
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-large sheet formation by 1D to 2D hierarchical self-assembly of a “rod–coil” graft copolymer with a polyphenylene backbone†
Yinjuan
Huang‡
a,
Rui
Yuan‡
a,
Fugui
Xu
a,
Yiyong
Mai
*a,
Xinliang
Feng
ab and
Deyue
Yan
a
aSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China. E-mail: mai@sjtu.edu.cn
bDepartment of Chemistry and Food Chemistry, Technische Universität Dresden, Mommsenstrasse 4, 01062 Dresden, Germany
First published on 4th January 2016
Abstract
This communication reports a unique ultra-large sheet formation through hierarchical self-assembly of a rod–coil graft copolymer containing a rigid polyphenylene backbone and flexible poly(ethylene oxide) (PEO) side chains. The hierarchical self-assembly process involved a distinctive morphological transition of 1D helical to 2D superstructures. The graft copolymer offers a new chance for the challenging bottom-up fabrication of ultra-large self-assembled nanosheets in solution, as well as a novel system for fundamental studies on 2D self-assembly of polymers.
“Rod–coil” graft copolymers are an important type of polymer containing a rigid backbone with densely tethered flexible polymeric chains.1–3 Their interesting one-dimensional (1D) brush-like structure leads to numerous potential applications, such as molecular actuators, templates for metal nanowires, etc.1,3 Conjugated polymers, e.g. polyphenylene and polythiophene, are the major category of rigid backbones in rod–coil graft copolymers, as introducing pendant moieties onto conjugated polymers to form graft copolymers is a general pathway to improve their solubility and tailor their optoelectronic properties.4–6
Self-assembly of rod–coil graft copolymers with conjugated polymer backbones and their linear counterpart, rod–coil block copolymers, has attracted much attention in recent decades, as rod–coil copolymers show distinct self-assembly features compared with flexible (coil–coil) copolymer systems due to the introduction of rigid blocks;6–20 moreover, the resultant supermolecular nanostructures of conjugated polymers generally exhibit unique thermal, optical or optoelectronic properties.6–20 In particular, the self-assembly of the rod–coil block copolymers has been studied extensively. Several key issues, including aggregate morphologies, morphological control approaches, etc., have been explored.7–16 For example, Winnik, Manners and coworkers prepared nanocylinders of controlled lengths by the crystallization-driven self-assembly of poly(3-hexylthiophene)-block-poly-(dimethylsiloxane) rod–coil block copolymers.8 Although the self-assembly of rod–coil graft copolymers has also been investigated in recent years,6,17–20 it remains much less understood compared with that of rod–coil block copolymers.
In the present study, we synthesized a rod–coil graft copolymer containing a laterally expanded poly-para-phenylene backbone (i.e. poly-para-phenylene with dendritic tetraphenylbenzene substituents) tethered with poly(ethylene oxide) (PEO) side chains. Interestingly, the graft copolymer exhibited hierarchical self-assembly behavior in a CHCl3–CH3OH organic cosolvent at room temperature (Fig. 1). Driven by the methanolphobic interaction of the polyphenylene and the crystallization of the PEO chains, the graft copolymer first self-organized into 1D nanowires, which bundled into 10–60 μm ultralong helices, then evolved to 2D raft-like nanostructures, and, eventually, to ultra-large multilayered nanosheets with remarkable lateral dimensions of ca. 10 μm × 10 μm to 100 μm × 100 μm, after the aging of the aggregate solution for ca. 2 days (Fig. 1). To the best of our knowledge, the bottom-up preparation of such large polymeric sheets by supramolecular self-assembly, without support from a planar interface, has been a severe challenge with only a few successful cases21,22 and has not been achieved for graft copolymers before, as planar polymer assemblies usually rolled or closed and formed tubes or vesicles in solution.23 In addition, the unprecedented hierarchical self-assembly process involving 1D helical to 2D superstructures provides a significant reference for the 2D self-assembly of polymers.
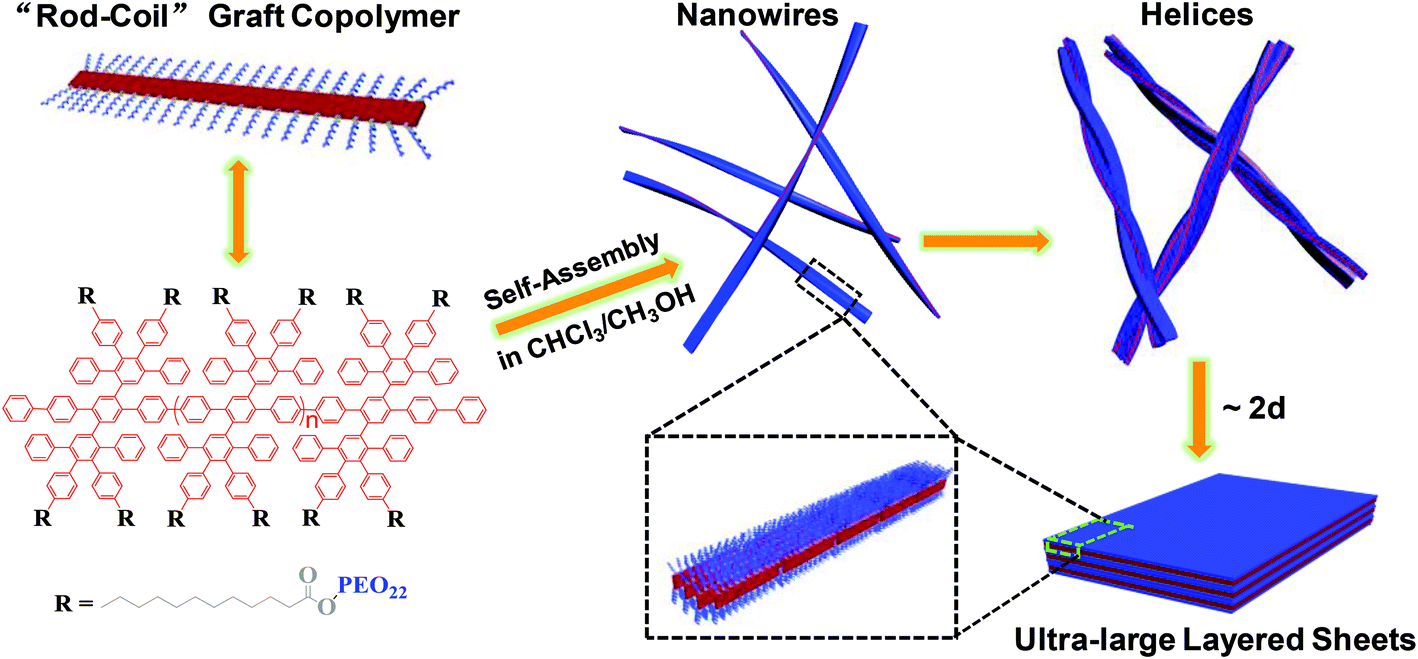 | ||
| Fig. 1 Schematic illustration of the formation of ultra-large multilayered sheets by 1D to 2D hierarchical self-assembly of the rod–coil graft copolymer at room temperature. | ||
The synthesis procedure of the rod–coil graft copolymer is illustrated in Scheme S1† and the experimental details are described in the ESI.† First, Yamamoto polymerization of a dichloro-substituted oligophenylene monomer produced a laterally expanded poly-para-phenylene modified with –C10H20COOCH3 (denoted as PP-COOCH3). Then, PP-COOCH3 was hydrolyzed to PP-COOH. Afterwards, the esterification of the carboxyl groups on PP-COOH with the hydroxyl groups on 1 kg mol−1 poly(ethylene oxide) monomethyl ether yielded the graft copolymer. Fourier transform infrared (FTIR) spectroscopy (Fig. S4A†) and nuclear magnetic resonance (NMR) (Fig. S5†) measurements validated the successful grafting of the PEO chains.
The grafting percentage (GP) of the copolymer was determined to be ∼92% by NMR, which was supported by the calculation based on elemental analyses (EA) (see pages S8 and S9†). Gel permeation chromatography (GPC) analysis against polystyrene standards in tetrahydrofuran (THF) gave a number-average molecular weight (Mn) of 136![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 and a polydispersity index (PDI) of 1.3 for the graft copolymer, as well as a Mn of 51700 g mol−1 and a PDI of 1.1 for PP-COOCH3 (their single-peak distribution GPC curves are presented in Fig. S4B†). The much larger Mn of the graft copolymer further confirmed the successful grafting of the PEO coils.
200 g mol−1 and a polydispersity index (PDI) of 1.3 for the graft copolymer, as well as a Mn of 51700 g mol−1 and a PDI of 1.1 for PP-COOCH3 (their single-peak distribution GPC curves are presented in Fig. S4B†). The much larger Mn of the graft copolymer further confirmed the successful grafting of the PEO coils.
The self-assembly of the rod–coil graft copolymer was carried out through a cosolvent method23 at room temperature (∼20 °C). First, a CHCl3 solution with a concentration of 0.01 mg mL−1 was prepared by dissolving the graft copolymer in CHCl3, which is a common solvent for both polyphenylene and PEO. Then, under gentle stirring, the CHCl3 solution was added dropwise (∼60 μL min−1) into a 9-fold amount of CH3OH, a selective solvent for PEO. Finally, a light blue mixed solution was obtained, suggesting the formation of polymer aggregates. The as-prepared solution was incubated at room temperature for a period of days.
The aggregation of the graft copolymer in the CHCl3–CH3OH (v/v 1:9) cosolvent was confirmed by ultraviolet-visible (UV-vis) and photoluminescence (PL) spectroscopy (Fig. 2). The maximum absorption of the copolymer in CHCl3–CH3OH red-shifted to ∼280 nm compared with ∼270 nm in CHCl3 (Fig. 2A), indicating π–π interactions associated with the aggregation of the polyphenylene backbones.9 The PL spectra showed the distinct quenching of the photoluminescence of the copolymer in CHCl3–CH3OH (Fig. 2B), further confirming polymer aggregation. In addition, the consecutive quenching of the photoluminescence with the aging of the aggregate solution over 2 days suggested possible progressive changes in the morphology of the polymer aggregates.
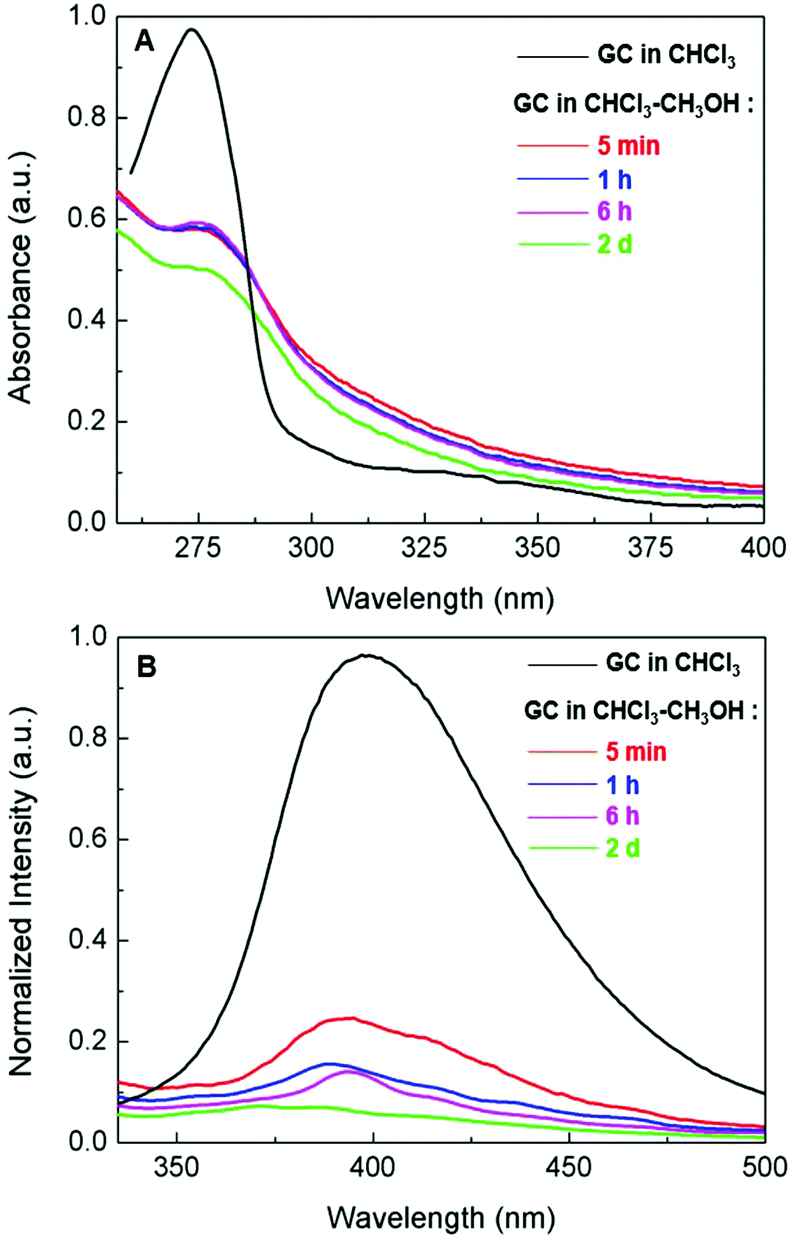 | ||
| Fig. 2 (A) UV-Vis and (B) photoluminescence spectra of the graft copolymer (GC) in CHCl3 and in CHCl3–CH3OH (v/v 1:9) with the aging (from 5 minutes to 2 days) of the aggregate solution at room temperature (concentration: 10−3 mg mL−1). | ||
The structures of the polymer aggregates formed in the CHCl3–CH3OH (v/v 1:9) solution were examined by transmission electron microscopy (TEM) and atomic force microscopy (AFM) (Fig. 3). TEM images revealed a 1D helical structure for the as-formed graft copolymer assemblies in CHCl3–CH3OH (Fig. 3A, B and Fig. S6A, B†). Left- and right-handed helices coexisted as no chiral moieties were employed during the self-assembly. The size statistics gave an average diameter of 17 ± 9 nm and lengths of 10–60 μm for the helices. The helices were formed by the twist of 1D aggregates of ∼4 nm width (Fig. 3B and the inset shows a typical high-resolution TEM image). The AFM profiles gave a thickness of ∼4 nm for the 1D aggregates and confirmed that they bundled into the helices (Fig. 3C and D). The combination of the 4 nm width and thickness manifests that the 1D aggregates are supermolecular nanowires. Since the thickness of the nanowires is close to the calculated width (∼3.9 nm) of a graft copolymer molecule while their width is much smaller than the length (∼36 nm) of the copolymer molecules (see the calculation in section 4.3.1 in page S12, ESI†), it is reasonable to speculate that the nanowires are assembled by an “end-to-end” and “side-by-side” combined alignment of the brush-like molecules, as illustrated in the middle of Fig. 1. The high-magnification TEM images (e.g.Fig. 3B) revealed that most of the helices (ca. 80%) were double- and triple-stranded in terms of their diameters and they coexisted with a number of multistranded helices (≥4 strands).
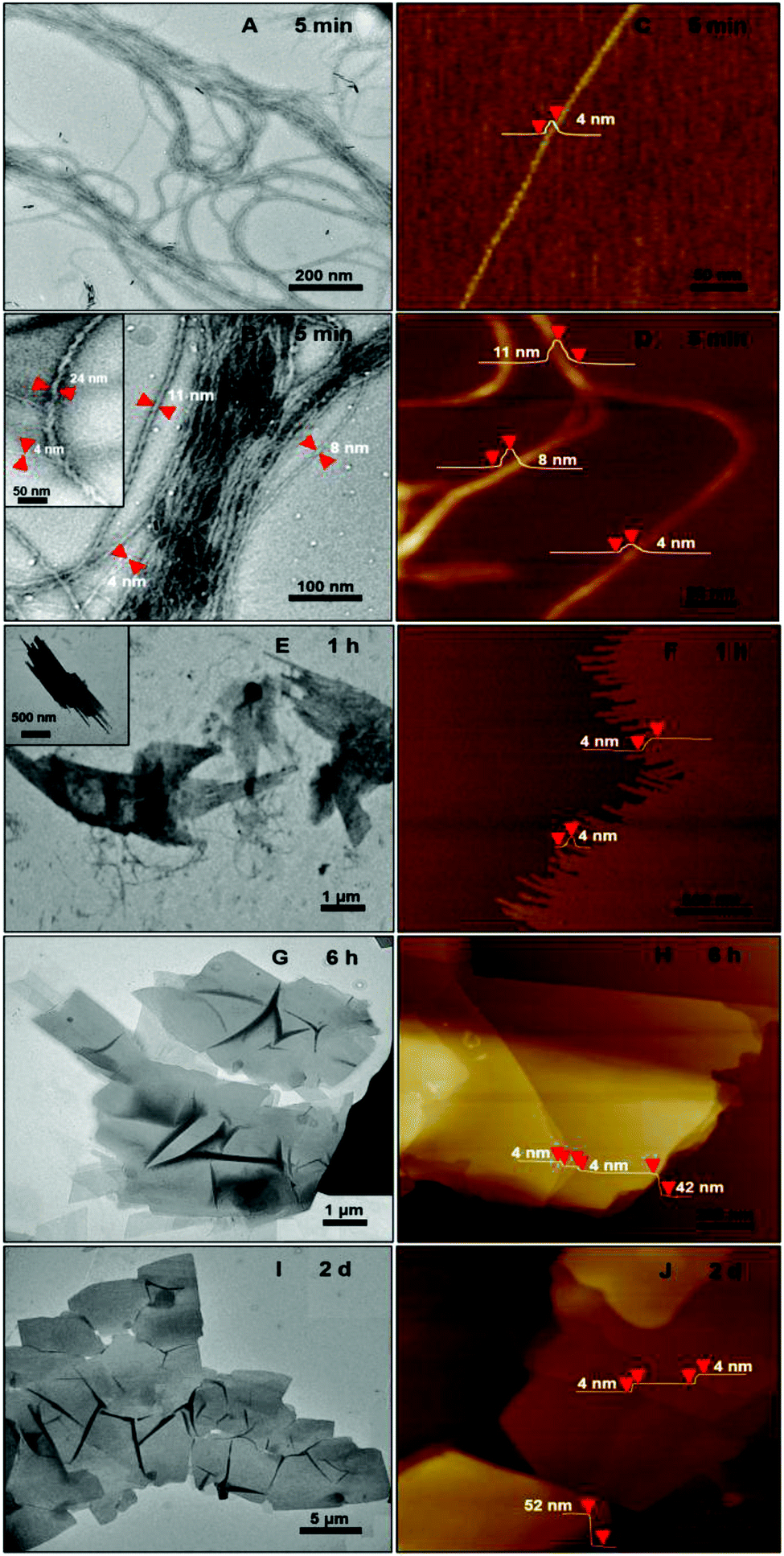 | ||
| Fig. 3 TEM and AFM images of the assemblies formed by the hierarchical self-assembly of the graft copolymer in the CHCl3–CH3OH (v/v 1:9) solution after 5 minutes (A–D), 1 hours (E, F), 6 hours (G, H), 2 days (I, J). The relevant dimensions of the assemblies are indicated in the corresponding TEM and AFM images. | ||
The formation of the helices was found to be kinetically controlled and they were thermodynamically unstable, namely their morphology was quenched temporarily in the mixed solvent with a large amount of CH3OH and also in pure methanol after the dialysis of the CHCl3–CH3OH solution. After one-hour aging, the helices evolved into “rafts” that consisted of 1D nanostructures (Fig. 3E and F). The thickness of the rafts was ∼4 nm (Fig. 3F), the same as that of the aforementioned nanowires. Therefore, it is believed that the rafts were single-layered and formed by the planar alignment of the nanowires. The unexpected transformation of 1D helical to 2D raft-like superstructures has never been documented before in 2D supramolecular self-assembly. With the aging of the aggregate solution for ∼2 days, the rafts further developed into ultra-large sheets with an average thickness of 56 ± 4 nm and lateral dimensions of ca. 10 μm × 10 μm to 100 μm × 100 μm (Fig. 3G–J and S6D and E†). The greater thickness than that of the single-layered rafts indicated a multilayered structure of the sheets. The wrinkles observed by TEM on the nanosheets reflected a flexible feature (Fig. 3G, I and S6D†). Afterwards, no big changes were observed on the dimensions of the sheets. The ultra-large sheets suspended in solution were also observed by optical microscopy (Fig. S7†), confirming that the sheets were formed and free-standing in solution. Dynamic light scattering (DLS) measurements revealed an increase in the hydrodynamic diameter of the graft copolymer assemblies with the aging of their solution (Fig. 4 and S8†), confirming the progressive growth of the copolymer assemblies in CHCl3–CH3OH.
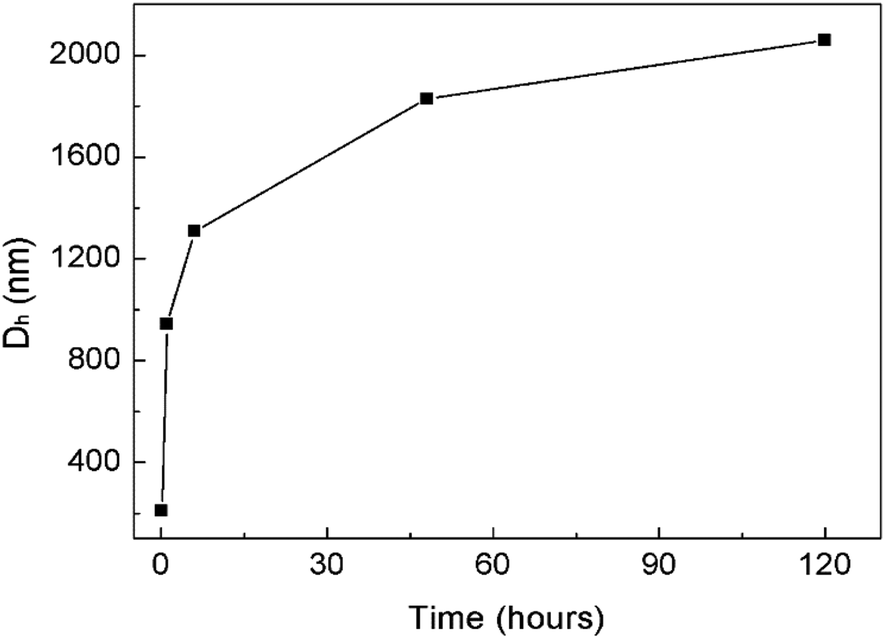 | ||
| Fig. 4 DLS results showing an increase in the hydrodynamic diameter (Dh) of the graft copolymer assemblies with the aging of their solution at room temperature. The corresponding DLS plots are presented in Fig. S8.† | ||
In light of previous studies on PEO crystallization-driven 2D self-assembly of polymers in solution,24–26 we consider that PEO crystallization in organic media contributed to the solution-growth of the ultra-large multilayered sheets. Micro differential scanning calorimetry (μDSC) analysis gave a crystallization temperature (Tc) of ∼26 °C for the PEO coils in the copolymer aggregates in CHCl3–CH3OH (Fig. S9†). Note that the 2D self-assembly of the graft copolymer occurred (at ∼20 °C) below the Tc of the PEO chains. In addition, a short average distance of ∼0.7 nm between neighboring PEO chains on the polyphenylene backbone would be favorable for the crystallization of the PEO coils if the graft copolymer aggregated into sheet-like structures, as the short distance resulted in a high number density of PEO chains at the sheet surfaces, which exceeded the onset density for the crystallization of the PEO chains below their Tc (see the calculations in section 4.3.2 in the ESI, pages S13 and S14†).26,27 Thus, it is reasonable to believe that the crystallization of the PEO chains acted as a key driving force for the formation of the ultra-large sheets. Two additional lines of evidence also support this point. First, the graft copolymer with a lower GP of ∼60% did not form multilayered sheets under similar experimental conditions, probably due to the larger average distance between adjacent PEO coils on the polyphenylene backbone. Second, at a temperature (∼30 °C) higher than the Tc of the PEO chains, the graft copolymer produced irregular aggregates rather than sheets in CHCl3–CH3OH.
Based on the above-mentioned results, a possible mechanism for the 1D to 2D hierarchical self-assembly of the graft copolymer is illustrated in Fig. 1. In the CHCl3–CH3OH solution, driven by the methanolphobic interaction, the graft copolymer first self-organized into 4 nm thin nanowires with a polyphenylene layer sandwiched by PEO coils on both up and down sides (middle of Fig. 1). Because of the relatively low PEO number density on the other two sides of the polyphenylene layer, the nanowires tend to associate to minimize the polyphenylene–methanol contact. Around the associated nanowires the PEO coils were crowded, as the estimated radius of gyration (Rg = ∼1.5 nm (ref. 28)) of a 1 kg mol−1 PEO chain at its end-free state in solution is much larger than the short average distance of 0.7 nm between neighboring PEO chains in the graft copolymer in the nanowires. Hence, the steric interaction among the PEO chains drove the twist of the associated nanowires to provide more peripheral space for the PEO coils, thus leading to the formation of 1D helices. At this point, it is worth mentioning that although helix formation represents a self-assembly motif for some chiral polymers, owing to the ordered helical arrangement of their chiral moieties,10,11,15 it rarely happens to achiral polymers;9,29,30 nevertheless, in the present work, long helices were frequently found in the self-organization of the achiral graft copolymers in the CHCl3–CH3OH cosolvent system, which can be attributed to their unique rod–coil brush-like structure with a short average distance between adjacent PEO chains on the rigid polyphenylene backbone. However, since the helix formation occurred below the Tc of the PEO coils, the crystallization and the resulting compact state of the PEO chains alleviated their steric repulsion and thus favored the transformation of the helices to the thermodynamically more stable sheet-like structures. During the transformation, an untwist process of the helices could be considered for their transition to the single-layered rafts which further grew to the ultra-large multilayered sheets driven by the PEO crystallization.
Soft nanosheets with lateral dimensions of over 10 μm × 10 μm are quite difficult to achieve by the self-assembly of polymers in solution without support from a planar interface; only a few successful cases were documented.21,22 In the present work, the rod–coil brush-like structure of the graft copolymer accounts for the ultra-large lateral sizes of the sheets, since the molecules of such an architecture incline to form assemblies with a sandwich structure, in which the laterally expanded poly-para-phenylene rigid backbones locate in the middle layer (Fig. 1). Such a sandwich structure along with the PEO crystallization is in favor of the growth of assemblies in 2D directions. Moreover, the rigid polyphenylene layer and the PEO crystallization can limit the rolling or closing of the sheets, which would generate tubes or vesicles as usually seen in the self-assembly of many flexible polymer systems.23,31,32
In summary, this work demonstrates a unique sheet formation by 1D to 2D hierarchical self-assembly of a rod–coil graft copolymer comprising a poly-para-phenylene backbone and PEO side chains. Driven by the methanolphobic interaction of the polyphenylene and the crystallization of the PEO chains in a CHCl3–CH3OH mixed organic solvent, the graft copolymer exhibited an unprecedented hierarchical self-assembly process from nanowires to 10–60 μm ultralong helices, then to single-layered “rafts”, and finally to ultra-large multilayered nanosheets with lateral dimensions of ca. 10 μm × 10 μm to 100 μm × 100 μm. The rod–coil graft polymers afford a new chance for the so far challenging preparation of ultra-large self-assembled nanosheets in solution, as well as a novel system for fundamental studies on 2D self-assembly of conjugated polymers, including the mechanism, factors affecting the morphology, potential applications, etc.
Acknowledgements
The authors acknowledge the financial support from the 973 Programs of China (2012CB933404 and 2013CBA01602), the Natural Science Foundation of China (21320102006, 21304057 and 51573091), the Natural Science Foundation of Shanghai (13ZR1421200), the Program for Eastern Scholar in Shanghai, and the MPI Partner Group Project for Polymer Chemistry of Graphene Nanoribbons. The authors also appreciate the Instrumental Analysis Center of Shanghai Jiao Tong University for some measurements.Notes and references
- J. Yuan, Y. Xu and A. H. E. Müller, Chem. Soc. Rev., 2011, 40, 640 RSC.
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146 CrossRef CAS.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405 RSC.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759 CrossRef CAS.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817 CrossRef CAS PubMed.
- Y. Huang, Y. Mai, X. Yang, U. Beser, J. Liu, F. Zhang, D. Yan, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 11602 CrossRef CAS PubMed.
- S. A. Jenekhe and X. L. Chen, Science, 1998, 279, 1903 CrossRef CAS PubMed.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842 CrossRef CAS PubMed.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390 CrossRef CAS PubMed.
- R.-M. Ho, Y.-W. Chiang, S.-C. Lin and C.-K. Chen, Prog. Polym. Sci., 2011, 36, 376 CrossRef CAS.
- C. Cai, Y. Li, J. Lin, L. Wang, S. Lin, X.-S. Wang and T. Jiang, Angew. Chem., Int. Ed., 2013, 52, 7732 CrossRef CAS PubMed.
- Y. Zheng, H. Zhou, D. Liu, G. Floudas, M. Wagner, K. Koynov, M. Mezger, H.-J. Butt and T. Ikeda, Angew. Chem., Int. Ed., 2013, 52, 4845 CrossRef CAS PubMed.
- A. Yassar, L. Miozzo, R. Gironda and G. Horowitz, Prog. Polym. Sci., 2013, 38, 791 CrossRef CAS.
- A. C. Kamps, M. H. M. Cativo, M. Fryd and S.-J. Park, Macromolecules, 2014, 47, 161 CrossRef CAS.
- J. F. Reuther, D. A. Siriwardane, R. Campos and B. M. Nova, Macromolecules, 2015, 48, 6890 CrossRef CAS.
- H.-C. Ho, Y.-H. Lee, C.-A. Dai, R. A. Segalman and W.-F. Su, Macromolecules, 2009, 42, 4208 CrossRef CAS; S.-H. Lin, S.-J. Wu, C.-C. Ho and W.-F. Su, Macromolecules, 2013, 46, 2725 CrossRef; C.-C. Ho, S.-J. Wu, S.-H. Lin, S. B. Darling and W.-F. Su, Macromol. Rapid Commun., 2015, 36, 1329 CrossRef PubMed.
- J. H. Yao, K. Y. Mya, L. Shen, B. He, L. Li, Z. Li, Z. Chen, X. Li and K. P. Loh, Macromolecules, 2008, 41, 1438 CrossRef CAS.
- C. Cai, J. Lin, T. Chen and X. Tian, Langmuir, 2010, 26, 2791 CrossRef CAS PubMed.
- G. M. Miyake, R. A. Weitekamp, V. A. Piunova and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 14249 CrossRef CAS PubMed.
- Y. Li, T. Jiang, S. Lin, J. Lin, C. Cai and X. Zhu, Sci. Rep., 2015, 5, 10137 CrossRef CAS PubMed.
- J.-K. Kim, E. Y. Lee, H. Jeong, J.-K. Lee, W.-C. Zin and M. Lee, J. Am. Chem. Soc., 2007, 129, 6082 CrossRef CAS PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- G. Rizis, T. G. M. van de Ven and A. Eisenberg, Angew. Chem., Int. Ed., 2014, 53, 9000 CrossRef CAS PubMed.
- J. R. Sun, X. S. Chen, C. L. He and X. B. Jing, Macromolecules, 2006, 39, 3717 CrossRef CAS.
- R. M. Van Horn, J. X. Zheng, H.-J. Sun, M.-S. Hsiao, W.-B. Zhang, X.-H. Dong, J. Xu, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2010, 43, 6113 CrossRef CAS.
- J. X. Zheng, H. Xiong, W. Y. Chen, K. Lee, R. M. Van Horn, R. P. Quirk, B. Lotz, E. L. Thomas, A.-C. Shi and S. Z. D. Cheng, Macromolecules, 2006, 39, 641 CrossRef CAS.
- The Rg for a PEO chain at its end-free state in solution is estimated using Rg2 = b2Nb/6 from: M. Rubenstein and R. H. Colby, Polymer Physics, Oxford University Press, New York, 2004 Search PubMed, where b = 0.8 nm for PEO.
- S. Zhang, H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2008, 4, 90 RSC.
- J. Dupont, G. Liu, K.-I. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144 CrossRef CAS PubMed.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, ESI figures, calculations, etc. See DOI: 10.1039/c5py01969a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |