
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ravynic acid, an antibiotic polyeneyne tetramic acid from Penicillium sp. elucidated through synthesis†
J. D.
Myrtle
,
A. M.
Beekman‡
and
R. A.
Barrow
*
Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. E-mail: rab@anu.edu.au
First published on 21st July 2016
Abstract
A new antibiotic natural product, ravynic acid, has been isolated from a Penicillium sp. of fungus, collected from Ravensbourne National Park. The 3-acylpolyenyne tetramic acid structure was definitively elucidated via synthesis. Highlights of the synthetic method include the heat induced formation of the 3-acylphosphorane tetramic acid and a selective Wittig cross-coupling to efficiently prepare the natural compounds carbon skeleton. The natural compound was shown to inhibit the growth of Staphylococcus aureus down to concentrations of 2.5 µg mL−1.
Introduction
The search for new antibiotic compounds that possess novel modes of action and inhibit new targets is an on-going challenge in the struggle against infectious disease. One class of natural products that has attracted much attention are those that bear the tetramic acid moiety.1 Some years ago we reported the isolation and elucidation of a tetramic acid containing natural product, ravenic acid, 1 (tetramic acid moiety highlighted in red, Fig. 1), from a Penicillium sp. collected from Ravensbourne National Park in south-east Queensland, Australia.2 Ravenic acid possessed moderate antibiotic activity against a clinical strain of methicillin resistant Staphylococcus aureus (MRSA). Re-examination of the fungal extract has revealed a very minor co-metabolite isolated from the Penicillium sp., that was shown to possess significantly greater antibiotic activity than ravenic acid, but was present in microgram quantities from large scale fermentation, effectively precluding study by NMR. However, a mass spectrum was recorded, and the CID ESI-MS fragmentation pattern displayed many similarities to 1, but with a m/z ratio of the deprotonated molecule (m/z 256, −ve ESI) along with those of some daughter ions two units lower. Given ravenic acid's structure, and following careful analysis of the mass spectrum we reasoned the most probable modification accounting for a loss of two mass units represents an oxidation of a double bond to afford a triple bond in the polyene side chain. As such, tetramic acids 2 and 3 were identified as synthetic targets to allow the definitive elucidation of the co-metabolite of ravenic acid (Fig. 1).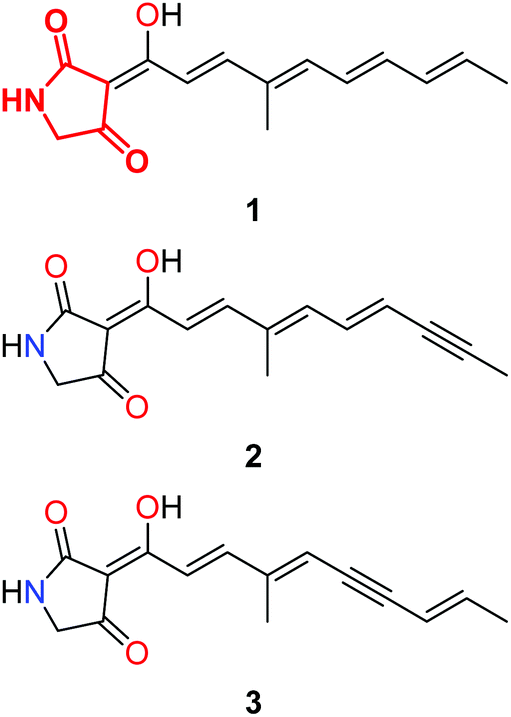 | ||
| Fig. 1 Ravenic acid (1), with the tetramic acid core highlighted in red, and possible structures for unknown co-metabolite (2 & 3). | ||
Results and discussion
Pathways presented previously by Ley3 and Moloney4 offered promise for the preparation of 2 and 3, but had been shown to be unsuccessful with unsubstituted amides and unsaturated side chains. During our studies of ravenic acid and its potential co-metabolites, Schobert and co-workers published an elegant synthesis of ravenic acid, representing a new synthetic procedure to prepare 3-acyl tetramic acids in a highly efficient manner.5 Thus, we modelled a retrosynthetic pathway on the procedure outlined by Schobert (Fig. 2). Late stage Wittig olefination would allow a divergent strategy to the preparation of 2 and 3 from the common intermediate 4 and aldehydes 5 and 6. Compound 4 could be prepared by the thermal coupling of tetramic acid 7 and phosphorane 8. The use of the Boc protecting group is notable, as it had been employed successfully by Schobert, in contrast to the di-methoxybenzyl protecting group which had been the downfall of our previous attempts to synthesize ravenic acid. Despite numerous attempts employing a variety of deprotection protocols starting material consistently decomposed.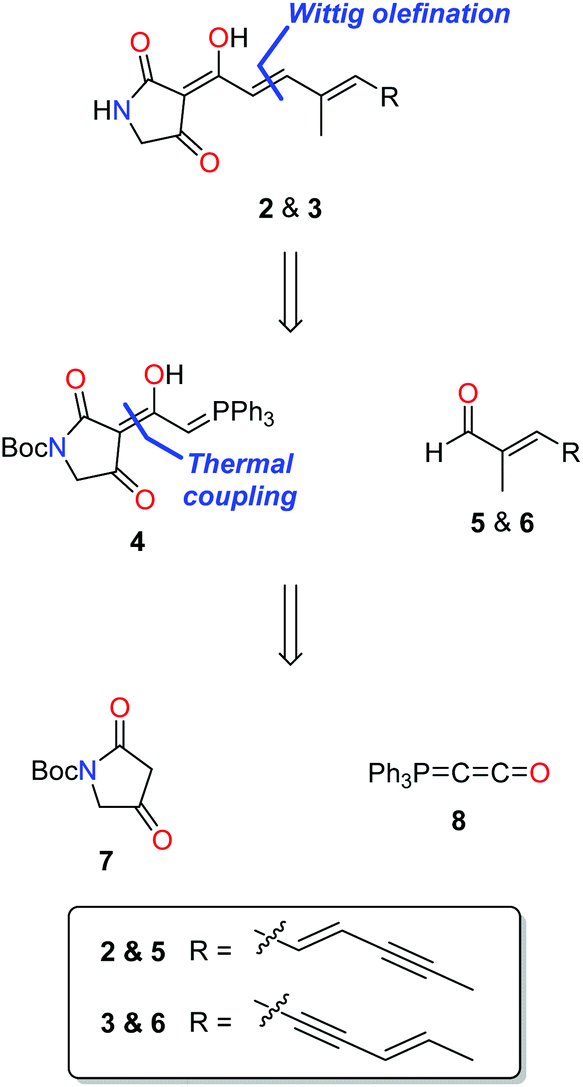 | ||
| Fig. 2 Retrosynthetic pathway for putative co-metabolites 2 and 3. | ||
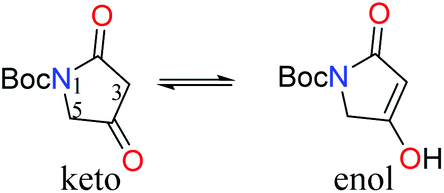
In a forward manner, tetramic acid 7 was prepared in one step from N-Boc-glycine (9) by coupling with Meldrum's acid mediated by EDC and DMAP, followed by heat induced rearrangement, in 83% yield (Scheme 1). Consistent with previously reported data,67 exists as a mixture of keto and enol tautomers in CDCl3 in the approximate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while in deuterated methanol, the enol form is exclusively observed.
:1, while in deuterated methanol, the enol form is exclusively observed.
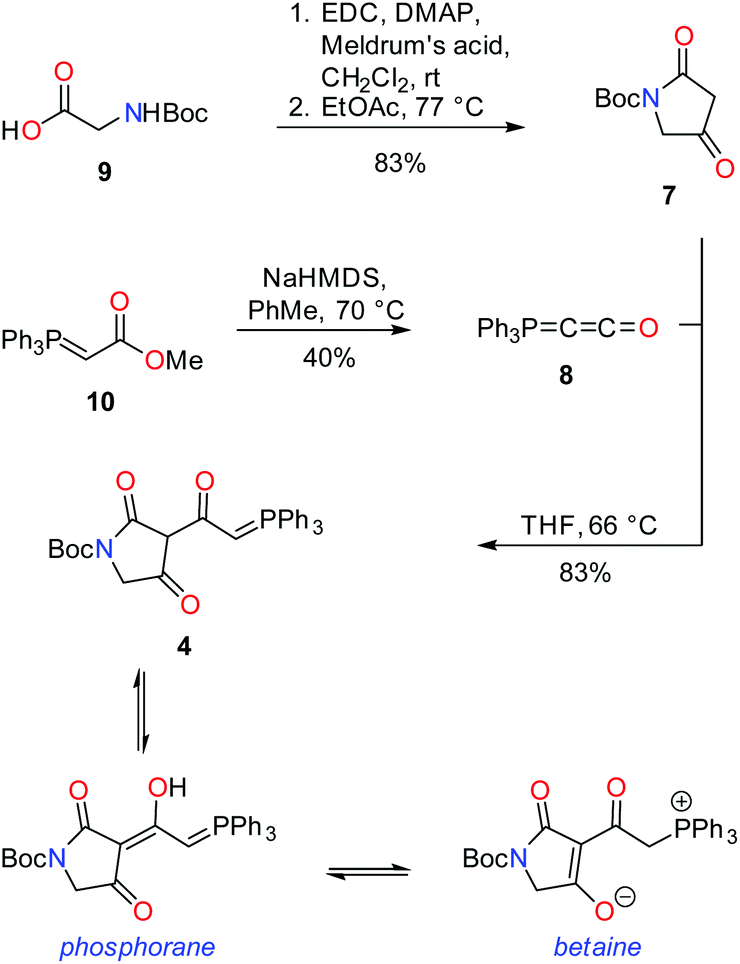 | ||
| Scheme 1 Synthesis of key intermediate tetramic acid 4. | ||
Phosphorane 8 was formed by treatment of phosphorane 10 with sodium hexamethyldisilazide.5 In spite of the structural similarity of 8 with ketenes, it is relatively stable, not displaying a tendency to dimerise.7 Phosphorane 8 and tetramic acid 7 were coupled under thermal conditions to form key intermediate 4. Heat facilitates nucleophilic addition of the enol tautomer of 7 on 8, with subsequent proton transfer and tautomerism leading to the formation of 4. In CDCl3, tetramic acid 4 exists in two forms, which have been assigned as the phosphorane and the tautomeric/resonance betaine form.
Preparation of the requisite aldehydes 5 and 6 was achieved in a rapid and efficient manner (Scheme 2). Propargyl alcohol was brominated by treatment with potassium hydroxide and bromine to form 11, and subsequent copper-mediated coupling with propyne led directly to alcohol 12. Stereoselective reduction of the triple bond proximal to the alcohol with LiAlH4 afforded an alcohol, where the stereochemistry of the newly created alkene was deduced from the coupling constant (3J = 15.6 Hz) between the olefinic protons at δ 5.68 and 6.15, indicative of the trans relationship of substituents. Oxidation employing Dess–Martin periodinane yielded aldehyde 13 which was employed directly in the following step. Sonogashira coupling of E-1-bromoprop-1-ene with propargyl alcohol successfully afforded alcohol 14, with subsequent oxidation using DMP providing aldehyde 15, with a yield of 77% over the 2 steps. Isomerization was not observed over either of the two steps, with the stereochemistry of alcohol 14 determined through analysis of the olefinic coupling constants (3J = 15.8 Hz). Examination of conditions for the Horner–Wadsworth–Emmons olefination between triethyl-2-phosphonopropionate and aldehydes 13 and 15 found that sodium hydride as the base increased the yield and selectively giving the desired E isomer. The proton NMR spectra and GCMS data indicated that one stereoisomer was formed preferentially (>95%), with literature comparisons,8 and the HWE reaction's known stereochemical outcomes, supporting the assignment of the E configuration.9 Attempts were made to proceed directly to aldehydes 5 and 6 in one step using DIBAL-H as the reducing agent. Treatment of the resulting esters 16–17 with one equivalent of DIBAL-H resulted in a 1:1 mixture of the alcohol and the starting ester, indicating that the intermediate aldehyde is reduced more rapidly than the ester. Thus three equivalents of DIBAL-H were used to ensure complete conversion of the esters to conjugated alcohols, and the alcohols subsequently oxidised to aldehydes 5 and 6 using DMP. With the synthesis of the target aldehydes complete, attention turned to completion of the final steps in the synthesis of the ravenic acid analogues.
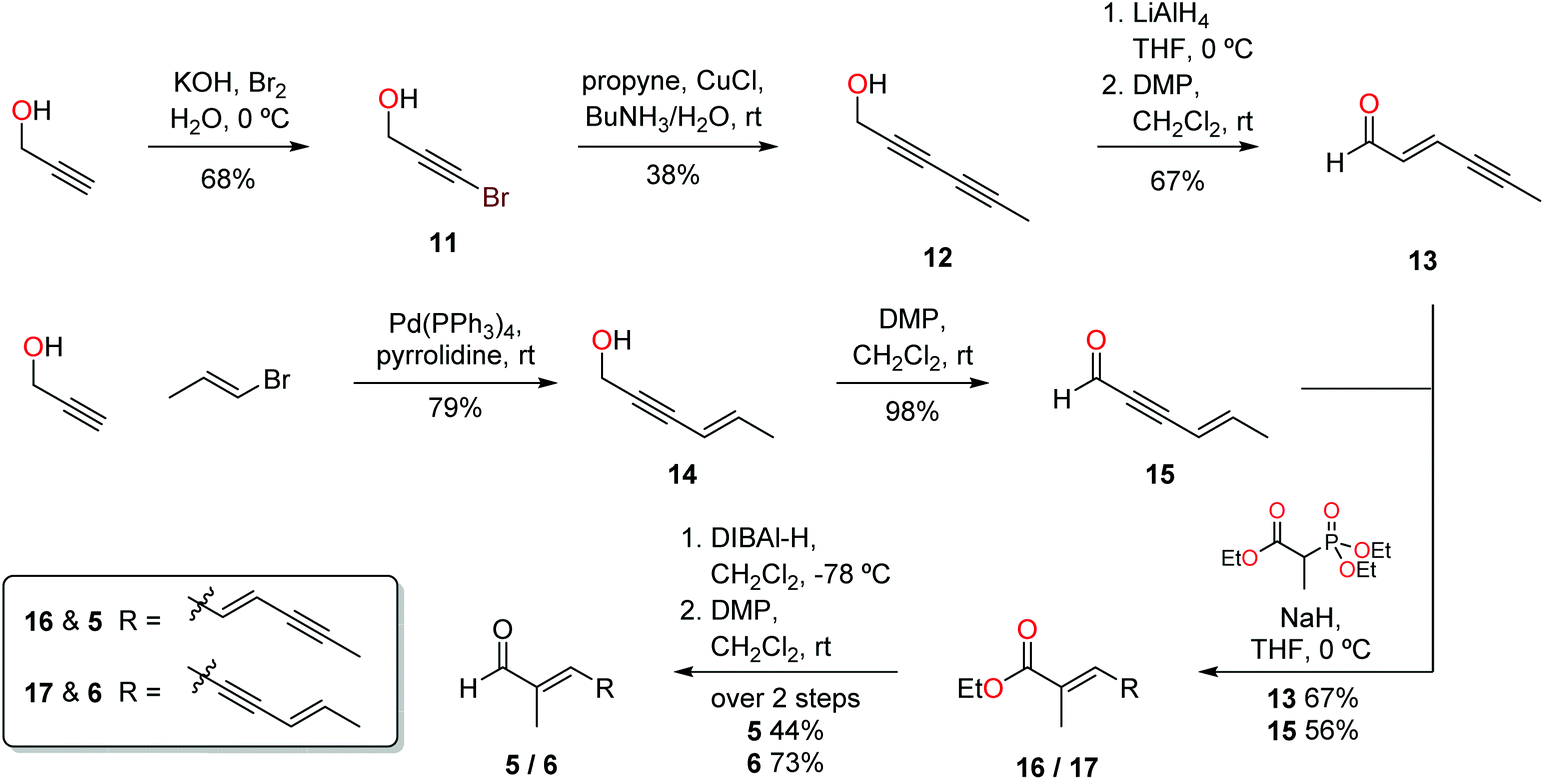 | ||
| Scheme 2 Synthesis of aldehydes 5 and 6. | ||
The common tetramic acid phosphorane intermediate 4 was coupled with aldehydes 5 and 6via the Wittig olefination to give Boc-protected ravenic acid analogues 18 and 19 in yields of 52% and 62%, respectively (Scheme 3). With either silica or alumina column chromatography resulting in decomposition, size exclusion chromatography (Sephadex LH-20) was employed to purify the polyene tetramic acids. In each case, the product with the E stereochemistry about the newly created olefinic double bond was formed as deduced by analysis of the olefinic coupling constants (3J = 15.6 Hz) in the 1H NMR spectrum. The concluding step involved treatment with TFA in dichloromethane resulting in the cleavage of the Boc protecting group, providing the ravenic acid analogues 2 and 3.
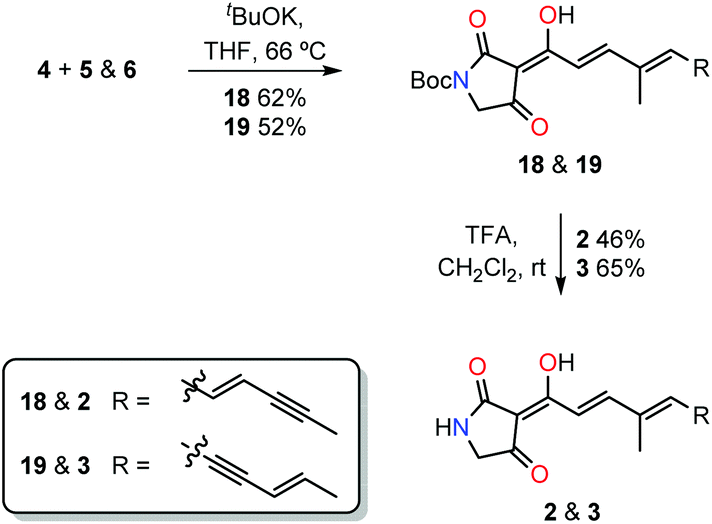 | ||
| Scheme 3 Final steps of the synthesis of 2 and 3. | ||
Finally, comparison of the mass spectral data as well as co-injection on a HPLC column allowed the assignment of the co-metabolite as structure 3, named here as ravynic acid. The synthesis of this newly discovered natural product was performed in a convergent manner with the longest linear sequence of steps counting nine. Ravenic acid had been shown to inhibit the growth of a methicillin-resistant Staphylococcus aureus strain down to 25 µg mL−1.2 Ravynic acid has been shown to inhibit the growth of the same strain down to approximately 2.5 µg mL−1. Further biological testing will allow for the assessment of the spectrum of activity and determination of the mode of action of this antibiotic.
Conclusions
In conclusion, we have isolated and unambiguously identified a new antibacterial tetramic acid natural product from a Penicillium sp. of fungus. Ravynic acid is a 3-acyltetramic acid, which has been synthesized from Boc-glycine and propargyl alcohol, to allow definitive elucidation of the structure. Notable features of the synthesis include the thermally induced formation of the 3-acylphosphorane tetramic acid and a selective Wittig cross-coupling to efficiently prepare the natural compounds carbon skeleton.Experimental
General information
1H, 13C NMR spectra were recorded at 298 K, at 300 MHz, 400 MHz or 500 MHz and 75 MHz, 100 MHz or 125 MHz respectively, on an Inova 300, Varian MR-300, Varian MR-400 or Inova 500 instruments. Chemical shifts are reported in ppm (δ). NMR experiments were run in deuterated chloroform (CDCl3), or methanol (CD3OD) as indicated; 1H NMR spectra are referenced to the resonance from residual CHCl3 at 7.26 ppm, or CHD2OD at 3.31 ppm, 13C NMR spectra are referenced to the central peak in the signal from CDCl3 at 77.0 ppm, or CD3OD at 49.0 ppm. The appearance and multiplicities of 1H resonances are expressed by the abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and combinations thereof for more highly coupled systems. 13C NMR were run as proton decoupled spectra. 1H signals and 13C signals, where appropriate, are described by chemical shift δ (multiplicity, |J| (Hz), integration, assignment). Where the spectral characteristics (1H and 13C NMR) agree with published data, a literature reference is cited. Where no literature is available, including known but insufficiently characterised compounds, data (1H NMR, 13C NMR, IR, MS, HRMS) are presented. Assignments were supported by 2D NMR analysis including HSQC, HMBC and NOESY data and by literature precedence. EI-MS and HREI-MS were recorded on a VG autospec mass spectrometer, operating at 70 eV. ESI-MS and HRESI-MS were recorded on a Bruker Apex 3. Positive ionisation was detected unless otherwise indicated. Mass/charge ratios (m/z) are reported and relative abundance of the ions is as percentage of base peak intensity. IR spectra were recorded on a Bruker Alpha-P as neat solid or film. Diagnostic peaks are presented in wavenumbers (cm−1). Peak strengths are expressed by the abbreviations: s (strong), m (medium), w (weak) and b (broad). Melting points were determined on a Reichert Thermogalen hot stage microscope and are uncorrected. Flash chromatography was performed under pressure using silica gel (230–400 mesh Scharlau 60, 40–60 micron). Analytical thin layer chromatography (TLC) was performed on Merck silica gel 60 F254 aluminium backed plates. Plates were viewed under UV light (254 nm) and/or by developing with ceric phosphomolybdic acid (100 mL water, 5 g phosphomolybdic acid, 0.6 g Ce(SO4)2, 6 mL conc. H2SO4) dip. In general, reagents were purchased from commercial suppliers and used without further purification unless otherwise stated. According to standard procedures, all solvents were dried and distilled either immediately prior to use or stored as appropriate. Diethyl ether and THF were refluxed under a static N2 atmosphere over sodium and benzophenone. Dichloromethane was distilled from calcium hydride. Toluene was distilled from sodium. Ethanol was distilled from magnesium. The petroleum ether fraction used was 60–80 °C.:petroleum ether yielded the title compound as an unstable white solid (644 mg, 38%). 1H NMR (300 MHz, CDCl3) δ 4.31 (s, 2H, H-1), 1.94 (s, 3H, H-6). 13C (75 MHz, CDCl3) δ 77.0, 72.9, 70.3, 63.5, 50.9, 4.0. IR: ν (cm−1) 3301 (s, br), 1445 (s), 1347 (s), 1021 (s), 625 (s). HREI-MS: calc 94.0419 (C6H6O) found 94.0419.
:petroleum ether as eluent afforded the title compound as a clear oil (198 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 6.15 (dt, J = 15.6, 5.6 Hz, 1H, H-2), 5.68 (dq, J = 15.6, 2.4 Hz, 1H, H-3), 4.16 (dd, J = 6.0, 5.6 Hz, 2H, H-1), 1.94 (d, J = 2.4 Hz, 3H, H-6), 1.44 (t, J = 6.0 Hz, 1H, O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ). 13C (75 MHz, CDCl3) δ 140.2 (C-2), 111.2 (C-3), 86.8 (C-5), 77.4 (C-4), 63.0 (C-1), 4.2 (C-6). IR: ν (cm−1) 3317 (br s), 2917 (w), 1432 (m), 1091 (s), 1008 (s), 951 (s). EI-MS m/z (%) 95 (28, M˙+), 84 (17), 81 (100), 67 (29), 53 (30). HREI-MS: calc 95.0497 (C6H7O) found 95.0497.
:petroleum ether as eluent gave the desired product as a pale yellow oil (1.78 g, 79%). 1H NMR (300 MHz, CDCl3) δ 6.16 (dq, J = 15.8, 6.9 Hz, 1H, H-5), 5.50 (dt, J = 15.8, 2.1 Hz, 1H, H-4), 4.36 (d, J = 2.1 Hz, 2H, H-1), 1.78 (d, J = 6.9, 3H, H-6). 13C (75 MHz, CDCl3) δ 140.1 (C-5), 110.0 (C-4), 85.4, 84.0, 51.0 (C-1), 18.4 (C-6). IR: ν (cm−1) 3345 (br s), 2380 (m), 1443 (m), 1163 (m), 1013 (s), 953 (s). EI-MS m/z (%) 96 (85, M˙+), 81 (100), 77 (35), 67 (31), 53 (45), 39 (52). HREI-MS: calc 96.0575 (C6H8O) found 96.0578.
2CH3), 3.02 (dq, 2JPH = 23.3 Hz, 3JHH = 7.2 Hz, 1H, H-2), 1.44 (dd, 3JPH = 18.2 Hz, 3JHH = 7.2 Hz, 3H, H-3), 1.25–1.28 (m, 9H, 3 × OCH2C3).
:petroleum ether as eluent to yield ester 16 as a clear oil (239 mg, 67% over 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.13 (d, J = 12.0 Hz, 1H, H-3), 6.76 (dd, J = 15.3, 12.0 Hz, 1H, H-4), 5.86 (dq, J = 15.3, 2.1 Hz, 1H, H-5), 4.18 (q, J = 7.2 Hz, 2H, OC2CH3), 1.99 (d, J = 2.1 Hz, 3H, H-8), 1.92 (s, 3H, C-2 Me), 1.27 (t, J = 7.2 Hz, 3H, OCH2C3). 13C (75 MHz, CDCl3) δ 167.9 (C-1), 136.9, 135.4, 128.5 (C-2), 118.4 (C-5), 92.3 (C-7), 79.1 (C-6), 60.6 (O
). 13C (75 MHz, CDCl3) δ 140.2 (C-2), 111.2 (C-3), 86.8 (C-5), 77.4 (C-4), 63.0 (C-1), 4.2 (C-6). IR: ν (cm−1) 3317 (br s), 2917 (w), 1432 (m), 1091 (s), 1008 (s), 951 (s). EI-MS m/z (%) 95 (28, M˙+), 84 (17), 81 (100), 67 (29), 53 (30). HREI-MS: calc 95.0497 (C6H7O) found 95.0497.
:petroleum ether as eluent gave the desired product as a pale yellow oil (1.78 g, 79%). 1H NMR (300 MHz, CDCl3) δ 6.16 (dq, J = 15.8, 6.9 Hz, 1H, H-5), 5.50 (dt, J = 15.8, 2.1 Hz, 1H, H-4), 4.36 (d, J = 2.1 Hz, 2H, H-1), 1.78 (d, J = 6.9, 3H, H-6). 13C (75 MHz, CDCl3) δ 140.1 (C-5), 110.0 (C-4), 85.4, 84.0, 51.0 (C-1), 18.4 (C-6). IR: ν (cm−1) 3345 (br s), 2380 (m), 1443 (m), 1163 (m), 1013 (s), 953 (s). EI-MS m/z (%) 96 (85, M˙+), 81 (100), 77 (35), 67 (31), 53 (45), 39 (52). HREI-MS: calc 96.0575 (C6H8O) found 96.0578.
2CH3), 3.02 (dq, 2JPH = 23.3 Hz, 3JHH = 7.2 Hz, 1H, H-2), 1.44 (dd, 3JPH = 18.2 Hz, 3JHH = 7.2 Hz, 3H, H-3), 1.25–1.28 (m, 9H, 3 × OCH2C3).
:petroleum ether as eluent to yield ester 16 as a clear oil (239 mg, 67% over 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.13 (d, J = 12.0 Hz, 1H, H-3), 6.76 (dd, J = 15.3, 12.0 Hz, 1H, H-4), 5.86 (dq, J = 15.3, 2.1 Hz, 1H, H-5), 4.18 (q, J = 7.2 Hz, 2H, OC2CH3), 1.99 (d, J = 2.1 Hz, 3H, H-8), 1.92 (s, 3H, C-2 Me), 1.27 (t, J = 7.2 Hz, 3H, OCH2C3). 13C (75 MHz, CDCl3) δ 167.9 (C-1), 136.9, 135.4, 128.5 (C-2), 118.4 (C-5), 92.3 (C-7), 79.1 (C-6), 60.6 (O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CH3), 14.2 (OCH2H3), 12.7 (C-2 Me), 4.6 (C-8). IR: ν (cm−1) 2980 (w), 1703 (s), 1368 (m), 1283 (s), 1227 (s), 1101 (m), 959(m). EI-MS m/z (%) 178 (98, M˙+), 163 (96), 149 (93), 135 (100), 121 (95). HREI-MS: calc 178.0994 (C11H14O2) found 178.0992.
:petroleum ether. Clear oil (129 mg, 56% over 2 steps) from alcohol 14 (113 mg, 1.20 mmol). 1H NMR (300 MHz, CDCl3) δ 6.72 (s, 1H, H-3), 6.25 (dq, J = 15.6, 6.9 Hz, 1H, H-7), 5.70 (dq, J = 15.6, 1.8 Hz, 1H, H-6), 4.20 (q, J = 6.9 Hz, 2H, OC2CH3), 2.04 (s, 3H, C-2 Me), 1.82 (dd, J = 6.9, 1.8 Hz, 3H, H-8), 1.29 (t, J = 6.9 Hz, 3H, OCH2C3). 13C (75 MHz, CDCl3) δ 167.1, 141.6, 137.9, 119.7, 110.7, 100.5, 84.6, 60.8, 18.8, 15.2, 14.1. EI-MS m/z (%) 178 (100, M˙+), 149 (80), 133 (91), 121 (66), 93 (52), 77 (95), 51 (67), 39 (57). HREI-MS: calc 178.0994 (C11H14O2) found 178.0994.
:petroleum ether as eluent to give aldehyde 5 as a highly unstable pale yellow oil (19 mg, 44%) which was used immediately in the next step. 1H NMR (300 MHz, CDCl3) δ 9.44 (s, 1H, H-1), 6.70–7.00 (m, 2H, H-3 and H-4), 6.02 (dq, J = 14.9, 2.4 Hz, 1H, H-5), 2.04 (d, J = 2.4 Hz, 3H, H-8), 1.86 (s, 3H, C-2 Me).
H2CH3), 14.2 (OCH2H3), 12.7 (C-2 Me), 4.6 (C-8). IR: ν (cm−1) 2980 (w), 1703 (s), 1368 (m), 1283 (s), 1227 (s), 1101 (m), 959(m). EI-MS m/z (%) 178 (98, M˙+), 163 (96), 149 (93), 135 (100), 121 (95). HREI-MS: calc 178.0994 (C11H14O2) found 178.0992.
:petroleum ether. Clear oil (129 mg, 56% over 2 steps) from alcohol 14 (113 mg, 1.20 mmol). 1H NMR (300 MHz, CDCl3) δ 6.72 (s, 1H, H-3), 6.25 (dq, J = 15.6, 6.9 Hz, 1H, H-7), 5.70 (dq, J = 15.6, 1.8 Hz, 1H, H-6), 4.20 (q, J = 6.9 Hz, 2H, OC2CH3), 2.04 (s, 3H, C-2 Me), 1.82 (dd, J = 6.9, 1.8 Hz, 3H, H-8), 1.29 (t, J = 6.9 Hz, 3H, OCH2C3). 13C (75 MHz, CDCl3) δ 167.1, 141.6, 137.9, 119.7, 110.7, 100.5, 84.6, 60.8, 18.8, 15.2, 14.1. EI-MS m/z (%) 178 (100, M˙+), 149 (80), 133 (91), 121 (66), 93 (52), 77 (95), 51 (67), 39 (57). HREI-MS: calc 178.0994 (C11H14O2) found 178.0994.
:petroleum ether as eluent to give aldehyde 5 as a highly unstable pale yellow oil (19 mg, 44%) which was used immediately in the next step. 1H NMR (300 MHz, CDCl3) δ 9.44 (s, 1H, H-1), 6.70–7.00 (m, 2H, H-3 and H-4), 6.02 (dq, J = 14.9, 2.4 Hz, 1H, H-5), 2.04 (d, J = 2.4 Hz, 3H, H-8), 1.86 (s, 3H, C-2 Me).
 :enol = 53:47 in CDCl3, only enol form in CD3OD. Note: H-3 and C-3 peaks not observed in spectra when run in CD3OD. 1H NMR (300 MHz, CDCl3) δ keto: 4.24 (s, 2H, H-5), 3.25 (s, 2H, H-3), 1.54 (s, 9H, C(CH3)3). enol: 5.13 (s, 1H, H-3), 4.22 (s, 2H, H-5), 1.51 (s, 9H, C(CH3)3). 1H NMR (300 MHz, CD3OD) δ 4.23 (s, 2H, H-5), 1.53 (s, 9H, C(CH3)3). 13C (100 MHz, CDCl3) δ keto: 200.5 (C-4), 173.1 (C-2), 148.9 (NO2tBu), 84.4 ((CH3)3), 57.1 (C-5), 43.6 (C-3), 27.9 (C(H3)3). enol: 175.5 (C-4), 167.4 (C-2), 149.3 (NO2tBu), 95.8 (C-3), 83.1 ((CH3)3), 50.3 (C-5), 28.1 (C(H3)3). 13C (100 MHz, CD3OD) δ 176.8 (C-4), 173.7 (C-2), 150.8 (C-6), 83.6 ((CH3)3), 51.1 (C-5), 28.3 (C(H3)3). IR: ν (cm−1) 2978 (m), 1755 (s), 1597 (s), 1363 (s), 1150 (s), 1076 (s), 800 (s). EIS-MS (+ve ion) m/z (%) 222 (52, [M + Na]+), 166 (100), 144 (66), 126 (80), 122 (80). HRESI-MS (+ve ion): calc 222.0742 (C9H13NO4Na, for [M + Na]+) found 222.0736.
:CH2Cl2 as eluent to yield the desired product as a white solid (3.80 g, 84%). In CDCl3: 2:1 mixture of phosphorane (A) and betaine (B). In CD3OD: 4:1 mixture of tautomers major (a) and minor (b). Note: peaks associated with C-7 and H-7 not observed in CD3OD. 1H NMR (500 MHz, CDCl3): δ Form A: 7.45–7.75 (m, 15H, PPh3), 5.44 (d, J = 19.5 Hz, 1H, P
:enol = 53:47 in CDCl3, only enol form in CD3OD. Note: H-3 and C-3 peaks not observed in spectra when run in CD3OD. 1H NMR (300 MHz, CDCl3) δ keto: 4.24 (s, 2H, H-5), 3.25 (s, 2H, H-3), 1.54 (s, 9H, C(CH3)3). enol: 5.13 (s, 1H, H-3), 4.22 (s, 2H, H-5), 1.51 (s, 9H, C(CH3)3). 1H NMR (300 MHz, CD3OD) δ 4.23 (s, 2H, H-5), 1.53 (s, 9H, C(CH3)3). 13C (100 MHz, CDCl3) δ keto: 200.5 (C-4), 173.1 (C-2), 148.9 (NO2tBu), 84.4 ((CH3)3), 57.1 (C-5), 43.6 (C-3), 27.9 (C(H3)3). enol: 175.5 (C-4), 167.4 (C-2), 149.3 (NO2tBu), 95.8 (C-3), 83.1 ((CH3)3), 50.3 (C-5), 28.1 (C(H3)3). 13C (100 MHz, CD3OD) δ 176.8 (C-4), 173.7 (C-2), 150.8 (C-6), 83.6 ((CH3)3), 51.1 (C-5), 28.3 (C(H3)3). IR: ν (cm−1) 2978 (m), 1755 (s), 1597 (s), 1363 (s), 1150 (s), 1076 (s), 800 (s). EIS-MS (+ve ion) m/z (%) 222 (52, [M + Na]+), 166 (100), 144 (66), 126 (80), 122 (80). HRESI-MS (+ve ion): calc 222.0742 (C9H13NO4Na, for [M + Na]+) found 222.0736.
:CH2Cl2 as eluent to yield the desired product as a white solid (3.80 g, 84%). In CDCl3: 2:1 mixture of phosphorane (A) and betaine (B). In CD3OD: 4:1 mixture of tautomers major (a) and minor (b). Note: peaks associated with C-7 and H-7 not observed in CD3OD. 1H NMR (500 MHz, CDCl3): δ Form A: 7.45–7.75 (m, 15H, PPh3), 5.44 (d, J = 19.5 Hz, 1H, P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 3.94 (s, 2H, H-5), 1.49 (s, 9H, C(C3)3). Form B: 7.45–7.75 (m, 15H, PPh3), 5.10 (d, J = 12.0 Hz, 2H, CH2P), 3.78 (s, 2H, H-5), 1.48 (s, 9H, C(C3)3). 13C (100 MHz, CD3OD) δ 192.2 (C-4), 182.3 (C-6), 171.2 (C-2), 149.9 (NO2tBu), 134.33, 134.29, 133.8 (d, J = 10.3 Hz), 133.3 (d, J = 2.6 Hz), 132.9 (d, J = 10.5 Hz), 132.3 (d, J = 2.4 Hz), 131.6 (d, J = 10.1 Hz), 129.6 (d, J = 12.8 Hz), 129.3 (d, J = 12.7 Hz), 128.5 (d, J = 12.2 Hz), 123.6 (d, J = 92.0 Hz), 119.6 (d, J = 88.0 Hz), 104.7 (C-3), 83.0 ((CH3)3), 53.6 (C-5), 28.4 (C(H3)3). IR: ν (cm−1) 1753 (m), 1620 (s), 1555 (s), 1436 (s), 1327 (s), 1153 (s), 1102 (s), 844 (s), 689 (s). EI-MS m/z (%) 501 (14, M˙+), 401 (65), 301 (90), 277 (100), 183 (61), 57 (74). HREI-MS: calc 501.1705 (C29H28NO5P) found 501.1710.
CH), 3.94 (s, 2H, H-5), 1.49 (s, 9H, C(C3)3). Form B: 7.45–7.75 (m, 15H, PPh3), 5.10 (d, J = 12.0 Hz, 2H, CH2P), 3.78 (s, 2H, H-5), 1.48 (s, 9H, C(C3)3). 13C (100 MHz, CD3OD) δ 192.2 (C-4), 182.3 (C-6), 171.2 (C-2), 149.9 (NO2tBu), 134.33, 134.29, 133.8 (d, J = 10.3 Hz), 133.3 (d, J = 2.6 Hz), 132.9 (d, J = 10.5 Hz), 132.3 (d, J = 2.4 Hz), 131.6 (d, J = 10.1 Hz), 129.6 (d, J = 12.8 Hz), 129.3 (d, J = 12.7 Hz), 128.5 (d, J = 12.2 Hz), 123.6 (d, J = 92.0 Hz), 119.6 (d, J = 88.0 Hz), 104.7 (C-3), 83.0 ((CH3)3), 53.6 (C-5), 28.4 (C(H3)3). IR: ν (cm−1) 1753 (m), 1620 (s), 1555 (s), 1436 (s), 1327 (s), 1153 (s), 1102 (s), 844 (s), 689 (s). EI-MS m/z (%) 501 (14, M˙+), 401 (65), 301 (90), 277 (100), 183 (61), 57 (74). HREI-MS: calc 501.1705 (C29H28NO5P) found 501.1710. 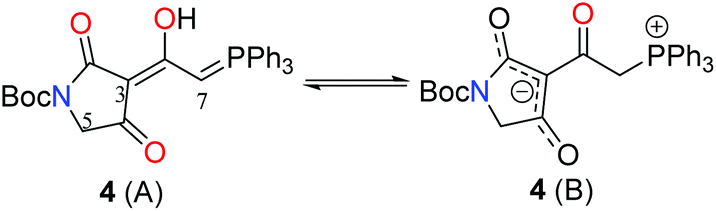 :MeOH to yield the target compound as an orange oil (74 mg, 52%). Tautomeric ratio Z:E = ∼2:1 in CDCl3. For 1H NMR spectrum the major tautomer is assigned, for the 13C NMR spectrum, both tautomers are assigned. 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 15.6 Hz, 1H, H-8), 7.31 (d, J = 15.6 Hz, 1H, H-7), 6.29 (dq, J = 15.6, 6.8 Hz, 1H, H-14), 6.10 (s, 1H, H-10), 5.74 (dq, J = 15.6, 2.0 Hz, 1H, H-13), 4.06 (s, 2H, H-5), 2.11 (s, 3H, C-9 Me), 1.86 (dd, J = 6.8, 2.0 Hz, 3H, H-15), 1.56 (s, 9H, C(C3)3). 13C (100 MHz, CDCl3) δ 197.7 (C-4 minor), 189.3 (C-4 major), 178.0 (C-6 minor), 176.2 (C-6 major), 173.6 (C-2 major), 164.8 (C-2 minor), 149.8 (NO2tBu minor), 149.4 (NO2tBu major), 148.7 (C-8), 144.8 (C-9), 142.1 (C-14), 122.4 (C-10), 118.6 (C-7 major), 118.0 (C-7 minor), 110.9 (C-13), 103.5 (C-3 minor), 102.9 (C-12), 101.5 (C-3 major), 86.7 (C-11), 84.0 ((CH3)3 major), 83.4 ((CH3)3 minor), 54.7 (C-5 major), 52.3 (C-5 minor), 28.0 (C(H3)3), 19.0 (C-15), 14.8 (C-9 Me). IR: ν (cm−1) 1743 (m), 1613 (s), 1551 (m), 1343 (s), 1310 (s), 1156 (s). EI-MS m/z (%) 357 (3, M˙+), 301 (7), 277 (52), 2257 (43), 126 (20), 71 (48), 56 (100). HREI-MS: calc 357.1576 (C20H23NO5) found 357.1563.
:E = ∼2:1 in CDCl3. For 1H NMR spectrum the major tautomer is assigned, for the 13C NMR spectrum, both tautomers are assigned. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 15.6 Hz, 1H, H-8), 7.32 (d, J = 15.6 Hz, 1H, H-7), 6.90 (dd, J = 15.2, 12.0 Hz, 1H, H-11), 6.51 (d, J = 12.0 Hz, 1H, H-10), 5.88 (dq, J = 15.2, 2.4 Hz, 1H, H-12), 4.06 (s, 2H, H-5), 2.04 (d, J = 2.4 Hz, 3H, H-15), 2.00 (s, 3H, C-9 Me), 1.56 (s, 9H, C(C3)3). 13C (100 MHz, CDCl3) δ 197.7 (C-4 minor), 189.2 (C-4 major), 178.1 (C-6 minor), 176.5 (C-6 major), 173.2 (C-2 major), 165.0 (C-2 minor), 151.9 (NO2tBu minor), 151.2 (NO2tBu major), 150.6 (C-8), 141.9 (C-9, C-10 and C-11), 136.1, 133.9, 118.7 (C-7 and C-12), 117.5, 103.0 (C-3 minor), 101.1 (C-3 major), 94.4 (C-14), 83.9 (O(CH3)3 major), 83.3 (O(CH3)3 minor), 79.7 (C-13), 54.6 (C-5 major), 52.3 (C-5 minor), 28.2 (C(H3)3), 12.6 (C-9 Me), 4.9 (C-15). IR: ν (cm−1) 1706 (m), 1615 (s), 1573 (s), 1310 (s), 1154 (s). EI-MS m/z (%) 357 (21, M˙+), 301 (22), 257 (97), 126 (54), 91 (100), 84 (53), 57 (100). HREI-MS: calc 357.1576 (C20H23NO5) found 357.1566.
:MeOH as eluent to afford the title compound as an orange solid (6 mg, 65%). Further purification was achieved using semi-preparative HPLC using a gradient eluent of 60% MeCN: H2O/TFA (0.1%) to 80% MeCN: H2O/TFA (0.1%). See S14 for detailed HPLC experimental. Tautomeric ratio Z:E – 4:1. 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 15.5 Hz, 1H, H-8), 7.28 (d, J = 15.5 Hz, 1H, H-7), 6.28 (dq, J = 16.0, 7.0 Hz, 1H, H-14), 6.06 (s, 1H, H-10), 5.96 (br s, 1H, NH), 5.76 (d, J = 16.0 Hz, 1H, H-13), 3.94 (s, 2H, H-5 minor), 3.84 (s, 2H, H-5 major), 2.14 (s, 3H, C-9 Me), 1.87 (d, J = 7.0 Hz, 3H, H-15). 13C (125 MHz, CDCl3) δ 192.4 (C-4), 176.2 (C-2), 174.2 (C-6), 146.8 (C-8), 144.9 (C-9), 141.7 (C-14), 121.0 (C-10), 119.0 (C-7), 110.9 (C-13), 101.9 (C-12), 100.5 (C-3), 86.7 (C-11), 51.6 (C-5), 19.0 (C-15), 14.9 (C-9 Me). EI-MS m/z (%) 257 (100, M˙+), 242 (10), 126 (48), 84 (56), 69 (67). HREI-MS: calc 257.1052 (C15H15NO3) found 257.1051. ESI-MS (−ve) m/z (%) 513 (3, [2M − H]−), 256 (19, [M − H]−), 140 (8), 124 (12), 98 (38), 69 (100), 42 (67). HRESI-MS: calc 256.0974 (for C15H14NO3, [M − H]−) found 256.0974.
:MeOH to yield the target compound as an orange oil (74 mg, 52%). Tautomeric ratio Z:E = ∼2:1 in CDCl3. For 1H NMR spectrum the major tautomer is assigned, for the 13C NMR spectrum, both tautomers are assigned. 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 15.6 Hz, 1H, H-8), 7.31 (d, J = 15.6 Hz, 1H, H-7), 6.29 (dq, J = 15.6, 6.8 Hz, 1H, H-14), 6.10 (s, 1H, H-10), 5.74 (dq, J = 15.6, 2.0 Hz, 1H, H-13), 4.06 (s, 2H, H-5), 2.11 (s, 3H, C-9 Me), 1.86 (dd, J = 6.8, 2.0 Hz, 3H, H-15), 1.56 (s, 9H, C(C3)3). 13C (100 MHz, CDCl3) δ 197.7 (C-4 minor), 189.3 (C-4 major), 178.0 (C-6 minor), 176.2 (C-6 major), 173.6 (C-2 major), 164.8 (C-2 minor), 149.8 (NO2tBu minor), 149.4 (NO2tBu major), 148.7 (C-8), 144.8 (C-9), 142.1 (C-14), 122.4 (C-10), 118.6 (C-7 major), 118.0 (C-7 minor), 110.9 (C-13), 103.5 (C-3 minor), 102.9 (C-12), 101.5 (C-3 major), 86.7 (C-11), 84.0 ((CH3)3 major), 83.4 ((CH3)3 minor), 54.7 (C-5 major), 52.3 (C-5 minor), 28.0 (C(H3)3), 19.0 (C-15), 14.8 (C-9 Me). IR: ν (cm−1) 1743 (m), 1613 (s), 1551 (m), 1343 (s), 1310 (s), 1156 (s). EI-MS m/z (%) 357 (3, M˙+), 301 (7), 277 (52), 2257 (43), 126 (20), 71 (48), 56 (100). HREI-MS: calc 357.1576 (C20H23NO5) found 357.1563.
:E = ∼2:1 in CDCl3. For 1H NMR spectrum the major tautomer is assigned, for the 13C NMR spectrum, both tautomers are assigned. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 15.6 Hz, 1H, H-8), 7.32 (d, J = 15.6 Hz, 1H, H-7), 6.90 (dd, J = 15.2, 12.0 Hz, 1H, H-11), 6.51 (d, J = 12.0 Hz, 1H, H-10), 5.88 (dq, J = 15.2, 2.4 Hz, 1H, H-12), 4.06 (s, 2H, H-5), 2.04 (d, J = 2.4 Hz, 3H, H-15), 2.00 (s, 3H, C-9 Me), 1.56 (s, 9H, C(C3)3). 13C (100 MHz, CDCl3) δ 197.7 (C-4 minor), 189.2 (C-4 major), 178.1 (C-6 minor), 176.5 (C-6 major), 173.2 (C-2 major), 165.0 (C-2 minor), 151.9 (NO2tBu minor), 151.2 (NO2tBu major), 150.6 (C-8), 141.9 (C-9, C-10 and C-11), 136.1, 133.9, 118.7 (C-7 and C-12), 117.5, 103.0 (C-3 minor), 101.1 (C-3 major), 94.4 (C-14), 83.9 (O(CH3)3 major), 83.3 (O(CH3)3 minor), 79.7 (C-13), 54.6 (C-5 major), 52.3 (C-5 minor), 28.2 (C(H3)3), 12.6 (C-9 Me), 4.9 (C-15). IR: ν (cm−1) 1706 (m), 1615 (s), 1573 (s), 1310 (s), 1154 (s). EI-MS m/z (%) 357 (21, M˙+), 301 (22), 257 (97), 126 (54), 91 (100), 84 (53), 57 (100). HREI-MS: calc 357.1576 (C20H23NO5) found 357.1566.
:MeOH as eluent to afford the title compound as an orange solid (6 mg, 65%). Further purification was achieved using semi-preparative HPLC using a gradient eluent of 60% MeCN: H2O/TFA (0.1%) to 80% MeCN: H2O/TFA (0.1%). See S14 for detailed HPLC experimental. Tautomeric ratio Z:E – 4:1. 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 15.5 Hz, 1H, H-8), 7.28 (d, J = 15.5 Hz, 1H, H-7), 6.28 (dq, J = 16.0, 7.0 Hz, 1H, H-14), 6.06 (s, 1H, H-10), 5.96 (br s, 1H, NH), 5.76 (d, J = 16.0 Hz, 1H, H-13), 3.94 (s, 2H, H-5 minor), 3.84 (s, 2H, H-5 major), 2.14 (s, 3H, C-9 Me), 1.87 (d, J = 7.0 Hz, 3H, H-15). 13C (125 MHz, CDCl3) δ 192.4 (C-4), 176.2 (C-2), 174.2 (C-6), 146.8 (C-8), 144.9 (C-9), 141.7 (C-14), 121.0 (C-10), 119.0 (C-7), 110.9 (C-13), 101.9 (C-12), 100.5 (C-3), 86.7 (C-11), 51.6 (C-5), 19.0 (C-15), 14.9 (C-9 Me). EI-MS m/z (%) 257 (100, M˙+), 242 (10), 126 (48), 84 (56), 69 (67). HREI-MS: calc 257.1052 (C15H15NO3) found 257.1051. ESI-MS (−ve) m/z (%) 513 (3, [2M − H]−), 256 (19, [M − H]−), 140 (8), 124 (12), 98 (38), 69 (100), 42 (67). HRESI-MS: calc 256.0974 (for C15H14NO3, [M − H]−) found 256.0974.
Isolation of fungus (MINAP-9902)2
A fruiting body of the myxomycete Lycogala epidendrum was surface sterilised by immersion in a 10% aqueous NaOCl solution for 90 seconds. The fruiting body was then washed twice with sterile water and ruptured onto a 110 mm potato dextrose agar (PDA) plate (an initially white culture developed which turned green upon sporulation). Yellow globules developed after 4 days. A sterile inoculating loop was used to transfer fungal material from the radius of the loop to a new PDA plate which grew to give a pure culture of the fungus sp. MINAP-9902, which was identified as a Penicillium species.Culturing of MINAP-9902
The fungus was grown on 110 mm malt extract agar plates at 25 °C for 4 days. Eight 250 mL conical flasks were prepared, four with 50 mL potato dextrose broth (PDB) and four with 50 mL of malt extract broth (MEB). Each flask was inoculated with two 1 cm2 blocks of fungal culture of the malt extract plate. These flasks were maintained at 25 °C, and agitated on an orbital shaker at 120 rpm for 4, 6, 8 and 10 days (two conical flasks, one each of PDB and MEB, were removed periodically every two days from the four day mark). For each of the conical flasks, the mycelia were mechanically separated from the broth by filtration and the mycelia were homogenised in a solution of CH2Cl2/EtOH (1:4). The crude extracts were concentrated, then suspended in 50 mL of deionised water, and extracted successively with hexane (2 × 50 mL), CH2Cl2 (2 × 50 mL) and EtOAc (2 × 50 mL). Previous studies1 indicated that the bioactive metabolites of interest were to be found in the CH2Cl2 layer. Thus, the CH2Cl2 layers were dried over MgSO4, filtered, and concentrated in vacuo to provide samples that were subjected to HPLC.
HPLC
Analytical HPLC analyses were conducted on the eight fungal extracts prepared. HPLC conditions:Analytical and semi-preparative HPLC was performed using an Agilent 1100 system, utilising a Phenomenex LUNA 5μ C-18(2) 250 × 4.60 mm column (analytical) or a Phenomenex LUNA 5μ C-18(2) 250 × 10.00 mm column (semi-preparative) and an Agilent 1100 series diode array detector for eluate detection. Elution was carried out using 0.1% v/v TFA in Milli-Q water and pre-filtered (0.45 μm nylon membrane filter) MeCN, with detection at 254 nm and 300 nm or 400 nm. Elution was carried out with 60% MeCN/H2O (0.1% TFA), increasing to 80% MeCN/H2O (0.1% TFA) over 30 min, with flow rates of 1.5 mL min−1 (analytical) or 2.5 mL min−1 (semi-preparative). In the first stage of HPLC work, analytical HPLC was carried out on synthetic analogues 2 and 3 to determine the retention times of these compounds. Subsequent analytical runs were conducted on the eight fungal extracts prepared from MINAP-9902, to probe for retention times similar to those for the prepared synthetic analogues. Our analysis of these runs revealed that the fungal extracts that were cultured for 6 days (both PDB and MEB) contained the metabolite of interest in a ratio that was yet to be overwhelmed by a suite of compounds produced by this fungus upon prolonged culture. This was based on the presence of peaks with similar retention times to our synthetic analogues and the comparison of the UV spectra of these peaks to our synthetic analogues. In particular, we decided to focus on the PDB broth that was cultured for 6 days (sample name: PDB6), as the signals of interest appeared stronger in the PDB HPLC trace, in comparison to that of the MEB HPLC trace. In PDB6, a strong peak observed at 14.4 min (analytical HPLC) was assigned to be ravenic acid. Peaks with retention times in the vicinity of analogue 3 (16.0 min) were also present (15.9 min and 17.6 min), but peaks with retention times close to that of 2 (13.2 min) were notably absent. Co-injection of 3 with PDB6 resulted in peak enhancement, strongly indicative of matching structures. At this stage semi-preparative HPLC was performed on PDB6 to isolate the metabolite of interest. Small quantities of the metabolite were isolated and mass spectral data recorded: EI-MS m/z (%) 257 (93, M˙+), 242 (10), 129 (48), 126 (46), 84 (75), 69 (76), 57 (100). HREI-MS: calc 257.1052 (C15H15NO3) found 257.1052. The EI-MS of the metabolite isolated from PDB6 closely resembled the EI-MS of 3, with matching molecular ions (m/z 257), as well as a number of daughter ions (m/z 126, 84, 69) observed in both spectra. Re-injection of the metabolite of interest to the HPLC returned a peak with a retention time matching that of synthetic analogue 3 (see ESI†), confirming that no decomposition or structural modification had taken place. The matching retention times and mass spectral data of the unknown metabolite with analogue 3, provide strong evidence that the structure of the metabolite corresponds to that of 3.
Determination of the sensitivity of compounds against Staphylococcus aureus
Kirby Bauer bioassays were performed routinely to identify fractions containing an active compound(s). This was achieved by inoculating an 83 mm nutrient agar plate with 200 µL suspension of Staphylococcus aureus cells in sterilised deionised water, and spreading this suspension over the agar surface with a flame sterilised glass spreader. The plate was dried for 10 min in a laminar flow hood, while 5 mm paper disks were soaked in a solution of the fractions or compounds of interest, dissolved in a volatile solvent. The disks were allowed to dry and then lightly pressed onto the agar surface. The plates were then incubated at 37 °C for 16 hours and examined for growth inhibition around the disks. The sensitivity of a clinical strain of Staphylococcus aureus was assessed by a turbidity assay OD600 (optical density at λ 600 nm) as described in a previous publication.17Acknowledgements
JDM acknowledges the ANU for an Australian Postgraduate Award. This research was undertaken with the support of the ANU and an NHMRC project grant (1028092).Notes and references
- (a) B. J. L. Royles, Chem. Rev., 1995, 95, 1981 CrossRef CAS; (b) X. Mo, Q. Li and J. Ju, RSC Adv., 2014, 4, 50566 RSC.
- A. P. Michael, E. J. Grace, M. Kotiw and R. A. Barrow, J. Nat. Prod., 2002, 65, 1360 CrossRef CAS.
- S. V. Ley, S. C. Smith and P. R. Woodward, Tetrahedron, 1992, 48, 1145 CrossRef CAS.
- Y.-C. Jeong and M. G. Moloney, J. Org. Chem., 2011, 76, 1342 CrossRef CAS PubMed.
- A. Schlenk, R. Diestel, F. Sasse and R. Schobert, Chem. – Eur. J., 2010, 16, 2599 CrossRef CAS PubMed.
- Y.-C. Jeong and M. G. Moloney, Synlett, 2009, 2487 CAS.
- H. J. Bestmann, Angew. Chem., Int. Ed., 1977, 16, 349 CrossRef.
- D. A. Burr, X. B. Chen and J. C. Vederas, Org. Lett., 2006, 9, 161 CrossRef PubMed.
- N. J. Lawrence, in Preparation of Alkenes, ed. J. M. J. Williams, Oxford University Press, New York, 1996, p. 19 Search PubMed.
- N. P. Bowling, N. J. Burrmann, R. J. Halter, J. A. Hodges and R. J. McMahon, J. Org. Chem., 2010, 75, 6382 CrossRef CAS PubMed.
- J. Robertson and S. Naud, Org. Lett., 2008, 10, 5445 CrossRef CAS PubMed.
- W. Boland, N. Schroer, C. Sieler and M. Feigel, Helv. Chim. Acta, 1987, 70, 1025 CrossRef CAS.
- G. Pohnert and W. Boland, Tetrahedron, 1994, 50, 10235 CrossRef CAS.
- Y. Jeong and M. G. Moloney, Synlett, 2009, 2487 CAS.
- R. Schobert, Org. Synth., 2005, 82, 140 CrossRef CAS.
- A. Schlenk, R. Diestel, F. Sasse and R. Schobert, Chem. – Eur. J., 2010, 16, 2599 CrossRef CAS PubMed.
- A. M. Beekman and R. A. Barrow, Aust. J. Chem., 2015, 68, 1583 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c6ob00938g |
| ‡ Current address: School of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, Norfolk, UK NR4 7TJ. |
| This journal is © The Royal Society of Chemistry 2016 |