
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelation-assisted CuAAC in star-shaped polymers enables fast self-healing at low temperatures†
S.
Neumann
,
D.
Döhler
,
D.
Ströhl
and
W. H.
Binder
*
Chair of Macromolecular Chemistry, Institute of Chemistry, Division of Technical and Macromolecular Chemistry, Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle D-06120, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de; Fax: +49 345 55 27392
First published on 9th February 2016
Abstract
The achievement of self-healing (SH) under ambient conditions (low temperature, no external input of energy) still presents a significant area of research, and is strongly linked to fast and efficient cross-linking reactions. We investigate here fast cross-linking reactions of star-shaped polymers containing copper chelating moieties (picolinazide) at the end of each arm, able to promote a “click”-reaction through chelation of the Cu(I)-catalyst and thus strongly increasing cross-linking rates. The synthetic preparation and cross-linking kinetics of a low molecular weight model system (p-carboxylic-acid-methylester-picolinazide and phenylacetylene) were investigated by utilizing different catalysts (CuBr, CuBr(PPh3)3, Cu(MeCN)4PF6 and CuOAc) and applying in situ NMR experiments. The most efficient catalyst systems (CuBr, CuBr(PPh3)3, CuF(PPH3)3) were used to monitor the cross-linking of three-arm star polymers bearing the carboxylic acid-methylester-picolinazide moiety on each arm via melt-rheology studies, in turn enabling self-healing. The complete cross-linking of the components can be observed within 71 minutes even at low temperatures (10 °C), thus generating a highly efficient low-temperature SH-system. Self-healing of a polymeric material at room temperature was demonstrated, consisting of a star-shaped picolinazido-telechelic PIB, an encapsulated multivalent alkyne embedded within a high molecular-weight PIB matrix, together with CuBr(PPh3)3 and a fluorogenic dye, the latter acting as sensing tool for the proceeding click network formation. A damage-induced increase in the fluorescence intensity due to the click activation of the fluorogenic dye at room-temperature and the formation of a polymer network was thus proven. We envision that this highly enhanced cross-linking speed will facilitate applications of self-healing polymers under low temperature conditions.
1. Introduction
The concept of self-healing polymers envisions everlasting materials that are able to repair damage autonomously.1 For the successful development of self-healing (SH) polymers, the diffusion and reaction of the reactants is critical, in turn filling a crack through a newly formed network, either via purely physical (“supramolecular”) forces,2–4 or by the action of chemical forces, in both cases requiring partially reversible5–7 or stable8,9 covalent bonds. Prominent examples of such chemical healing processes include Diels–Alder reactions,5,10,11 epoxide chemistry,12–22 “click-based” chemistry,8,23–30 isocyanate chemistry,31 olefin metathesis9,32–34 and thiol chemistry.35,36 Although the topic of SH has been investigated for more than a decade, there are still major drawbacks preventing crack-formation via SH concepts, especially at low temperature. Among the many reported SH systems, a significant number work at relatively high temperatures (e.g. recent examples heal at 100 °C (ref. 37) or even 160 °C (ref. 38)), so in these cases SH seems to be more of a mending process via thermoplastic shape reformation rather than a self-healing process acting autonomously. Additionally, external stimuli such as photochemical activation are often required to improve the kinetics of healing.39,40 Therefore many of the few fast self-healing systems are directed towards click-reactions, especially the most prominent example (CuAAC)23 which has been used to create SH-polymers that work at room-temperature. Thus, the copper-catalyzed azide/alkyne “click” reaction (CuAAC)41–44 is a potentially useful chemical cross-linking reaction, as high thermodynamic gains tend to drive the reaction to completion, resulting in a complete and dense network formation. Through the use of copper44 in the +1 oxidation state as catalyst, the 1,3-dipolar Huisgen reaction fulfils the criteria for a click41 candidate. CuAAC accelerates the process by a factor of 107 resulting in high yields (<99%) within short timescales.26,27,42,45,46 Therefore, CuAAC is an excellent candidate for self-healing systems and is consequently already used widely: namely, bis-azides, bis-alkynes30,47 and azido-star-shaped polymers or monomers and oligomers8,28,29,48,49 have been demonstrated to generate self-healing systems that are highly efficient at room-temperature within hours, even minutes in some cases. Further investigations have improved the efficiency of such systems by modifying the catalyst,8,49 increasing the density of functional groups,28,48 decreasing the starting viscosity of the polymer mixture and using different molecular weight polymers.28 To further accelerate CuAAC mediated SH, often the addition of bases (mainly amine bases) and external ligands such as tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), 2-[4-({bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}methyl)-1H-1,2,3-triazol-1-yl]ethyl hydrogen sulfate (BTTES) or 3-[4-({bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}amethyl)-1H-1,2,3-triazol-1-yl]propyl hydrogen sulfate (BTTPS) have been investigated.50–53 It is generally assumed that amine bases promote the formation of the copper acetylide and protect copper(I) from oxidation and disproportionation, further avoiding the formation of toxic reactive oxygen species (ROS) of Cu(I) in the presence of oxygen, and simultaneously keeping the amount of catalytically potent species at a high level for a longer period of time. Thus, the combination of a click-based SH system with an internal ligand,54,55 able to accelerate the “click” reaction without the need of an added amine, represents a simplified concept where the number of required components is reduced. We report here for the first time a self-healing system containing star-shaped picolinazido- and alkyne-telechelic polymers based on poly(isobutylene) (PIB), able to form a network with the aid of chelation-assisted CuAAC which heals within minutes at low temperatures (10 °C) (see Fig. 1).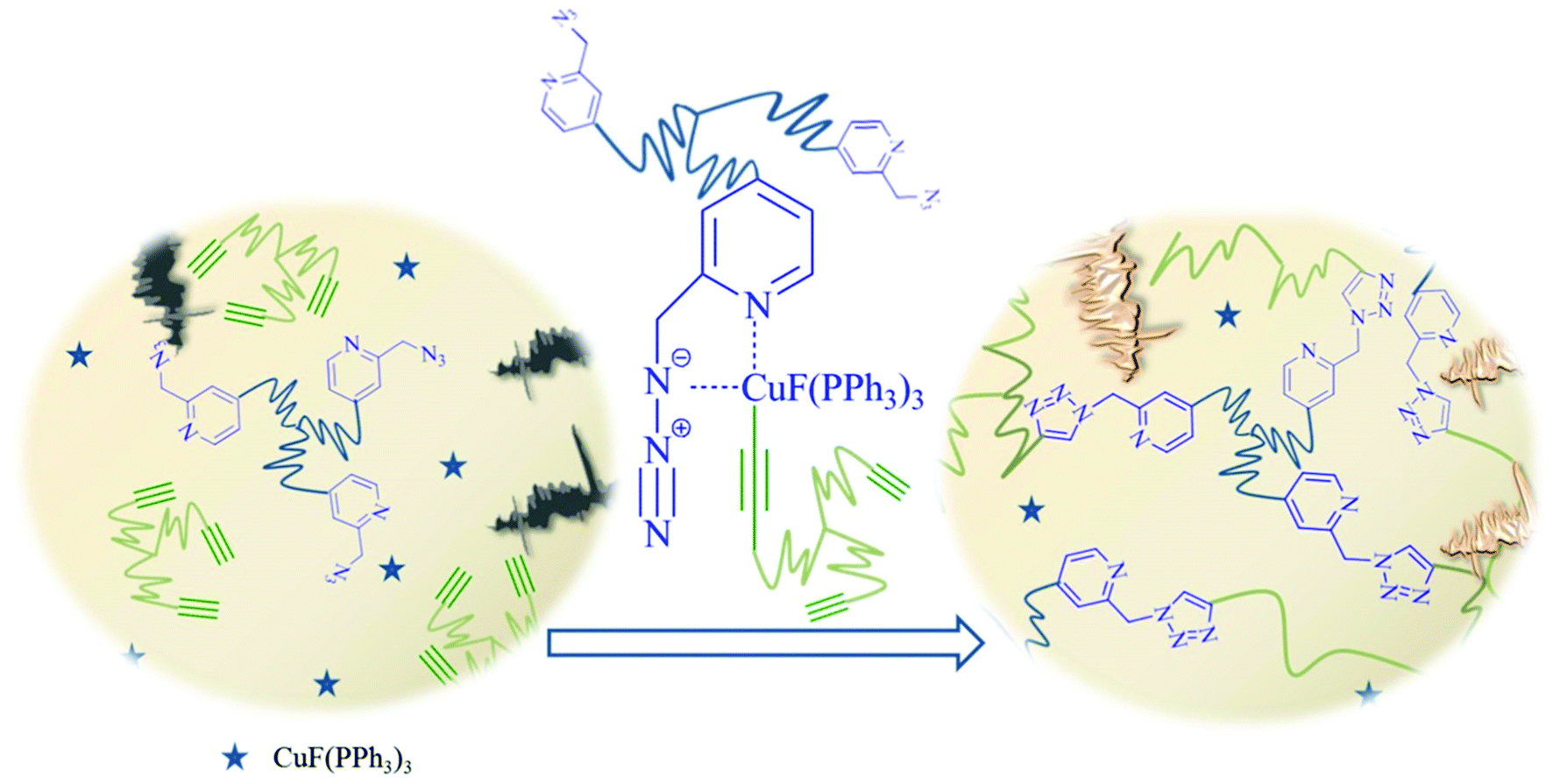 | ||
| Fig. 1 Proposed healing mechanism. Using star-shaped polymers functionalized with copper chelating azides suitable for chelation-assisted CuAAC enables fast network-formation, even at lower temperatures. | ||
As the click-based ligand is now designed to effect chelation of the Cu(I)-catalyst prior to cross-linking via CuAAC, we expect a significantly enhanced cross-linking rate at lower temperature, thus approaching a low temperature SH-system in the absence of externally added components. We explore the synthesis of star-shaped polymers, together with the optimization of the catalyst as well as the final cross-linking in the melt state. Furthermore, we investigate a fluorogenic dye as a sensing tool that is activated via the chelation-assisted click reaction, in turn visualizing the cross-linking reaction within a bulk material after physical damage.
2. Experimental section
2.1 Materials
2,4-Pyridinecarboxylic acid hydrate was purchased from TCI and used without further purification. Calcium chloride (ultra dry 99.9%) was purchased from Alfa Aesar, lithium hydroxide from Lachema, oxalyl chloride from Merck, high molecular weight poly(isobutylene) (250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) from BASF and 4-(dimethylamino)pyridine from Fluka, which were used without further purification. Copper(I)bromide was obtained from Sigma-Aldrich and washed with glacial acetic acid followed by ethanol and finally by diethyl ether, before it was dried in high vacuum prior to use. Copper(I)acetate, copper in charcoal (3 wt%), bromotris(triphenylphosphine)copper(I), fluorotris(triphenylphosphine)copper(I) and tetrakis(acetonitrile)copper(I)hexafluorophosphate were purchased from Sigma Aldrich and used without further purification. All other materials were obtained from Sigma-Aldrich and used as received if not mentioned otherwise. N,N,N-Triethylamine and methanol were freshly distilled over CaH2 under a nitrogen atmosphere prior to use. n-Hexane was pre-dried over KOH and freshly distilled over sodium and KOH under a nitrogen atmosphere prior to use. Tetrahydrofuran was pre-dried over KOH and CuCl and freshly distilled over sodium and benzophenone under a nitrogen atmosphere prior to use. Dichloromethane was pre-dried over CaCl2 and freshly distilled over CaH2 under a nitrogen atmosphere prior to use.
000 g mol−1) from BASF and 4-(dimethylamino)pyridine from Fluka, which were used without further purification. Copper(I)bromide was obtained from Sigma-Aldrich and washed with glacial acetic acid followed by ethanol and finally by diethyl ether, before it was dried in high vacuum prior to use. Copper(I)acetate, copper in charcoal (3 wt%), bromotris(triphenylphosphine)copper(I), fluorotris(triphenylphosphine)copper(I) and tetrakis(acetonitrile)copper(I)hexafluorophosphate were purchased from Sigma Aldrich and used without further purification. All other materials were obtained from Sigma-Aldrich and used as received if not mentioned otherwise. N,N,N-Triethylamine and methanol were freshly distilled over CaH2 under a nitrogen atmosphere prior to use. n-Hexane was pre-dried over KOH and freshly distilled over sodium and KOH under a nitrogen atmosphere prior to use. Tetrahydrofuran was pre-dried over KOH and CuCl and freshly distilled over sodium and benzophenone under a nitrogen atmosphere prior to use. Dichloromethane was pre-dried over CaCl2 and freshly distilled over CaH2 under a nitrogen atmosphere prior to use.
The synthesis and encapsulation of (2,2-bis(prop-2-2-ynyloxymethyl)-1-(prop-2-ynyloxy)butane (4) in urea–formaldehyde microcapsules (6–8 μm) using an oil-in-water emulsion technique was done according to literature.8,29,56,57 3-Azido-7-hydroxy-coumarin was synthesized according to known procedures.58
2.2 Measurements
NMR spectra were recorded on a Varian Gemini 2000 (400 MHz) or on a Varian Unity Inova 500 (500 MHz) at 27 °C. Deuterated chloroform (CDCl3) or deuterated tetrahydrofuran (THF-d8) were used as solvents. All chemical shifts are given in ppm. MestReNova software (version 6.0.2-5475) was used for interpretation of the NMR spectra. For the kinetic investigations, 2-(6-azidomethyl)-pyridine-4-carboxylic acid methyl ester (15 mg, 1.0 equivalent) was dissolved in THF-d8 (0.35 mL), while phenylacetylene (10.3 μL, 1.2 eq.), the catalyst (0.05 or 0.1 eq.) and DIPEA (0.01, 0.1 or 1.2 eq.) (Table 1, entries 5–8) were separately dissolved in THF-d8 (0.40 mL). When using 0.01 eq. of DIPEA, a stock solution in THF-d8 was prepared. Mixing the azide-containing solution and the alkyne-containing solution represents the starting point for the in situ NMR experiments. The first NMR-experiment was recorded directly after mixing the solutions. Further experiments were conducted at different defined periods of time while the corresponding conversion was calculated for each NMR-experiment by comparing the integrals of the CH2-moiety attached to the azide (educt) and the CH2-moiety attached to the triazol ring (product) (for detailed information see ESI†).| Entry | Catalyst | Time | DIPEA | Conv. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 0.1 equivalents of catalyst were added. b 0.05 equivalents of catalyst were added. c First NMR-spectrum showed complete conversion. d c = 125 mM. e c = 10.4 mM. f c = 1.04 mM. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Cu/charcoala | 9 h | — | <1% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | [Cu(CH3CN)4]PF6a |
24 h | — | 32% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Cu(OAc)a | 16 h | — | 82% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | CuBra | 9 h | — | >99% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CuBra | <5 minc | 1.2 eq.d | >99% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | CuBra | <5 minc | 0.1 eq.e | >99% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | CuBra | 17 min | 0.01 eq.f | >99% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | CuBr(PPh3)3b |
2 h | 0.01 eq.f | 10% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For inline FTIR-measurements a Bruker Vertex 70 MIR spectrometer equipped with an ATR-FTIR diamond probe was used. Opus 6.5 was used for analyzing the data.
Rheology experiments were performed on an Anton Paar (Physica) MCR 101/SN 80753612 at either 20 °C or 10 °C. For regulating the sample temperature, thermoelectric cooling/heating in a Peltier chamber under a dry oxygen atmosphere was applied. For all measurements parallel plates with a diameter of 8 mm were used. Frequency measurements were performed within the linear viscoelastic range. For cross-linking experiments a picolin-azide-functionalized star-shaped PIB (30.0 mg) and an alkyne-functionalized star-shaped PIB (26.0 mg) were put in a vial and dissolved in CHCl3. After mixing both solutions and evaporating the solvent, the obtained polymer mixture was dried under high vacuum. The catalyst (CuBr, CuBr(PPh3)3 or CuF(PPh3)3) (0.1 eq. per functional group) was dissolved in CHCl3 (20 μL) and added as a stock solution to the polymer blend. In the cases of CuBr and CuBr(PPh3)3, N,N-diisopropylethylamine (DIPEA) (0.01 or 0.1 equivalents per functional group) was additionally added as a stock solution. Subsequently, the reaction mixture was mixed with a spatula and was immediately put on the rheometer plate. Cross-linking experiments were performed with a strain γ of 0.1% and with an angular frequency ω ranging from 100 to 1 rad s−1. Gelation times28,48,59 were determined as a crossover of the storage (G′) and loss modulus (G′′) at 10 rad s−1. Each measurement was stopped when the values of loss and storage modulus stayed constant (to two decimal places) for at least one hour. This time is considered as the total time. The determined plateau moduli correspond to the storage moduli measured at this total time at 100 Hz. For the evaluation of this data, RheoPlus/32 software (V 3.40) and OriginPro8 were used.
Gel permeation chromatography (GPC) measurements were performed on a Viscotek GPCmax VE 2002 using a HHRH Guard-17369 and a GMHHR-N-18055 column in THF at 40 °C and the detection of the refractive index was performed with a VE 3580 RI detector of Viscotek. For external calibration, PIB-standards (320 g mol−1 to 578000 g mol−1) from Viscotek were used. The concentration of all samples was 3 mg mL−1 and the flow rate was 1 mL min−1.
Fluorescence measurements were performed on a Cary Eclipse Fluorescence Spectrophotometer from Agilent Technologies. The excitation wavelength was set to 330 nm while detecting the fluorescence emission from 350 to 650 nm. Therefore, solid samples were fixed with quartz glass plates and measured within a solid sample holder. A measurement was performed every 5 minutes over the course of 24 hours. In the case of the control experiments (unscratched specimen, scratched specimen without CuBr(PPh3)3) measurements were performed every 5 minutes over the course of 4 hours. For the evaluation of the data, Cary Eclipse Scan Applications Software (v. 1.2 (147)) and Origin Pro8G (v. 8.0951) were used.
2.3 Synthesis
2,4-Pyridinedicarboxylic acid dimethyl ester (3.8 mmol, 750 mg) and ultra dry calcium chloride (99.99%, 17.1 mmol, 1.9 g) were dissolved in anhydrous tetrahydrofuran (6.0 mL) and anhydrous methanol (12.0 mL). The solution was cooled to −5 °C and subsequently sodium borohydride was added in small portions (5.6 mmol, 225 mg, (3 × 75 mg)). The reaction was accomplished after ∼2 hours 40 minutes and quenched with ice-cold water (15.0 mL). The solution was extracted with chloroform (3 × 40.0 mL) and the combined organic layers were dried over magnesium sulphate. The solvent was removed under reduced pressure to afford 2-(6-hydroxymethyl)-pyridine-4-carboxylic acid methyl ester. Rf (EtOAc) = 0.41. 1H-NMR (CDCl3, 400 MHz): δ = 8.70 (d, 1H, 3JH,H = 5.1 Hz, CH), 7.83 (s, 1H, CH), 7.75 (dd, 1H, 3JH,H = 5.1 Hz, 4JH,H = 0.7 Hz, CH), 4.83 (s, 2H, CH2), 3.96 (s, 3H, CH3) ppm; 13C-NMR (CDCl3, 100 MHz): δ = 165.5, 160.4, 149.4, 138.1, 121.6, 119.8, 64.2, 52.7 ppm.
2-(6-Hydroxymethyl)-pyridine-4-carboxylic acid methyl ester (0.3 mmol, 50 mg) was dissolved in anhydrous dichloromethane (6.6 mL), followed by the addition of N,N,N-triethylamine (1.5 mmol, 207 μL) and para-toluenesulfonyl chloride (0.5 mmol, 87 mg). After stirring for two hours the solvent was removed under reduced pressure. The residue was dissolved in anhydrous tetrahydrofuran (3.3 mL) and sodium azide (3.0 mmol, 193 mg) was added. The reaction was stirred for a further 24 hours at room temperature, after which time it was diluted with ethyl acetate (30.0 mL) and water (30.0 mL). After extraction of the aqueous layer with ethyl acetate (three times 30.0 mL), the combined organic layers were washed with a saturated solution of sodium chloride and dried over magnesium sulphate. The crude product was purified by silica chromatography (n-Hex:EtOAc, 4:1, Rf = 0.65 in n-Hex:EtOAc, 1:1) providing 2-(6-azidomethyl)-pyridine-4-carboxylic acid methyl ester. 1H-NMR (CDCl3, 400 MHz): δ = 8.74 (d, 1H, 3JH,H = 5.0 Hz, CH), 7.89 (s, 1H, CH), 7.79 (dd, 1H, 3JH,H = 5.0 Hz, 4JH,H = 1.4 Hz, CH), 4.56 (s, 2H, CH2), 3.96 (s, 3H, CH3) ppm; 13C-NMR (CDCl3, 100 MHz): δ = 165.3, 157.0, 150.5, 138.4, 122.1, 121.1, 55.4, 52.8 ppm.
2-(6-Azidomethyl)-pyridine-4-carboxylic acid methyl ester (2.6 mmol, 500 mg) was dissolved in methanol (10.0 mL), followed by the addition of a 1.0 M aqueous solution of lithium hydroxide (7.8 mmol, 7.8 mL). The reaction was stirred for 25 minutes at room temperature. Neutralization was achieved with the addition of a 1.0 M solution of hydrogen chloride. The solvent was removed under reduced pressure and the product was dried under high vacuum until constant weight to obtain 2-(6-azidomethyl)-pyridine-4-carboxylic acid (1a). 1H-NMR (DMSO-d6, 400 MHz): δ = 8.51 (d, 1H, 3JH,H = 4.9 Hz, CH), 7.74 (s, 1H, CH), 7.66 (dd, 1H, 3JH,H = 4.9 Hz, 4JH,H = 1.1 Hz, CH), 4.48 (s, 2H, CH2) ppm; 13C-NMR (DMSO-d6, 100 MHz): δ = 167.2, 155.7, 149.5, 149.2, 123.1, 122.4, 55.1 ppm.
To provide 2-(6-azidomethyl)-pyridine-5-carboxylic acid (1b) the same protocol was used, beginning with step II and using 2,5-pyridinedicarboxylic acid dimethyl ester as starting material. 1H-NMR (DMSO-d6, 400 MHz): δ = 8.99 (s, 1H, CH), 8.18 (dd, 1H, 3JH,H = 7.9 Hz, 4JH,H = 2.0 Hz, CH), 7.34 (d, 1H, 3JH,H = 7.9 Hz, CH), 4.49 (s, 2H, CH2) ppm; 13C-NMR (DMSO-d6, 100 MHz): δ = 167.6, 156.0, 151.0, 137.9, 134.7, 121.7, 54.8 ppm.
2-(6-Azidomethyl)-pyridine-4-carboxylic acid (1a) (674 μmol, 120 mg, 11.6 equiv.) was dissolved in anhydrous dichloromethane (5.0 mL), followed by the addition of oxalyl chloride (674 μmol, 56 μL, 11.6 equiv.). The reaction was heated under reflux for five hours and then allowed to cool to room temperature. Subsequently star-shaped PIB-OH (58 μmol, 350 mg, 1 equiv.) and 4-(dimethylamino)-pyridine (88 μmol, 11 mg, 1.5 equiv.) dissolved in dichloromethane (5.0 mL) were added to the solution. Finally N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (263 μmol, 50 mg, 4.5 equiv.) was added to the ice-cooled reaction mixture, which was then heated under reflux for three days. In the next step the solution was diluted with dichloromethane (30.0 mL) and filtered. The organic layer was washed with a saturated solution of ammonium chloride and dried over sodium sulphate. The solvent was evaporated under reduced pressure to afford the crude product, which was purified by dissolving in n-hexane and precipitated with an excess of methanol. The final polymer was dried under high vacuum until a constant weight was achieved to provide star-shaped azido-telechelic PIB (2a). (Mn(GPC) = 6520 g mol−1, Mw/Mn = 1.3). 1H-NMR (CDCl3, 500 MHz): δ = 8.76 (d, 3H, 3JH,H = 5.0 Hz, CH), 7.90 (s, 3H, CH), 7.80 (dd, 3H, 3JH,H = 5.0 Hz, 4JH,H = 1.1 Hz, CH), 7.13 (s, 3H, CH of initiator), 4.57 (s, 6H, CH2), 4.33 (t, 6H, 3JH,H = 6.8 Hz, CH2), 1.85 (s, 6H, CH2), 1.41 (s, CH2 of repetitive unit), 1.11 (s, CH3 of repetitive unit), 0.80 (s, 18H, CH3 of initiator) ppm.
The synthetic route to obtain 2b was the same as described for 2a, with the only exception that instead of using the esterification agent 1a its stereo-analogue 1b was used instead. (Mn(GPC) = 5780 g mol−1, Mw/Mn = 1.3). 1H-NMR (CDCl3, 400 MHz): δ = 9.20 (s, 3H, CH), 8.34 (dd, 3H, 3JH,H = 8.1 Hz, 4JH,H = 2.1 Hz, CH), 7.46 (d, 3H, 3JH,H = 8.1 Hz, CH), 7.14 (s, 3H, CH of initiator), 4.58 (s, 6H, CH2), 4.34 (t, 6H, 3JH,H = 6.7 Hz, CH2), 1.86 (s, 6H, CH2), 1.43 (s, CH2 of repetitive unit), 1.12 (s, CH3 of repetitive unit), 0.81 (s, 18H, CH3 of initiator) ppm.
000 g mol−1, 2.49 g) was dissolved in n-hexane (40.0 mL) overnight. To this highly viscous mixture, star-shaped picolinazido-telechelic PIB (2b) (5 wt%, 146 mg) dissolved in n-hexane (1.0 mL), UF-capsules containing (2,2-bis(prop-2-2-ynyloxymethyl)-1-(prop-2-ynyloxy)butane (4) (10 wt%, 300 mg), CuBr(PPh3)3 (2 wt%, 60 mg) and 3-azido-7-hydroxy-coumarin (2.5 wt% of 2b,4 mg) were added. Afterwards all components were homogeneously mixed by using a VORTEX-GENIE® touch mixer. To get rid of air-bubbles, the pressure was reduced carefully to 150 mbar and kept at this level for at least ten minutes. Finally, the viscous mixture was poured into a mould and kept at 50 °C overnight. A specimen of approximate dimensions of 5 mm × 13 mm × 0.8 mm was cut out with a razor blade.
For control experiments a second specimen was synthesized without any catalyst (CuBr(PPh3)3), therefore the amount of high molecular weight PIB was increased to 2.55 g.
3. Results and discussion
Reaction rates within the chelation-assisted mechanism are increased by several orders in comparison to classical CuAAC, caused by the use of internal ligands which are able to form a complex with copper as previously reported.55 Due to the pre-organization of the corresponding reactants (azide/alkyne) close to each other caused by the copper-chelating moiety, the chelation-assisted CuAAC is the fastest version of this click-type reaction so far. To utilize this concept to improve the healing kinetics of polymers, we attached the chelating 2-(6-azidomethyl)-pyridine-4-carboxylic acids (1a, 1b) to star-shaped hydroxy-telechelic PIB (see Fig. 2), in turn acting as a cross-linking reagent for SH.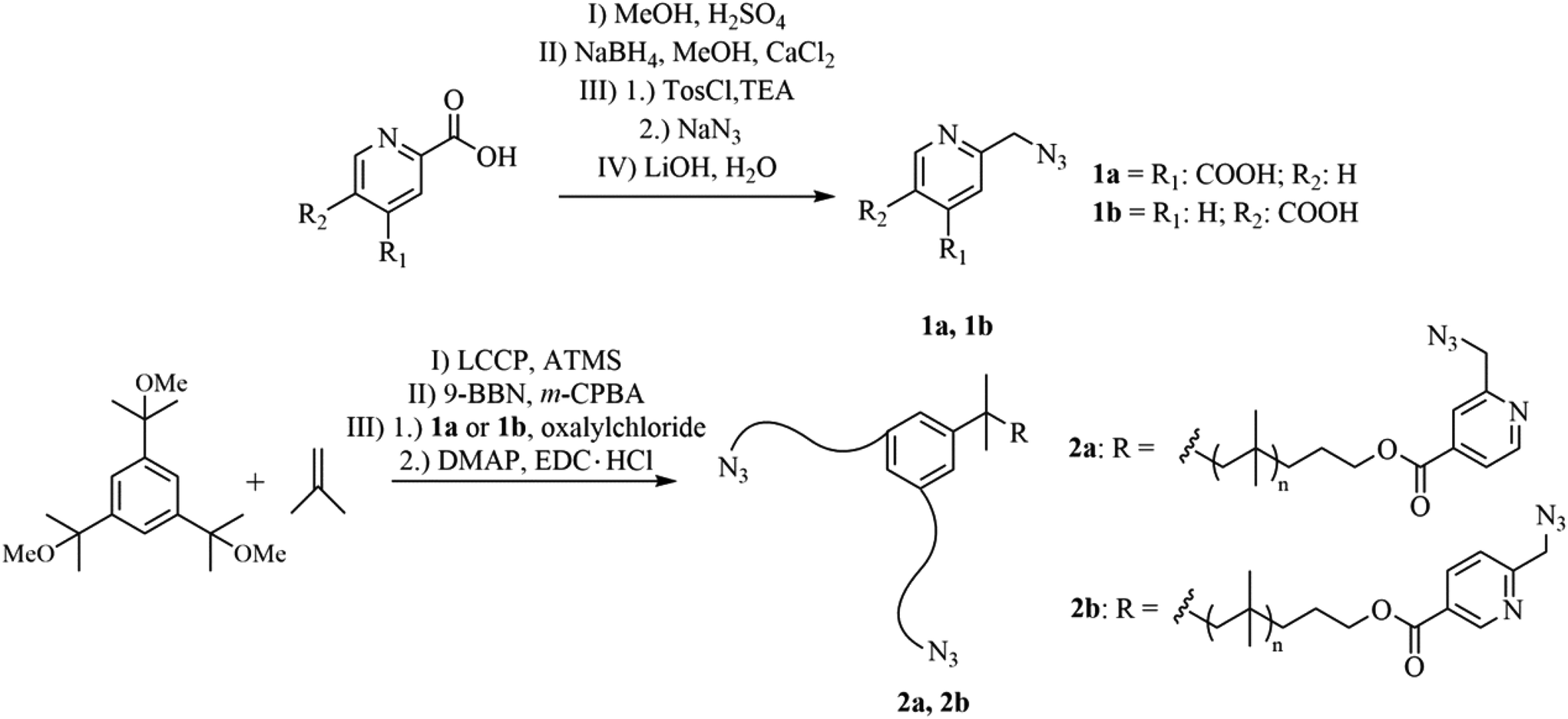 | ||
| Fig. 2 Synthetic route to obtain star-shaped azido-telechelic PIBs (2a, 2b) with a picolin-azide moiety suitable for chelation-assisted CuAAC. | ||
1a has been synthesized over four steps (I–IV), starting with esterification (I), followed by selective reduction (II) and substitution (III) of the corresponding alcohol to the azide group. Further hydrolysis (IV) opens the possibility to attach 1a to the corresponding polymeric alcohol via an esterification reaction. For the synthesis of 1b just three steps (II–IV) are required, due to the commercial availability of 2,5-pyridinedicarboxylic acid dimethyl ester.
The required hydroxy-telechelic PIB has been synthesized starting with living carbocationic polymerization (LCCP) of isobutylene and quenching with ATMS, consequently followed by complete end group transformation to the corresponding alcohol.63–65
Accordingly, we explored the “click”-kinetics of chelation-assisted CuAAC in solution for low molecular weight structures (3.1) as well as in the melt-state for polymeric structures (3.2).
3.1 In situ model NMR-investigations of the chelation-assisted CuAAC of low molecular weight 2-(6-azidomethyl)-pyridine-4-carboxylic acid methyl ester and phenylacetylene
To achieve a highly efficient and fast self-healing process via chelation-assisted CuAAC, different reaction setups including the change of the copper source and the addition of DIPEA as a base were investigated via in situ NMR measurements. 2-(6-azidomethyl)-pyridine-4-carboxylic acid methyl ester and phenylacetylene served as ideal low-molecular substrates for in situ NMR-studies, containing the azide, the alkyne, the copper(I) source and N,N-diisopropylethylamine (DIPEA) (Table 1, entries 5–8) separately dissolved in deuterated THF. Mixing all components together in a NMR-tube defined the starting point of every single experiment (see Table 1), in turn allowing the quantification of the kinetics of the reaction.First of all a commercially available Cu/charcoal catalyst was tested, resulting in poor conversion within nine hours (Table 1, entry 1). Changing the catalyst to [Cu(CH3CN)4]PF6 (Table 1, entry 2) and Cu(OAc) (Table 1, entry 3) resulted in enhanced conversions of 32% and 82% within one day, turning out to be too slow for fast self-healing applications. Upon testing several Cu(I)-catalysts, CuBr delivered the best results, driving the reaction to completion within nine hours (Table 1, entry 4). Thus, CuBr was chosen to run the reaction in the presence of DIPEA, which can promote the formation of a Cu-acetylide species in the first step and therefore “initially” the “click” reaction. In the first NMR spectrum measured five minutes after mixing all components no NMR resonances of the starting material can be seen (Table 1, entries 5 and 6). Even with just 1 mol% DIPEA the reaction went to completion within 17 minutes (Table 1, entry 7). CuBr(PPh3)3 was also tested as catalyst due to its increased solubility in THF,47 due to the attached triphenylphosphin ligands. Surprisingly, CuBr(PPh3)3 showed a lower conversion in the presence of 1 mol% DIPEA (Table 1, entry 8) in comparison to pure CuBr. Thus, we claim that DIPEA is not just promoting the formation of the initially required Cu-acetylide by acting as a base, but also further disintegrates unreactive CuBr clusters,42 delivering an ideal catalyst system for fast click reactions in solution.
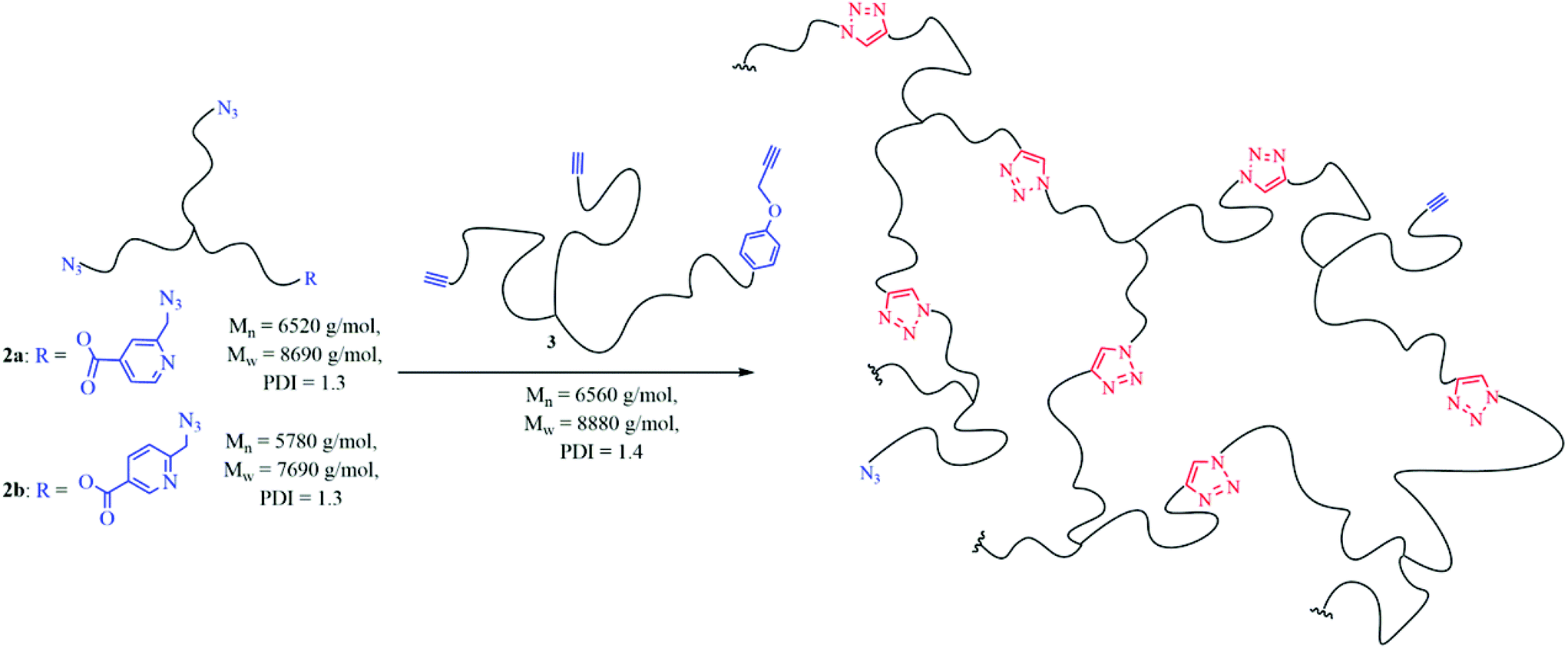
3.2 Cross-linking of star-shaped telechelic PIBs (2a + 3 and 2b + 3) via chelation-assisted CuAAC monitored via melt rheology
To adapt the concept of chelation-assisted CuAAC in order to facilitate fast cross-linking even at lower temperatures, it is necessary to use multivalent polymers, which ensure network formation. Thus, we have functionalized three arm-star poly(isobutylene) with 2-(6-azidomethyl)-pyridine-(4 or 5)-carboxylic acid (1a, 1b) via esterification. The as-obtained polymers (2a, 2b) have been consequently analyzed by GPC, NMR- and IR-spectroscopy, showing the successful introduction of the chelation-capable end group (see ESI†). We further investigated the cross-linking behaviour at different conditions (change of catalyst, temperature) by reacting azido-telechelic PIBs (2a, 2b) with the star-shaped PIB-alkyne (3), monitored by melt rheology measurements (see Table 2) similar to conventional CuAAC based systems.28,29,48,68 As the use of CuBr in solution delivered full conversion in the shortest period of time it was firstly tested in a melt rheology experiment. Although 1 mol% of DIPEA was added, the network formation took 870 minutes (gelation time, see Table 2, entry 1) which is mainly attributed to oxidation during the rheology experiment. Thus, we decided to test CuBr(PPh3)3 as an alternative catalyst despite its relatively poor performance in the initial low molecular weight compound tests.8,28,29,48 Consequently, this system showed significantly shorter gelation times (Table 2, entries 2 and 3); changing the counterion of the catalyst to fluoride, thus additionally improved the observed gelation-times tremendously. Accordingly, cross-linking took place within just 15 minutes at room-temperature (Table 2, entry 5). Thus, this system is even faster than cross-linking hyperbranched PIB-polymers (20 °C, 33 minutes) via conventional CuAAC.48 By changing from the 2,4- (2a) to the 2,5-isomer (2b) the cross-linking time is nearly doubled to 29 minutes at 20 °C (Table 2, entry 6). Although lowering the temperature below room temperature slows down the click-kinetics, cross-linking is still fast – even at 10 °C the gel point was reached within 71 minutes (Table 2, entry 7).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Mixture | Catalystc | T [°C] | Geltd [min] |
c M [M] | c Cu [M] (10−2) | k 0 [M−3 min−1] | k crossover [M−3 min−1] | r 0 [M min−1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ′ Values are reprinted with permission of ref. 29.a = 0.01 eq. of DIPEA was added.b = 0.1 eq. of DIPEA was added.c = 0.1 eq. of catalyst was added.d Determined via melt rheology, G′′ = G′ at ω = 10 Hz.e Reaction was too fast for determination. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
2a + 3a |
CuBr | 20 | 870 | 0.254 | 2.54 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
2a + 3a |
CuBr(PPh3)3 | 20 | 255 | 0.254. | 2.54 | 2200 | 7300 | 0.092 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
2a + 3b |
CuBr(PPh3)3 | 20 | 266 | 0.254 | 2.54 | 400 | 1000 | 0.017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4′ | 29 | CuBr(PPh3)3 | 20 | 290 | 0.238 | 2.38 | 700 | 2700 | 0.023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 2a + 3 | CuF(PPh3)3 | 20 | 15 | 0.254 | 2.54 | —e | —e | —e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 2b + 3 | CuF(PPh3)3 | 20 | 29 | 0.200 | 2.00 | 3100 | 9100 | 0.050 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 2b + 3 | CuF(PPh3)3 | 10 | 71 | 0.200 | 2.00 | 2500 | 7500 | 0.041 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Referring to Ampudia69 and Barton70 the rate constants near the gel point have been calculated according to eqn (1):
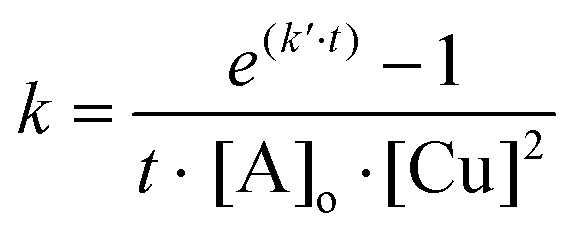 | (1) |
Thus, for all performed cross-linking reactions an acceleration of the reaction rates with progressing time can be observed (see Fig. 3).
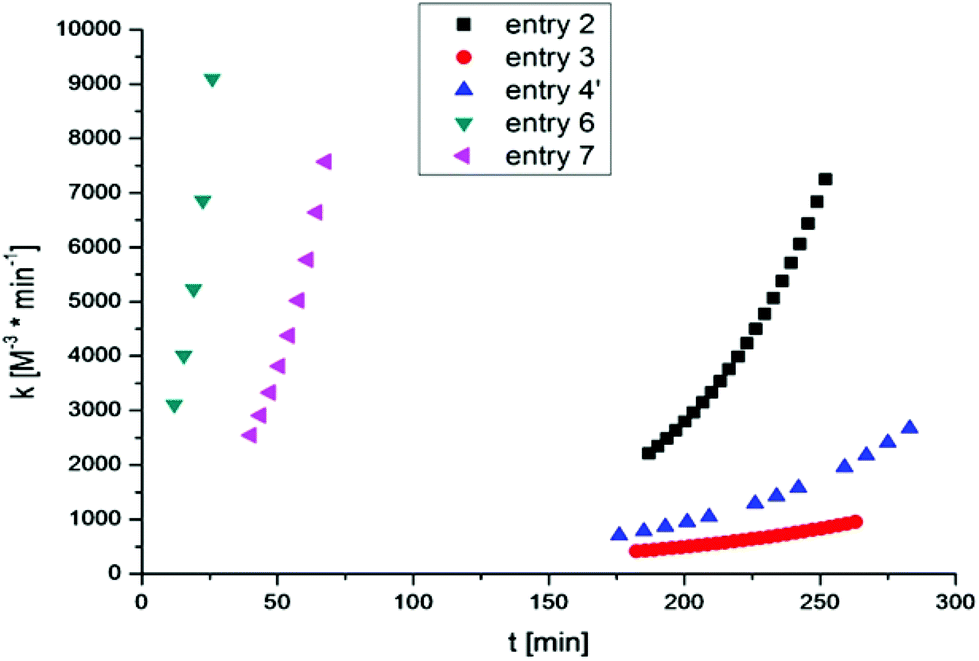 | ||
| Fig. 3 Development of the rate constant k vs. time t for the cross-linking of 2a + 3 and 2b + 3. | ||
This autocatalytic effect is caused by an increasing concentration of the formed triazole rings during the proceeding cross-linking reactions, as these heterocyclic rings can act as internal ligands, capable of chelating copper. Further auto-acceleration of chelation-assisted CuAAC by the newly formed triazole rings should not be as strong as for classical CuAAC, due to the presence of the internal pyridinium ligand already at the very beginning of each reaction. Indeed, the acceleration factors for the chelation-assisted CuAAC vary from 2.9–3.3 (Table 2 and Fig. 3, entries 2, 6 and 7) and are therefore lower compared to the classical CuAAC approach utilizing polymers with comparable molecular weights (Table 2, entry 4′, acceleration factor of 3.4). Although there is only a slight difference in the acceleration of chelation-assisted CuAAC and classical CuAAC, there are tremendous differences in the calculated rate constants. While classical CuAAC starts with a rate constant of k0 ∼ 700 M−3 min−1 (Table 2, entry 4′), up to fourfold higher k0 values (2200–3100 M−3 min−1) are observed for the cross-linking reactions following the chelation-assisted CuAAC mechanism from the very beginning (Table 2, entries 2, 6 and 7). In entry 3, the rate constant k0 is ∼400 M−3 min−1 and is therefore just 1/5 of k0 in entry 2, which is in contrast to identical gelation times of ∼260 minutes for both experiments. Nevertheless, for cross-linking 2b + 3 in the presence of the most active catalyst (CuF(PPh3)3), k0-values of ∼2500–3100 M−3 min−1 (Table 2, entries 6 and 7) are observed already at the very beginning of the reaction, resulting in strongly reduced gelation times (29–71 minutes) even at 10 °C.
By using eqn (2)71,72 network strand densities νx were determined for the final cross-linked materials:
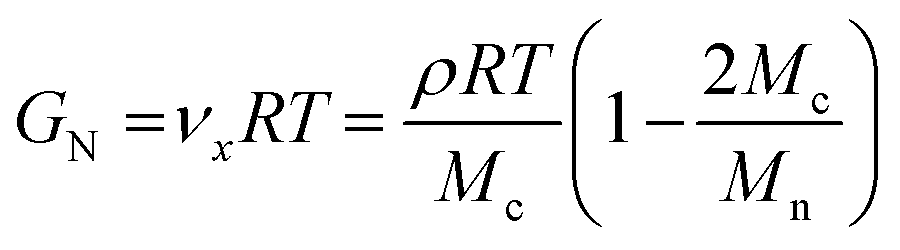 | (2) |
| Entry | Mixture |
G
Ne [Pa] |
ν x ,max [mol m−3] | ν x ,exp [mol m−3] | ν x ,exp/νx,max | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Applying CuBr at 20 °C. b Applying CuBr(PPh3)3 at 20 °C. c Applying CuF(PPh3)3 at 20 °C. d Applying CuF(PPh3)3 at 10 °C. e Constant value of ω = 100 Hz. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
2a + 3a |
1 × 104 | 254 | 4 | 1.6% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
2a + 3b |
1.03 × 105 | 254 | 43 | 17.0% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
2a + 3b |
1.13 × 105 | 254 | 46 | 18.5% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
2a + 3c |
2.48 × 105 | 254 | 91 | 40.2% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
2b + 3c |
8.73 × 104 | 181 | 36 | 19.9% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
2b + 3d |
1.07 × 105 | 181 | 45 | 24.9% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Using CuBr as catalyst (Table 3, entry 1), both the plateau modulus GN and the calculated network density νx,exp are low (1.6%), which is in agreement with only slight changes in the viscosity during the cross-linking reaction, indicating that this system is not suitable for efficient and fast cross-linking in the melt state. Applying CuBr(PPh3)3 as catalyst resulted in increased network densities of up to 18.5% (Table 3, entries 2 and 3). By changing to the most active catalyst, namely CuF(PPh3)3, the network density is doubled to 40.2% (Table 3, entry 5), which is in accordance with the observed short gelation time (15 minutes), whereas cross-linking of isomer 2b, generated decreased network strand densities of about 20–25% (Table 3, entries 6 and 7). IR-measurements of the finally obtained networks showed complete disappearance of the azide vibration at ν ∼ 2100 cm−1 (see ESI†).
3.3 “Click”-induced self-healing in polymeric materials
Autonomous self-healing within polymer materials composed of high molecular weight PIB was probed directly via a fluorogenic dye, able to measure the damage induced “click” reaction directly within the material. The specimen contained the star-shaped picolinazido-telechelic PIB (2b, 5 wt%), micro-sized UF-capsules filled with a trivalent alkyne (2,2-bis(prop-2-2-ynyloxymethyl)-1-(prop-2-ynyloxy)butane (4), 10 wt%), the Cu(I)-source (CuBr(PPh3)3, 2 wt%) and the fluorogenic azido-coumarin dye (2.5 wt% of 2b). To evoke the rupture of the capsules, in turn inducing the healing reaction, the specimen has been damaged by several scratches. Subsequently, the fluorescence emission of the fluorogenic dye, generated in the subsequent “click” reaction was measured (see Fig. 4). The chelation-assisted fluorogenic click reaction was investigated for 24 hours at room temperature monitoring the in situ fluorescence (excitation: 330 nm, emission: 350–650 nm) (see Fig. 4). The observed increase in the fluorescence emission is thus related to the fluorogenic click reaction indicative of the healing. In turn, the alkyne (4) undergoes click reactions with both the star-shaped picolinazido-telechelic PIB 2b and the azido-coumarin-dye within the PIB-matrix, which is directly visualized by the formed fluorogenic coumarin-triazol product. As a reference experiment the unscratched specimen was measured, showing no increase of the fluorescence as the incorporated capsules are not broken and therefore no fluorescent click product can be formed (see ESI, Fig. S25†). In a second control experiment a scratched specimen without copper(I) catalyst was investigated, showing no increase of the fluorescence and thus no triggered click reactions (see ESI, Fig. S26†).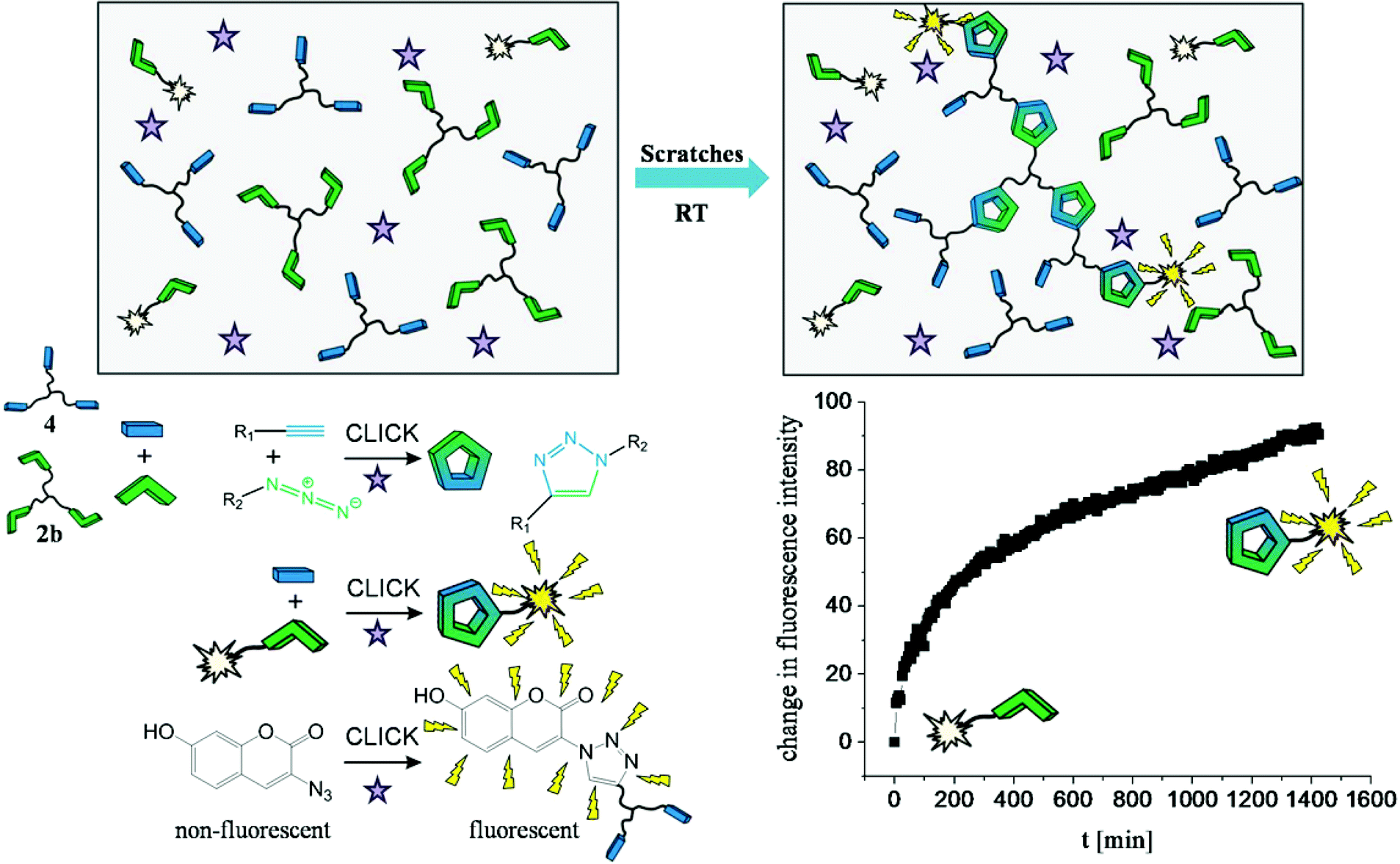 | ||
| Fig. 4 Fluorogenic click reaction in SH-PIB-specimen. Star-shaped picolinazido-telechelic PIB (2b), UF-capsules filled with a trivalent alkyne (the capsules are not pictured in this scheme to keep it clearly arranged), CuBr(PPh3)3 and the fluorogenic azido-coumarin dye are incorporated into a high molecular weight PIB matrix. Micron-sized capsule breaking was evoked by scratches, in turn increasing the fluorescence intensity of the fluorogenic dye. | ||
4. Conclusions
Within this work we have overcome one major drawback of the most self-healing systems known so far – efficient fast healing kinetics, especially at temperatures below room temperature. By applying the concept of chelation-assisted CuAAC to self-healing star-shaped PIBs we have successfully demonstrated a cross-linked system working fast even under low temperature conditions (10 °C).We have investigated a synthetic route towards star-shaped picolinazido-telechelic PIBs (2a, 2b) applicable for subsequent cross-linking via chelation-assisted CuAAC aimed at the development of low temperature self-healing polymers. Therefore, a model click reaction between 2-(6-azidomethyl)-pyridine-4-carboxylic acid methyl ester and phenylacetylene was investigated via in situ NMR measurements in solution varying the catalyst and adding different amounts of base. For the model click reaction CuBr turned out to be the best catalyst in the presence of a catalytic amount of DIPEA, ensuring complete conversion within five minutes at room-temperature.
Due to the pre-organization of the copper acetylide via the donating nitrogen-atom of α-picolin-azide, herein acting as an internal ligand and therefore enabling close proximity of both reactants, the reaction rate of chelation-assisted CuAAC is increased in contrast to the conventional Cu(I)-promoted “click” reaction. Thus, while transferring this concept for the first time to self-healing polymers, we functionalized star-shaped PIBs with the corresponding 2-(6-azidomethyl)-pyridine-(4 or 5)-carboxylic acid (1a, 1b) end group. Further cross-linking experiments of star-shaped azido-telechelic PIBs (2a, 2b) and PIB-alkyne (3) were conducted by applying melt rheology measurements. In contrast to the obtained results in solution, CuBr was not suitable for efficient cross-linking in the melt state, presumably due to its decreased solubility and oxidation stability. Therefore by using its more stable analogue CuBr(PPh3)3 network formation was achieved at room temperature within 255 minutes. Furthermore, cross-linking was accomplished within 71 minutes even at hampered conditions as lower temperatures (10 °C) utilizing the more active CuF(PPh3)3 as catalyst. We successfully demonstrated the self-healing of embedded star-shaped picolinazido-telechelic PIB 2b with encapsulated alkyne 4, using CuBr(PPh3)3 as catalyst and small amounts of a fluorogenic dye within the polymer matrix (PIB). Scratch experiments with subsequent fluorescence measurements of the generated fluorogenic dye have proven the subsequent crosslinking reaction via the formation of the highly fluorescent click product within the PIB matrix at room temperature.
Acknowledgements
We gratefully acknowledge Grant DFG BI 1337/8-2 (within the SPP 1568 “Design and Generic Principles of Self-Healing Materials”).Notes and references
- Self-Healing Polymers. From Principles to Applications, ed. W. H. Binder, Wiley-VCH, Weinheim, 2013 Search PubMed.
- W. H. Binder and R. Zirbs, in Hydrogen Bonded Polymers, 2007, pp. 1–78 Search PubMed.
- B. A. Blight, C. A. Hunter, D. A. Leigh, H. McNab and P. I. T. Thomson, Nat. Chem., 2011, 3, 244–248 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519–1521 RSC.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- M. Gragert, M. Schunack and W. H. Binder, Macromol. Rapid Commun., 2011, 32, 419–425 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–817 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- E. N. Brown, S. R. White and N. R. Sottos, Compos. Sci. Technol., 2005, 65, 2466–2473 CrossRef CAS.
- M. M. Caruso, D. A. Delafuente, V. Ho, N. R. Sottos, J. S. Moore and S. R. White, Macromolecules, 2007, 40, 8830–8832 CrossRef CAS.
- T. S. Coope, U. F. J. Mayer, D. F. Wass, R. S. Trask and I. P. Bond, Adv. Funct. Mater., 2011, 21, 4624–4631 CrossRef CAS.
- L. Guadagno, P. Longo, M. Raimondo, C. Naddeo, A. Mariconda, A. Sorrentino, V. Vittoria, G. Iannuzzo and S. Russo, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 2413–2423 CrossRef CAS.
- M. A. M. Rahmathullah and G. R. Palmese, J. Appl. Polym. Sci., 2009, 113, 2191–2201 CrossRef CAS.
- M. Z. Rong, M. Q. Zhang and W. Zhang, Adv. Compos. Lett., 2007, 16, 167–172 Search PubMed.
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289–1296 RSC.
- G. O. Wilson, J. S. Moore, S. R. White, N. R. Sottos and H. M. Andersson, Adv. Funct. Mater., 2008, 18, 44–52 CrossRef CAS.
- T. Yin, M. Z. Rong, M. Q. Zhang and G. C. Yang, Compos. Sci. Technol., 2007, 67, 201–212 CrossRef CAS.
- Y. C. Yuan, M. Z. Rong, M. Q. Zhang and G. C. Yang, eXPRESS Polym. Lett., 2011, 5, 47–59 CrossRef CAS.
- M. Zako and N. Takano, J. Intell. Mater. Syst. Struct., 1999, 10, 836–841 CrossRef.
- W. H. Binder and F. Herbst, in McGraw-Hill Yearbook of Science & Technology, ed. D. Blumel, McGraw-Hill, New York, 2011, pp. 46–49 Search PubMed.
- W. H. Binder and C. Kluger, Curr. Org. Chem., 2007, 10, 1791 CrossRef.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- W. H. Binder and R. Zirbs, Encyclopedia of Polymer Science and Technology, 2009, DOI:10.1002/0471440264.pst0471440565.
- D. Döhler, P. Michael and W. H. Binder, Macromolecules, 2012, 45, 3335–3345 CrossRef.
- M. Schunack, M. Gragert, D. Döhler, P. Michael and W. H. Binder, Macromol. Chem. Phys., 2012, 213, 205–214 CrossRef CAS.
- X. Sheng, T. C. Mauldin and M. R. Kessler, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4093–4102 CrossRef CAS.
- J. Yang, M. W. Keller, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2008, 41, 9650–9655 CrossRef CAS.
- G. E. Larin, N. Bernklau, M. R. Kessler and J. C. DiCesare, Polym. Eng. Sci., 2006, 46, 1804–1811 CAS.
- J. K. Lee, X. Liu, S. H. Yoon and M. R. Kessler, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1771–1780 CrossRef CAS.
- X. Sheng, J. K. Lee and M. R. Kessler, Polymer, 2009, 50, 1264–1269 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634–1638 CrossRef CAS PubMed.
- R. K. Bose, J. Kötteritzsch, S. J. Garcia, M. D. Hager, U. S. Schubert and S. van der Zwaag, J. Polym. Sci., Part A: Polym. Chem., 2014, 32, 1669–1675 CrossRef.
- X. J. Ye, Y. X. Song, Y. Zhu, G. C. Yang, M. Z. Rong and M. Q. Zhang, Compos. Sci. Technol., 2014, 104, 40–46 CrossRef CAS.
- X. J. Ye, J.-L. Zhang, Y. Zhu, M. Z. Rong, M. Q. Zhang, Y. X. Song and H.-X. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 3661–3670 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- W. H. Binder and R. Sachenshofer, in Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, Wiley-Blackwell, 2009, pp. 119–175 Search PubMed.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- X. Sheng, D. M. Rock, T. C. Mauldin and M. R. Kessler, Polymer, 2011, 52, 4435–4441 CrossRef CAS.
- D. Döhler, P. Zare and W. H. Binder, Polym. Chem., 2014, 5, 992–1000 RSC.
- A. Shaygan-Nia, S. Rana, D. Döhler, X. Noirfalise, A. Belfiore and W. H. Binder, Chem. Commun., 2014, 50, 15374–15377 RSC.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- D. Soriano del Amo, W. Wang, H. Jiang, C. Besanceney-Webler, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef CAS PubMed.
- W. Wang, S. Hong, A. Tran, H. Jiang, R. Triano, Y. Liu, X. Chen and P. Wu, Chem. – Asian J., 2011, 6, 2796–2802 CrossRef CAS PubMed.
- C. Uttamapinant, M. I. Sanchez, D. S. Liu, J. Z. Yao and A. Y. Ting, Nat. Protocols, 2013, 8, 1620–1634 Search PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 124, 5954–5958 CrossRef.
- K. Banert, J. Wutke, T. Rüffer and H. Lang, Synthesis, 2008, 2603–2609 CrossRef CAS.
- E. N. Brown, M. R. Kessler, N. R. Sottos and S. R. White, J. Microencapsulation, 2003, 20, 719–730 CrossRef CAS PubMed.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed.
- T. G. Metzger, The Rheology Handbook: For users of rotational and oscillatory rheometers, Vincentz Network, Hannover, 2006 Search PubMed.
- K. G. Hull, M. Visnick, W. Tautz and A. Sheffron, Tetrahedron, 1997, 53, 12405–12414 CrossRef CAS.
- X.-B. Wang, J. E. Dacres, X. Yang, K. M. Broadus, L. Lis, L.-S. Wang and S. R. Kass, J. Am. Chem. Soc., 2002, 125, 296–304 CrossRef PubMed.
- M. Mishra, B. Wang and J. Kennedy, Polym. Bull., 1987, 17, 307–314 CrossRef CAS.
- W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749–1759 CrossRef CAS.
- D. L. Morgan, N. Martinez-Castro and R. F. Storey, Macromolecules, 2010, 43, 8724–8740 CrossRef CAS.
- D. L. Morgan and R. F. Storey, Macromolecules, 2009, 42, 6844–6847 CrossRef CAS.
- B. Iván and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 89–104 CrossRef.
- M. Schulz, S. Tanner, H. Barqawi and W. H. Binder, J. Polym. Sci., Part A: Polym Chem., 2010, 48, 671–680 CrossRef CAS.
- S. Vasiliu, B. Kampe, F. Theil, B. Dietzek, D. Döhler, P. Michael, W. H. Binder and J. Popp, Appl. Spectrosc., 2014, 68, 541–548 CrossRef CAS PubMed.
- J. Ampudia, E. Larrauri, E. M. Gil, M. Rodríguez and L. M. León, J. Appl. Polym. Sci., 1999, 71, 1239–1245 CrossRef CAS.
- J. M. Barton, D. C. L. Greenfield and K. A. Hodd, Polymer, 1992, 33, 1177–1186 CrossRef CAS.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2009, 42, 9072–9081 CrossRef CAS.
- M. Müller, U. Seidel and R. Stadler, Polymer, 1995, 36, 3143–3150 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01818h |
| This journal is © The Royal Society of Chemistry 2016 |