
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copolymerization of zinc-activated isoindigo- and naphthalene-diimide based monomers: an efficient route to low bandgap π-conjugated random copolymers with tunable properties†
Yevhen
Karpov
a,
Jatindranath
Maiti
b,
Roman
Tkachov
a,
Tetyana
Beryozkina
c,
Vasiliy
Bakulev
c,
Wei
Liu
a,
Hartmut
Komber
a,
Uwe
Lappan
a,
Mahmoud
Al-Hussein
d,
Manfred
Stamm
a,
Brigitte
Voit
ae and
Anton
Kiriy
*ae
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: kiriy@ipfdd.de
bGachon University, Department of Chemical Engineering, 461-701, Republic of Korea
cUral Federal University, Mira str., 28, 620002, Yekaterinburg, Russia
dThe University of Jordan, Physics Department, Amman 11942, Jordan
eTechnische Universität Dresden, Center for Advancing Electronics Dresden (CFAED), 01062, Dresden, Germany
First published on 22nd March 2016
Abstract
The present work aims at the extension of the scope of a recently discovered polycondensation of AB-type anion-radical monomers. To this end, we investigate the polymerization of isoindigo-based monomer and its copolymerization with the naphthalenediimide-based monomer. Although polycondensations of parent naphthalenediimide- and perylenediimide-based monomers involve chain-growth mechanism, we found that the corresponding isoindigo-based monomer polymerizes in a step-growth manner under the same reaction conditions. In contrast to Stille, Suzuki and direct arylation polycondensations which require prolonged stirring at high temperatures, the polymerization approach we employed in this study proceeds fast at room temperature. It was found that near statistical copolymerization of isoindigo-based anion-radical monomers with corresponding naphtalenediimide-based monomers proceeds smoothly resulting in a library of copolymers with varying composition and properties depending on the ratio of the monomers.
Introduction
π-Conjugated polymers have attracted considerable attention as promising materials for organic electronics because they allow the production of low-cost, light-weight, large-area, flexible devices through ink-jet printing and roll-to-roll solution processing.1 Alternate polymerization of electron-rich and electron-deficient aromatic building blocks is an efficient approach for engineering the energy levels and light absorption properties of π-conjugated copolymers which is essential for the use of these materials in solar cells light emitting diodes and transistors.2 Statistical copolymerization of several aromatic monomers was recently introduced for the preparation of a library of copolymers with continuously varied composition and, hence, properties.3,4 Traditionally, step-growth polycondensation methods, such as Stille, Suzuki and direct arylation are used for the preparation of both alternate and statistical copolymers.3,4 Chain-growth catalyst-transfer polycondensation is relatively new but rapidly developing synthetic approach which provides an access to polymers with controlled molecular weight and desirable architecture such as block copolymers and polymer brushes.5–9 Although chain-growth polymerization of simple, yet relatively electron-rich monomers is well-developed, only limited number of electron-deficient monomers can be polymerized on the chain-growth manner.10–16N-Type (or electron-conducting) polymers are essential components in organic devices such as ambipolar and n-channel field-effect transistors and organic photovoltaics.1 Particularly, naphthalene diimide (NDI) and perylene diimide (PDI) alternating main chain copolymers,17 as well as diketopyrrolopyrrole-18 and isoindigo-19 based copolymers constitute an intriguing class of electron-conducting materials with excellent charge transport properties. Recently, we discovered Ni-catalyzed polymerization of highly unusual monomer – a complex formed upon reaction of activated Zn powder and 2,6-bis(2-bromothien-5-yl) naphtalene-1,4,5,8-tetracarboxylic-N,N′-bis(2-octyldodecyl) diimide (Br-TNDIT-Br).11 It was particularly found that addition of activated Zn powder to equimolar amounts of Br-TNDIT-Br resulted in an immediate color change from red-orange (inherent to Br-TNDIT-Br) to deep-green and accompanied by dissolution of the solid phase. Interestingly, the acidic work-up of the thus-prepared Br-TNDIT-Br/Zn complex resulted in a quantitative recovering of Br-TNDIT-Br but not of Br-TNDIT-H indicating that Br-TNDIT-ZnBr was not formed under these conditions. We proposed that either single- or two-electron transfer process occurs between electron-deficient Br-TNDIT-Br and electron-rich Zn. Consequently, anion-radical or dianion species may be responsible for the formation of complex Br-TNDIT-Br/Zn (Scheme 1). Electron paramagnetic resonance (EPR) measurements of Br-TNDIT-Br/Zn revealed the presence of paramagnetic species (signal at g = 2.0035) suggesting the formation of anion-radicals. Despite of the fact that Zn is not inserted in any of the two C–Br bonds of Br-TNDIT-Br, polymerization is going on in the presence of Ni and Pd catalysts resulting into corresponding n-type copolymer PTNDIT. Importantly, under Ni(dppe)Cl2 catalysis, the polycondensation involves the chain-growth catalyst-transfer mechanism and leads to P(TNDIT) with controlled molecular weights and relatively low dispersity (Đ = Mw/Mn) in a range of 1.3–1.7. However, molecular weight (MW) achievable with this catalyst is limited to only about 100 kg mol−1.11a It was further found that Pd complex having a bulky, electron-rich t-butylphosphine ligand is much more efficient catalyst and P(TNDIT) with MW up to 1000 kg mol−1 can be routinely obtained.11c Unfortunately, Pd/PtBu3-catalyzed polycondensation is not controlled and it involves the formation of shorter chains being formed on the chain-growth manner which further polymerize via the step-growth mechanism.
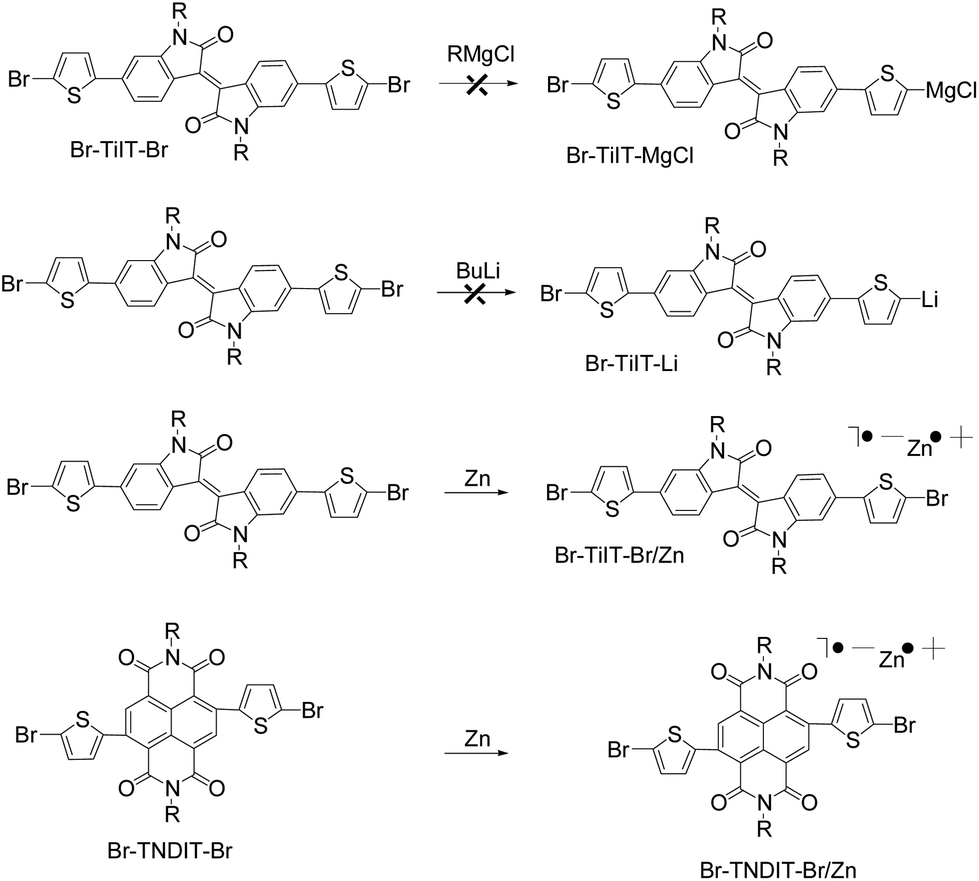 | ||
| Scheme 1 Preparation of monomers Br-TiIT-Br/Zn and Br-TNDIT-Br/Zn. | ||
Very recently, we found that another rylene-diimide derivative-2,6-bis(2-bromothien-5-yl)perylene-1,4,5,8-tetra-carboxylic-N,N′-bis(2-octyldodecyl) diimide (Br-TPDIT-Br) – behaves similarly to Br-TNDIT-Br/Zn and forms, upon reaction with Zn, the charge-transfer complex Br-TPDIT-Br/Zn.15 Furthermore, Br-TPDIT-Br/Zn was shown to undergo chain-growth polycondensation in the presence of Pd/PtBu316 into corresponding polymer PPDIT2 with moderate molecular weight and moderate polydispersity although it appears to be unreactive in the presence of Ni catalysts. In the present work, aiming at extension of the scope of new polycondensation, we attempted polymerization of isoindigo-based monomers, as they were shown to be useful building blocks for construction of highly performing semiconducting polymers. We supposed that isoindigo,19–22 being an aromatic lactam by its chemical structure, may react with active zinc similarly to rylene-diamides forming charge-transfer complexes even though that isoindigo contains two times less amount of electron-accepting carbonyl groups.
Results and discussion
Isoindigo-based dibromide Br-TiIT-Br having two 2-octyldodecyl solubilizers was prepared as described previously,22 coupled with two molecules of thiophenestannane under Stille cross-coupling conditions and brominated with NBS giving (E)-6,6′-bis(5-bromothiophen-2-yl)-1,1′-bis(2-octyldodecyl)-[3,3′-biindolinylidene]-2,2′-dione (see ESI†). In general, chain-growth catalyst-transfer polycondensations involve AB-monomers (i.e., building blocks in which metalorganic (nucleophilic) and halide (electrophilic) functions are present in the same molecule) whereas polycondensation of AA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +BB monomers proceeds via a step-growth mechanism. In the case of electron-rich aromatics, AB-monomers can be prepared via metal/halogen exchange reactions (e.g., by reaction of aryldihalide precursors with alkyl magnesium halides or other metalorganic molecules). In this work we found that treatment of isoindigo-based dibromide Br-TiIT-Br with alkyl magnesium halides also did not lead to the corresponding AB-type monomer Br-TiIT-MgBr (Scheme 1). Similarly, the treatment of Br-TiIT-Br with n-BuLi failed to give the corresponding lithiation product Br-TiIT-Li. Such a behavior was previously observed during attempts of activation of structurally similar naphthalene- and perylene-diimide derivatives.11 We explained these results by a concurrent single-electron transfer from electron-rich alkylmetals to electron-deficient dibromo-arylimide which occurs faster than the halogen-metal exchange. An alternative Zn-based route for activation of electron-deficient naphthalene- and perylene-diimide-based dihalides was reported in our previous works. An attractive feature of this method is that the polycondensation of thus-prepared monomers may proceed via the chain-growth catalyst-transfer mechanism, at least in the case of Br-TNDIT-Br/Zn and Br-TPDIT-Br/Zn. In the present work we found that Br-TiIT-Br reacts with activated Zn powder within minutes at room temperature resulting into zinc–organic complex Br-TiIT-Br/Zn soluble in THF. Titration experiments with iodine revealed the 1/1 ratio between Br-TiIT-Br and Zn in Br-TiIT-Br, irrespective of whether an equimolar amount or excess of Zn was added. The acidic workup of Br-TiIT-Br/Zn led to a quantitative recovering of Br-TiIT-Br but not to a hydrogen-terminated product Br-TiIT-H formation of which is expected upon hydrolysis of “normal” zinc–organic compound Br-TiIT-ZnBr (Scheme 1).
+BB monomers proceeds via a step-growth mechanism. In the case of electron-rich aromatics, AB-monomers can be prepared via metal/halogen exchange reactions (e.g., by reaction of aryldihalide precursors with alkyl magnesium halides or other metalorganic molecules). In this work we found that treatment of isoindigo-based dibromide Br-TiIT-Br with alkyl magnesium halides also did not lead to the corresponding AB-type monomer Br-TiIT-MgBr (Scheme 1). Similarly, the treatment of Br-TiIT-Br with n-BuLi failed to give the corresponding lithiation product Br-TiIT-Li. Such a behavior was previously observed during attempts of activation of structurally similar naphthalene- and perylene-diimide derivatives.11 We explained these results by a concurrent single-electron transfer from electron-rich alkylmetals to electron-deficient dibromo-arylimide which occurs faster than the halogen-metal exchange. An alternative Zn-based route for activation of electron-deficient naphthalene- and perylene-diimide-based dihalides was reported in our previous works. An attractive feature of this method is that the polycondensation of thus-prepared monomers may proceed via the chain-growth catalyst-transfer mechanism, at least in the case of Br-TNDIT-Br/Zn and Br-TPDIT-Br/Zn. In the present work we found that Br-TiIT-Br reacts with activated Zn powder within minutes at room temperature resulting into zinc–organic complex Br-TiIT-Br/Zn soluble in THF. Titration experiments with iodine revealed the 1/1 ratio between Br-TiIT-Br and Zn in Br-TiIT-Br, irrespective of whether an equimolar amount or excess of Zn was added. The acidic workup of Br-TiIT-Br/Zn led to a quantitative recovering of Br-TiIT-Br but not to a hydrogen-terminated product Br-TiIT-H formation of which is expected upon hydrolysis of “normal” zinc–organic compound Br-TiIT-ZnBr (Scheme 1).
EPR measurements of Br-TiIT-Br/Zn reveal the presence of paramagnetic species with an intense signal at g = 2.0031 (Fig. 1). This observation is consistent with electron transfer from Zn to the electron-deficient Br-TiIT-Br which leads to the radical-anion (Scheme 1). As such, Br-TiIT-Br behaves similarly to rylene-diimide-based compounds in the presence of Zn.
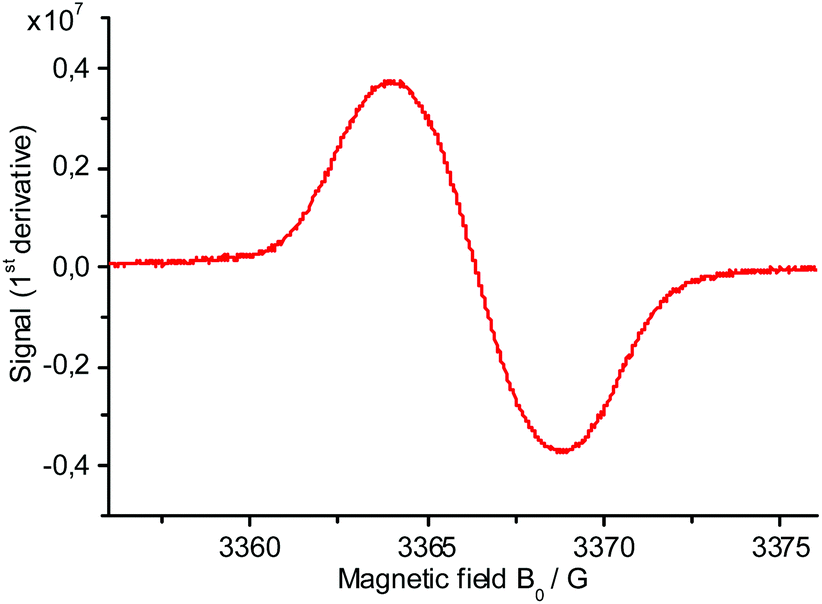 | ||
| Fig. 1 EPR spectrum of the anion-radical Br-TiIT-Br/Zn in THF. | ||
Polymerization
Ni catalysts (Ni(dppe)Cl2 and Ni(dppp)Cl2) failed to polymerize Br-TiIT-Br/Zn at various reaction conditions and starting dibromide Br-TiIT-Br was recovered after quenching of the reaction mixture with acidic water in all cases. It was previously shown that Pd/PtBu3 displays higher catalytic activity than their Ni catalysts in a variety of cross-coupling reactions of small molecules and related polycondensations.23 In this work, the Pd catalyst was prepared in situ by mixing of 1 equivalent of Pd(CH3CN)2Cl2 and 1 equivalent of PtBu3. Addition of Br-TiIT-Br/Zn to the freshly prepared Pd/PtBu3 at room temperature led to the formation of a dark-blue polymer within several hours (Scheme 2).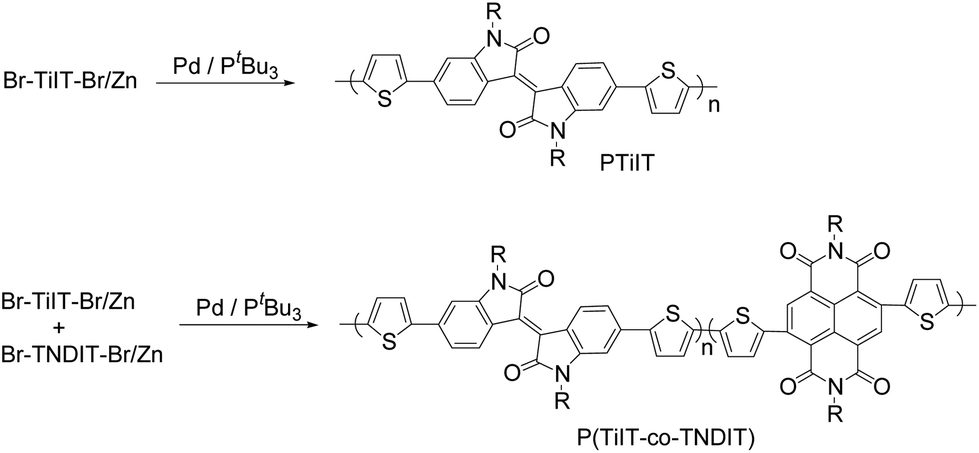 | ||
| Scheme 2 Polymerization of zinc-activated isoindigo-based anion-radical monomer Br-TiIT-Br/Zn and its copolymerization with Br-TNDIT-Br/Zn. | ||
Polymerization course was monitored at [Br-TiIT-Br/Zn]/[Pd/PtBu3] ratio of 100/1 (Table 1). As seen from the evolution of GPC traces which correspond to crude reaction mixtures sampled-out at different polymerization times (Fig. 2), the monomer peak almost disappears already after one hour polymerization when only short isoindigo-oligomers are formed. During the polymerization, the oligomers’ peak gradually shifts to higher molecular weights and levels at Mw ≈ 200 kg mol−1 (Table 1). Importantly, polydispersities Đ of sampled polymers are large and increase with the increase of the polymerization time. These data are not consistent with the chain-growth propagation mechanism observed earlier in polycondensations of Br-TNDIT-Br/Zn and Br-TPDIT-Br/Zn conducted under similar conditions. Rather, these data suggest that the polycondensation involves the step-growth mechanism (see ESI, Fig. S6 and Table S1†).
 | ||
| Fig. 2 Evolution of GPC curves of crude reaction mixtures obtained upon the polymerization of Br-TiIT-Br/Zn at the [Br-TiIT-Br/Zn]/[Pd/PtBu3] ratio of 100/1. | ||
| Polymerization time | M n (kg mol−1) | M w (kg mol−1) | Đ |
|---|---|---|---|
| 1 h | 4.5 | 16 | 3.7 |
| 2 h | 9 | 51 | 5.9 |
| 4 h | 12 | 125 | 10.2 |
| 6 h | 16 | 150 | 9.3 |
| 8 h | 17 | 220 | 13.0 |
| 16 h | 18 | 370 | 20.5 |
Although exact reason of the suppressed chain-growth behavior (compared to rylene-based monomers) requires further elucidation, it can be proposed that the presence of a double bond in the TiIT monomer may cause a strong coordination of Pd(0) catalyst formed after the reduction elimination step. It is known that olefins are much stronger ligands for transition metals than aryls.24 This complexation process may prohibit the intramolecular transfer of the catalyst toward the growing chain end (ring-walking)25 and thus, prevent the intramolecular oxidative addition of Pd(0) to C–Br. In this case, an alternative intermolecular oxidative addition may be a more favoured process because it involves a direct attack of arylhalides (i.e., another monomer or oligomer having C–Br bonds) onto Pd(0) and thus it does not require for Pd(0) to be mobile.
As such, preparation of complex polymer architectures via chain-extension route, such as of all-conjugated block copolymers, is problematic by using this polymerization route as it requires clean chain-growth propagation. Nevertheless, we assumed that synthesis of random-copolymers, such as of Br-TNDIT-Br/Zn and Br-TiIT-Br/Zn, might be possible because polymerization of both these monomers occurs at the same polymerization conditions and in the presence of the same catalyst. To verify this idea, polymerization of Br-TiIT-Br/Zn and Br-TNDIT-Br/Zn was attempted at different monomer ratios of 75:25; 50:50 and 25:75, respectively. We found that the monomer mixtures smoothly polymerize at room temperature giving dark-blue polymeric products. To suppress possible aggregations, their molecular weights were determined by GPC at different temperatures (at 40 °C in chloroform and 60 and 150 °C in trichlorobenzene, Fig. S8†). The GPC traces are more broadened and exhibit multimodal distributions at lower temperatures which may reflect an aggregation of chains. The measurements at 150 °C provided much sharper and monomodal peaks indicating that aggregation is suppressed at this temperature. The latter measurements gave number-average Mn in 15–22 kg mol−1 range and Đ in 2.1–2.5 range.
To elucidate chemical structure of the polymerization products, they were further investigated by 1H NMR spectroscopy and their spectra were compared with the spectra of corresponding homopolymers (Fig. 3). Both the homopolymers and the copolymers tend to form aggregates in solution at lower temperatures. However, well resolved 1H NMR spectra could be obtained in C2D2Cl4 at 120 °C for samples with medium molecular weight. The spectrum of P(TNDIT) (Fig. 3a) shows a low-field-shifted signal of the NDI proton and an AB spin system for the thiophene protons having almost the same chemical shift. A significantly low-field-shifted signal is also observed for PTiIT representing H8 which is located within the deshielding region of the carbonyl group (Fig. 3d). Again, the chemical shift difference between both thiophene protons is small (0.1 ppm) but a ROESY effect between H5 and H6 allows to distinguish both thiophene proton signals. It is obvious from the spectra of the copolymers (Fig. 3b and c) that the isolated polymeric products contain both NDI and isoindigo monomers (marked in Fig. 3 as “N” and “iI”, respectively). The monomer ratio was determined from signal integrals of H3 (TNDIT) and H8 (TiIT), respectively. Copolymerization is proved by composition-dependent signal splitting observed for H1 of NDI and H6 of iI units. As indicated in Fig. 3c, these splittings are due to NDI-centred triads and TiIT–TiIT and TiIT–TNDIT diads, respectively. Unfortunately, only the diads’ content could be determined with good accuracy by signal deconvolution. Relating these values to those expected for random copolymerization reveals that for both copolymer compositions the content of the TiIT–TiIT diad is increased in the copolymers (47% vs. 27% for P(TiIT27-co-TNDIT73) b) and 65% vs. 53% for P(TiIT53-co-TNDIT47) c). Hence, the copolymers show a certain degree of blockiness. Moreover, the increased content of homodiads points to somewhat preferred homopropagation in the copolymerization process. Nevertheless, compositions of the obtained copolymers, as determined by integration of TiIT and TNDIT units, are close to the feed ratios of respective comonomers (compare experimentally found compositions and feed ratios, respectively: 47:53 versus 50:50 and 27:73 versus 25:75).
 | ||
| Fig. 3 1H NMR spectra (regions of aromatic and thiophene protons) of (a) P(TNDIT) and (d) P(TiIT) in comparison to two P(TiIT-co-TNDIT) samples with 73 mol% (b) and 47 mol% (c), respectively, TNDIT comonomer content. Triad signals observed for H3 and diad signals observed for H6 are assigned (N – TNDIT; iI – TiIT). # – thiophene end group signals. Solvent: C2D2Cl4 at 120 °C. | ||
Films of PTiIT and PTNDIT homopolymers as well as P(TiIT-co-TNDIT) copolymers were prepared on electrodes and their redox behavior was studied by cyclic voltammetry in acetonitrile in the presence of 0.1 M NBu4PF6 as electrolyte (Fig. S5†). The polymers oxidize irreversibly so that determination of their HOMO levels was problematic. In contrast, the polymers exhibited reversible reduction behavior and reduction of PTiIT homopolymer occurred at 0.25 eV more electronegative potential than that of PTNDIT. In general, our data for homopolymers are in accordance with previously reported data for the same polymers obtained by Stille polycondensation which report LUMO levels of −3.7 eV (ref. 19 and 20) and −3.95 eV for PTiIT and PTNDIT, respectively. As expected, the volagramms of the P(TiIT-co-TNDIT) copolymers with near equal TiIT/TNDIT composition contains features inherent to both homopolymers. On the other hand, redox behaviour of the copolymers with the major TiIT (80%) or TNDIT (73%) components resembles the behavior of the respective homopolymers.
To further verify the successful copolymerization of TiIT and TNDIT units, UV-vis absorption and fluorescence spectra of the homopolymers and copolymers were recorded in chloroform solutions. As seen from Fig. 4a, the UV-vis spectrum of PTiIT has an absorption of a moderate intensity around 400 nm assignable to the π–π* transition and two low-energy absorption maxima at 650 nm and 725 nm corresponding to the charge transfer (CT) excitations. Further evidence of successful copolymerization comes from fluorescent spectroscopy. As seen from Fig. 4b, incorporation of TNDIT units leads to almost complete quenching of the TiIT fluorescence even in the sample with the lowest TNDIT content. The fluorescence quenching is likely due to energy and/or electron transfer from TiIT to TNDIT. Since the energy and electron transfer processes may proceed when the donor and acceptor units are located closely to each other, the quenching occurring in solution confirms the covalent connection of the TiIT and TNDIT units.
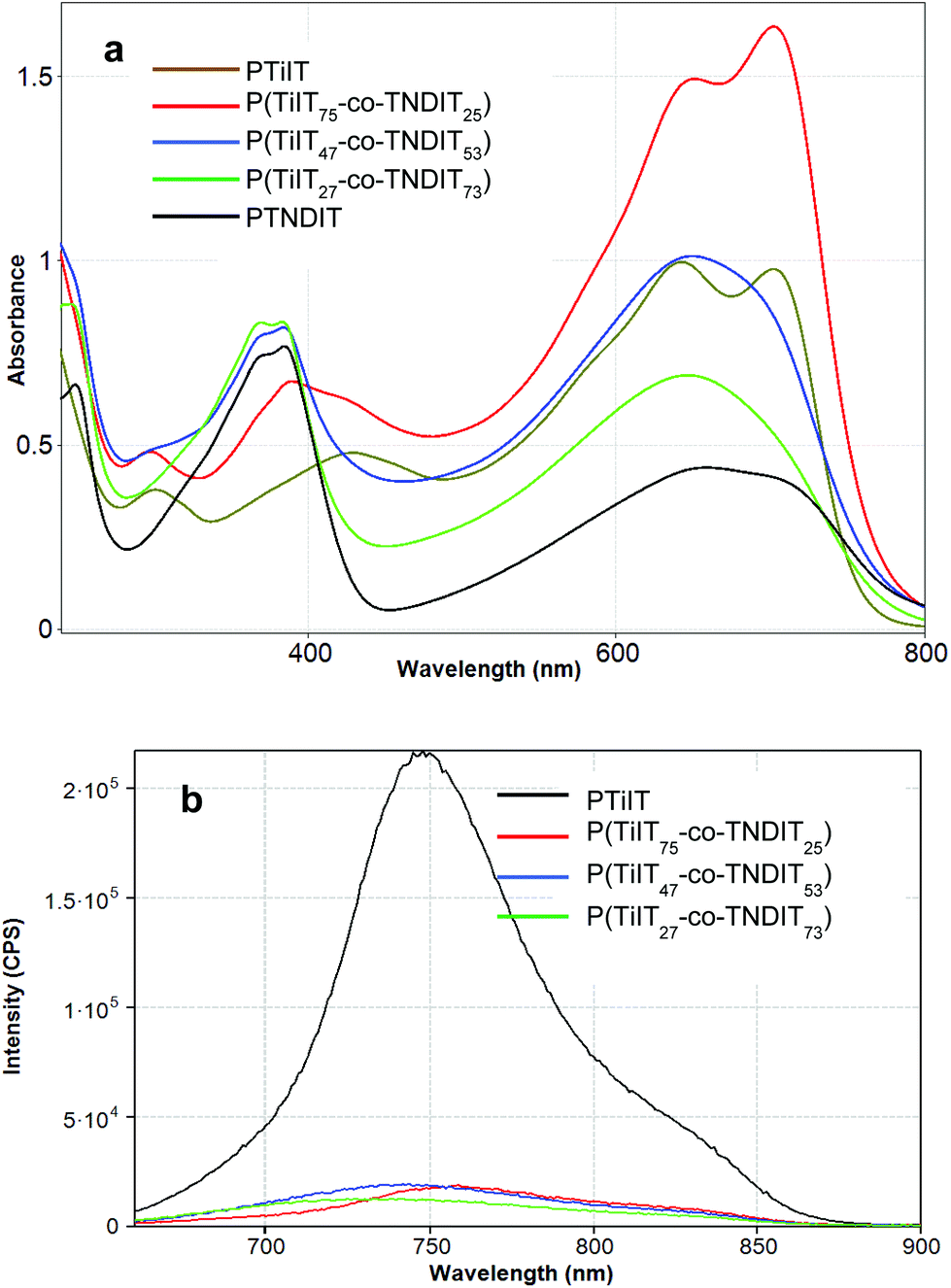 | ||
| Fig. 4 UV-vis (a) and luminescence (b) spectra of the homopolymers PTiIT and PTNDIT and copolymers in chloroform solutions (concentrations: for UV-vis – 0.05 g L−1; for luminescence – 0.001 g L−1). | ||
X-ray diffraction
The molecular packing and morphology of PTiIT were probed using X-ray diffraction (XRD). Free-standing PTiIT films with a thickness of about several hundreds of nanometers were prepared by drop-casting and investigated with X-rays incident slightly tilted to the films. Fig. 5a and b show two-dimensional (2D) XRD patterns of the polymer film before and after thermal annealing for 20 min at 300 °C, respectively. The corresponding radially averaged diffraction patterns are shown in Fig. 5c and d, respectively. Evidently, the sample before annealing is essentially amorphous. After annealing, PTiIT shows some crystallinity,19,20 however it is lower than that in other donor/acceptor copolymers, such as PTNDIT26 or some of diketopyrrolopyrrole-based copolymers.27 For the annealed sample, the (100) lamellar stacking peak position lies at q ∼ 0.29 Å−1 (lamellar spacing d ∼ 22 Å). The broad hump from q ∼ 1.1 to 2.0 Å−1 is attributed to amorphous scattering from disordered side chains. Also discernable is a weak (010) peak at q ∼ 1.68 Å−1 corresponding to π–π stacking with 3.7 Å spacing. | ||
| Fig. 5 2D X-ray diffraction patterns obtained with the beam slightly tilted to (a) as-prepared PTiIT film and (b) thermally annealed PTiIT film at 300 °C for 20 min; (c) and (d) plot of the respective radially averaged curves. | ||
In conclusion, reaction of isoindigo-based dibromide Br-TiIT-Br with activated Zn leads to anion-radical species without insertion of Zn into C–Br bonds similarly to the case of rylenediimide-based dibromides. Thus-activated Br-TiIT-Br/Zn polymerizes in the presence of Pd/PtBu3 catalyst giving PTiIT with a relatively high molecular weight of Mw = 370 kg mol−1. Unlike the polymerization of rylenediimide-based monomers which involves the chain-growth mechanism, isoindigo-based monomer polymerizes on the step-growth manner under the same reaction conditions. X-ray diffraction measurements reveal a semicrystallinity of PTiIT. Statistical copolymerization of isoindigo-based anion-radical monomers with corresponding naphtalenediimide-based monomers proceeds smoothly giving a library of copolymers composition and properties of which can be varied depending on ratio of the monomers. Particularly, reduction of PTiIT homopolymer occurred at a 0.25 eV more electronegative potential than that of PTNDIT (LUMO levels of −3.7 and −3.95 eV, respectively); increase of the content of isoindigo-based units in the copolymers enhances intensity of more electronegative reduction peak. An important feature of the developed polycondensation is that in contrast to Stille, Suzuki and direct arylation methods for preparation of polyisoindigos, polycondensation studied herein proceeds fast at room temperature. We believe that findings of this work are useful in a view of very promising performance of isoindigo-based polymers in solar cells and transistors, reported previously.
Experimental part
Materials
All reagents and starting materials were purchased from Aldrich and TCI and used without further purification. 6,6-Dibromoisoindigo was prepared according to the literature methods.22Polymerization
All operations were done in glovebox under inert gas atmosphere. The activated monomer was prepared as follows: monomer precursor Br-TiIT-Br (300 mg, 0.262 mmol) was placed in a flask equipped with a magnetic stirrer and a septum. Dry THF (30 mL) and active Zn (2.62 mmol) were added and the mixture was stirred for 1 h. Afterwards, mixture was filtrated through 0.2 μm PTFE filter and Pd catalyst (1 mg, 0.01 mol%) (1 equivalent of Pd(CH3CN)2Cl2 and 1 equivalent of PtBu3) in 1 mL THF was added rapidly. The reaction mixture was quenched with methanol and extracted with CHCl3. Organic layer was washed with water, dried over anhydrous MgSO4, evaporated under reduced pressure to give the crude polymer. P(TiIT-co-TNDIT) copolymers were prepared by using the same protocol with the only difference that mixture of Br-TiIT-Br/Zn and Br-TNDIT-Br/Zn taken at a desired ratio was used instead of a single monomer. In this case, the monomer Br-TNDIT-Br/Zn was prepared as described previously.11aAcknowledgements
We gratefully acknowledge support from the German Excellence Initiative via the Cluster of Excellence EXC 1056 “Center for Advancing Electronics Dresden” (cfAED), International Helmholtz Research School (IHRS) NanoNet and DFG (grant KI 1094/9-1). T. Beryozkina thanks Government of the Russian Federation, Act 211, Agreement no, 02.A03.21.0006 for financial support. V. Bakulev thanks RFBR (14-03-01033) for financial support. M. Al-Hussein thanks The University of Jordan and Leibniz-Institut für Polymerforschung, Dresden (IPF) for financial support. Maiti had DAAD-Leibniz fellowship. We thank Josef Brandt for making high-temperature GPC and Dr Ulrich Oertel for UV-vis measurements.Notes and references
- (a) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS; (b) A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed; (c) P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed.
- (a) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed; (b) I. Osaka, Polym. J., 2015, 47, 18–25 CrossRef CAS.
- (a) J. Li, K.-H. Ong, S.-L. Lim, G.-M. Ng, H.-S. Tan and Z.-K. Chen, Chem. Commun., 2011, 47, 9480–9482 RSC; (b) C. B. Nielsen, R. S. Ashraf, B. C. Schroeder, P. D'Angelo, S. E. Watkins, K. Song, T. D. Anthopoulos and I. McCulloch, Chem. Commun., 2012, 48, 5832–5834 RSC.
- (a) Y. J. Hwang, T. Earmme, B. A. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed; (b) A. F. Sousa, A. C. Fonseca, A. C. Serra, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, Polym. Chem., 2016, 7, 1049–1058 RSC.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed.
- Y. H. Geng, L. Huang, S. P. Wu and F. S. Wang, Sci. China Chem., 2010, 53, 1620–1633 CrossRef CAS.
- (a) A. Kiriy, V. Senkovskyy and M. Sommer, Macromol. Rapid Commun., 2011, 32, 1503–1517 CrossRef CAS PubMed; (b) R. Tkachov, V. Senkovskyy, M. Horecha, U. Oertel, M. Stamm and A. Kiriy, Chem. Commun., 2010, 46, 1425–1427 RSC; (c) R. Tkachov, V. Senkovskyy, T. Beryozkina, K. Boyko, V. Bakulev, A. Lederer, K. Sahre, B. Voit and A. Kiriy, Angew. Chem., Int. Ed., 2014, 53, 2402–2407 CrossRef CAS PubMed.
- (a) E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573–16579 CrossRef CAS PubMed; (b) K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC; (c) N. Marshall, S. K. Sontag and J. Locklin, Chem. Commun., 2011, 47, 5681–5689 RSC.
- E. Elmalem, A. Kiriy and W. T. S. Huck, Macromolecules, 2011, 44, 9057–9061 CrossRef CAS.
- (a) V. Senkovskyy, R. Tkachov, H. Komber, M. Sommer, M. Heuken, B. Voit, W. T. S. Huck, V. Kataev, A. Petr and A. Kiriy, J. Am. Chem. Soc., 2011, 131, 19966–19970 CrossRef PubMed; (b) V. Senkovskyy, R. Tkachov, H. Komber, A. John, J.-U. Sommer and A. Kiriy, Macromolecules, 2012, 45, 7770–7777 CrossRef CAS; (c) G. C. Schmidt, D. Höft, K. Haase, A. C. Hübler, E. Karpov, R. Tkachov, M. Stamm, A. Kiriy, F. Haidu, D. Zahn, H. Yane and A. Facchetti, J. Mater. Chem. C, 2014, 2, 5149–5154 RSC.
- Y. Nanashima, A. Yokoyama and T. Yokozawa, Macromolecules, 2012, 45, 2609–2613 CrossRef CAS.
- F. Pammer and U. Passlack, ACS Macro Lett., 2014, 3, 170–174 CrossRef CAS.
- C. Bridges, H. Yan, A. Pollit and D. Seferos, ACS Macro Lett., 2014, 3, 671–674 CrossRef CAS.
- W. Liu, R. Tkachov, H. Komber, V. Senkovskyy, M. Schubert, D. Neher, W. Zhao, A. Facchetti and A. Kiriy, Polym. Chem., 2014, 5, 3404–3411 RSC.
- (a) R. Tkachov, H. Komber, S. Rauch, A. Lederer, U. Oertel, L. Häußler, B. Voit and A. Kiriy, Macromolecules, 2014, 47, 4994–5001 CrossRef CAS; (b) R. Tkachov, Y. Karpov, V. Senkovskyy, I. Raguzin, J. Zessin, A. Lederer, M. Stamm, B. Voit, T. Beryozkina, V. Bakulev, W. Zhao, A. Facchetti and A. Kiriy, Macromolecules, 2014, 47, 3845–3851 CrossRef CAS.
- (a) X. Zhan, Z. Tan, B. An, Z. Domercq, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed; (b) Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- (a) J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed; (b) T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed; (c) G. Zhang, Y. Fu, Zh. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414–1420 CrossRef CAS.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- (a) C. Dai and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 2719 CrossRef CAS PubMed; (b) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed; (c) M. R. Biscoe, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7232 CrossRef CAS PubMed.
- F. R. Hartley, Chem. Rev., 1969, 69, 799 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803 CrossRef CAS PubMed.
- R. Steyrleuthner, R. Di Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. W. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245–4256 CrossRef CAS PubMed.
- J. Li, Y. Zhao, H. Sh. Tan, Y. Guo, Ch. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754–763 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer preparation procedure. See DOI: 10.1039/c6py00055j |
| This journal is © The Royal Society of Chemistry 2016 |