
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenetic incorporation of 1,2-aminothiol functionality for site-specific protein modification via thiazolidine formation†
Xiaobao
Bi
,
Kalyan Kumar
Pasunooti
,
Ahmad Hussen
Tareq
,
John
Takyi-Williams
and
Chuan-Fa
Liu
*
Division of Structural Biology and Biochemistry, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore. E-mail: cfliu@ntu.edu.sg; Fax: +65 67953021
First published on 12th May 2016
Abstract
Here we report a new site-specific conjugation strategy to modify proteins via thiazolidine ligation. Proteins harbouring a 1,2-aminothiol moiety introduced by amber codon suppression technology could be modified chemoselectively with aldehyde-functionalized reagents, such as a biotin-labeled peptide or ubiquitin, under mild conditions to yield homogeneous biotinylated or ubiquitinated products.
Introduction
Genetic incorporation of unnatural amino acids into proteins provides an important tool to manipulate proteins with diverse tailor-made functionalities. So far many functional groups, such as azides, alkynes, ketones, anilines, alkenes, tetrazoles and pentafluoro azobenzene, have been introduced into proteins in a site-specific or residue-specific manner for various bioconjugation studies.1 These orthogonal functional groups don't exist in natural proteins and are used to modify protein by chemoselectively reacting with another probe bearing a complementary functionality. Naturally occurring 1,2-aminothiol (i.e., from an N-ter cysteine residue) can also be introduced into recombinant proteins in response to amber stop codon using amber codon suppression technology. By using native chemical ligation (NCL) of ubiquitin (G75) Cα–thioester with a genetically installed 1,2-aminothiol group on the side chain of a lysine residue, Chan and coworkers developed a strategy for site-specific protein ubiquitination.2 Chin and coworkers reported a robust site-specific protein labeling strategy which relies on the condensation reaction between 1,2-aminothiol and cyanobenzothiazole in aqueous media.3 Besides, 1,2-aminothiol can also react with various aldehydes under weakly acidic condition to form a stable thiazolidine ring.4,5 This facile chemistry has been utilized in different applications through selectively modifying peptides or proteins.6 However, these reported methods are only limited to N- or C-terminal protein modification. The strategy for labeling proteins at internal sites via thiazolidine ligation has not been explored yet. Here, using the method for unnatural amino acid incorporation, we aim to develop a new strategy to site-specifically modify proteins harbouring genetically incorporated 1,2-aminothiol via thiazolidine ring formation with aldehyde-functionalized labeling reagents (Scheme 1).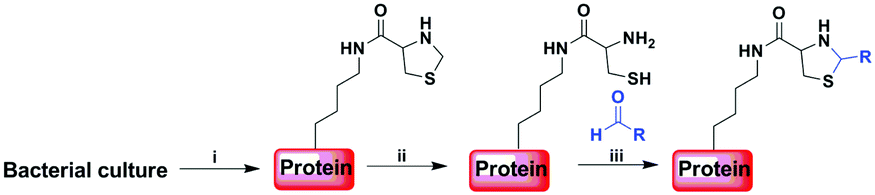 | ||
| Scheme 1 Strategy for site-specific modification of protein by combining amber codon suppression and thiazolidine ligation. (i) Genetic incorporation of 1,2-aminothiol into protein in E. coli cells transfected with genes for the ThzKRS/tRNA pair and a target protein containing an amber codon mutation. (ii) Deprotection of thiazolidine ring on the ThzK residue. (iii) Ligation via thiazolidine ring formation reaction between the 1,2-aminothiol and an aldehyde-functionalized reagent. | ||
Results and discussion
To demonstrate our strategy, we first synthesized the unnatural amino acid 2 carrying the masked 1,2-aminothiol group according to previously described method (Fig. 1A).3 The reason for the esterification of the carboxyl group of the lysine derivative 1 or ThzK is that the use of the ester form 2 can increase its cellular uptake and intracellular concentration and that the ester will be hydrolyzed in situ to regenerate 1.7 To test the genetic incorporation of ThzK into proteins via amber codon technology, we used the reported mutant Methanosarcina barkeri (Mb) Pyrrolysyl-tRNA synthetase/tRNACUA pair (ThzKRS/pylT) originally identified by Chin and coworkers, who found that this pair can efficiently recognize the unnatural amino acid 1.3 In our study, the gene encoding ThzKRS was inserted into a pEVOL-derived plasmid, which was known to be an optimized system that can afford higher yield of mutant proteins incorporated with the unnatural amino acid.8 Therefore, we constructed a plasmid pEVOL-pylT-ThzKRS bearing the genes encoding ThzKRS and pylT, in which two copies of ThzKRS were placed under the control of arabinose-inducible araBAD promoter and constitutive glnS′ promoter, respectively, and the pylT gene was flanked by the proK promoter and the proK terminator. The expression plasmid bearing a gene encoding ubiquitin with an amber mutation at position K48 or K63 and a 6XHis tag at the C-terminus was constructed from pETduet-1 vector (ESI†). The cotransformation of these two plasmids into E. coli BL21(DE3) cells and subsequent growth in LB medium supplemented with 1 mM 2 and 1 mM IPTG afforded full-length ubiquitin 3, which was confirmed by SDS-PAGE analysis (Fig. 1B). The ubiquitin 3 was obtained after purification by Ni-NTA chromatography in yields of 4–5 mg L−1. The deconvoluted electrospray ionization mass spectrometry (ESI-MS) spectrum of ubiquitin 3 revealed a single peak corresponding to the expected mass of the full-length protein incorporating ThzK (Fig. 1C). To deprotect ThzK in the recombinant protein, the above ubiquitin 3 was treated with 200 mM methoxylamine in 6 M guanidinium chloride at pH 4 for 4 h, 37 °C. After HPLC purification, ES-MS analysis showed that thiazolidine ring on ThzK was quantitatively cleaved to afford ubiquitin 4 (Fig. 1D).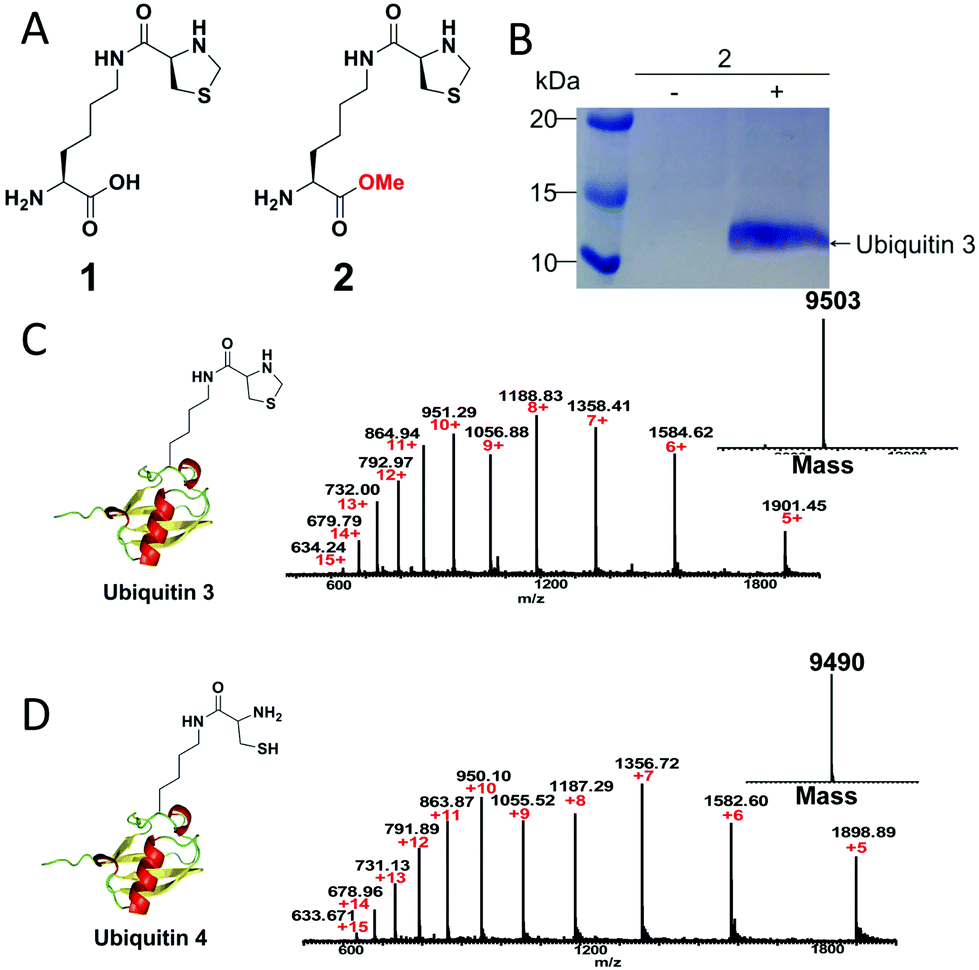 | ||
| Fig. 1 (A) Chemical structure of unnatural amino acid 1 and 2. 1 is reported by Chin and coworkers3 and 2 is studied in our work. (B) SDS-PAGE analysis of the expression of ubiquitin 3 with amber mutation at K48 in E. coli cells containing the ThzKRS/pylT pair in the presence of 2. (C) ESI-MS analysis of ubiquitin 3. Expected mass: 9502.91; observed mass: 9503. (D) ESI-MS analysis of ubiquitin 4. Expected mass: 9490.9; observed mass: 9490. | ||
Next, to test whether the 1,2-aminothiol of ubiquitin 4 can be modified by an aldehyde-functionalized reagent, we synthesized a short biotin-labeled peptide with an aldehyde group at its N-terminus (Fig. 2A). It is known that the reaction between an aldehyde and 1,2-aminothiol proceeds best at weakly acidic condition.4 But one study shows that thiazolidine ring formation can also be performed at neutral condition if aniline is added as the catalyst, which is good for proteins that are unstable under acidic conditions.9 So in our test, thiazolidine formation was performed in 200 mM phosphate buffer (pH 7) containing 10 mM aniline with 5, 10 and 20 equivalents of the aldehyde-functionalized peptide, respectively. The reaction was monitored by C4 analytic HPLC analysis. Results showed that a higher concentration of the aldehyde peptide reagent led to an increased yield of the desired product and ubiquitin 4 was near quantitatively converted into the desired product ubiquitin 5 after 8 h using 20 equivalents of the reagent (Fig. 2B). Furthermore, considering the very low concentrations of the reactants used in our experiments: 25 μM for the ubiquitin 4 and 0.5 mM for the aldehyde reagent (at the 20 fold excess), it is rather remarkable that the conjugation reaction could be completed in 8 h at neutral pH. At the same time, a small amount of ubiquitin dimer via disulfide bond formation was also observed. This problem could be prevented in the presence of a reducing reagent such as TCEP, and the yield of thiazolidine product could increase as shown in a previous study.6e After semi-preparative HPLC purification of the biotin labeled ubiquitin 5, it was subjected to western blot analysis. Results showed that only the biotin labeled ubiquitin 5 could be detected by the anti-biotin antibody (Fig. 2C). ESI-MS analysis confirmed the mass of the conjugate (Fig. 2D). The site-specific labelling of ubiquitin at position K48 was further confirmed by tandem mass spectral analysis of the tryptic ThzK-containing fragment IFAGK*QLEDGR (K* denotes modified lysine). The mass difference between y7 and y6 ions corresponds to the mass of K* (ESI, Fig. S1†). Overall, these data show that thiazolidine ligation with aniline as catalyst is a good conjugation method to label proteins under neural pH conditions.
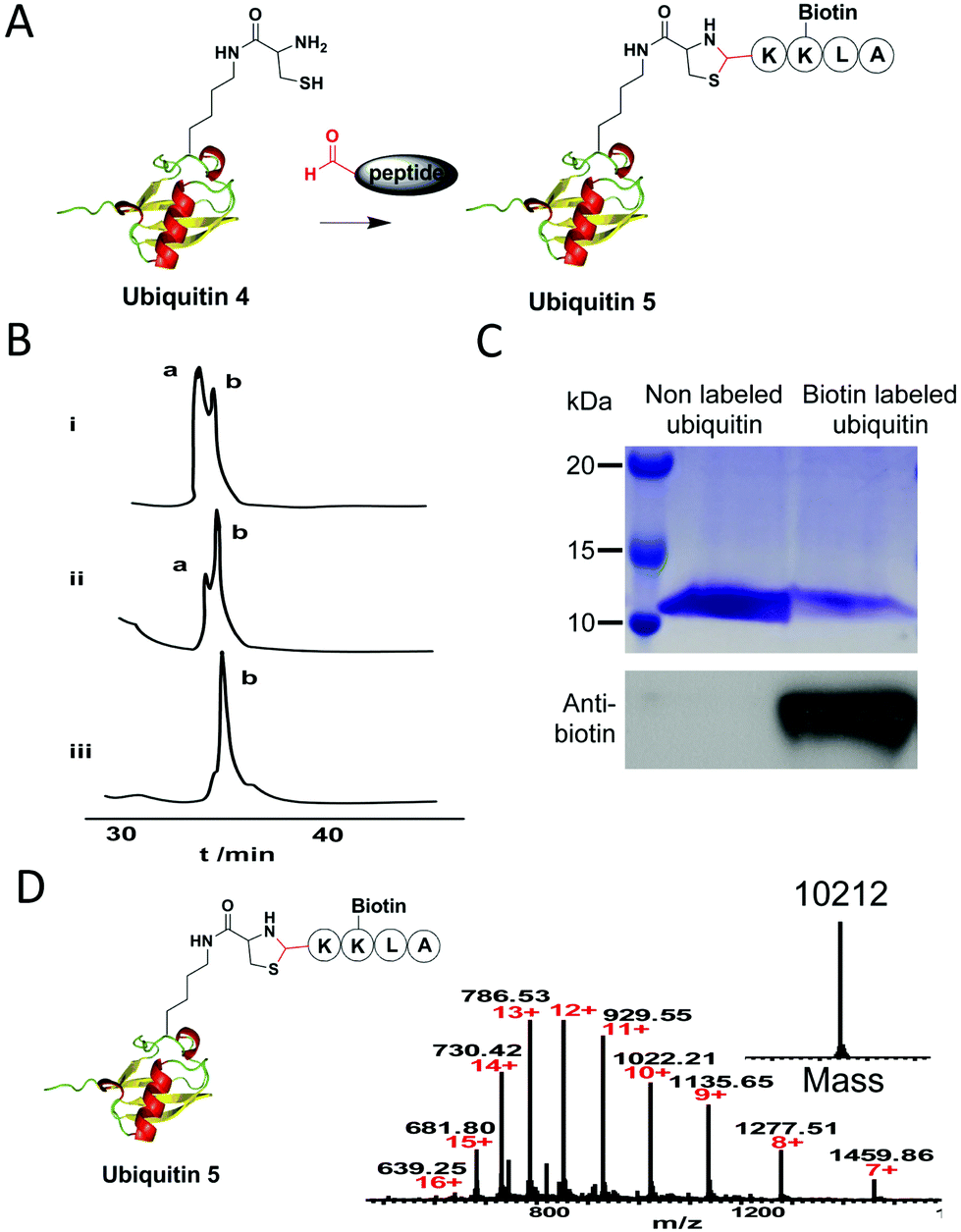 | ||
Fig. 2 (A) Schematic illustration of site-specific biotinylation of ubiquitin via thiazolidine ligation of aldehyde-functionalized biotin labeled peptide with 1,2-aminothiol moiety. (B) Analytic C4 HPLC analysis of thiazolidine ring formation between different equivalents of aldehyde-functionalized peptide with ubiquitin 4. Peak a: non-modified ubiquitin 4; peak b: biotin labeled ubiquitin 5. i: 5 equivalent of peptide; ii: 10 equivalent of peptide; iii: 20 equivalent of peptide. (C) SDS-PAGE (up panel) and western blot (down panel) analysis of biotin labeled ubiquitin 5. Only biotin labeled ubiquitin 5 can be detected by anti-biotin antibody. (D) ESI-MS analysis of biotin labelled ubiquitin 5. Expected mass: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 212.84; observed mass: 10212. 212.84; observed mass: 10212. | ||
Several methods have been developed so far to prepare ubiquitin conjugates.10 However, the strategy to prepare ubiquitin conjugates linked through thiazolidine ring has not been reported. To this end, we decided to semisynthesize thiazolidine-linked analogs of ubiquitin dimer as shown in Fig. 3A. The distal ubiquitin 6 with an aldehyde moiety at its C-terminus was prepared as described previously (ESI, Fig. S2†).11 The resulting ubiquitin 6 could be obtained with yields of 5 mg L−1. For the chemical conjugation between aldehyde and aminothiol, ubiquitin 6 was mixed with ubiquitin 4 in a 1:1 molar ratio in 0.2 M sodium acetate buffer containing 6 M guanidinium chloride and 1 mM TCEP (pH 5.4). After overnight reaction, SDS-PAGE analysis of the reaction mixtures revealed ubiquitin dimer formation with yield of about 15–20% (Fig. 3B). The desired thiazolidine linked ubiquitin dimer 7 could be purified by HPLC. ESI-MS analysis showed that thiazolidine-tethered ubiquitin dimer 7 has a good purity profile (Fig. 3C). The di-ubiquitin conjugation reaction also worked in a buffer without the Gdn–HCl denaturing agent, albeit with a slightly slower rate. By using this strategy, we were able to produce several milligrams of linkage-specific ubiquitin dimer 7 at K48 or K63 position.
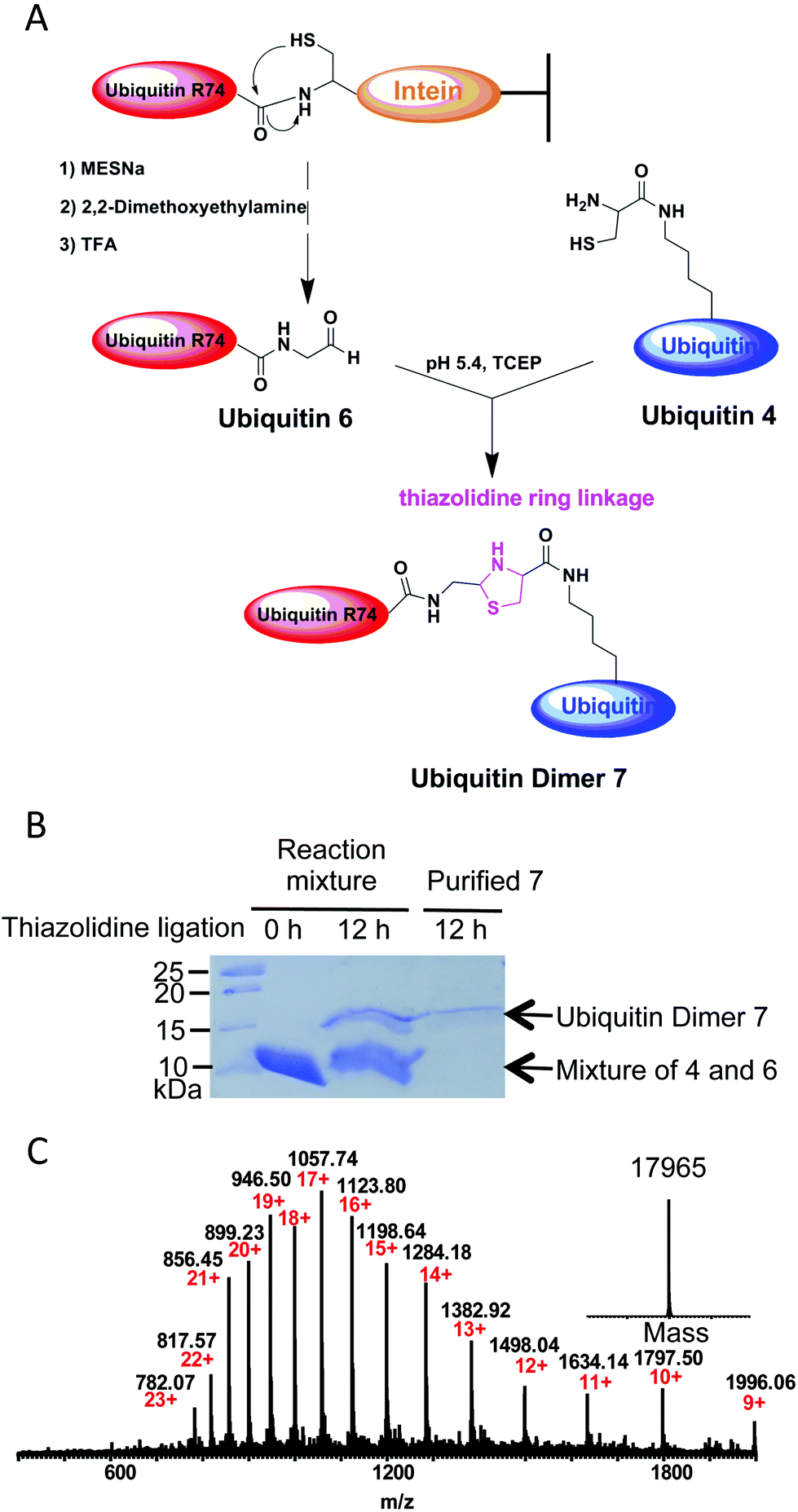 | ||
| Fig. 3 (A) Schematic illustration for preparation of thiazolidine conjugated ubiquitin dimer analog. (B) SDS-PAGE analysis of ubiquitin dimer 7 formation at 0 h and 12 h. (C) ESI-MS analysis of thiazolidine conjugated ubiquitin dimer 7. Expected mass: 17964.75; observed mass: 17965. | ||
Crosas and coworkers showed that yeast Rpn10 can be monoubiquinated in vivo at four different lysine residues (K71, K84, K99, K268) and the predominant K84 monoubiquitination reduced the capability of Rpn10 to interact with the substrate by inhibiting Rpn10's ubiquitin interacting motif (UIM).12 However, the function of monoubiquitination at different sites, such as K99, of Rpn10 is not known yet and elucidation of their function is mainly hampered by the lack of homogeneous ubiquitinated samples. Therefore, to further expand the utility of our method, we also semisynthesized monoubiquitinated Rpn10 at K99 as another demonstration. We expressed and purified Rpn10 containing the residue of 1 at position 99 in good yield (1.5 mg L−1) from cells harbouring the ThzKRS/pylT pair and a Rpn10 gene with an amber codon at position K99 and demonstrated the quantitative removal of thiazolidine ring from protein (ESI, Fig. S3†). The thiazolidine ligation between ubiquitin-aldehyde and Rpn10 containing 1,2-aminothiol was monitored by C4 analytic HPLC. Results show that about 30 percent of ubiquitinated Rpn10 (ubRpn10) can be observed after overnight reaction. ESI-MS analysis confirmed the conjugation of ubiquitin to Rpn10 (ESI, Fig. S3†). All these data indicated that our method can be a good alternative strategy to prepare ubiquitin conjugates.
Conclusions
In summary, we have presented two examples (biotin and ubiquitin conjugation) to demonstrate our new strategy for site-specific protein modification by combining thiazolidine ligation chemistry and amber codon suppression technology. In comparison to previous work on thiazolidine formation, our method does not require the use of periodate oxidation of N terminal serine or threonine to introduce aldehyde into a protein, which potentially can damage the protein.6a,13 Second, the 1,2-aminothiol functionality can be installed easily into a protein at any site via amber codon suppression technology, which makes it possible to conjugate protein through thiazolidine formation at any site. Moreover, with the aniline as the catalyst, thiazolidine formation can occur efficiently under neutral condition and work more efficiently than oxime ligations at the same condition.9 We envision that thiazolidine ligation will also benefit from newly discovered more efficient catalyst for oxime ligation.14 One limitation in our study is that the thiazolidine ring deprotection was performed under acidic condition (pH 4), which may not be applicable to acid-sensitive proteins. However, previous studies showed that the thiazolidine ring deprotection can also be performed at neutral pH in phosphate buffer with the help of methoxylamine or water-soluble palladium complex.3,15 These indicate that the whole scheme of our strategy can be done under neutral conditions for site-specific protein modification. Compared to native chemical ligation which generates a Cys residue at the ligation site, an advantage of the thiazolidine conjugation method is that the thiol group is blocked in the five-member thiazolidine ring, which will prevent the possible side reactions involving the highly nucleophilic thiol. Overall, we believe that our method will also be of use to site-specifically introduce other biochemical and biophysical probes into proteins, including fluorescent labels, NMR and EPR probes, for the elucidation of protein structure and function in future.Acknowledgements
This work is supported by the Agency for Science, Technology and Research (A*STAR) of Singapore (SERC 112 120 2017 and ETPL-QP-19-06 to CFL) and Nanyang Technological University.Notes and references
- (a) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed; (b) M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC; (c) A. Tuley, Y. J. Lee, B. Wu, Z. U. Wang and W. R. Liu, Chem. Commun., 2014, 50, 7424–7426 RSC; (d) S. A. Kularatne, V. Deshmukh, J. Ma, V. Tardif, R. K. V. Lim, H. M. Pugh, Y. Sun, A. Manibusan, A. J. Sellers, R. S. Barnett, S. Srinagesh, J. S. Forsyth, W. Hassenpflug, F. Tian, T. Javahishvili, B. Felding-Habermann, B. R. Lawson, S. A. Kazane and P. G. Schultz, Angew. Chem., Int. Ed., 2014, 53, 11863–11867 CrossRef CAS PubMed; (e) R. Yang, X. Bi, F. Li, Y. Cao and C. F. Liu, Chem. Commun., 2014, 50, 7971–7974 RSC; (f) T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssinger, Bioconjugate Chem., 2015, 26, 802–806 CrossRef CAS PubMed; (g) C. Hoppmann, I. Maslennikov, S. Choe and L. Wang, J. Am. Chem. Soc., 2015, 137, 11218–11221 CrossRef CAS PubMed; (h) J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed; (i) R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed; (j) A. Yamaguchi, T. Matsuda, K. Ohtake, T. Yanagisawa, S. Yokoyama, Y. Fujiwara, T. Watanabe, T. Hohsaka and K. Sakamoto, Bioconjugate Chem., 2016, 27, 198–206 CrossRef CAS PubMed; (k) X. Bi, R. Yang, X. Feng, D. Rhodes and C. F. Liu, Org. Biomol. Chem., 2016, 14, 835–839 RSC; (l) A. Mahdavi, G. D. Hamblin, G. A. Jindal, J. D. Bagert, C. Dong, M. J. Sweredoski, S. Hess, E. M. Schuman and D. A. Tirrell, J. Am. Chem. Soc., 2016, 138, 4278–4281 CrossRef CAS PubMed.
- X. Li, T. Fekner, J. J. Ottesen and M. K. Chan, Angew. Chem., Int. Ed., 2009, 121, 9348–9351 CrossRef.
- D. P. Nguyen, T. Elliott, M. Holt, T. W. Muir and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 11418–11421 CrossRef CAS PubMed.
- (a) C. F. Liu and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 4149–4153 CrossRef CAS; (b) C. F. Liu and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6584–6588 CrossRef CAS PubMed; (c) C. Rao and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 6975–6976 CrossRef CAS.
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- (a) L. Zhang and J. P. Tam, Anal. Biochem., 1996, 233, 87–93 CrossRef CAS PubMed; (b) M. N. Mathieu, J. D. Wade, Y. Y. Tan, R. J. Summers and G. W. Tregear, J. Pept. Sci., 2000, 6, 235–242 CrossRef CAS PubMed; (c) Z. G. Zhao, J. S. Im, K. S. Lam and D. F. Lake, Bioconjugate Chem., 1999, 10, 424–430 CrossRef CAS PubMed; (d) J. D. Wade, T. Domagala, J. Rothacker, B. Catimel and E. Nice, Lett. Pept. Sci., 2002, 8, 211–220 CrossRef PubMed; (e) M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS PubMed; (f) G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS PubMed.
- J. K. Takimoto, Z. Xiang, J. Y. Kang and L. Wang, ChemBioChem, 2010, 11, 2268–2272 CrossRef CAS PubMed.
- (a) T. S. Young, I. Ahmad, J. A. Yin and P. G. Schultz, J. Mol. Biol., 2010, 395, 361–374 CrossRef CAS PubMed; (b) J. W. Chin, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11020–11024 CrossRef CAS PubMed.
- (a) A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed; (b) M. Mühlberg, PhD thesis, Freie Universität, Berlin, 2014.
- (a) N. D. Weikart, S. Sommer and H. D. Mootz, Chem. Commun., 2012, 48, 296–298 RSC; (b) S. Eger, M. Scheffner, A. Marx and M. Rubini, J. Am. Chem. Soc., 2010, 132, 16337–16339 CrossRef CAS PubMed; (c) R. E. Morgan, V. Chudasama, P. Moody, M. E. B. Smith and S. Caddick, Org. Biomol. Chem., 2015, 13, 4165–4168 RSC.
- K. D. Wilkinson, T. Gan-Erdene and N. Kolli, Methods Enzymol., 2005, 399, 37–51 CAS.
- M. Isasa, E. J. Katz, W. Kim, V. Yugo, S. González, D. S. Kirkpatrick, T. M. Thomson, D. Finley, S. P. Gygi and B. Crosas, Mol. Cell, 2010, 38, 733–745 CrossRef CAS PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS PubMed.
- M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00854b |
| This journal is © The Royal Society of Chemistry 2016 |