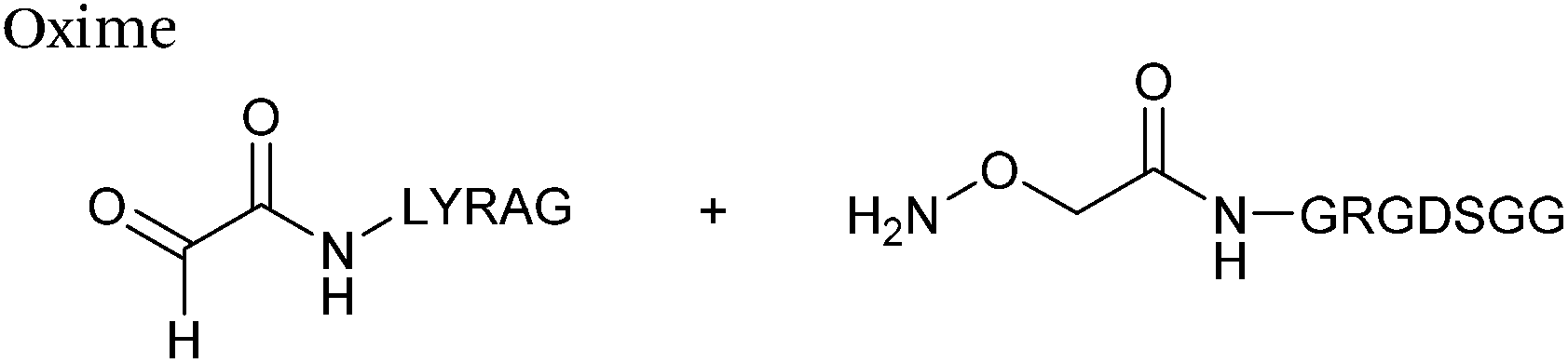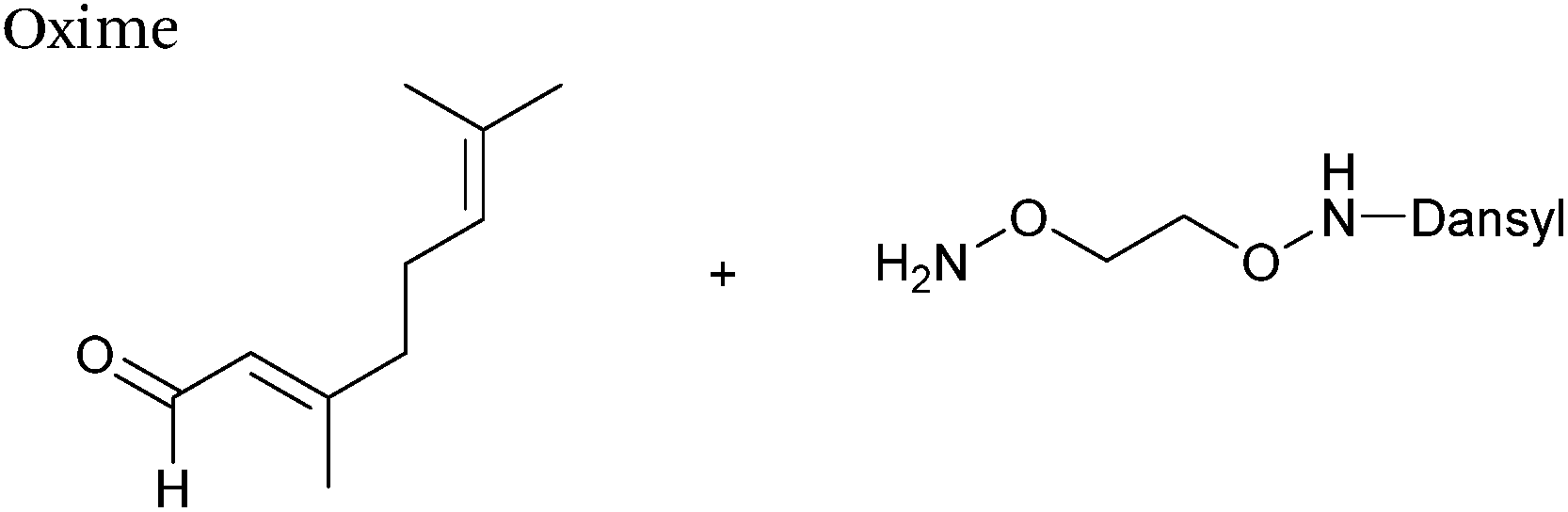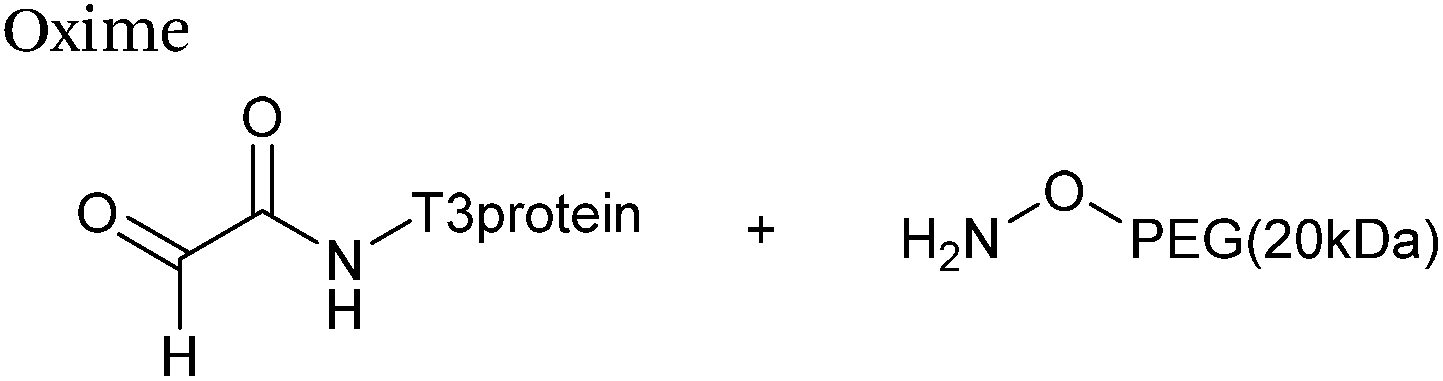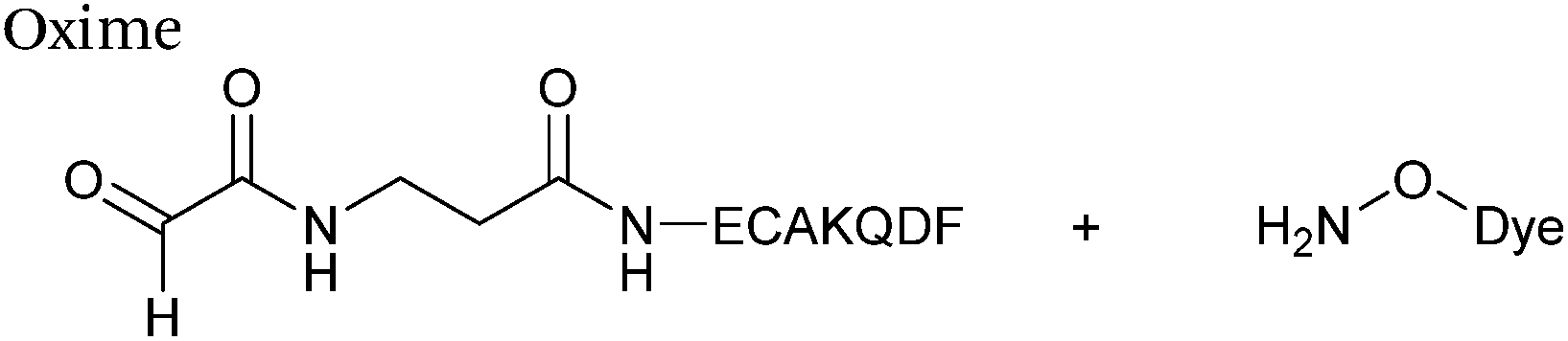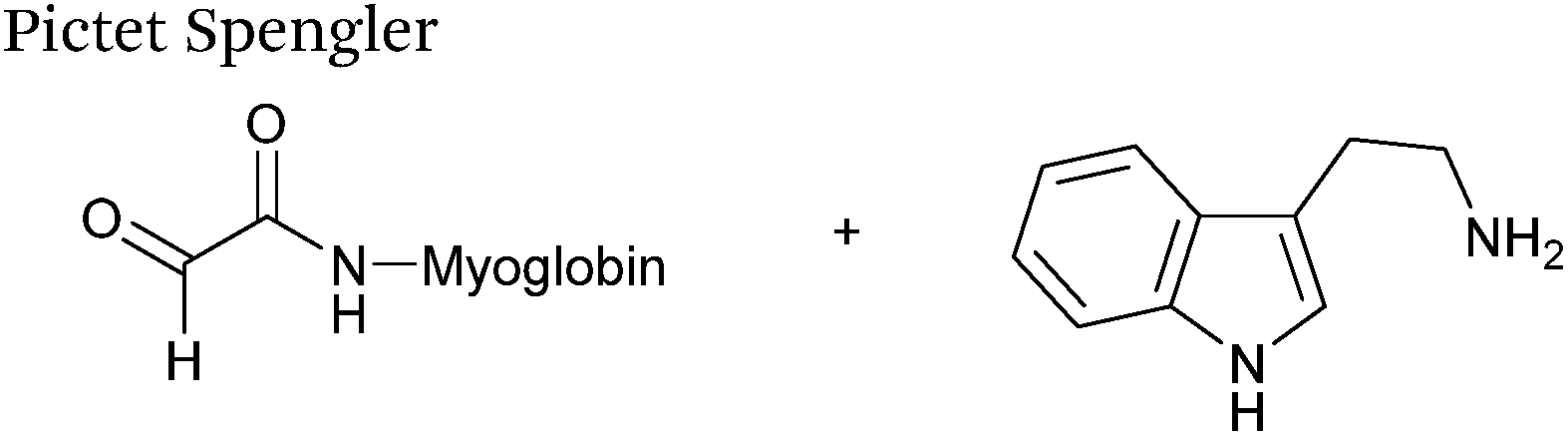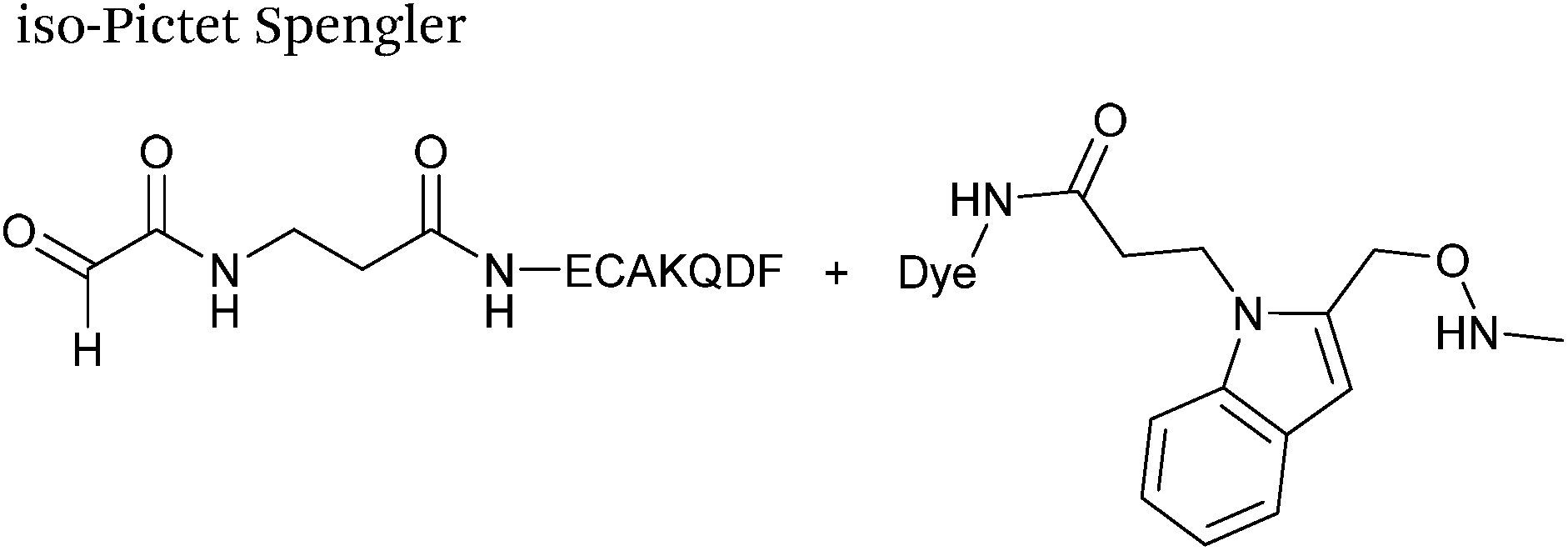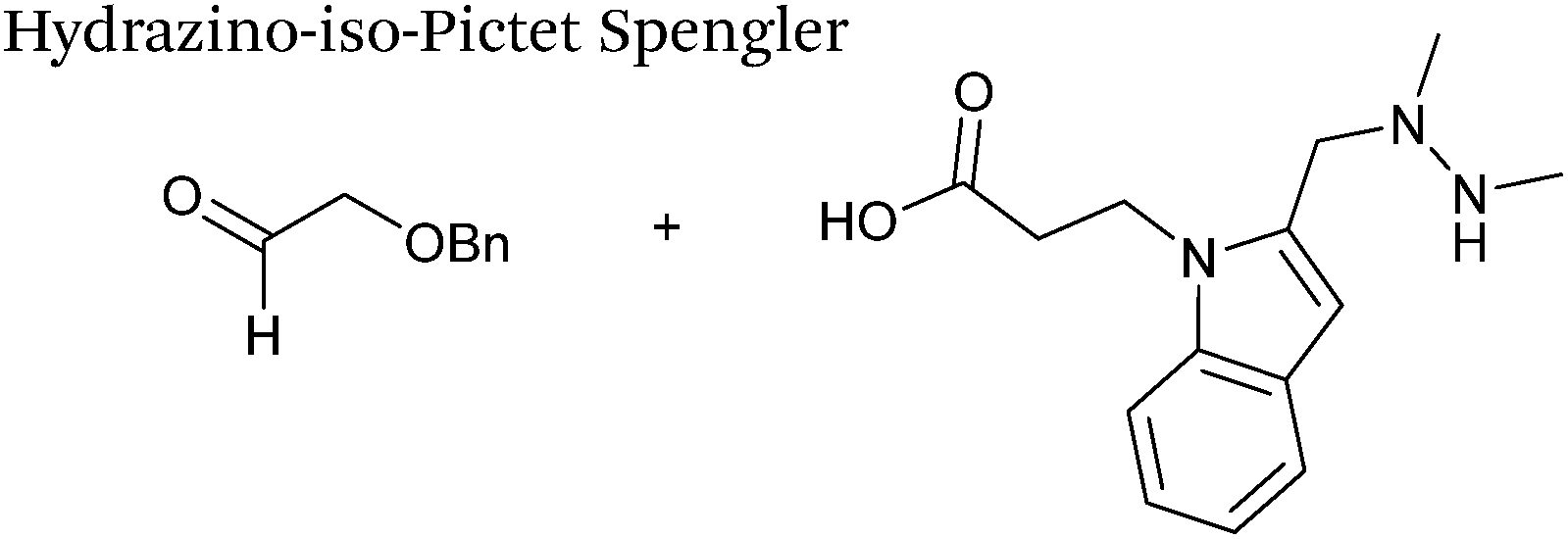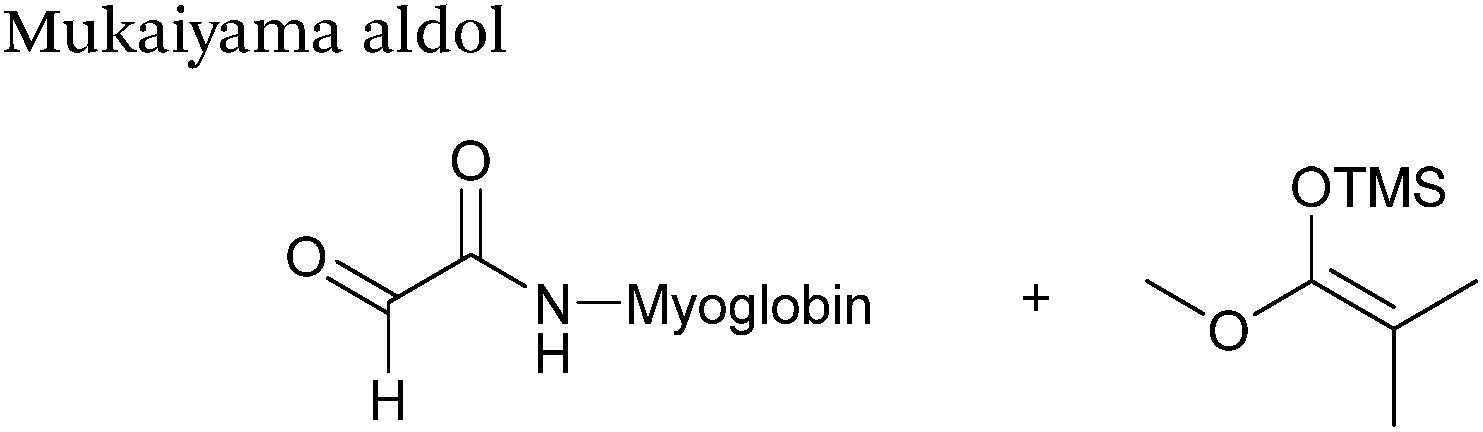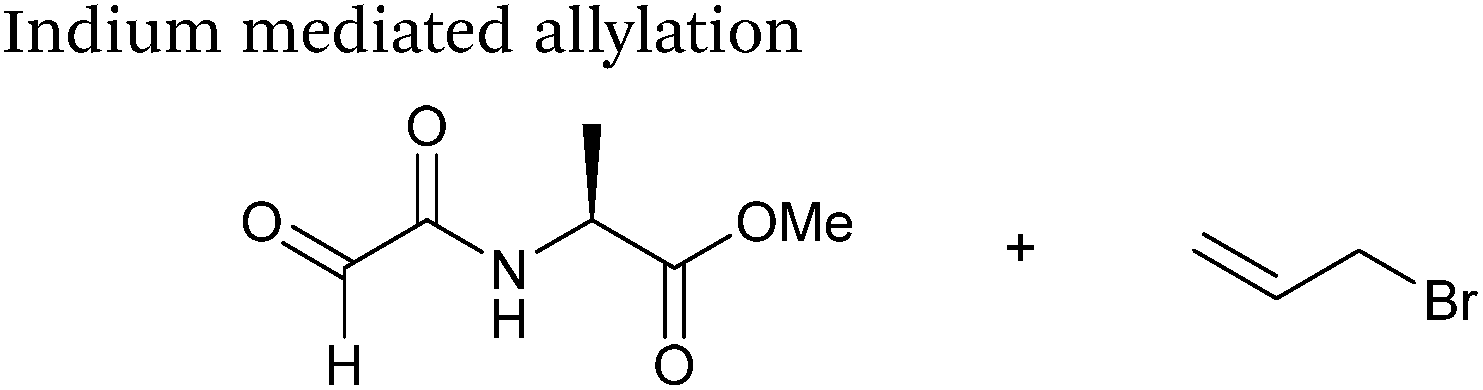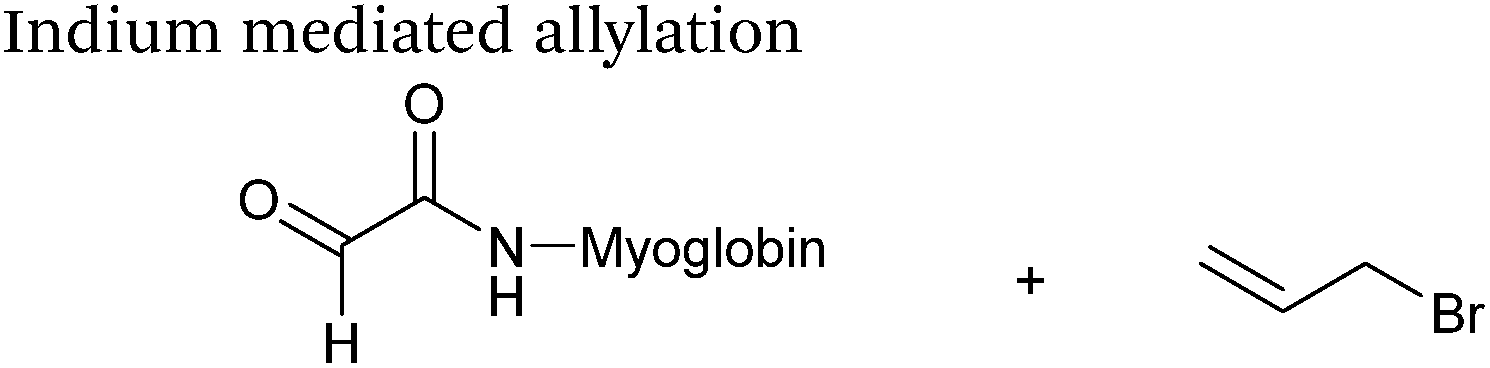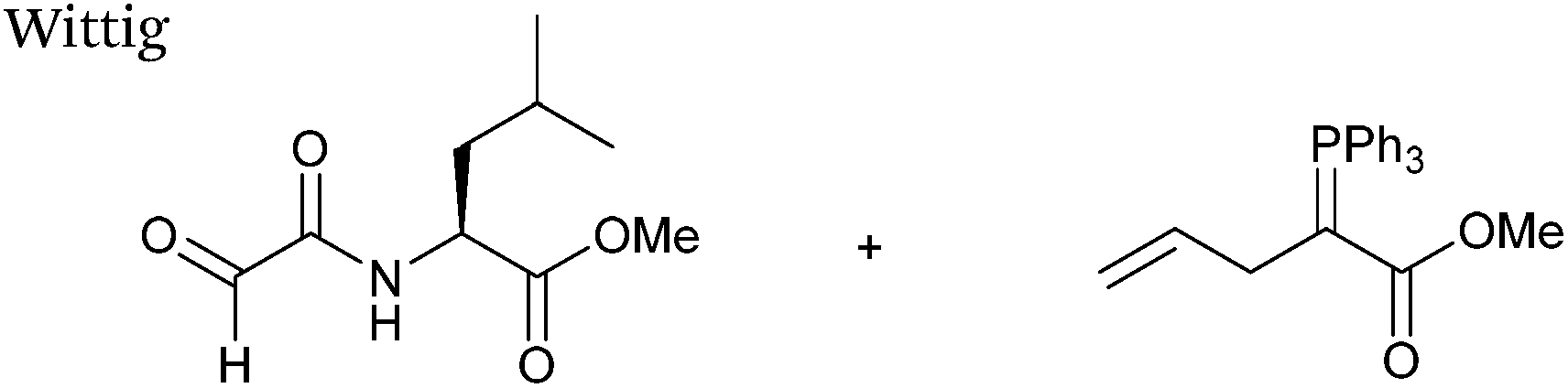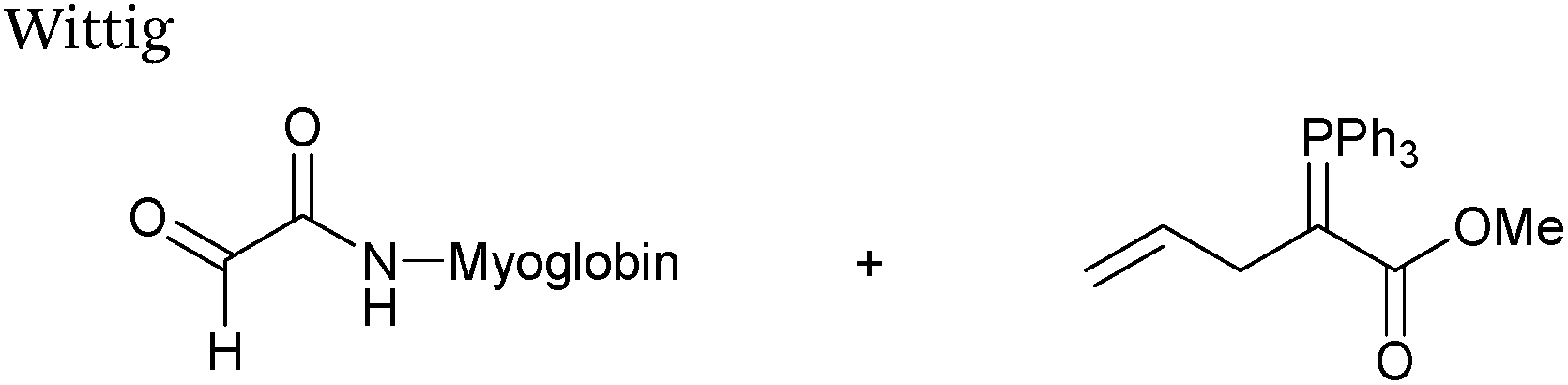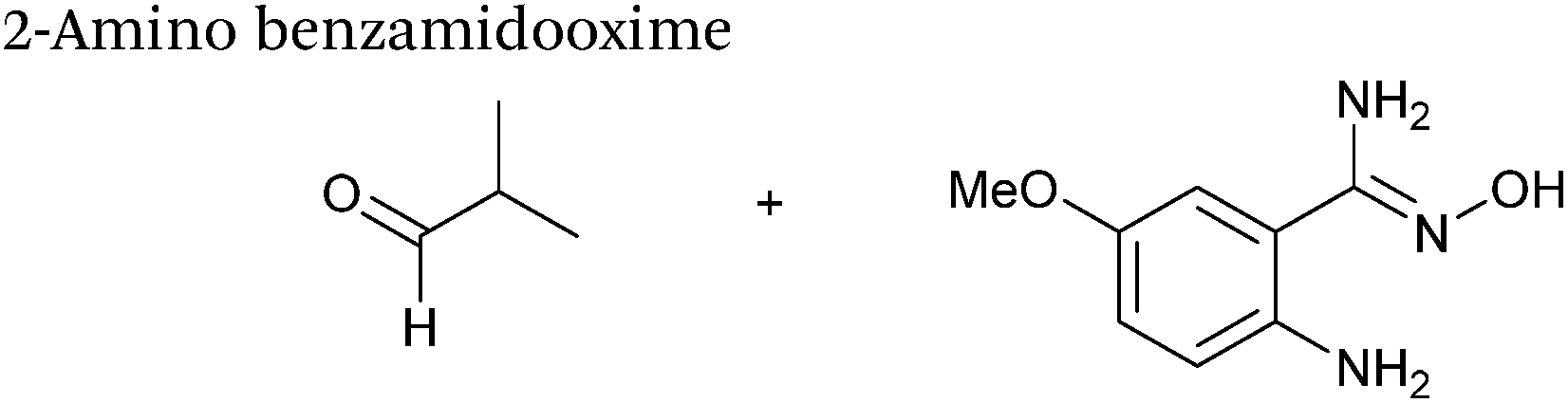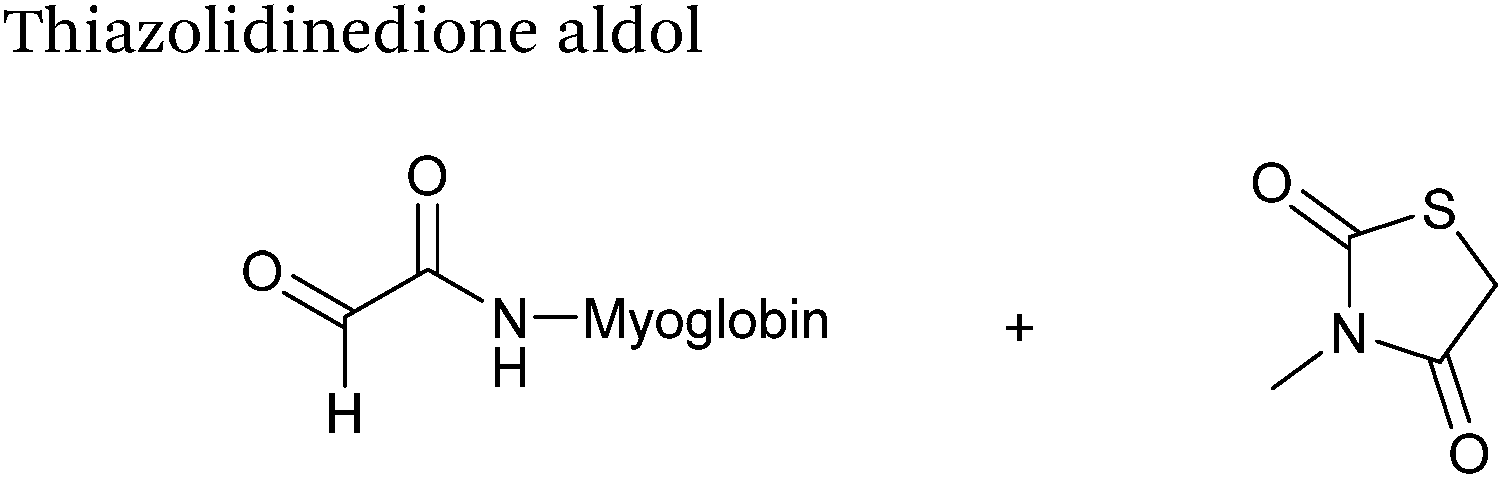DOI:
10.1039/C6OB00778C
(Review Article)
Org. Biomol. Chem., 2016,
14, 7622-7638
Site-selective incorporation and ligation of protein aldehydes
Received
11th April 2016
, Accepted 30th June 2016
First published on 30th June 2016
Abstract
The incorporation of aldehyde handles into proteins, and subsequent chemical reactions thereof, is rapidly proving to be an effective way of generating homogeneous, covalently linked protein constructs that can display a vast array of functionality. In this review, we discuss methods for introducing aldehydes into target proteins, and summarise the ligation strategies for site-selective modification of proteins containing this class of functional handles.
 Richard J. Spears | Richard Spears was born in Leeds, UK, in 1992 and received his MChem degree in Chemistry from the Department of Chemistry at The University of York in 2014. Following his Master's research in structural and mechanistic studies of α-mannosidases and α-mannanases, he commenced his PhD in Dr Martin Fascione's group at the York Structural Biology Laboratory, within the Department of Chemistry in 2014. His current research is focused on developing new methodologies for the site-selective modification of proteins for wide ranging applications. |
 Martin A. Fascione | Martin Fascione received his Ph.D. from the University of Leeds in 2009, working under the tutelage of W. Bruce Turnbull on the stereoselective synthesis of 1,2-cis-glycosides. Following a postdoctoral period in Leeds, he was then awarded a Marie Curie International Outgoing Fellowship to study the mechanisms of carbohydrate-processing enzymes with Professor Steve Withers, FRS, at the University of British Columbia in Vancouver, Canada (2012–2013) and Professor Gideon Davies, FRS, FMedSci, at the University of York, UK (2013–2014). In August 2014 he took up a lectureship in the York Structural Biology Laboratory, within the Department of Chemistry. His research interests include chemical glycobiology, and bioorthogonal chemistry. |
I. Introduction
The ability to site-selectively modify proteins with functional moieties is of major interest within the field of chemical biology, where the chemical ligation of functional molecules to proteins can vastly enhance their properties. For example, conjugation of compounds such as polyethyleneglycol (PEG) can improve the half-life of protein probes and therapeutics,1 while the ligation of fluorescent/spectroscopic probes has been utilised for in vivo imaging and tracking of proteins and other biomolecules.2,3 Additionally, pioneering advances in translational personalised medicine have been achieved through chemical ligation strategies, including the construction of homogeneous antibody-drug conjugates (ADCs) that are robust, effective, and display selective targeting potential.4 From a cell biology perspective, the ability to functionalise proteins at specific sites also shows particular promise in the design of new tools for probing post-translational modifications (PTMs).5
Many chemical protein modification strategies involve reaction of an amino acid side chain, such as the ε-amino group of a lysine6 and/or the thiol group of a cysteine,7 whilst tyrosine8 and tryptophan9 side chains can also be exploited for chemical functionalisation of proteins. All of these methodologies share one common drawback however, in that site-selectivity cannot be achieved when more than one of these amino acid side chains is present within a protein. In recent years more nuanced protein labelling strategies have emerged; these strategies primarily focus around incorporating a unique functional ‘handle’ into a protein that is not naturally present within the proteinogenic amino acid side chains. The tagged protein is then subjected to a substrate that bears a reactive moiety that specifically reacts only with the handle, and does not interact with other functional groups, generating homogenous protein constructs. Revolutionary examples include the Staudinger ligation,10,11 azide–alkyne click reactions,12 and metal mediated metathesis reactions.13 These strategies have also been utilised in labelling of glycoproteins in complex biological environments, including live Zebrafish embryos,2,3 without perturbing the native environment; this is known as ‘bioorthogonal chemistry’. A general review covering this rapidly evolving field has recently been published.14
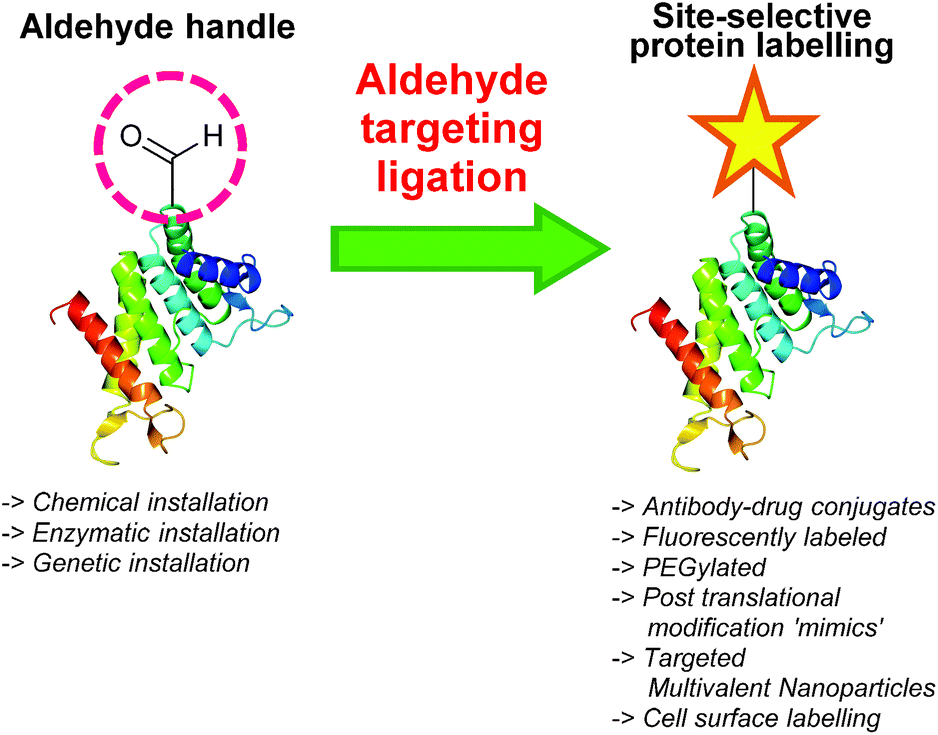
One functional group not traditionally found within proteins is the aldehyde functionality. This reactive carbonyl moiety has wide ranging applications in traditional organic synthesis and can participate in a variety of synthetically useful reactions, including synthesis of native peptide bonds.15–17 Aldehydes also feature notably in protein semisynthesis protocols, including serine/threonine ligations at C-terminal salicylaldehyde esters,18,19 and a variant of the α-ketoacid-hydroxylamine (KAHA) ligation where incorporated aldehyde side chains can act as purification handles.20 It is therefore of no surprise that the aldehyde functionality has seen frequent use within the field of site-selective protein modification. Upon installation of an aldehyde handle, it is possible to perform chemistry that reacts with the aldehyde group to site-selectively label a target protein. Many strategies exist for the incorporation an aldehyde handle into a desired protein, along with an increasing selection of ligation protocols for subsequent site-selective labelling. Of particular note is the significant number of chemical probes targeting these handles that are now available commercially. Numerous examples of aldehyde-based ligations have been reported in the literature, including synthesis of antibody-drug conjugates (ADCs), design of proteins bearing PTM ‘mimics’, and labelling of specific proteins within crude cell lysates (Scheme 1). This review aims to provide an overview of the strategies currently reported for incorporating aldehydes into proteins, alongside reported ligation methods exploited in their subsequent chemical modification.
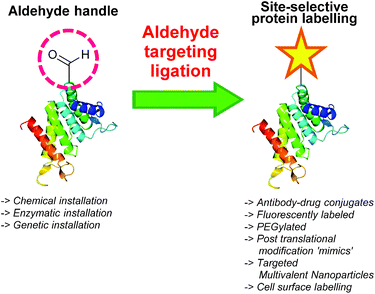 |
| | Scheme 1 Overview site-selective ligation of an aldehyde handle incorporated into a generic protein. | |
II. Incorporating aldehydes into proteins
A wide variety of strategies can be employed for the incorporation of aldehyde handles into proteins. It is worth noting at this point that, although all strategies described incorporate an aldehyde functionality, the electronic properties of the aldehyde installed can vastly differ depending on which strategy is used. These range from α,β-unsaturated aldehydes to aryl aldehydes, as well as electron rich to electron deficient aldehydes. The tag can also differ in location and distance from the protein it is attached to. Some of the methods described for installing aldehyde handles into proteins can also be used to install ketone handles into proteins; these will be briefly considered where appropriate.
Oxidation of β amino-alcohols via periodate cleavage
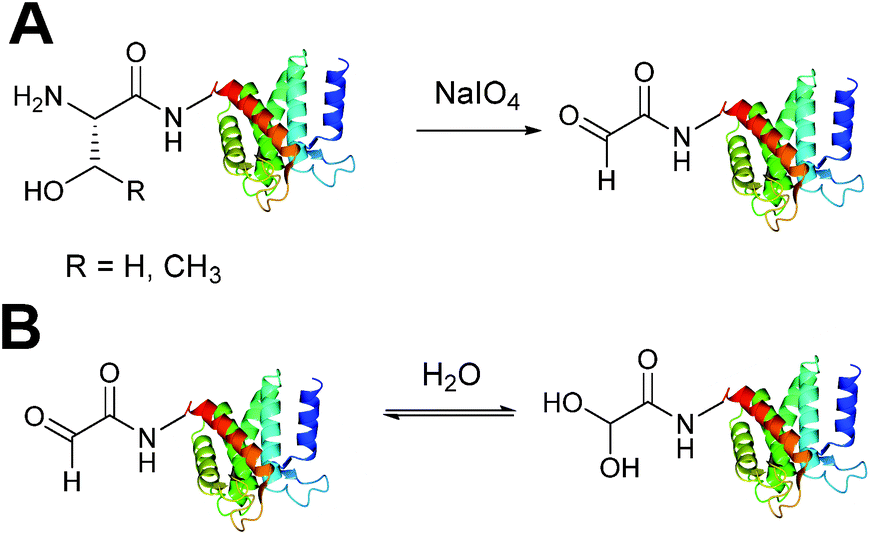
Aldehydes can be installed into proteins via oxidative cleavage of serine and threonine residues located on the N-terminus of a protein (Scheme 2).21 This can be achieved through reaction with sodium periodate (NaIO4) and is analogous to vicinal diol cleavage (also known as the Malaprade reaction).22 The resulting electron-poor N-terminal aldehyde is referred to as an α-oxo aldehyde or glyoxylamide (or glyoxylylamide). An extensive review on glyoxylamides, along with their carboxylic acid counterpart (glyoxylic acids), has recently been published.23 A particular advantage of generating an aldehyde via this method is that the reaction is rapid; oxidation of β amino alcohols is 100–1000 times faster than the oxidation of vicinal diols encountered in carbohydrates.24 Care must be taken when using this method however as other amino acids can be oxidised under certain conditions (cysteine and methionine residues in particular).23 Therefore, the reaction is usually performed with or quenched by another component such as excess methionine or ethylene glycol, followed by purification of the oxidised protein,25,26 which exists in an equilibrium as a mixture of aldehyde and hydrate form (Scheme 2B).
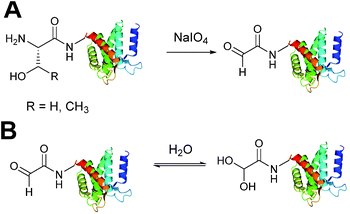 |
| | Scheme 2 (A) Oxidation of serine/threonine N-terminated proteins to generate a glyoxylamide (glyoxyl) species. (B) The aldehyde and hydrate form of the glyoxyl species. | |
N-Terminal oxidation strategies
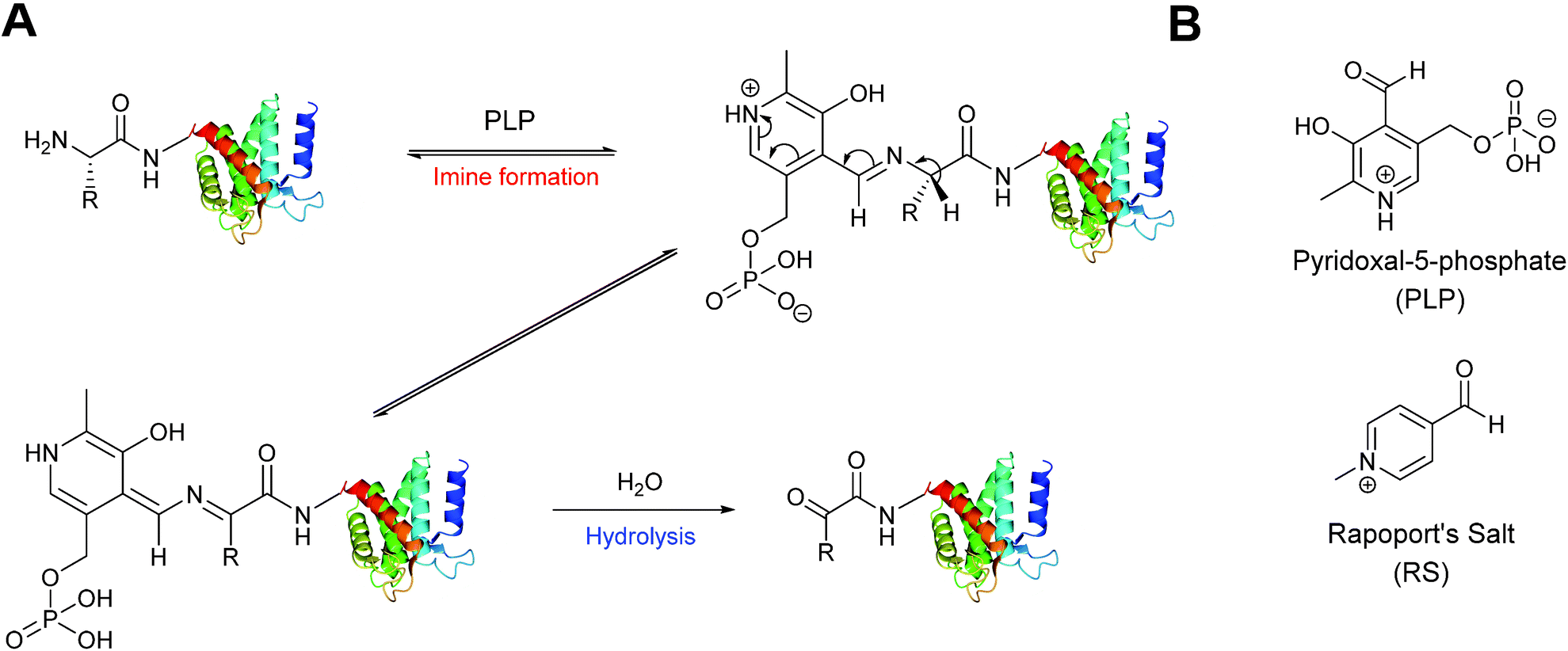
A biomimetic transamination reaction for the installation of both aldehydes and ketones into proteins has also been described in the literature (Scheme 3). An initial screen of aldehyde reagents on the peptide angiotensin I identified pyridoxal 5-phosphate (PLP) as a successful reagent for the oxidation of the N-terminal aspartate residue (generating a ketone) under mild conditions (pH 6.5 PB, 18 hours, 37 °C). The N-terminal ketone generated from this biomimetic transformation can then be site-selectively modified further. PLP-mediated transamination was successfully demonstrated on a number of proteins bearing different amino acids at their N-terminus, including conversion of N-terminal glycine to a glyoxylamide in horse heart myoglobin, and oxidation of N-terminal lysine to a ketone in RNAase A.27 The transamination is specific for the amino group of the N-terminal amino acid, and does not modify other amine bearing residues such as lysine side chains. Optimisation of PLP-mediated transamination has since undergone extensive investigation utilising solid phase peptide synthesis (SPPS)28 and combinatorial peptide libraries.29 PLP mediated transamination of resin bound peptides identified that sequences containing alanine, glycine, aspartate, glutamate, or glutamine residues at the N-terminus were transformed into the desired transamination product in the highest yields. Histidine, lysine, tryptophan and proline N-termini containing peptides gave rise to PLP-adduct-type byproducts, whereas isoleucine and valine N-terminated peptides displayed very poor reactivity in PLP-mediated transamination.28 The observation that the valine N-termini are unsuitable for transamination is contradictory to the previously reported successful transamination of valine N-terminal green fluorescent protein (GFP1 V);27 this was rationalised on the basis that the internal sequence of a peptide/protein may also affect the success of PLP-mediated transamination. Additional screening of resin bound lysine and valine N-terminal tetrapeptides demonstrated that the identity of the amino acid at position 2 and 3 can dramatically affect the outcome of the transamination reaction.28 Combinatorial peptide libraries screens have subsequently revealed that alanine N-terminated peptides/proteins, directly followed by a lysine residue, are highly reactive towards PLP-mediated transamination.29 Transamination of short isoleucine, leucine, and valine N-terminal peptides using another pyridoxal derivative, along with the effects of using an imidazole-based buffer system on the transamination reaction, have also been recently reported.30
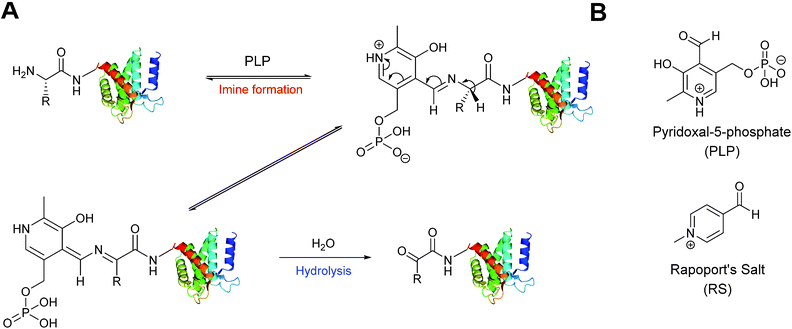 |
| | Scheme 3 (A) Oxidation of the N-terminus using pyridoxal-5-phosphate (PLP) to generate a reactive aldehyde (or ketone). (B) The structure PLP and Rapoport's Salt (RS). | |
An alternative reagent for transamination-mediated reactions on proteins has been reported more recently.31 Using previously described combinatorial peptide libraries,29N-methylpyridinium-4-carboxaldehyde (Rapoport's Salt, ‘RS’, Scheme 3) has since been identified as an effective reagent for transamination-type modification of proteins; in particular, RS is highly suitable for glutamate N-terminated peptides/proteins.31 Optimisation of the transamination using SPPS has demonstrated that RS is a poor transamination reagent for glycine, serine, threonine, or proline N-terminated peptides, but is very suitable for alanine, and glutamate N-terminated peptides. Additionally, it is worth noting that for all N-terminated amino acids discussed (with the exception of proline), improved yields of the desired transamination product were obtained when using RS under more basic conditions (pH 7.5–8.5 as opposed to pH 6.5).32
Genetic incorporation of a formylglycine tag
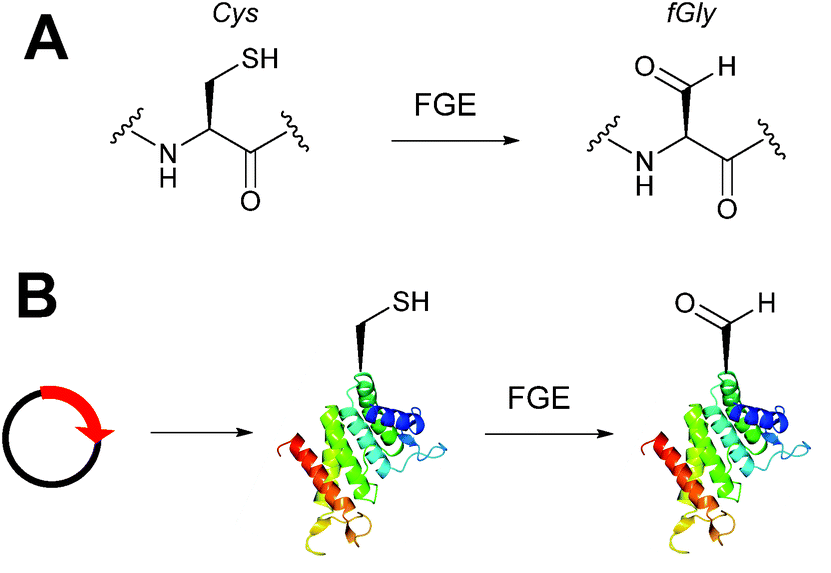
Alongside the chemical methods described earlier for installing aldehydes into proteins, enzymatic methods, such as the formylglycine tag, have also been exploited and reported (Scheme 4).33 Formylglycine (fGly) is an electron-rich aldehyde side chain derived from the chemoenzymatic oxidation and subsequent hydrolysis of a cysteine residue that is located within a consensus sequence of CxPxR (where x = any amino acid). Conversion of the thiol to the aldehyde is carried out by the Formylglycine Generating Enzyme (FGE). In nature, this transformation is essential for the enzymatic turnover in the active site of sulfatase enzymes; fGly is the active site residue that is responsible for the catalytic hydrolysis of sulfate esters.34 Use of the fGly residue for selective modifications of proteins was first described in 2007.33 Short tags containing motifs that are targeted by sulfatases and subjected to fGly conversion in nature were installed at the N or C-termini of human growth hormone, mycobacterial sulfotransferase, and maltose-binding protein. Co-expression of the recombinant target proteins in E. coli alongside M. tuberculosis FGE resulted in >85% conversion of the CxPxR-containing proteins to the desired fGly-tagged proteins (confirmed by MS and subsequent tryptic digest/modification experiments). The fGly tag has since been utilised to install monosaccharides35 (which can be used as glycosyl acceptors to build N-glycans via endoglycosynthase enzymatic derivatisation),36 biotinylated probes,37 and therapeutically relevant small molecules.37–40 The fGly handle has also been used in the specific labelling of tagged proteins in cell extracts.41 In addition to prokaryotic cell lines, this methodology has also been utilised successfully in mammalian expression systems.42 Investigation into preactivation of FGE to improve turnover rates using copper has also been reported.43 For a more detailed discussion on FGEs and the fGly motif, readers are directed to a recent review in the field.44
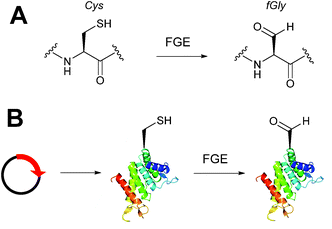 |
| | Scheme 4 (A) Outline of generating the formylglycine (fGly) residue on peptide/proteins bearing a cysteine within a CxPxR sequence. (B) Expression of a plasmid encoding for the desired protein containing the CxPxR sequence, followed by treatment with the formylglycine generating enzyme (FGE) leads to fGly-proteins. | |
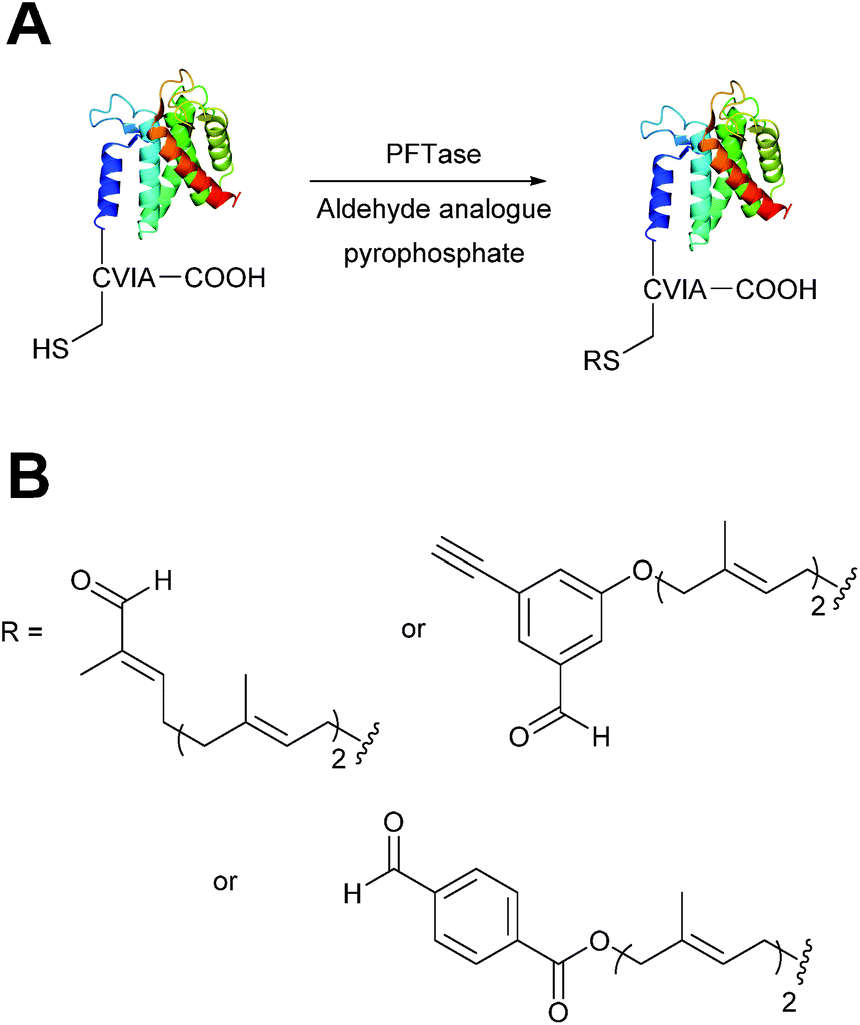
Enzymatic incorporation of farnesyl pyrophosphate analogues
Protein farnesyl-transferase (PFTase) is an enzyme that catalyses the farnesylation of thiol side chains of cysteine residues in proteins. PFTase utilises farnesyl pyrophosphate (FPP) for this transformation and is specific for cysteine residues within a tetrapeptide domain at the C-terminus of a peptide/protein. This tetrapeptide domain is commonly referred to as a CAAX-box, where A = aliphatic amino acid, and X = alanine, serine, methionine, or valine (usually cysteine-valine-isoleucine-alanine).45,46 Along with its natural substrate FPP, PFTase can also incorporate FPP analogues such as farnesyl aldehyde pyrophosphate (FAPP), allowing for installation of an α,β-unsaturated aldehyde tag into proteins bearing a CAAX-box motif (Scheme 5A).47 FAPP installation has been demonstrated on N-dansyl peptides and CAAX-box tagged GFP. Following aldehyde installation, tagged proteins can then be specifically isolated via modification of the installed aldehyde.48
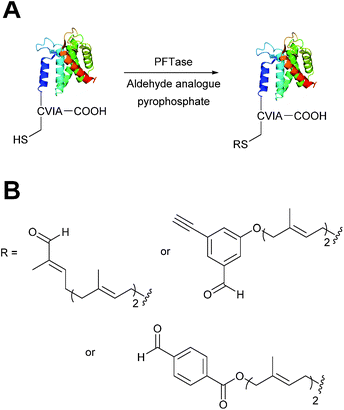 |
| | Scheme 5 (A) Protein farnesyl-transferase (PFTase) mediated installation of farnesyl-derived aldehydes into a protein bearing a C-terminal CAAX box. (B) Aldehyde analogues that can be incorporated. | |
PFTase strategies for aldehyde tag incorporation have since been extended to incorporation of an aryl aldehyde analogue (Scheme 5B),48 and modification of other proteins such as ciliary neurotrophic factor protein (CNTF).49 Other aryl aldehyde handles that bear alkyne functionality can also be incorporated using PFTase, allowing for selective, dual functionalisation of GFP.49 Mutant variants of PFTase have also been explored in a desire to improve catalytic efficiency of FPP aldehyde analogues; a 43 fold increase, for example, is observed for the incorporation of aryl aldehyde FPP into CAAX-box-bearing CNTF when using a W102A β subunit PFTase variant (as opposed to using wild type PFTase).50
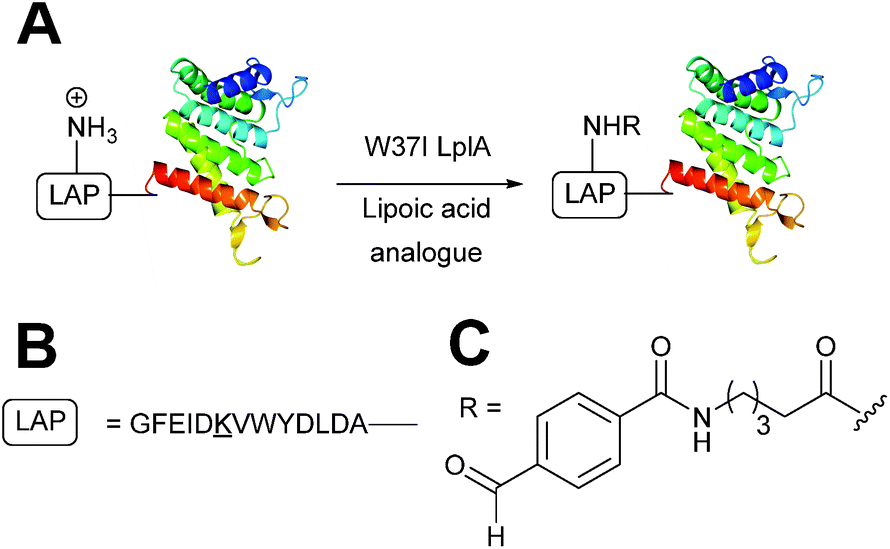
Enzymatic incorporation using lipoic acid ligase
In Escherichia coli, lipoic acid ligase (LplA) is responsible for linking lipoic acid to various proteins involved in oxidative metabolism.51 LplA specifically targets lysine side chains located within a GFEID![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) VWYDLDA sequence known as the LplA acceptor peptide (LAP). The incorporation of aldehyde tags utilising this enzymatic strategy has recently been described (Scheme 6). W37I mutants of LplA could be used to incorporate aryl aldehydes into a LAP peptide in a 500
VWYDLDA sequence known as the LplA acceptor peptide (LAP). The incorporation of aldehyde tags utilising this enzymatic strategy has recently been described (Scheme 6). W37I mutants of LplA could be used to incorporate aryl aldehydes into a LAP peptide in a 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :150:1 ratio of lipoic acid-analogue:LAP-peptide:LplA, with a maximum of 62% conversion to the aldehyde-tagged LAP peptide observed after 5 minutes of incubation. Additionally, proteins bearing LAP tags at the N-terminus, including LAP-tagged streptavidin and LAP-tagged alkaline phosphatase, were also successfully modified.52 More recently, this methodology has been extended to the labelling of LAP-tagged nanobodies.53
:150:1 ratio of lipoic acid-analogue:LAP-peptide:LplA, with a maximum of 62% conversion to the aldehyde-tagged LAP peptide observed after 5 minutes of incubation. Additionally, proteins bearing LAP tags at the N-terminus, including LAP-tagged streptavidin and LAP-tagged alkaline phosphatase, were also successfully modified.52 More recently, this methodology has been extended to the labelling of LAP-tagged nanobodies.53
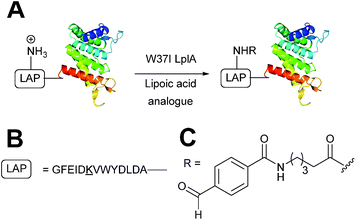 |
| | Scheme 6 (A) Outline of using Lipoic acid ligase (LplA) mutants for installing aldehydes into LplA accepter peptide (LAP) bearing proteins. (B) Amino acid sequence of the LAP. (C) Structure of the incorporated aldehyde. | |
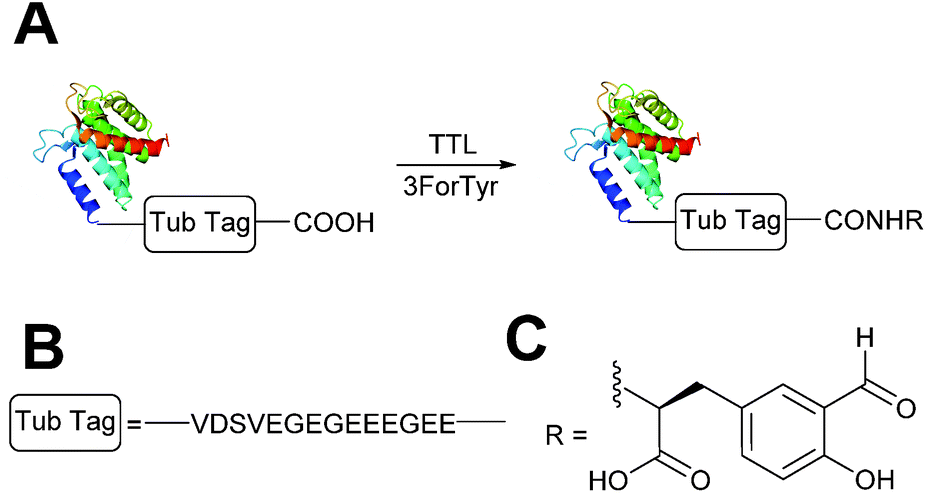
Enzymatic incorporation of 3-formyl-L-tyrosine
A very recently described chemoenzymatic method for installation of aldehyde tags into proteins is through enzymatic transformation using tubulin tyrosine ligase (TTL, Scheme 7).54 Although the natural function of TTL is to post-translationally modify the C-terminus of α-tubulin with tyrosine, TTL can also accept tyrosine analogues, such as 3-formyl-L-tyrosine (denoted as ‘3ForTyr’ in this review), in ligation of C-terminal peptides/proteins bearing a VDSVEGEGEEEGEE tag (known as a Tub tag). Mixing of Tub-tagged peptides with TTL in a 200:1 ratio afforded a 63% incorporation of 3ForTyr after 2 hours (monitored by HPLC, with >90% incorporation observed after 6 hours). This methodology has also been utilised in the tagging of Tub-tagged GFP, ubiquitin, and GFP-specific nanobodies. Incubation of TTL:Tub-tagged-protein in an optimal ratio of 1:5 for 3 hours at 37 °C gave rise to successful labelling with 3ForTyr (confirmed by tryptic digestion experiments). Conversions of 99% were achieved under these conditions (based on SDS-PAGE analysis and observations of other incorporated tyrosine analogues).54
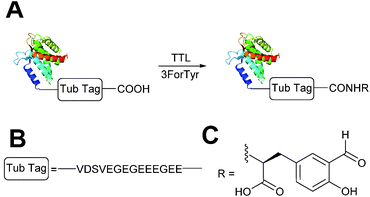 |
| | Scheme 7 (A) Outline of using Tubulin Tyrosine Ligase (TTL) to for the incorporation of 3-formyl-L-tyrosine (3ForTyr) into proteins bearing a ‘Tub Tag’. (B) The amino acid sequence of the Tub Tag. (C) Structure of the incorporated 3ForTyr. | |
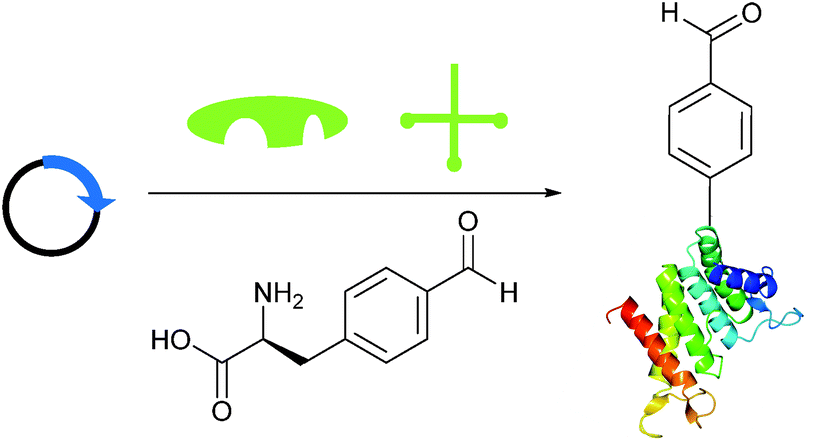
Genetic incorporation of non-native aldehyde containing amino acids
Briefly, another well-publicised strategy55,56 for installing an aldehyde (or ketone) tag into a protein is to utilise the ever-increasing number of evolved tRNA/tRNA synthetase pairs to perform unnatural amino acid mutagenesis (Scheme 8).57–60 The aldehyde (ketone) side chain can then be modified with a range of functional handles, including fluorescent probes,57,60,61 biotinylated probes,57,58,61 and carbohydrates.62 ADCs have also recently been developed utilising this methodology.63,64
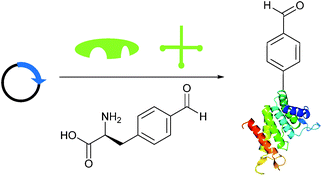 |
| | Scheme 8 Installation of aldehyde into proteins via unnatural amino acid mutagenesis. Expression of a plasmid encoding for the desired protein containing an amber STOP codon in conjunction with an orthogonal tRNA synthetase/tRNA pair and unnatural amino acid allows for incorporation of an aldehyde-containing amino acid at the aforementioned STOP position. | |
III. Chemical strategies for the modification of proteins bearing aldehyde handles
There now exist a plethora of strategies for the site-selective modification of proteins following installation of one of the aforementioned aldehyde tags. These range from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond formation to C–C bond formation, and from metal mediated ligations to intramolecular cyclisation ligations. The choice of aldehyde handle, along with the conditions for aldehyde modification (such as pH or use of additional co-solvent) ranges from strategy to strategy. The rate of ligation has also been profiled for the majority of strategies described; often using small molecule aldehydes mimics and, on some occasions, proteins bearing an aldehyde handle. Selected rate constants (and the conditions used) for each ligation strategy discussed herein can be found in Table 1.
N bond formation to C–C bond formation, and from metal mediated ligations to intramolecular cyclisation ligations. The choice of aldehyde handle, along with the conditions for aldehyde modification (such as pH or use of additional co-solvent) ranges from strategy to strategy. The rate of ligation has also been profiled for the majority of strategies described; often using small molecule aldehydes mimics and, on some occasions, proteins bearing an aldehyde handle. Selected rate constants (and the conditions used) for each ligation strategy discussed herein can be found in Table 1.
Table 1 Comparison of selected rate constants for strategies used in the modification of aldehydes under various conditions
| Entry |
Reaction |
Conditions |
Rate (M−1 s−1) |
Ref. |
|
No rate/yield/conversion given.
No rate studies performed, reaction time and obtained yield quoted as an alternative.
No rate studies performed, reaction time and conversion as judged by LC-MS quoted as an alternative.
|
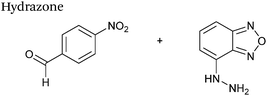
| 1 |
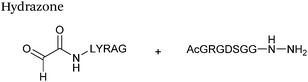
|
1 mM aldehyde |
0.0031 (uncatalysed) |
65
|
| 1 mM hydrazine |
0.21 (10 mM aniline) |
| pH 5.7, NH4OAc |
|
| 2 |
|
1 mM aldehyde |
0.0013 (uncatalysed) |
66
|
| 18 μM hydrazine |
0.11 (1 mM 5-methoxyanthralic acid) |
| pH 7.4, PBS (10% DMF) |
|
| 3 |
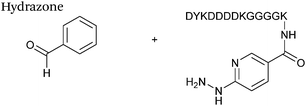
|
10 μM aldehyde |
170 (100 mM aniline) |
67
|
| 10 μM hydrazine |
|
| pH 7.0, PB |
|
| 4 |
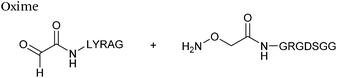
|
1 mM aldehyde |
0.057 (pH 4.5, uncatalysed) |
68
|
| 1 mM aminooxy |
0.68 (pH 4.5, 10 mM aniline) |
| pH 4.5, NH4OAc or pH 7.0, PB |
0.015 (pH 7.0, uncatalysed) |
|
|
0.061 (pH 7.0, 100 mM p-methoxyaniline) |
| 5 |
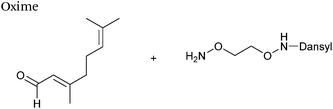
|
50 μM aldehyde |
24 (25 mM aniline) |
69
|
| 100 μM aminooxy |
41 (25 mM m-phenylenediamine) |
| pH 7.0, PB |
|
| 6 |
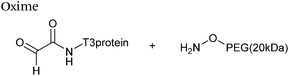
|
91 μM aldehyde |
0.22 (pH 4.0, uncatalysed) |
70
|
| 455 μM aminooxy |
4.3 (pH 4.0, 10 mM p-phenylenediamine) |
| pH 4.0, Na citrate or pH 7.0, PB |
0.006 (pH 7.0, uncatalysed) |
|
|
0.29 (pH 7.0, 10 mM p-phenylenediamine) |
| 7 |
|
10 mM aldehyde |
0.0013 |
71
|
| 10 mM aminooxy |
| pH 7.0, PB:MeOH (1:1) |
| 8 |

|
60 μM aldehyde |
N/Aa |
72
|
| 50 mM indole |
|
| pH 6.5, PB |
|
| 9 |
|
500 μM aldehyde |
10.6 (pH 4.5) |
37
|
| 500 μM indole |
0.26 (pH 7.0) |
| pH 4.5, NaOAc or pH 7.0, PB |
|
| 10 |
|
2.5 mM aldehyde |
0.015 |
71
|
| 2.5 mM aldehyde |
| pH 4, NaOAc:MeOH (1:1) |
| 11 |
|
50 μM aldehyde |
4.2 |
38
|
| 50 μM indole |
| pH 6, PB |
| 12 |
|
300 μM nitrone |
12.8 |
73
|
| 330 μM alkyne |
| H2O:MeCN (9:1) |
| 13 |
|
1:100 molar ratios (nitrone:alkyne) |
58.8 |
74
|
| MeCN |
| 14 |
|
200 μM aldehyde |
24 hours, 55% yield (obtained)b |
75
|
| 400 μM silyl ketene acetal |
| pH 7.0, PB |
| 15 |
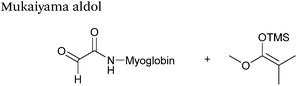
|
37.5 μM |
5 hours, 90% (conversion)c |
75
|
| 7.13 mM silyl ketene acetal |
| H2O:t-BuOH (5:1) |
| 16 |
|
21 mM aldehyde |
16 hours, 56% yield (obtained)b |
76
|
| 42 mM indium |
| 63 mM allyl bromide |
| PB pH 7.0 |
| 17 |

|
37.5 μM aldehyde |
20 hours, 54% (conversion)c |
76
|
| 29 mM indium |
| 836 μM allyl bromide |
| H2O:t-BuOH (6:4) |
| 18 |
|
10 mM aldehyde |
90 min, 67% yield (obtained)b |
77
|
| 20 mM ylide |
| H2O:t-BuOH (1:1) |
| 19 |

|
15 μM aldehyde |
30 min, 65% (conversion)c |
77
|
| 750 μM ylide |
| H2O:t-BuOH (3:1) |
| 20 |
|
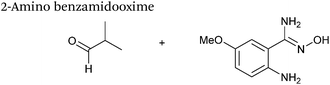
15 mM aldehyde |
0.0086 |
78
|
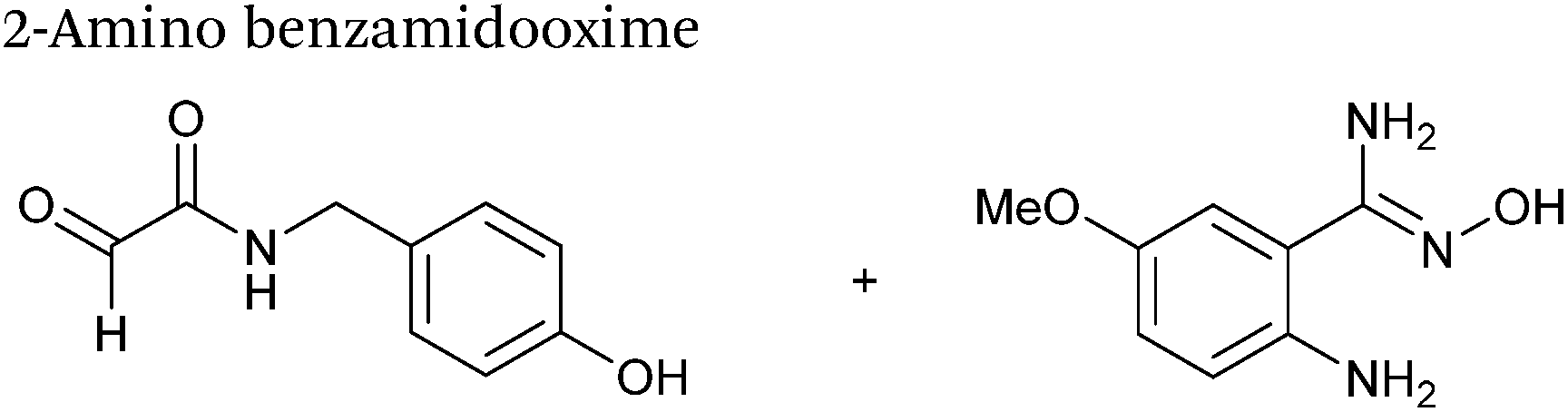
| 3 mM 2-aminobenzamidoxime |
| pH 4.5, NaOAc |
| 21 |
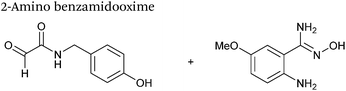
|
2 mM aldehyde |
1 |
78
|
| 1 mM 2-amino benzamidooxime |
| pH 4.5, NaOAC |
| 22 |
|
2 mM aldehyde |
40 |
78
|
| 1 mM 2-amino benzamidooxime |
| pH 4.5, NaOAC |
| 23 |
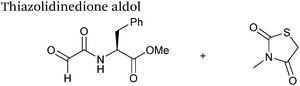
|
10 mM aldehyde |
0.0078 |
79
|
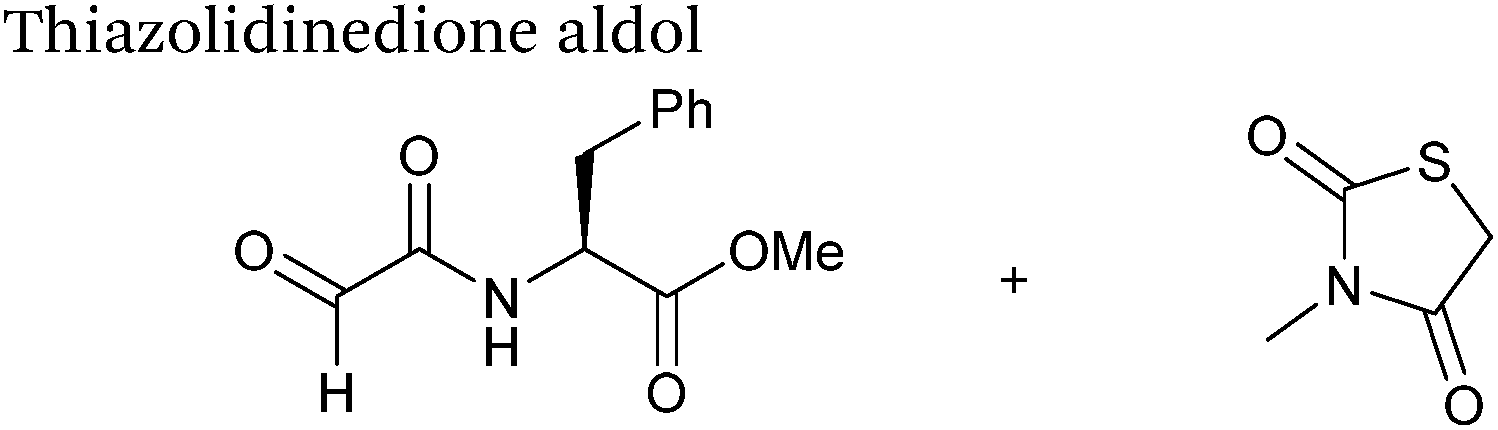
| 10 mM thiazolidinedione |
| pH 7.0, PB (25% DMSO) |
| 24 |
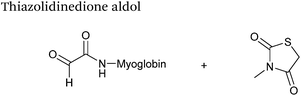
|
37.5 μM aldehyde |
3 hours, 83% (conversion)c |
79
|
| 37.5 mM thiazolidinedione |
| pH 7.0, PB |
| 25 |
|
25 μM aldehyde |
0.387 |
40
|
| 1 mM thiopyrazolone |
| pH 7.0, Na citrate |
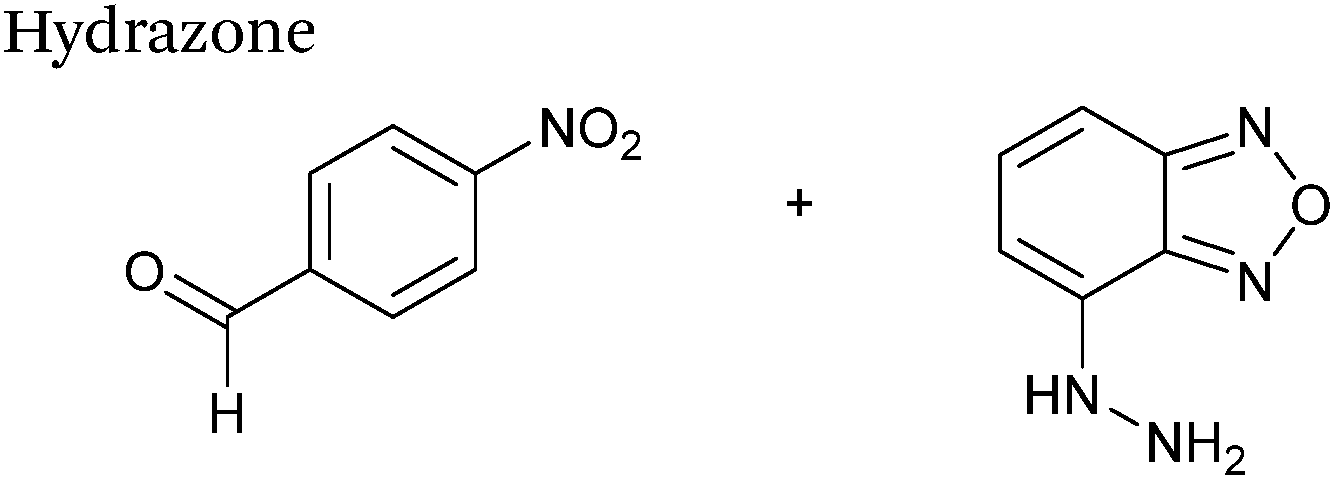
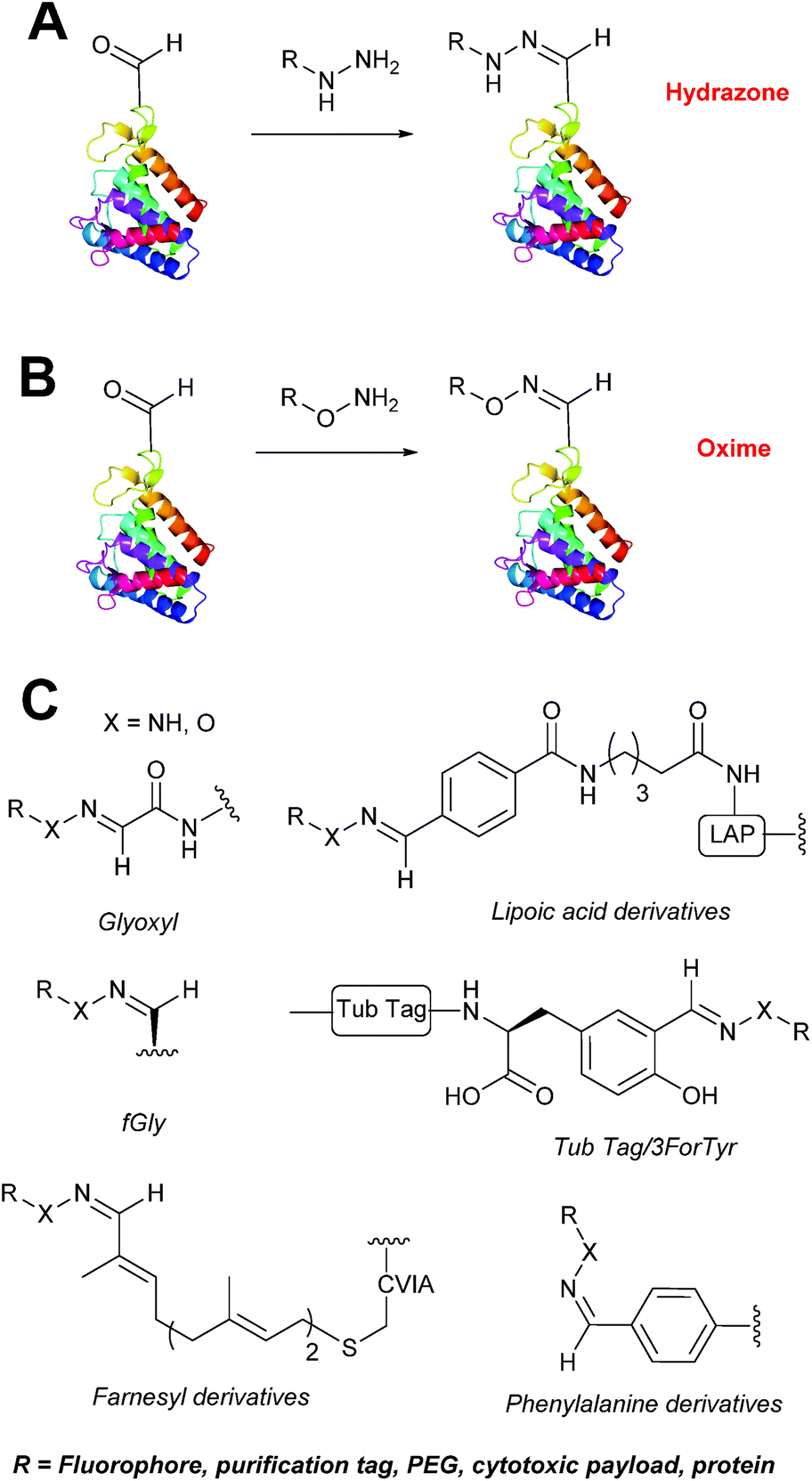
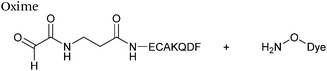
Hydrazone/oxime ligations
The reaction of hydrazine and hydroxylamine/aminooxy reagents with proteins bearing aldehydes (or ketones) to form hydrazone and oxime bonds, respectively, are among the best established methods for generating homogenous ligation products (Scheme 9A and B). These ligation strategies proceed via an iminium-type intermediate, and are by far the most popular method of modifying aldehydes installed into proteins. Indeed, a standard method for validating installation of an aldehyde handle into a protein is to perform hydrazone or oxime ligation with small molecule substrates, and then analyse the anticipated ligation product by protein mass spectrometry (protein MS).21,27,31,33,48,57 Uniquely, to date, they are the only ligation strategies reported in literature that have been demonstrated on all aldehyde handles that can be incorporated into proteins (Scheme 9C, Table 1, entries 1–7). More complex, functionalised hydrazine/hydroxylamine reagents are frequently employed for the purpose of protein bioconjugations. For example, labelling with hydrazine/hydroxylamine functionalised biotin facilitates isolation of the modified protein via binding with streptavidin.33,61,80 Use of aminooxy functionalised resins can also be applied to isolate aldehyde-tagged proteins via oxime ligation from crude cell lysates.47,48 Successful incorporation of an aldehyde handle into a protein is also frequently showcased through the reaction with hydrazine or hydroxylamine functionalised fluorescent probes.27,31,52,54,60 Analysis of these ligation products via SDS PAGE analysis with fluorescence emission scanning can be used to determine the level of success of the initial aldehyde incorporation method.36 In a similar manner, hydroxylamine functionalised PEG reagents can also be used to label incorporated aldehyde handles; these ligation products can then be visualised via SDS PAGE analysis.28,32,49,81 As discussed earlier, carbohydrates have also been installed into proteins via oxime ligation.35,36,62 In an alternate approach, glycosylated proteins have also been modified by initial oxidative cleavage of carbohydrate diols with NaIO4, followed by hydrazone ligation of the resultant aldehyde.82,83 Oxime linked ADCs have also been reported;53,84 these constructs display both in vitro63 and in vivo81 cytotoxicity. More unconventional uses of oxime ligations strategies involve linking protein species together for the design of new therapeutics.26,85 A review on oxime chemistry within the field of aldehyde handle labelling has very recently been published.86
 |
| | Scheme 9 (A) Outline of hydrazone ligation on a generic protein bearing an aldehyde handle. (B) Outline of oxime ligation. (C). Structure of hydrazone/oxime bond on aldehyde handles that can be incorporated into proteins. | |
Hydrazone and oxime ligations are undoubtedly the most frequently used method for modifying protein aldehyde/ketone handles; indeed, the aforementioned commercially available probes for modifying aldehyde handles are all hydrazine and hydroxylamine-based. However, these strategies are not without their drawbacks. Both ligation strategies result in formation of a CN bond between protein and substrate, and thus both are susceptible to hydrolysis over time. Of the two, oxime bonds display greater hydrolytic stability.87 Reductive amination of hydrazone bonds using sodium cyanoborohydride to form a more stable amine bond can be employed; however, this strategy is not commonly used within the context of modifying protein aldehydes (sodium cyanoborohydride is a water sensitive reagent and its use can lead to undesirable reduction of disulfide bonds88). Additionally, the optimal pH for hydrazone and oxime ligations is pH 4.5, with poor reactivity observed at neutral pH, typically displaying rate constants of 10−3 M−1 s−1.70,89 To address this, several catalysts for performing hydrazone and oxime ligation at neutral pH have been investigated; aniline in particular has received a great deal of attention in the area of hydrazone/oxime ligation catalysis. For hydrazone ligations of peptide substrates in 10 mM anilinium buffer, a 20 fold increase in ligation rate is observed at pH 4.5, and a 70 fold increase in ligation rate is observed at pH 5.7 (Table 1, entry 1).65 Similarly, for oxime ligations of peptide substrates in 100 mM anilinium buffer solutions, a 400 fold increase in ligation rate is observed at pH 4.5, and a 40 fold increase in ligation rate is observed at pH 7.0.68 Ligations between aryl aldehydes and 6-hydrazinopyridyl reagents are of particular note, displaying rate constants within the range of 102–103 M−1 s−1 (Table 1, entry 3).67 The use of aniline in protein bioconjugation at a neutral pH has subsequently been reported.26,60 Other aniline-based catalysts have also been reported, with meta-substituted anilines (Table 1, entry 5)69 and para-substituted anilines (Table 1, entry 6). displaying improved kinetics to that of aniline for oxime ligations at pH 7.0.70 In all cases, the catalysis is hypothesised to proceed via a ‘transimination’ type mechanism.68 In very particular cases, aniline does not appear to improve conjugation yields of oxime ligations.85 Additionally, it has been reported that typical concentrations of aniline used in oxime ligation strategies (i.e. 100 mM aniline)26,68 exhibit toxicity in live cell labelling experiments.60 Other catalysts include boronic acids90 and 2-aminophenols/2-(aminomethyl)benzimidazoles.91
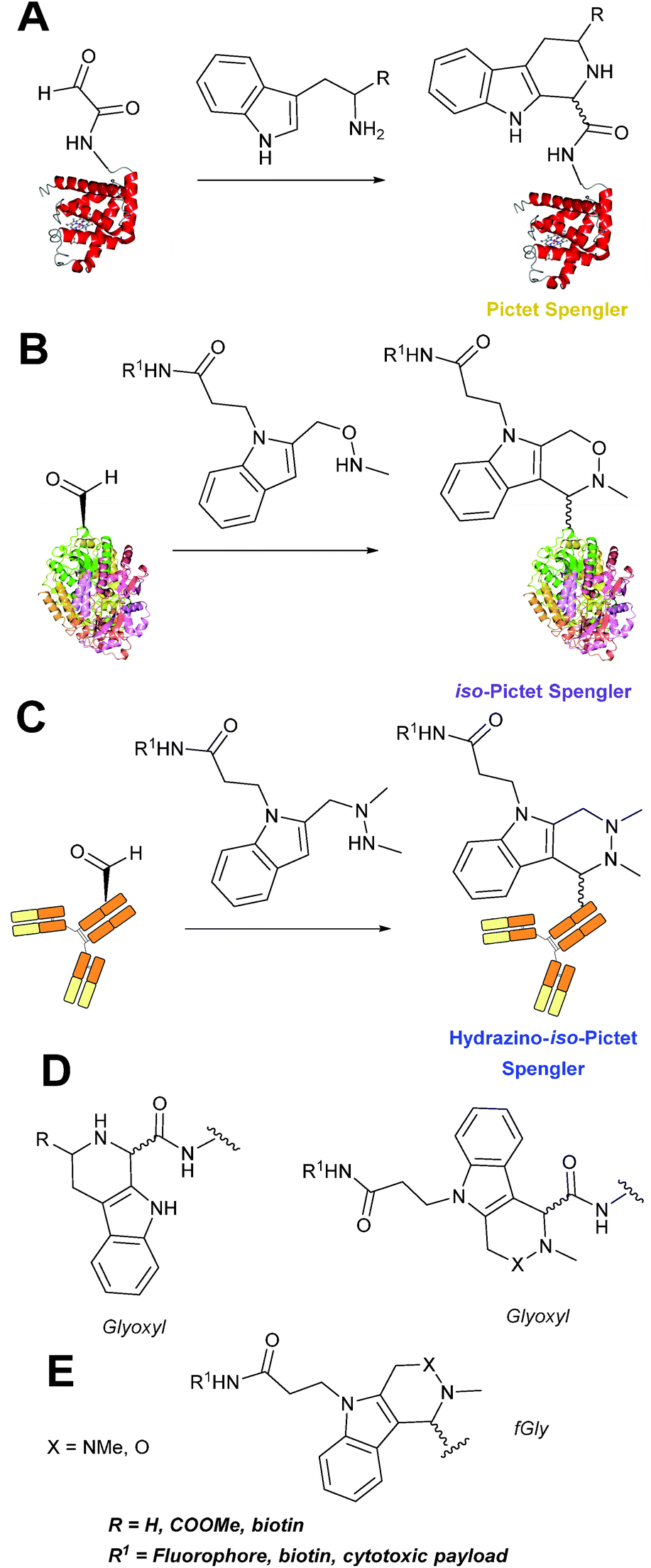
Pictet-Spengler
The cyclisation reaction between tryptamines and aldehydes is known as the Pictet-Spengler reaction. Formation of the Pictet-Spengler product occurs through initial iminium formation between the tryptamine amine and aldehyde, followed by intramolecular cyclisation. In the context of protein modification, an N-terminal Pictet-Spengler ligation on myoglobin has been reported (Scheme 10A, Table 1, entry 8).72 Following transamination with PLP,27 the Pictet-Spengler ligation between glyoxylamide-containing myoglobin (glyoxyl-myoglobin), and tryptamine, tryptophan methyl ester, and tryptamine-functionalised-biotin was successfully carried out at pH 6.5 (confirmed by MS and subsequent proteolytic digest experiments), leading to protein constructs ligated to substrates via a C–C bond.72
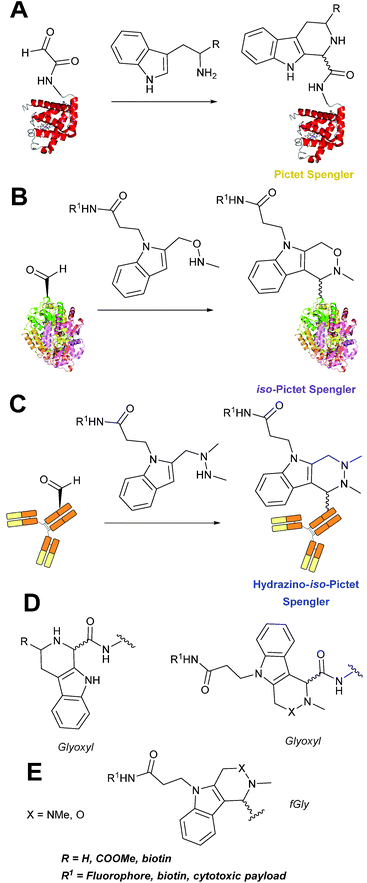 |
| | Scheme 10 (A) The Pictet-Spengler ligation on glyoxyl-myoglobin. (B) The iso-Pictet Spengler ligation on fGly-tagged maltose binding protein. (C). The Hydrainzo-iso-Pictet Spengler ligation on an fGly-tagged antibody. (D). The Pictet Spengler (left) and iso-Pictet/Hydrazino-iso-Pictet Spengler ligation at a glyoxyl handle. (E). The iso-Picet Spengler/Hydrazino-iso-Pictet Spengler ligation at an fGly handle. | |
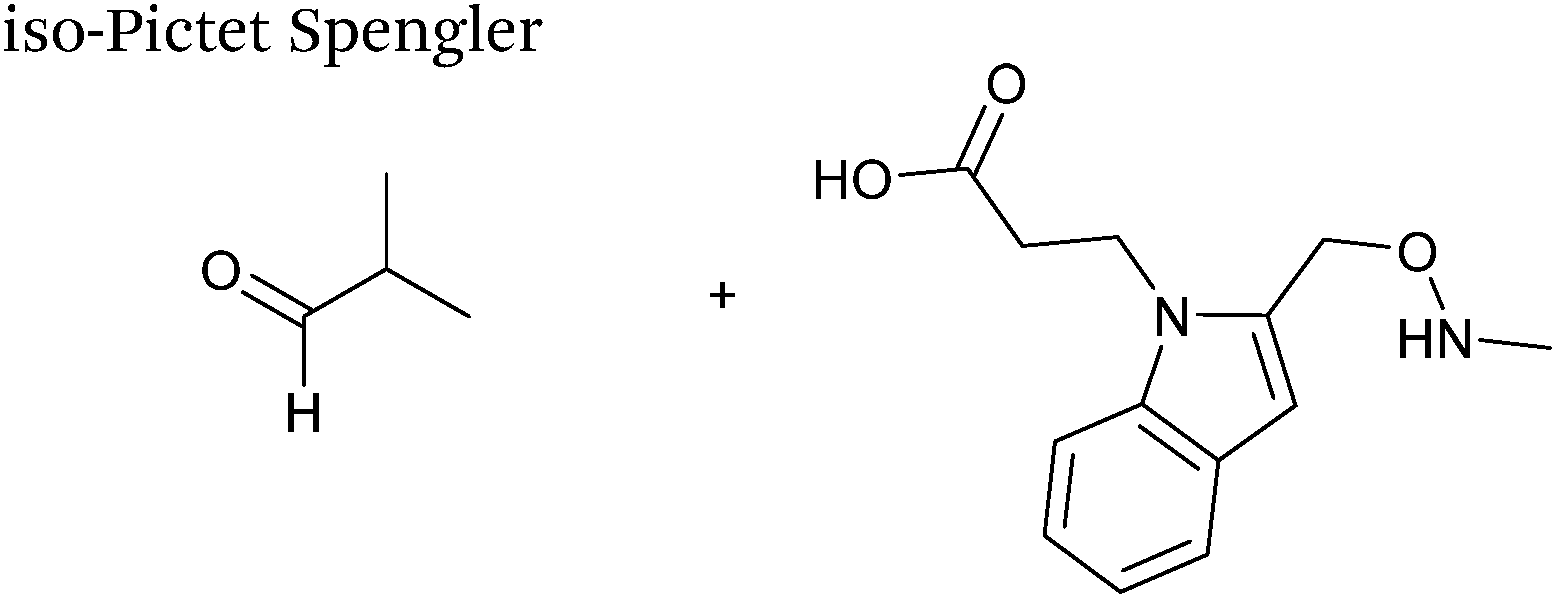
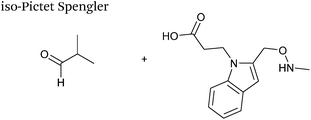
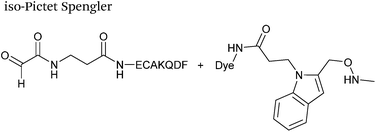
A variation of the Pictet-Spengler ligation, known as the iso-Pictet-Spengler ligation, has recently been reported for chemical modification of aldehyde handles incorporated into proteins (Scheme 10B).37 Analysis of the reaction kinetics of the traditional Pictet-Spengler ligation was used as the basis for the design of the iso-Pictet-Spengler ligation. Aminooxy-functionalised indoles (as opposed to tryptamines) were utilised to accelerate the initial iminium formation between the indole and the aldehyde tag. Additionally, the aminooxy side chain was located on the 2-position of the indole ring (as opposed to the 3 position which is the case for tryptamine reagents) to accelerate the final cyclisation step. Based on an isobutyraldehyde system (Table 1, entry 9), the optimal pH for the reaction was found to be pH 4.5 (rate constant of 10.6 M−1 s−1), although product formation is still observed at pH 6.0–7.0 (observed rate constants of 0.76 M−1 s−1 at pH 6.0 and 0.26 M−1 s−1 at pH 7.0).37 For glyoxyl systems, reduced rate constants for the iso-Pictet Spengler ligation have since been reported (Table 1, entry 10).71 The use of an aniline catalyst had no effect on the rate of reaction (and was noted to be inhibitory at pH 5.0–6.5). Iso-Pictet-Spengler ligations with PEGylated, biotinylated, and fluorescent probes were successfully demonstrated on glyoxyl-myoglobin, fGly-tagged maltose binding protein (MBP), and fGly-tagged Herceptin antibodies.37 Compared to the original Pictet-Spengler ligation,72 both reagent concentrations and reaction time are vastly reduced in the iso-Pictet-Spengler ligation.37
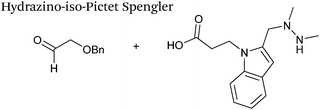
A minor alteration to the iso-Pictet-Spengler ligation, known as the Hydrazino-iso-Pictet-Spengler (HIPS) ligation, has also been described in the literature (Scheme 10C).38 The concept of the ligation strategy is nearly identical to that of the iso-Pictet-Spengler ligation – except an N-methyl hydrazine-functionalised indole is used instead of an aminooxy functionalised indole. This facilitates the ligation at a higher pH of 6.0 (as hydrazines are more reactive than aminooxy compounds at a higher pH).92 The HIPS ligation shows improved ligation kinetics to that of the iso-Pictet-Spengler ligation at pH 6.0 (observed rate constants of 4.2 M−1 s−1 using a benzyloxyacetaldehyde system, Table 1, entry 11), and has subsequently been utilised in the synthesis of ADCs.39
Strain-promoted alkyne-nitrone cycloaddition
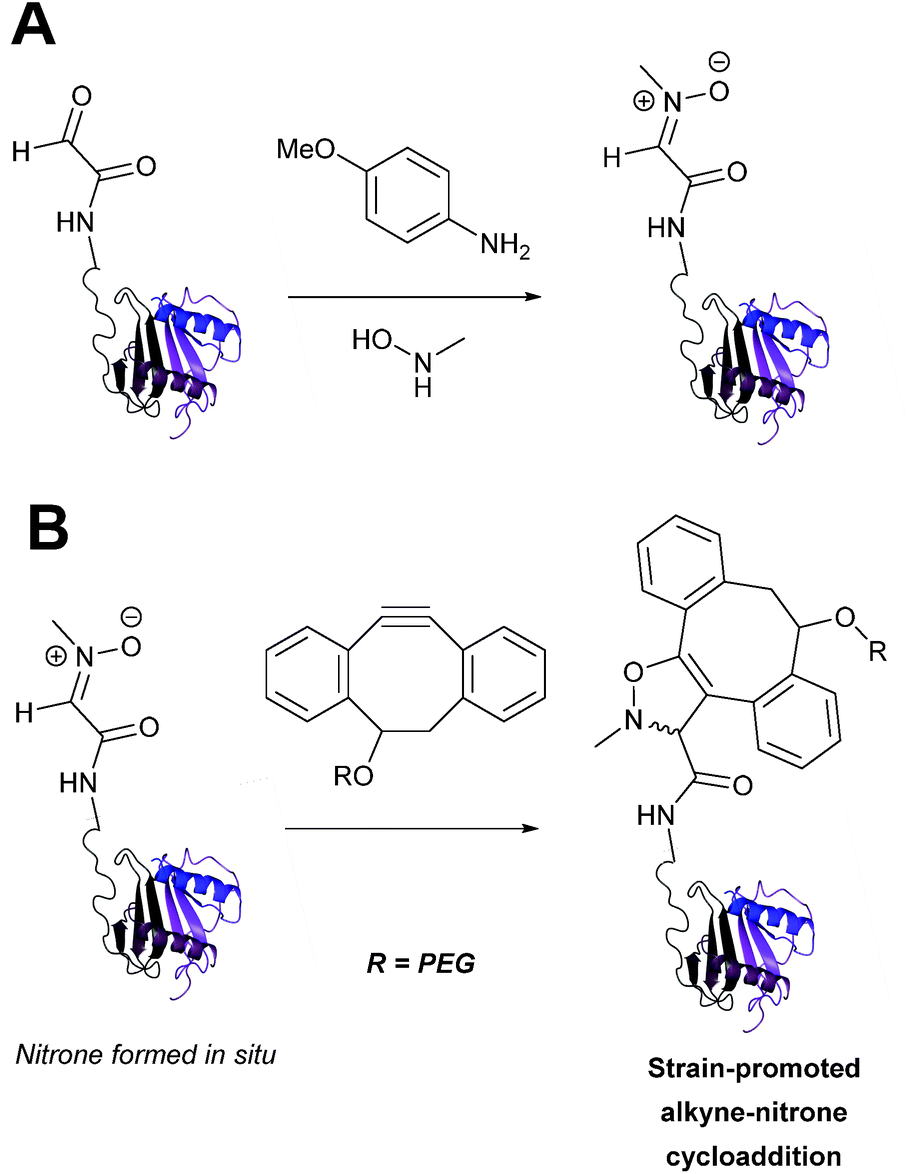
Alkyne reagents are frequently employed in strategies to selectively modify proteins.93 Typically, an azide handle is installed into a protein, and then subsequently reacted with the alkyne species to give the corresponding triazole in a click reaction. Reactions can be performed either using standard alkyne compounds with a copper catalyst, or can be performed without a catalyst by using strained alkyne systems such as cyclooctyne in a ‘strain promoted’ reaction (known as the Strain-Promoted Azide–Alkyne Cycloaddition, or SPAAC, ligation).12 The latter is more applicable to biological systems, since it does not require the use of copper which exhibits cytotoxicity in cells at high concentrations. Alkyne mediated ligation strategies are commonly utilised in metabolic labelling experiments due to their bioorthogonality.3
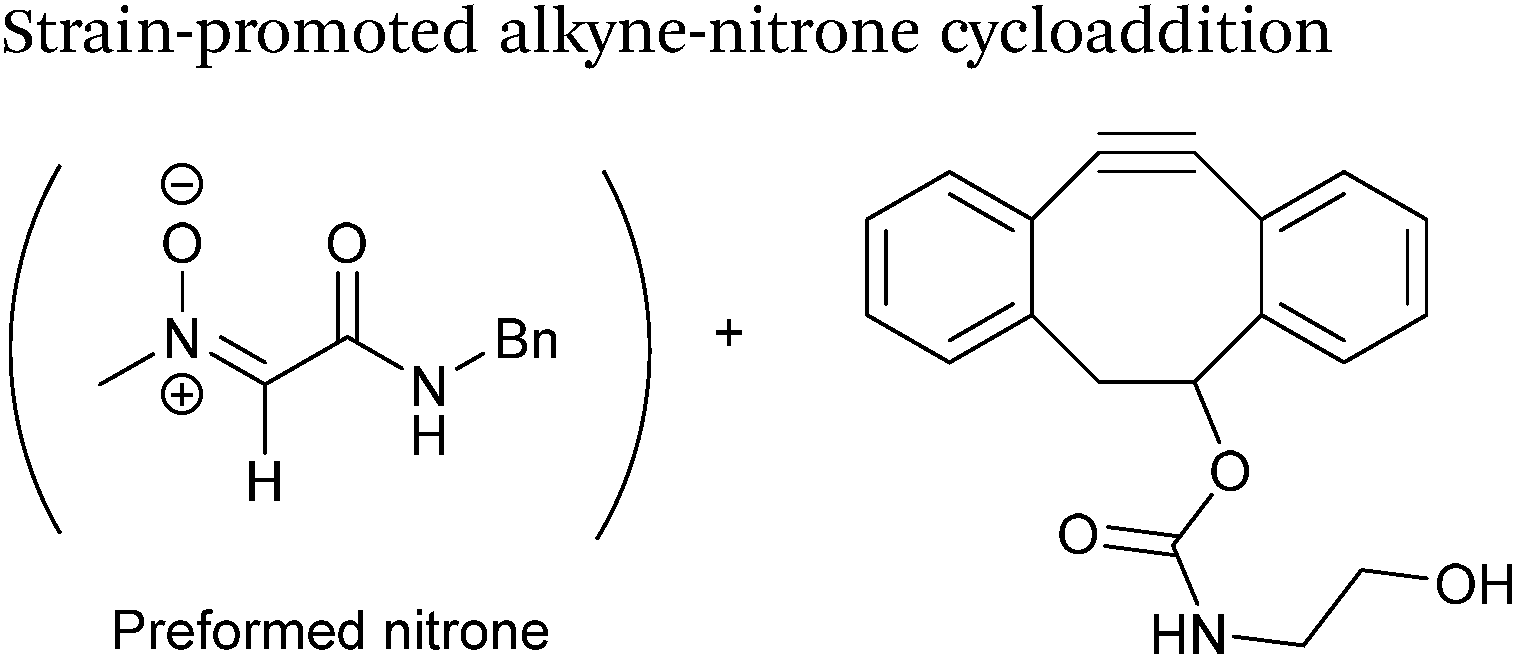
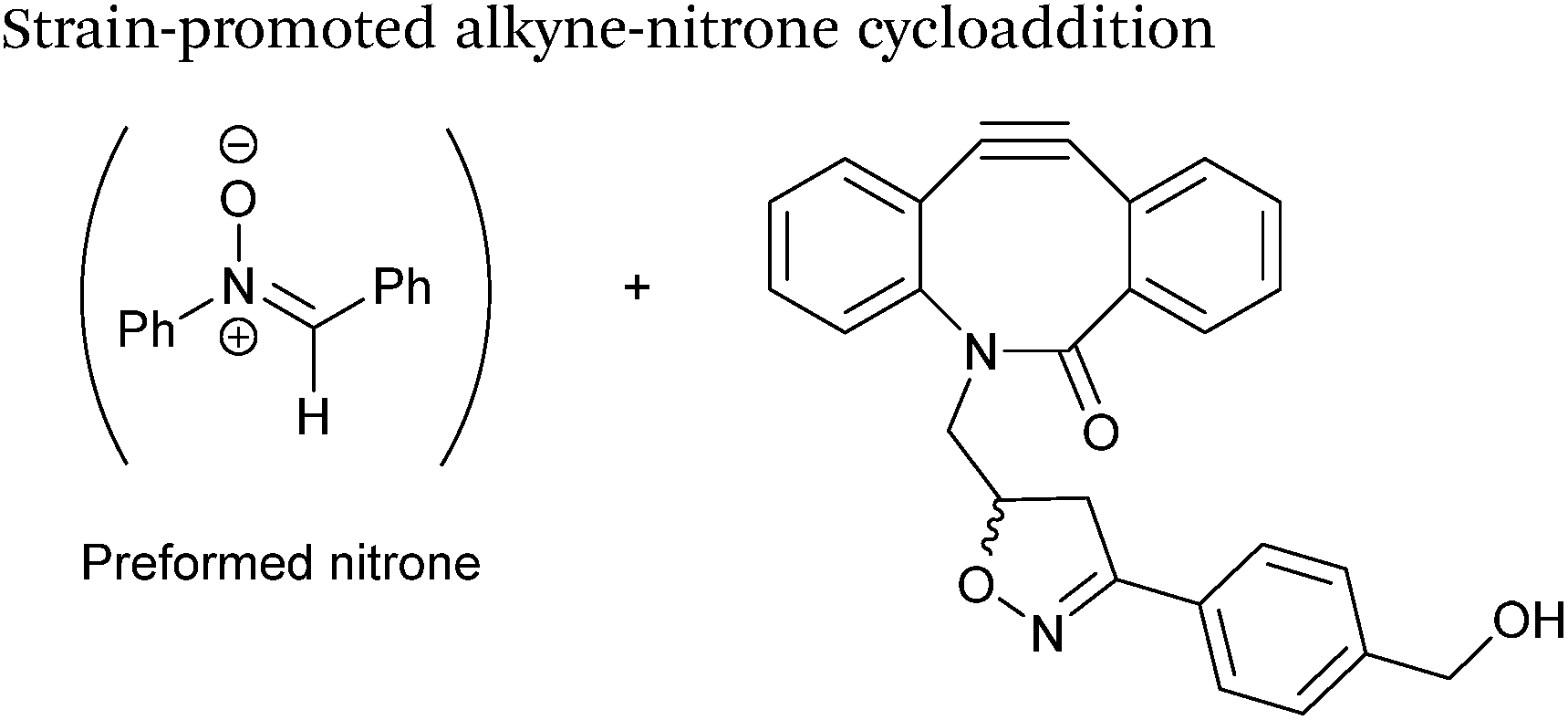
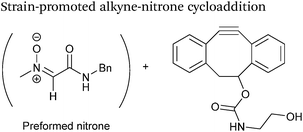
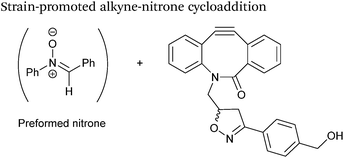
Alkynes can also be used to site-selectively modify glyoxyl-proteins via the Strain-Promoted Alkyne-Nitrone Cycloaddition (SPANC) ligation (Scheme 11). The SPANC ligation is a one-pot modification strategy that involves formation of a nitrone species between the aldehyde and N-methylhydroxylamine, followed by a cycloaddition reaction between the nitrone and strained cyclooctyne (see Table 1, entries 12 and 13 for examples with small molecules).73 The addition of p-anisidine greatly enhances the rate of the SPANC ligation – presumably by a mechanism that is akin to catalysis of hydrazone and oxime ligations. The SPANC ligation is usually performed at pH 6.8 (NH4OAc buffer) and has been used to site-selectively modify the oxidised chemokine interleukin 8 (glyoxyl-IL-8) with a 2 kDa PEG unit.73 Dual functionalisation of glyoxyl-peptides/proteins incorporating both azide–alkyne click chemistry and the SPANC ligation has also been reported.94 Additionally, SPANC ligation has been utilised in the design of antibody-nanoparticle constructs to design multivalent nanoparticles (MNPs) using N-terminal serine variant of an anti-HER2 scFv antibody; these MNPs can then be used to selectively, fluorescently label HER2 membrane receptors in breast cancer cells.95 A variation of the SPANC ligation using preformed nitrones has also been utilised in metabolic labelling of live cells (although these nitrones are susceptible to hydrolysis in aqueous systems).96 Preformed nitrones have also been the subject of various kinetic studies, identifying biaryl-aza-cycloctyne73 and acyclic nitrones systems74 as yielding the highest rate constants for the SPANC (58.8 M−1 s−1). A review for the SPANC ligation has recently been published.97
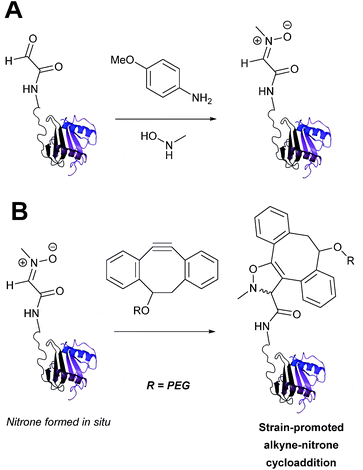 |
| | Scheme 11 (A) Glyoxyl-interleukin 8, and formation of a nitrone species. (B) Reaction of an alkyne with the pre-formed nitrone via a cycloaddition. | |
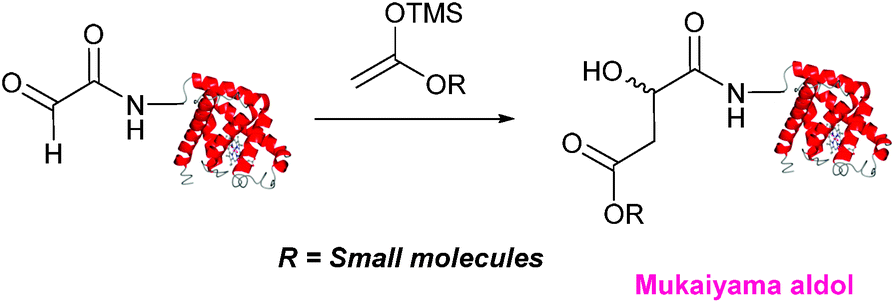
Mukaiyama aldol
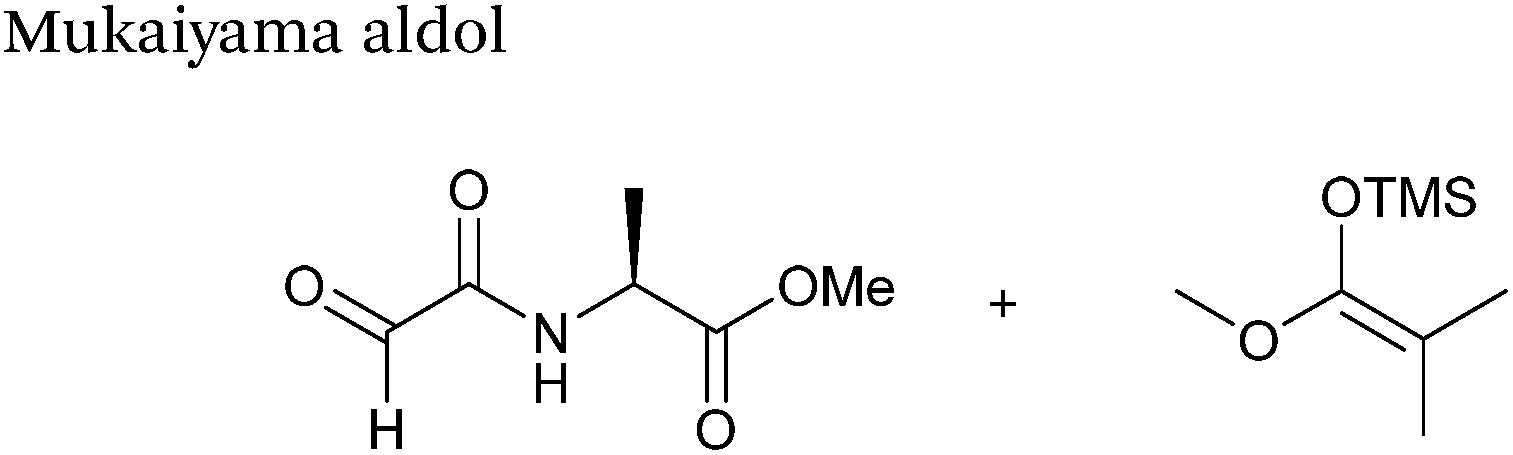
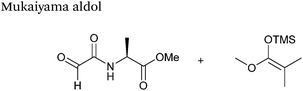
The Mukaiyama aldol reaction involves the reaction of an aldehyde group and silyl enol ether to give β-hydroxy ketone products. The reaction forms a C–C bond between the aldehyde and silyl enol ether, is catalysed by Lewis acids (such as TiCl4) and can be performed in aqueous solvent systems (see ref. 98 for a review on the subject). A Mukaiyama aldol ligation for peptide and protein modification was first reported in 2010 (Scheme 12).75 Glyoxyl-peptides were functionalised with various silyl enol ethers in aqueous conditions to afford the anticipated Mukaiyama aldol ligation products after 24 hours (see Table 1, entry 14 for an example). Products bearing alkene functionality could then be modified further by cross metathesis using a Hoveyda-Grubbs second generation catalyst. The Mukaiyama aldol ligation has been utilised to modify glyoxyl-myoglobin, with a maximum of 90% conversion observed as (judged by LC-MS) in a H2O:t-BuOH (5:1) solvent system (Table 1, entry 15). Choice of solvent system for the Mukaiyama aldol ligation is of particular importance; 1:1 systems such as H2O:t-BuOH, pH 7 PB:EtOH, and H2O:MeCN all lead to homogenous product formation (76–90% conversion as judged by LC-MS), whereas other solvent systems such as H2O:glycerol and H2O:etheylene glycol lead to no observed conversion to the Mukaiyama aldol ligation product, or solvent systems such as pH 7 PB:EtOH (4:1), pH 7 PB:t-BuOH (1:1), and H2O:t-BuOH (6:1) lead to heterogeneous product mixtures. UV-vis analysis and circular dichroism (CD) analysis of unmodified and modified myoglobin samples confirmed that the tertiary structure of the protein was unaffected by the Mukaiyama aldol ligation.75
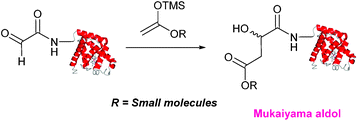 |
| | Scheme 12 Outline of a Mukaiyama ligation on glyoxyl-myoglobin. | |
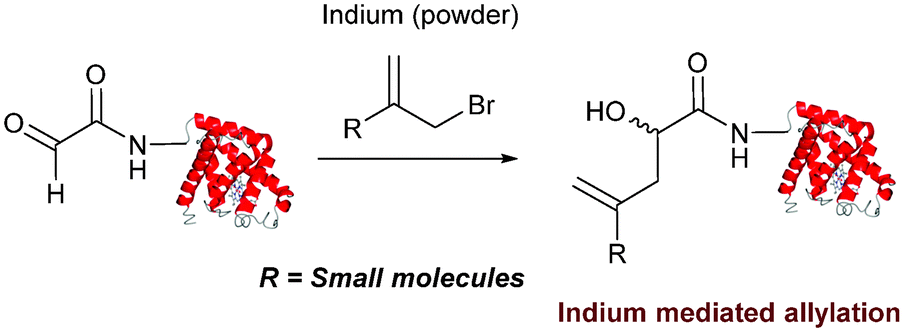
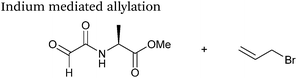
Indium mediated allylation
An indium mediated allylation within the context of modifying glyoxyl peptides and proteins with simple allyl bromides has also recently been reported (Scheme 13).76 Glyoxyl-peptides were modified by indium mediated allylation with a number of allyl bromides to give racemic mixtures of the desired products after 18 hours (Table 1, entry 16). The ligation results in formation of a C–C bond and has also been demonstrated on glyoxyl-myoglobin, affording a maximum of 54% conversion after 20 hours to the desired product (judged by LC-MS analysis, Table 1, entry 17). A 6:4 H2O:t-BuOH solvent system and sonication of the reaction mixture was utilised to achieve conversion to the desired product (these conditions cause partial dissociation of the haem group of myoglobin, although it is possible to reconstitute the protein following modification).76
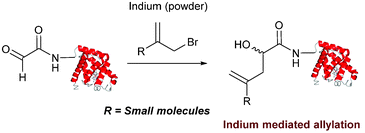 |
| | Scheme 13 Outline an indium mediated allylation of glyoxyl-myoglobin. | |
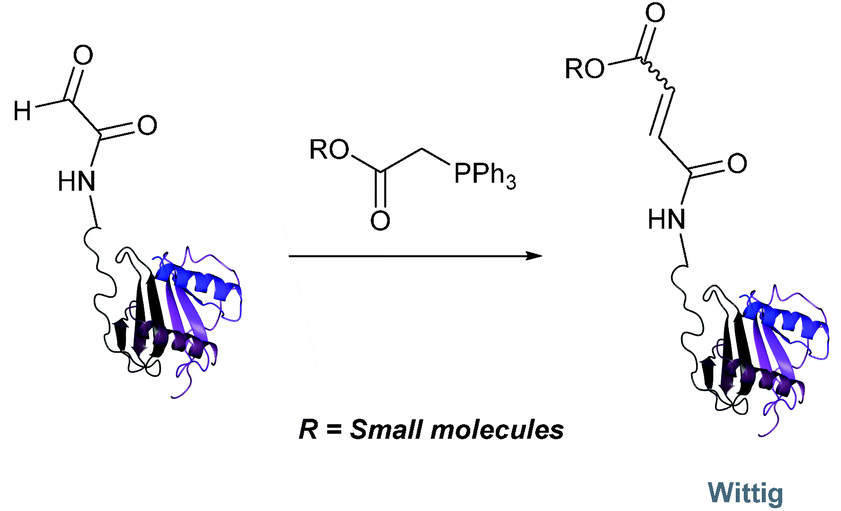
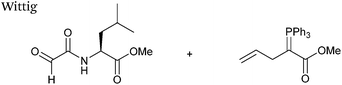
Wittig
The Wittig reaction, first reported in 1954, is the reaction between aldehydes and ylide-type reagents to give alkene products.99 Modification of glyoxyl peptides and proteins via a Wittig ligation have been recently demonstrated (Scheme 14).77 Glyoxyl-peptides were successfully modified with a number of small molecule ylides in a 1:1 H2O:t-BuOH solvent system, generating geometric isomers of the desired Wittig products (see Table 1, entry 18 for an example). Glyoxyl-myoglobin and glyoxyl-IL-8 were also modified using this chemistry in a 3:1 H2O:t-BuOH solvent system. The ligation can also be performed in pH 7–10 buffer:t-BuOH systems, but does not occur in buffer systems below pH 6. The Wittig ligation results in formation of a C–C bond, and gives a 65% conversion complete within 30 minutes on glyoxyl-myoglobin (judged by LC-MS analysis), (Table 1, entry 19). UV-vis analysis, CD analysis, and analysis of the proteins ability to store and release oxygen were performed on unmodified and modified myoglobin samples, confirming that the tertiary structure of the protein was unaffected.77 Other applications of Wittig olefinations within a bioorthogonal context include the labeling of the FK506 macrolide in aqueous media with biotin and fluorophores (the modified macrolide can then be used to label F506-binding protein FKBP12 with biotin and fluorophores),100 and the synthesis of alkenes in living tissue.101
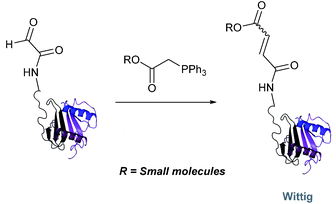 |
| | Scheme 14 Outline of Wittig ligation of glyoxyl-interleukin 8. | |
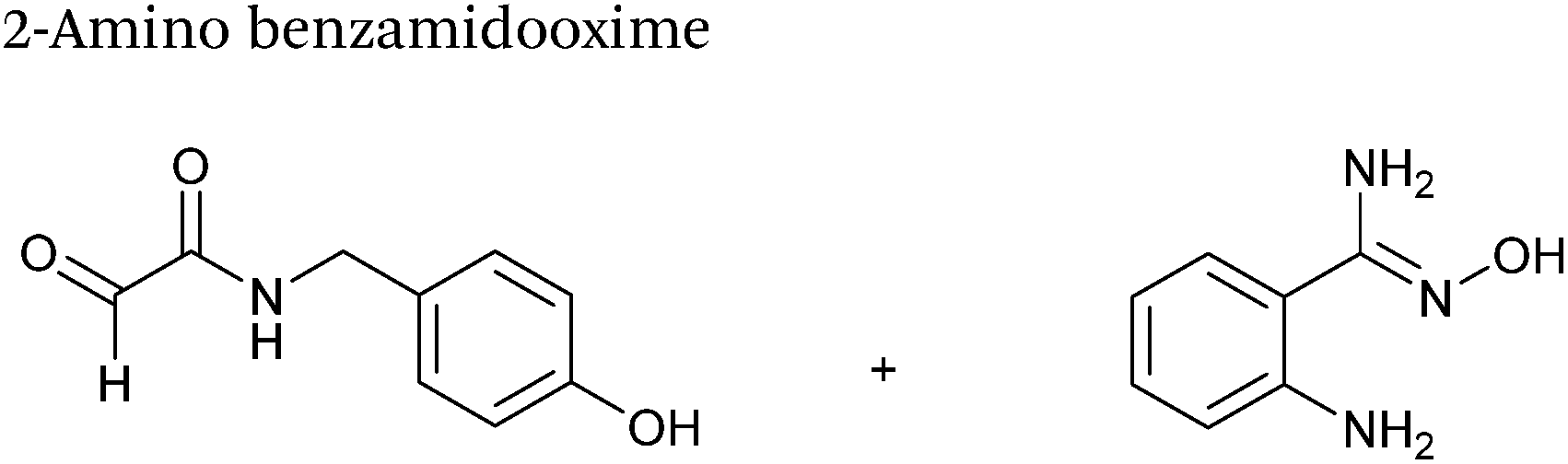
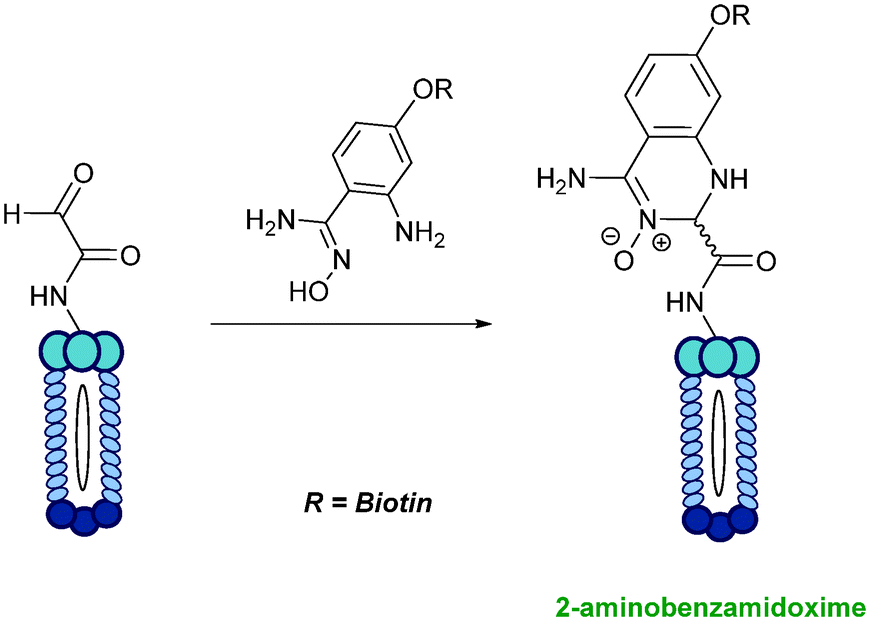
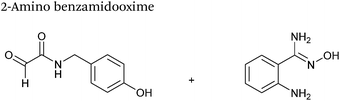
2-Amino benzamidoxime
The use of 2-amino benzamidoxime (ABAO) compounds in the modification of peptides and proteins bearing aldehyde handles has been very recently described (Scheme 15).78 Initial screening of a variety of substituted aniline compounds against model glyoxyl substrates identified ABAO compounds as suitable reagents for site-selective modification, giving rise to single, hydrolytically stable dihydroquinazoline products which exhibit fluorescence at 490 nm. The ligation is performed under acidic conditions (pH 4.5), and is thought to proceed through via imine formation between the aldehyde substrate and aniline component. This is followed by intramolecular cyclisation via attack of the ABAO ortho amidoxime group at the newly formed imine, completing the ligation. The rate of ABAO ligation can vary depending on the both the choice of aldehyde and ABAO derivative; in particular, higher rate constants are observed in isobutyraldehyde systems compared to glyoxyl systems (40 M−1 s−1 compared to 1 M−1 s−1, Table 1, entries 21 and 22), and also in para-methoxy ABAO systems (Table 1, entry 20). Acetate (from the NaOAc buffer) can also act as a weak catalyst in the ABAO ligation (as observed from altering buffer concentrations). Notably, ABAO ligation is slow when the aldehyde substrate in question is glucose (in a ring opened form), suggesting that ABAO ligation could be expanded to other selective labelling applications in more complex biological mixtures. The ABAO ligation has been used in the biotinylation of horseradish peroxidase (HRP) bearing aldehydes derived from oxidised glycans, and additionally in the biotinylation of glyoxyl-peptide bearing M13 phage libraries.78
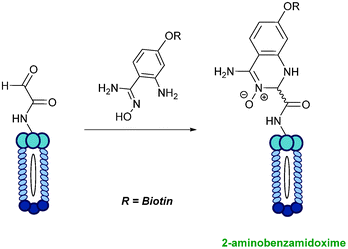 |
| | Scheme 15 Outline of 2-amino benzamidoxime ligation on M13 phage bearing glyoxyl-peptides. | |
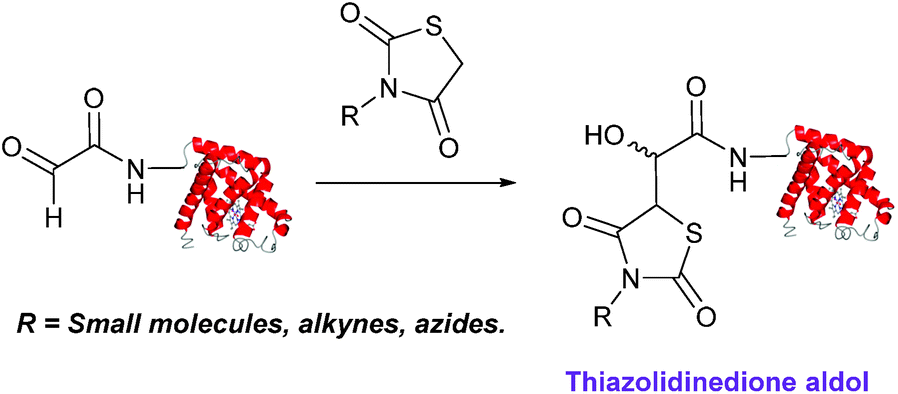
Thiazolidinedione aldol
Thiazolidinedione compounds have been described as acting as donor compounds in uncatalysed aldol reactions.102 This methodology has very recently been extended to peptide and protein modification (Scheme 16).79 The ligation was demonstrated on C-protected glyoxyl-dipeptides with a number of simple thiazolidinediones bearing various functionality such as alkyne, azide and short chain PEG moieties. Additionally, modification of glyoxyl-myoglobin can be achieved in a 5:1 pH 6.5 PB:t-BuOH solvent system with conversions of 66–83% observed within 3 hours at 37 °C (judged by LC-MS analysis, Table 1, entry 24). In all cases, high concentrations of thiazolidinedione reagent were utilised (1000 molar equivalents with respect to glyoxyl-myoglobin concentration). UV-vis analysis, CD analysis, and analysis of the proteins ability to store and release oxygen were performed on unmodified and modified myoglobin samples, confirming that the tertiary structure of the protein was unaffected.79 Observed reaction kinetics for the Thiazolidinedione aldol ligation were 0.0078 M−1 s−1 (Table 1, entry 23),79 which are comparable to that of uncatalysed hydrazone and oxime ligations at pH 7.0–7.4.66,70
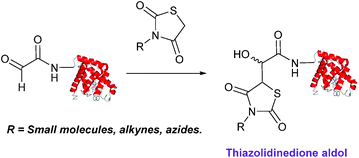 |
| | Scheme 16 Outline of modification of glyoxyl-myoglobin with thiazolidinediones. | |
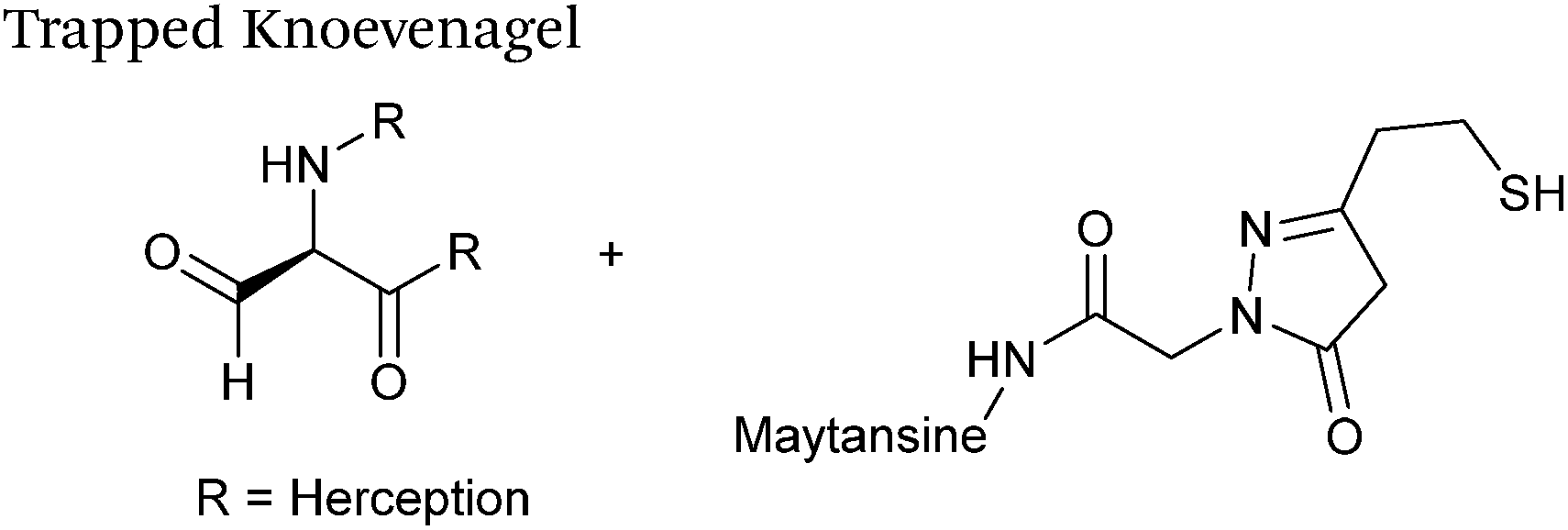
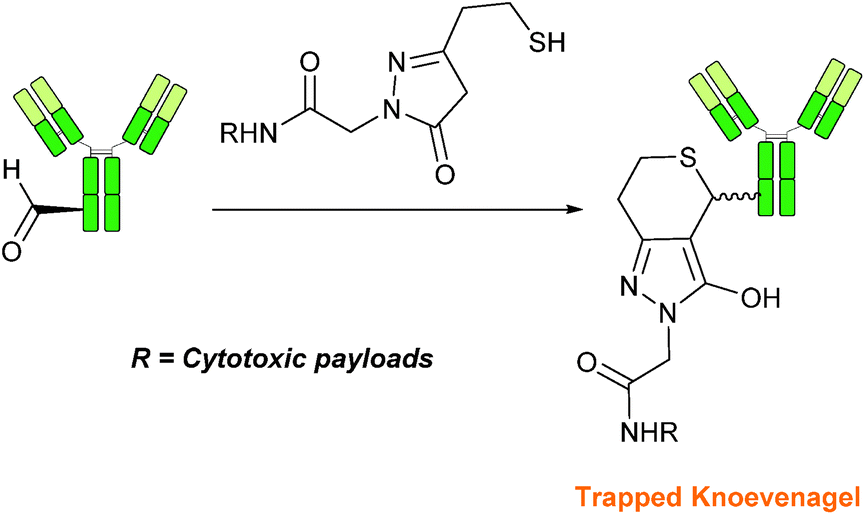
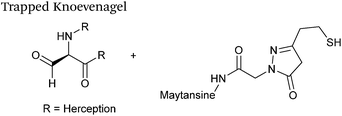
Trapped-Knoevenagel
A Knoevenagel condensation reaction between fGly peptides/antibodies and compounds containing enolisable protons has very recently been described (Scheme 17). In this ligation, thiopyrazolone reagents are subjected to the fGly species to afford C–C linked tetrahydrothiopyranopyrazolone products.40 The reaction occurs via initial enone formation between the aldehyde and thiopyrazolone, followed by intramolecular cyclisation through Michael-type addition of the thiol side chain onto a transiently generated enone (for this reason, the ligation is also referred to as a ‘trapped’ Knoevenagel ligation). This ligation strategy proceeds at pH 7.0–7.2 and has been validated on benzyloxyacetaldehyde, benzaldehyde and fGly functionalised peptides. The Trapped-Knoevenagel ligation has also been explored in the synthesis of ADCs through the tagging of fGly functionalised Herceptin with thiopyrazole-functionalised maytansine; ligation of these species exhibits a rate constant of 0.387 M−1 s−1 (Table 1, entry 25), and the resulting constructs display both in vitro and in vivo cytotoxicity.40
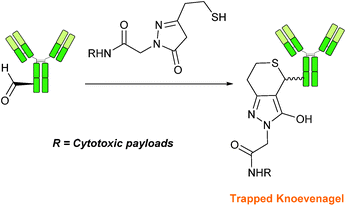 |
| | Scheme 17 Outline of the Trapped-Knoevenagel ligation on fGly-tagged Herceptin. | |
IV. Conclusions and outlooks
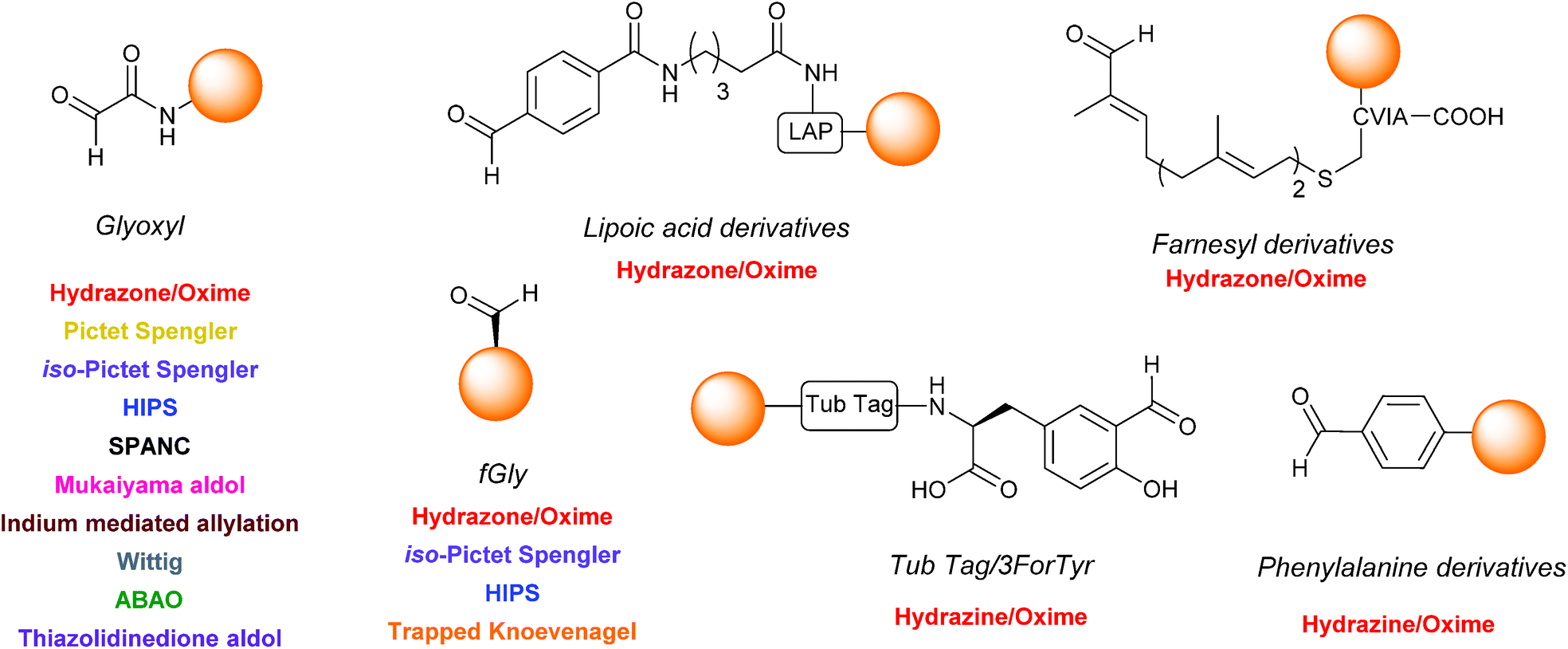
Over the past 25 years (and particularly within the last decade), the site-selective modification of aldehyde handles incorporated into proteins has been at the forefront of the field of chemical biology, with aldehydes proving to be a highly versatile method of generating homogenous protein conjugates with wide ranging applications. Although the aldehyde (and by extension, ketone) handle is sometimes referred to as a ‘biorestricted’ (due to difficulties in achieving site-selective modification in biological environments where other reactive carbonyls such as cofactors are present, or where acidic conditions cannot be used),103,104 this innovation drive has also lead to the development of new methods for labelling protein aldehyde which are functional in complex biological mixtures.41,48
As the number of methods available for both incorporation and modification of protein aldehydes continues to increase, it is therefore beneficial to consider potential areas for development in the field to further augment the existing ‘toolbox’. Firstly it would be advantageous to establish a hierarchy of privileged reaction partners which would allow non-specialists in the field to easily determine which ligation strategy is most appropriate for a specific experimental requirement i.e. which ligations afford either highest conversions (most important if pure protein conjugate is required), or fastest conversions (more important in in vivo studies) with each individual aldehyde partner e.g. glyoxyl, fGly, aryl aldehydes etc. To unequivocally establish these formulations, it is necessary for ligations to be trialled experimentally with a number of diverse aldehyde handles. Although oxime/hydrazine ligations have been widely deployed (see Table 1, entries 1–7), many other ligation strategies in the literature have, as yet, only been described for one type of aldehyde – typically a glyoxyl functionality (see Scheme 18), which currently makes effective comparison and profiling of the merits of each individual method a difficult proposition. On a related note, it is also important to continue on-going screening efforts to accelerate the rate of many of the ligations described. Although, the traditional hydrazone/oxime ligation is considered slow in comparison to alternative ‘click’ based bioorthogonal conjugation methods, especially at neutral pH, recent aniline catalyst explorations and rate profiling with aminooxy/hydrazine partners have facilitated significant rate enhancements, vastly increasing the general applicability of the ligation at neutral pH. Similarly, extensive screening of buffer conditions and reaction probe structure (as seen in the Pictet Spengler, SPANC and ABAO ligations) has also lead to increases in reaction rate and efficiency.
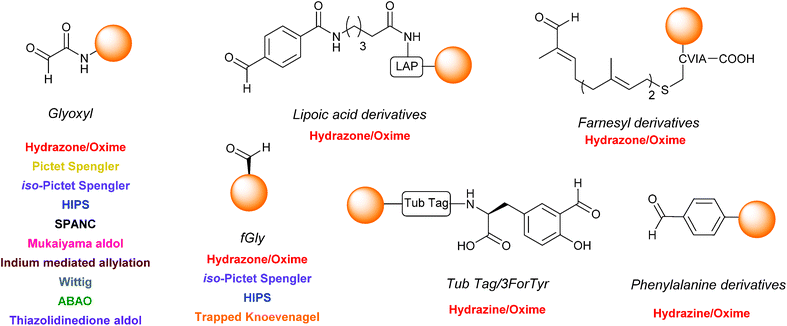 |
| | Scheme 18 The structures of the different aldehyde handles described in the literature that can be incorporated into proteins, and the ligation strategies which have (to date) been demonstrated upon a particular aldehyde handle. | |
It is also essential that efforts to explore new strategies for forming C–C bonds in aldehyde ligations continue. Although the efficiency of hydrazone/oxime ligations with diverse partners has justifiably established their position as the premier method for modifying protein aldehydes, the CN bond formed can be hydrolytically unstable in biological conditions. This is not the case for the C–C bond formed during other ligations described within this review, such as the Pictet Spengler series. These ligations can potentially generate more stable protein conjugates with longer half-lives, which potentially have more general biotechnological and translational applicability, as highlighted by recent syntheses of antibody-drug conjugates using Hydrazino-iso-Pictet Spengler ligations.105
Acknowledgements
The authors thank The University of York, and the European Commission for the award of a Marie–Curie Fellowship to M. A. F. (FP7-PEOPLE-2011-IOF-302246). This work was supported by an Engineering and Physical Sciences Research Council DTG Award to R. J. S.
References
- C. J. Fee, Biotechnol. Bioeng., 2007, 98, 725–731 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- P. Agarwal, B. J. Beahm, P. Shieh and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 11504–11510 CrossRef CAS PubMed.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CAS.
- A. Moosmann, J. Blath, R. Lindner, E. Müller and H. Böttinger, Bioconjugate Chem., 2011, 22, 1545–1558 CrossRef CAS PubMed.
- Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef CAS PubMed.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Brase, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- T. Groth and M. Meldal, J. Comb. Chem., 2001, 3, 45–63 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 6932–6940 CrossRef CAS PubMed.
- P. M. Levine, T. W. Craven, R. Bonneau and K. Kirshenbaum, Org. Lett., 2014, 16, 512–515 CrossRef CAS PubMed.
- C. L. Lee and X. Li, Curr. Opin. Chem. Biol., 2014, 22, 108–114 CrossRef CAS PubMed.
- C. E. Murar, F. Thuaud and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 18140–18148 CrossRef CAS PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS PubMed.
- B. Sklarz, Q. Rev., Chem. Soc., 1967, 21, 3–28 RSC.
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- C. B. Barlow, R. D. Guthrie and A. M. Prior, Chem. Commun., 1966, 268–269 RSC.
- J. Chen, W. Zeng, R. Offord and K. Rose, Bioconjugate Chem., 2003, 14, 614–618 CrossRef CAS PubMed.
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323–8327 CrossRef CAS PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- R. A. Scheck, M. T. Dedeo, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 11762–11770 CrossRef CAS PubMed.
- L. S. Witus, T. Moore, B. W. Thuronyi, A. P. Esser-Kahn, R. A. Scheck, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 16812–16817 CrossRef CAS PubMed.
- M. Zhang, X. Zhang, J. Li, Q. Guo and Q. Xiao, Chin. J. Chem., 2011, 29, 1715–1720 CrossRef CAS.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- K. S. Palla, L. S. Witus, K. J. Mackenzie, C. Netirojjanakul and M. B. Francis, J. Am. Chem. Soc., 2015, 137, 1123–1129 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- G. Lukatela, N. Krauss, K. Theis, T. Selmer, V. Gieselmann, K. von Figura and W. Saenger, Biochemistry, 1998, 37, 3654–3664 CrossRef CAS PubMed.
- J. E. Hudak, H. H. Yu and C. R. Bertozzi, J. Am. Chem. Soc., 2011, 133, 16127–16135 CrossRef CAS PubMed.
- E. L. Smith, J. P. Giddens, A. T. Iavarone, K. Godula, L.-X. Wang and C. R. Bertozzi, Bioconjugate Chem., 2014, 25, 788–795 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS PubMed.
- P. M. Drake, A. E. Albers, J. Baker, S. Banas, R. M. Barfield, A. S. Bhat, G. W. de Hart, A. W. Garofalo, P. Holder, L. C. Jones, R. Kudirka, J. McFarland, W. Zmolek and D. Rabuka, Bioconjugate Chem., 2014, 25, 1331–1341 CrossRef CAS PubMed.
- R. Kudirka, R. M. Barfield, J. McFarland, A. E. Albers, G. W. de Hart, P. M. Drake, P. G. Holder, S. Banas, L. C. Jones, A. W. Garofalo and D. Rabuka, Chem. Biol., 2015, 22, 293–298 CrossRef CAS PubMed.
- X. Shi, Y. Jung, L.-J. Lin, C. Liu, C. Wu, I. K. O. Cann and T. Ha, Nat. Methods, 2012, 9, 499–503 CrossRef CAS PubMed.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS PubMed.
- P. G. Holder, L. C. Jones, P. M. Drake, R. M. Barfield, S. Bañas, G. W. de Hart, J. Baker and D. Rabuka, J. Biol. Chem., 2015, 290, 15730–15745 CrossRef CAS PubMed.
- M. J. Appel and C. R. Bertozzi, ACS Chem. Biol., 2015, 10, 72–84 CrossRef CAS PubMed.
- C. Gauchet, G. R. Labadie and C. D. Poulter, J. Am. Chem. Soc., 2006, 128, 9274–9275 CrossRef CAS PubMed.
- U. T. T. Nguyen, J. Cramer, J. Gomis, R. Reents, M. Gutierrez-Rodriguez, R. S. Goody, K. Alexandrov and H. Waldmann, ChemBioChem, 2007, 8, 408–423 CrossRef CAS PubMed.
- M. Rashidian, J. K. Dozier, S. Lenevich and M. D. Distefano, Chem. Commun., 2010, 46, 8998–9000 RSC.
- M. Rashidian, J. M. Song, R. E. Pricer and M. D. Distefano, J. Am. Chem. Soc., 2012, 134, 8455–8467 CrossRef CAS PubMed.
- M. Rashidian, S. C. Kumarapperuma, K. Gabrielse, A. Fegan, C. R. Wagner and M. D. Distefano, J. Am. Chem. Soc., 2013, 135, 16388–16396 CrossRef CAS PubMed.
- J. K. Dozier, S. L. Khatwani, J. W. Wollack, Y.-C. Wang, C. Schmidt-Dannert and M. D. Distefano, Bioconjugate Chem., 2014, 25, 1203–1212 CrossRef CAS PubMed.
- S. Puthenveetil, D. S. Liu, K. A. White, S. Thompson and A. Y. Ting, J. Am. Chem. Soc., 2009, 131, 16430–16438 CrossRef CAS PubMed.
- J. D. Cohen, P. Zou and A. Y. Ting, ChemBioChem, 2012, 13, 888–894 CrossRef CAS PubMed.
- M. A. Gray, R. N. Tao, S. M. DePorter, D. A. Spiegel and B. R. McNaughton, ChemBioChem, 2016, 17, 155–158 CrossRef CAS PubMed.
- D. Schumacher, J. Helma, F. A. Mann, G. Pichler, F. Natale, E. Krause, M. C. Cardoso, C. P. R. Hackenberger and H. Leonhardt, Angew. Chem., Int. Ed., 2015, 54, 13787–13791 CrossRef CAS PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34–66 CrossRef CAS PubMed.
- L. Wang, Z. Zhang, A. Brock and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 56–61 CrossRef CAS PubMed.
- Y. Huang, W. Wan, W. K. Russell, P.-J. Pai, Z. Wang, D. H. Russell and W. Liu, Bioorg. Med. Chem. Lett., 2010, 20, 878–880 CrossRef CAS PubMed.
- A. Tuley, Y.-S. Wang, X. Fang, Y. Kurra, Y. H. Rezenom and W. R. Liu, Chem. Commun., 2014, 50, 2673–2675 RSC.
- A. Tuley, Y.-J. Lee, B. Wu, Z. U. Wang and W. R. Liu, Chem. Commun., 2014, 50, 7424–7426 RSC.
- H. Zeng, J. Xie and P. G. Schultz, Bioorg. Med. Chem. Lett., 2006, 16, 5356–5359 CrossRef CAS PubMed.
- H. Liu, L. Wang, A. Brock, C.-H. Wong and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 1702–1703 CrossRef CAS PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Tran, Y. Sun, T. Phuong, R. Barnett, B. Hehli, F. Song, M. J. DeGuzman, S. Ensari, J. K. Pinkstaff, L. M. Sullivan, S. L. Biroc, H. Cho, P. G. Schultz, J. DiJoseph, M. Dougher, D. Ma, R. Dushin, M. Leal, L. Tchistiakova, E. Feyfant, H.-P. Gerber and P. Sapra, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1766–1771 CrossRef CAS PubMed.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- P. Crisalli and E. T. Kool, J. Org. Chem., 2013, 78, 1184–1189 CrossRef CAS PubMed.
- A. Dirksen and P. E. Dawson, Bioconjugate Chem., 2008, 19, 2543–2548 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS PubMed.
- T. Sasaki, K. Kodama, H. Suzuki, S. Fukuzawa and K. Tachibana, Bioorg. Med. Chem. Lett., 2008, 18, 4550–4553 CrossRef CAS PubMed.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G.-J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065–3068 CrossRef CAS PubMed.
- C. S. McKay, M. Chigrinova, J. A. Blake and J. P. Pezacki, Org. Biomol. Chem., 2012, 10, 3066–3070 CAS.
- J. Alam, T. H. Keller and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 9546–9548 CrossRef CAS PubMed.
- J. Alam, T. H. Keller and T.-P. Loh, Chem. Commun., 2011, 47, 9066–9068 RSC.
- M.-J. Han, D.-C. Xiong and X.-S. Ye, Chem. Commun., 2012, 48, 11079–11081 RSC.
- P. I. Kitov, D. F. Vinals, S. Ng, K. F. Tjhung and R. Derda, J. Am. Chem. Soc., 2014, 136, 8149–8152 CrossRef CAS PubMed.
- P. Wang, S. Zhang, Q. Meng, Y. Liu, L. Shang and Z. Yin, Org. Lett., 2015, 17, 1361–1364 CrossRef CAS PubMed.
- D. Chelius and T. A. Shaler, Bioconjugate Chem., 2003, 14, 205–211 CrossRef CAS PubMed.
- S. A. Kularatne, V. Deshmukh, J. Ma, V. Tardif, R. K. V. Lim, H. M. Pugh, Y. Sun, A. Manibusan, A. J. Sellers, R. S. Barnett, S. Srinagesh, J. S. Forsyth, W. Hassenpflug, F. Tian, T. Javahishvili, B. Felding-Habermann, B. R. Lawson, S. A. Kazane and P. G. Schultz, Angew. Chem., Int. Ed., 2014, 53, 11863–11867 CrossRef CAS PubMed.
- D. J. O'Shannessy, M. J. Dobersen and R. H. Quarles, Immunol. Lett., 1984, 8, 273–277 CrossRef.
- C. A. C. Wolfe and D. S. Hage, Anal. Biochem., 1995, 231, 123–130 CrossRef CAS PubMed.
- P. Thompson, B. Bezabeh, R. Fleming, M. Pruitt, S. Mao, P. Strout, C. Chen, S. Cho, H. Zhong, H. Wu, C. Gao and N. Dimasi, Bioconjugate Chem., 2015, 26, 2085–2096 CrossRef CAS PubMed.
- J. E. Hudak, R. M. Barfield, G. W. de Hart, P. Grob, E. Nogales, C. R. Bertozzi and D. Rabuka, Angew. Chem., Int. Ed., 2012, 51, 4161–4165 CrossRef CAS PubMed.
- S. M. Agten, P. E. Dawson and T. M. Hackeng, J. Pept. Sci., 2016, 22, 271–279 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329–3333 RSC.
- D. Larsen, M. Pittelkow, S. Karmakar and E. T. Kool, Org. Lett., 2015, 17, 274–277 CrossRef CAS PubMed.
- T. A. Nigst, A. Antipova and H. Mayr, J. Org. Chem., 2012, 77, 8142–8155 CrossRef CAS PubMed.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS PubMed.
- R. P. Temming, L. Eggermont, M. B. van Eldijk, J. C. M. van Hest and F. L. van Delft, Org. Biomol. Chem., 2013, 11, 2772–2779 CAS.
- M. Colombo, S. Sommaruga, S. Mazzucchelli, L. Polito, P. Verderio, P. Galeffi, F. Corsi, P. Tortora and D. Prosperi, Angew. Chem., Int. Ed., 2012, 51, 496–499 CrossRef CAS PubMed.
- C. S. McKay, J. A. Blake, J. Cheng, D. C. Danielson and J. P. Pezacki, Chem. Commun., 2011, 47, 10040–10042 RSC.
- D. A. MacKenzie, A. R. Sherratt, M. Chigrinova, L. L. W. Cheung and J. P. Pezacki, Curr. Opin. Chem. Biol., 2014, 21, 81–88 CrossRef CAS PubMed.
- T. Kitanosono and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 3095–3118 CrossRef CAS PubMed.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318–1330 CrossRef.
- K. M. Lum, V. J. Xavier, M. J. H. Ong, C. W. Johannes and K.-P. Chan, Chem. Commun., 2013, 49, 11188–11190 RSC.
- D. McLeod and J. McNulty, Eur. J. Org. Chem., 2012, 6127–6131 CrossRef CAS.
- S. Paladhi, A. Chauhan, K. Dhara, A. K. Tiwari and J. Dash, Green Chem., 2012, 14, 2990–2995 RSC.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- A. E. Albers, A. W. Garofalo, P. M. Drake, R. Kudirka, G. W. de Hart, R. M. Barfield, J. Baker, S. Banas and D. Rabuka, Eur. J. Med. Chem., 2014, 88, 3–9 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence