
Hexagonal Co6 and zigzag Co4 cluster based magnetic MOFs with a pcu net for selective catalysis†
Ru-Xin
Yao
,
Xiao-Hui
Qiao
,
Xin
Cui
,
Xiao-Xia
Jia
and
Xian-Ming
Zhang
*
School of Chemistry & Material Science, Shanxi Normal University, Linfen 041004, P. R. China. E-mail: zhangxm@dns.sxnu.edu.cn
First published on 4th November 2015
Abstract
Two hydrophobic 3D cobalt-cluster based magnetic MOFs, [Co3(pimdc)2(H2O)6]·5H2O (1) and Co2(Hpimdc)2 (2) (H3pimdc = 2-propyl-imidazole-4,5-dicarboxylic acid), have been hydrothermally synthesized and magnetically characterized. Both of them exhibit a regular pcu net with hexagonal Co6 and zigzag Co4 clusters as 6-connected nodes, respectively. The magnetic studies indicate that 1 displays predominant antiferromagnetic interactions within the Co3(pimdc) triangle, while 2 shows spin-canting antiferromagnetic behaviour with larger canting angles. Furthermore, after heating under vacuum conditions, activated 1 exhibits an excellent catalytic selectivity in the allylic oxidation reaction of cyclohexene to form an α,β-unsaturated ketone, which indicates that the opened Co(II) sites play a significant role in the heterogeneous catalytic process.
1. Introduction
Metal–organic frameworks (MOFs) have attracted considerable attention due to their charming topologies and potential applications in molecular magnetism, chemical sensors, heterogeneous catalysis and drug delivery.1–3 Modular chemistry has become an effective strategy to construct multifunctional magnetic materials,4 because transition metal clusters as secondary building blocks (SBUs) could not only efficiently reduce the coordination number and geometric limitation, but also modulate the properties of MOFs as functional carriers. Hexagonal rings and zigzag clusters are relatively scarce in various cobalt clusters bridged by vertex-sharing or edge-sharing. Only a few complexes with a hexagonal M6 cluster have been documented, such as discrete Pd6 molecules,5 homochiral Fe6 wheels,6 a binodal (3,18) connected 3D net7 constructed from the cyclo-cluster [Co6(COO)6(H2O)12]6+ and two Co6 cluster based 3D polymers with a 2-position aromatic substituent of the imdc3− ligand.8On the other hand, the oxidation of cyclic olefins, especially selective formation of α,β-unsaturated ketones, is important in the chemical industry as well as value-added fine chemicals.9 However, heterogeneous transition-metal catalysts are still limited and most reports about the oxidation of cyclohexene by MOF catalysts mainly produce tert-butyl-2-cyclohexenyl-1-peroxide.10 MOFs provide a great platform to develop heterogeneous catalysis, due to their high catalyst loadings and more accessible and identifiable catalytic centers. However, it is still a great challenge to assemble metal cluster-based multifunctional magnetic materials, especially a selective catalyst for the oxidation of cyclohexene.
In our previous research on the coordinated diversity of the cadmium 4,5-imidazoledicarboxylate (H3imdc) framework,11 we found that H3imdc is a rigid ligand with versatile coordination modes and acidity-dependent properties, and has successfully assembled high dimension structures and zeolite frameworks as the M–im–M angle is similar to the average Si–O–Si angle (145°).12 Recently, this system has been extended by simple modification of the 2-position imidazole ring with alkyl (Me, Et, Pr) and aromatic phenyl, which may generate a significant effect on the final structure, but a search of the CSD database reveals that only a few low dimension polymers have been reported, such as square tetranuclear complexes, one-dimensional chains and two-dimensional layers.13 Using methyl or ethyl substituent imidazole ligands, Chen's group synthesized MAF-6 with exceptional hydrophobicity, which readily adsorbs organic molecules; MAF-X8 thin films can solid-phase micro-extract non-polar volatile organic compounds from polar phenols.14 Inspired by that work, herein we adopt 2-propyl imidazole-4,5-dicarboxylic acid (H3pimdc) and present two hydrophobic 3D cobalt cluster based magnetic MOFs with a regular pcu net, namely [Co3(pimdc)2(H2O)6]·5H2O (1) and Co2(Hpimdc)2 (2), constructed by using hexagonal Co6 and zigzag Co4 clusters, respectively. Magnetic studies indicated that 1 shows antiferromagnetic interactions within the Co3(pimdc) triangle, while 2 displays spin-canting antiferromagnetic behavior with larger canting angles. Furthermore, after heating under vacuum conditions, activated 1 exhibits an excellent catalytic selectivity in the allylic oxidation reaction of cyclohexene to form an α,β-unsaturated ketone with tert-butyl hydroperoxide (t-BuOOH) as the oxidant.
2. Experimental section
2.1 Materials and physical measurements
All chemicals were analytically pure from commercial sources and used without further purification, unless otherwise noted. 1,2,4-Trichlorobenzene was chromatographically pure. Cyclohexane and cyclohexene were dried with anhydrous CaCl2. t-BuOOH was dried with anhydrous MgSO4 in an ice bath. Elemental analyses were performed on a Vario EL-II analyzer. FTIR spectra were recorded using KBr pellets in the range 4000–400 cm−1 on a Nicolet 5DX spectrometer. Powder X-ray diffraction (PXRD) data were collected on a Bruker D8 advance diffractometer (CuKα, λ = 1.5418 Å). The thermogravimetric analysis (TG) was carried out under air using SETARAM LABSYS equipment at a heating rate of 10 °C min−1. The magnetic measurements were performed with Quantum Design SQUID MPMS XL-5 instruments. The magnetic data were corrected for the diamagnetism of the samples using Pascal constants. N2 adsorption measurement was performed using a Micromeritics ASAP 2020HD88 surface area and pore size analyzer, and was conducted up to a relative pressure (p/p0) of 1.0 at STP.2.2 Synthesis of [Co3(pimdc)2(H2O)6]·5H2O (1)
A mixture of Co(NO3)2·6H2O (0.030 g, 0.10 mmol), H3pimdc (0.029 g, 0.20 mmol), [N(CH3)4]Cl (0.012 g, 0.1 mmol) and H2O (6 mL) was stirred, and then sealed in a 15 mL Teflon-lined stainless autoclave at 160 °C for 5 days. After it was cooled to room temperature and subjected to filtration, red block crystals of 1 in a yield of 35.8% (based on pimdc) were obtained. Anal. calcd (%) for C16H29Co3N4O17 (1): C, 26.46; H, 4.02; N, 7.71. Found: C, 26.51; H, 3.97; N, 7.68. IR data (KBr, cm−1, Fig. S1†): 3433(b), 2964(s), 1656(s), 1426(s), 1324(s), 1121(m), 1054(w), 875(w), 826(w), 767(w), 542(w).2.3 Synthesis of Co2(Hpimdc)2 (2)
A mixture of Co(NO3)2·6H2O (0.031 g, 0.15 mmol), H3pimdc (0.041 g, 0.20 mmol), CH3OH (1 mL) and H2O (5 mL) was stirred, adjusted to pH = 6 by the addition of NaOH (0.008 g, 0.20 mmol), and then sealed in a 15 mL Teflon-lined stainless autoclave at 160 °C for 5 days. After it was cooled to room temperature and subjected to filtration, red cubic crystals of 2 in a yield of 67.5% (based on pimdc) were obtained. Anal. calcd (%) for C16H14Co2N4O8 (2): C, 37.82; H, 2.78; N, 11.03. Found: C, 37.78; H, 2.81; N, 11.01. IR data (KBr, cm−1, Fig. S1†): 3427(b), 2957(s), 2928(w), 2869(w), 1606(s), 1446(s), 1243(m), 1115(m), 1042(w), 869(w), 781(w), 544(w).2.4 Crystallography studies
Single-crystal X-ray diffraction measurements were carried out at room temperature on an Agilent Gemini Eos diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). CrysAlisPro (Agilent Technologies) was used for absorption correction.15 All the structures were solved by direct methods and refined by full-matrix least-squares analysis with SHELXL-97 software.16 All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms of the organic ligand were generated theoretically onto the specific carbon and refined isotropically with fixed thermal factors. Further details for structural analysis are summarized in Table 1, the corresponding bond lengths and bond angles are shown in Table S3.†| Compound | 1 | 2 |
|---|---|---|
| a R 1 = ∑||Fo| ( |Fc||/∑|Fo|. b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | ||
| Empirical | C16H29Co3N4O17 | C16H14Co2N4O8 |
| F w | 726.47 | 508.17 |
| Crystal system | Trigonal | Orthorhombic |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
Pbam |
| a (Å) | 24.4314(6) | 15.9765(17) |
| b (Å) | 24.4314(6) | 15.0880(15) |
| c (Å) | 27.9400(9) | 15.9616(16) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 120 | 90 |
| V (Å3) | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 442.9(9) 442.9(9) |
3847.6(7) |
| Z | 18 | 8 |
| ρ calc (g cm−3) | 1.503 | 1.755 |
| μ (mm−1) | 1.603 | 1.778 |
| F(000) | 6664 | 2048 |
| Crystal size (mm) | 0.20 × 0.16 × 0.10 | 0.22 × 0.16 × 0.12 |
| Reflections | 11498/3288 |
22442/4089 |
| T max/Tmin | 0.994/1.000 | 0.703/1.000 |
| Data/parameters | 3288/213 | 4089/312 |
| S | 1.046 | 1.048 |
| R 1,a wR2b[I > 2σ(I)] | 0.0319, 0.0912 | 0.0568, 0.1641 |
| R 1, wR2 (all data) | 0.0398, 0.0948 | 0.0715, 0.1769 |
| Δρmax/Δρmin (e A−3) | 0.61/−0.36 | 1.22/−0.96 |
2.5 Catalytic oxidation of cyclohexene
Compound 1 was immersed in methanol for 3 days to remove the guest water molecules, and fresh methanol was changed at least three times a day after the extract was decanted. Then the sample was dried under vacuum at 100 °C overnight to obtain activated 1, during which the crystal color changed from red to purple (Fig. S2†). Activated 1 was added to a solution of cyclohexene (3.8 mmol), t-BuOOH (14 mmol) and 1,2,4-trichlorobenzene (1 mmol, as the internal standard). The reaction mixture was stirred at 70 °C. Aliquots of the reaction mixture (0.1 mL) were removed after the time interval, and diluted with cyclohexane (1 mL) and filtered through a 0.25 mm Acrodisc nylon filter. Then, the sample was identified by GC-MS and quantified by GC. For investigations on catalyst activity, the catalyst was separated from the reaction mixture by centrifugation and rinsed five times with cyclohexane before reuse. The reaction progress was monitored by GC analysis, which was performed using an Agilent 7820 GC with cross-linked (95%)-dimethyl–(5%)-diphenylpolysiloxane (HP-5, 30 m × 0.32 mm × 0.25 μm), injector temperature 280 °C, FID detector temperature 300 °C, and oven temperature program 40 °C (3 min)–20 °C min−1–250 °C (2 min). GC/MS analysis was performed using a Thermo Fisher Polaris Q (Ion Trap) gas chromatograph/mass spectrometer.3. Results and discussion
3.1 Description of structure
Single-crystal X-ray diffraction analysis reveals that compound 1 crystallizes in the trigonal space group Rc, and the asymmetric unit consists of two crystallographic independent Co2+ ions, one pimdc3− ligand, three terminal coordinated water molecules, and two and a half crystal lattice water molecules (Fig. 1a). All of the atoms, except for C7, C8 and Co2, localize in general positions. Both Co centers adopt octahedral coordination environments with [CoN2O4] and [CoO6] modes, respectively. The Co1 is coordinated by two nitrogen atoms from two imidazole groups, two carboxylate oxygen atoms and two terminal water molecules. Comparatively, the Co2 is located at the center of inversion with a site occupancy of 0.5, its equatorial plane is occupied by four carboxylate oxygen atoms from two different pimdc3− ligands, and the axial positions are occupied by two terminal water molecules. Additionally, the carbon atoms of the propyl group (C7 and C8) are disordered, located on the c-position glide plane. The Co–L (L = N or O) bond lengths are in the range of 2.038(2)–2.182(3) Å. The cis and trans L–Co–L (L = N or O) bond angles are within the range of 78.32(9)–106.77(9)° and 159.47(10)–180.00(13)°, respectively. Based on the unique triangle bridging mode of the pimdc3− ligand i.e. μ3-kN,O:kO′,O′′:kN′,O′′′ (Fig. S3†), six Co(1) atoms form a regular hexagonal ring [Co6(pimdc)6] with the neighboring Co⋯Co distance being 6.2715(9) Å. Meanwhile, Co(2) atoms alternately point up and down the hexagonal plane, looking like a royal crown [Co(1)6Co(2)6(pimdc)6] (Fig. 1b). Moreover, countless hexagonal rings constitute the honeycomb-like 3D framework with a distinct hydrophobic void, which is filled by flexible propyl groups (Fig. 1c). Calculation with PLATON reveals that the free volume per unit cell is 1828.8 Å3 (12.7% of the total volume). The solvent contact angle measurement is carried out to assess the particle surface properties of 1. Interestingly, a water droplet can maintain its shape and the powder of 1 can float on the water surface (Fig. S4a†). But the powder of 1 exhibits a wet crystal surface with a smaller contact angle for ethanol and can't float on the ethanol surface (Fig. S4b†), which confirms the higher affinity for polar solvents and the hydrophobic nature of the pore surface of 1. From a topological point of view, the hexagonal ring [Co6(pimdc)6] can be considered as a 6-connected node which links with six neighboring [Co6(pimdc)6] units via Co(2) linkers (Fig. S5a†). Based on this simplification, the overall 3D framework of 1 can be described as a uninodal 6-connected pcu topology with the short Schläfli symbol of (412·63). To the best of our knowledge, the pcu topology is one of the five regular prototypal nets, and diversified vertex-sharing/edge-sharing hexanuclear cobalt clusters have been reported as 6-connected nodes,17,18 but few of pcu frameworks with hexagonal Co6 clusters.
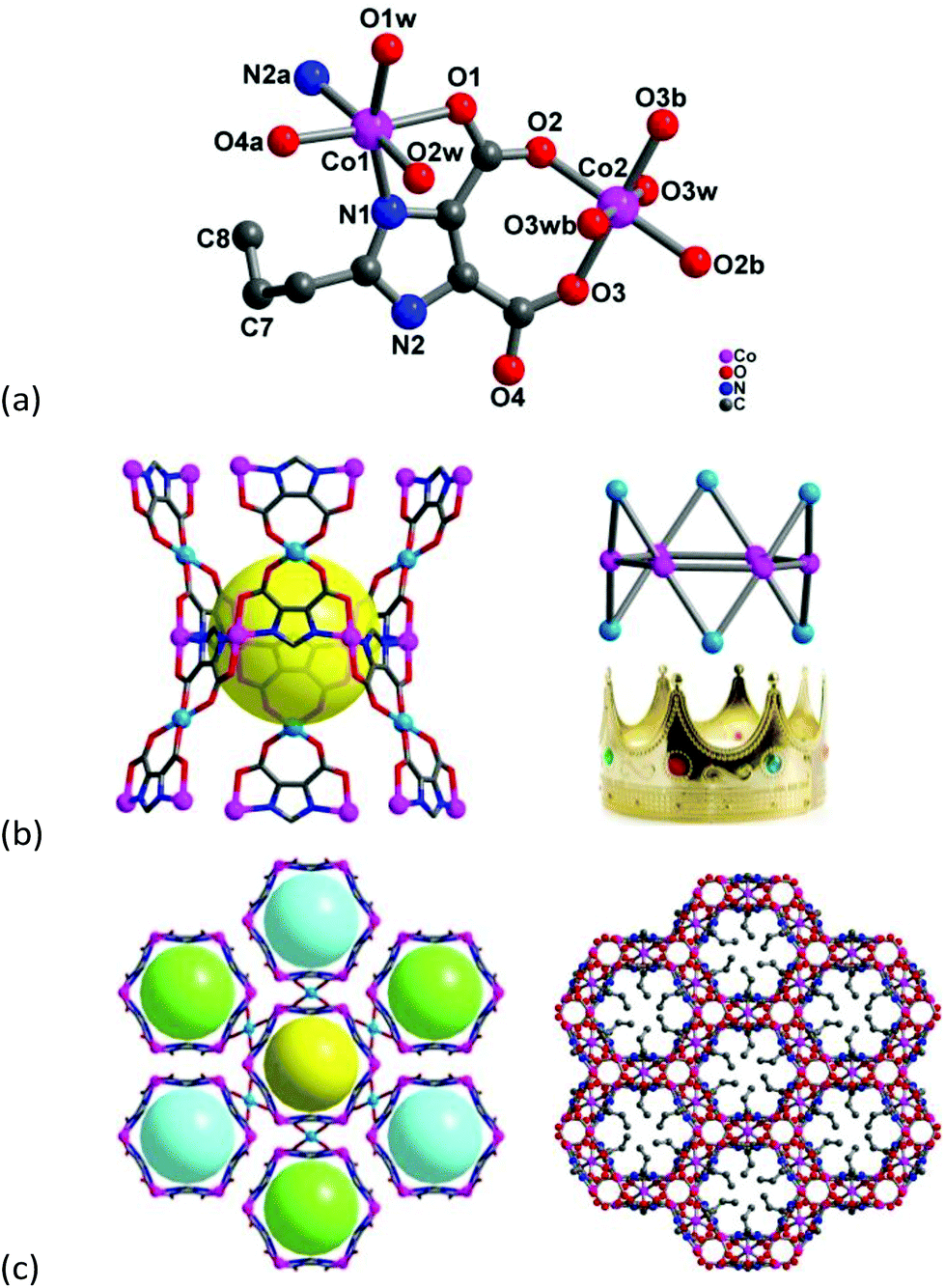 | ||
| Fig. 1 (a) Coordination environment of the Co2+ ions, (b) the hexagonal Co6 unit looks like a royal crown, (c) Co6 cluster is linked by six clusters along the a-axis direction and the honeycomb-like 3D framework with a pcu topology in 1 (note: the propyl group is deleted for clarity in plot (b)). | ||
Compound 2 crystallizes in the orthorhombic space group Pbam, and the asymmetric unit consists of two crystallographic independent Co2+ ions and three Hpimdc2− ligands, of which two ligands are mirror symmetric along the propyl group (Fig. 2a). The propyl group is also disordered, and carbon atoms of the propyl group (C12, C13, C14, C18, C19, C20), C9 and C15 locate on the mirror plane with a site occupancy of 0.5. Both Co2+ centers adopt octahedral geometry with [CoN2O4] mode, and they are coordinated by two nitrogen atoms from two imidazole groups and four carboxylate oxygen atoms from four different Hpimdc2− ligands. The Co–O distances are in the range of 2.101(3)–2.222(4) Å and Co–N lengths are 2.051(3)–2.092(3) Å. The cis and trans L–Co–L (L = N or O) angles are within the range 75.75(11)–119.49(13)° and 149.17(13)–173.18(13)°, respectively. The Hpimdc2− ligand adopts μ4-kN,O:kO′:kO′′:kN′,O′′ and μ4-kN,O:kO:kO′: kN′,O′ modes bridging Co(1) and Co(2) ions (Fig. S3†). It is to be noted that the edge-sharing Co(2) dimer is linked with one Co(1) atom through edge-sharing mode by carboxylate oxygen atoms (O5 and O8), thus forming a zigzag Co4 cluster with a Co⋯Co distance of 3.33–3.35 Å, and the angle of Co1–Co2–Co2a is 107.949(18)° (Fig. 2b). The Co4 cluster is centrosymmetric with the inversion center located at the midpoint of two Co(2) ions, and only a few complexes with zigzag Co4O6 cores have been reported may be because it is apt to form an infinite cobalt chain.19 Each Co4 SBU is surrounded by four Hpimdc2− ligands, meanwhile each Hpimdc2− ligand is connected by two tetramers to yield a 2D (4,4) rhombic grid (Fig. 2c and S5b†). The adjacent layers are further linked by μ4-kN,O:kO:kO′:kN′,O′ modes of Hpimdc2− ligands to form a 3D framework (Fig. 2d). It is also a uninodal 6-connected pcu topology with a short Schläfli symbol of (412·63), in which the zigzag Co4 clusters are regarded as 6-connected nodes and Hpimdc2− ligands as linkers.
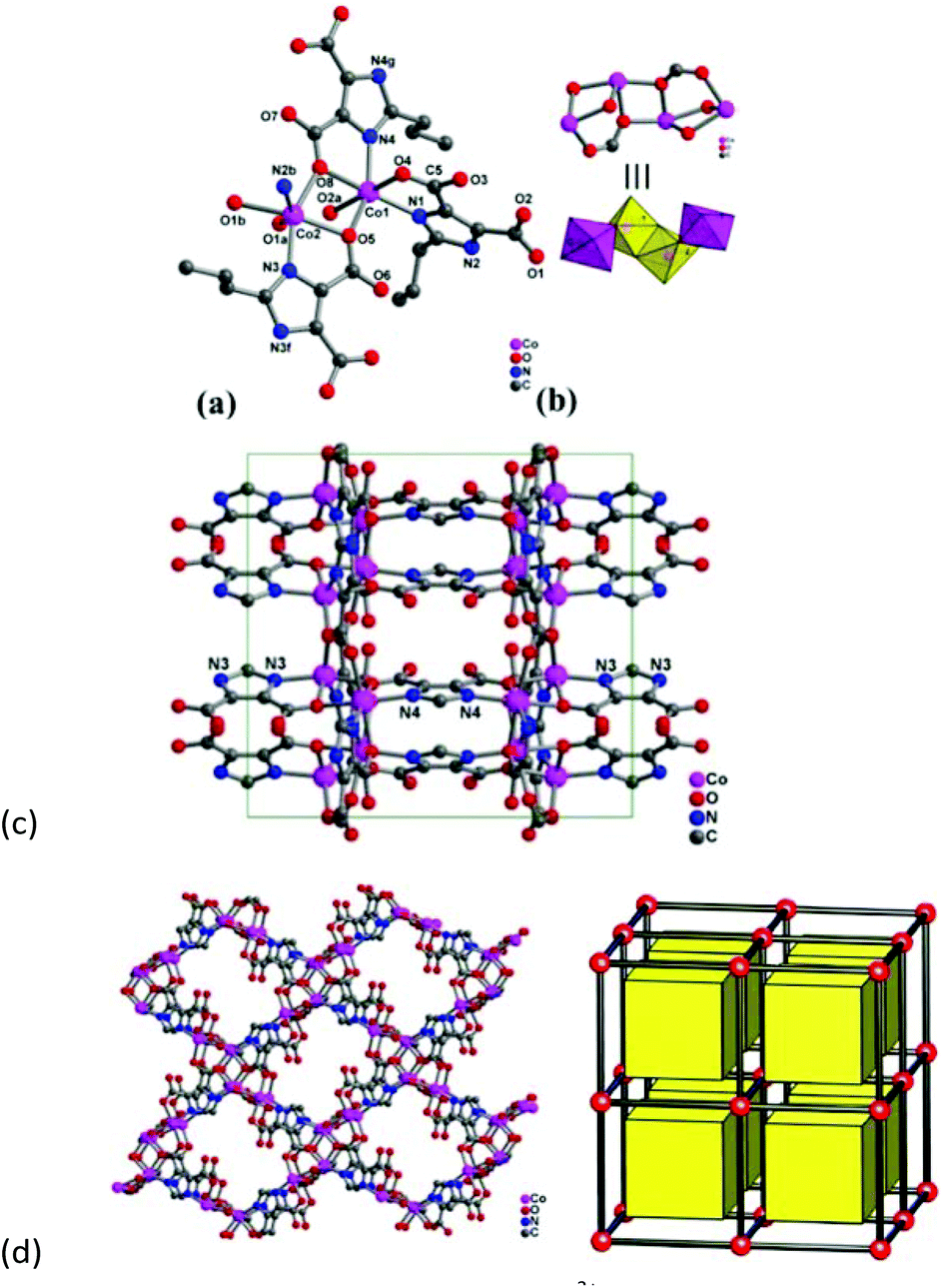 | ||
| Fig. 2 (a) Coordination environment of Co2+ atoms, (b) the tetranuclear Co4O6 unit, (c) 2D rhombic grid along the a-axis direction, (d) 3D framework with a uninodal 6-connected pcu topology in 2 (note: the propyl group is deleted for clarity). | ||
3.2 Thermal properties and PXRD
TG/DTA analyses of 1 and 2 were performed to examine thermal stabilities on polycrystalline samples under air at the heating rate of 10 °C min−1. Compounds 1 and 2 have the same pcu networks but different coordinated environments, which results in different thermal stabilities (Fig. S6†). For 1, the initial weight loss of about 24.41% (calcd 26.06%) in the temperature range of 20–130 °C corresponds to the removal of free and coordinated water molecules. The framework is stable up to around 300 °C in air. The second weight loss in the temperature range of 300–600 °C is in agreement with the removal of pimdc3− groups, accompanied by the decomposition of the framework. The final residues of 30.70% (calcd 31.71%) for 1 are close to the percentage of Co3O4. On the contrary, the TG curve shows that 2 is stable up to 350 °C (Fig. S6†), which may be ascribed to the lack of terminal coordinated water molecules of Co2+ in 2. The DTA plot of 1 presents one endothermic peak at 130 °C and two exothermic peaks ca. 360 °C, while that of 2 exhibits two endothermic peaks at 393 and 478 °C. The phase purity of the bulk samples 1 and 2 is confirmed by PXRD. Most peak positions of the simulated and experimental patterns are in good agreement with each other. The dissimilarities in the intensity may be due to the preferred orientation of the crystalline powder samples (Fig. S7†).3.3 Magnetic studies
Magnetic susceptibility was measured on the phase-pure samples of 1 and 2 in the temperature range of 300–2 K at a field of 1000 Oe. For 1, the χmT value per Co3 unit is 8.26 cm3 K mol−1 at room temperature, which is much higher than the expected value of 5.625 cm3 K mol−1 for three magnetically isolated high-spin Co2+ ions with S = 3/2 and g = 2.00, owing to the significant orbital contribution of Co2+ ions in an octahedral environment.20 Upon cooling, the χmT value gradually decreases to reach a minimum of 0.82 cm3 K mol−1 at 2 K. Such a decrease is a characteristic of dominant antiferromagnetic (AF) exchange interactions as well as the effect of spin–orbit coupling in Co2+ ions. Between 300 and 10.0 K, the susceptibility obeys the Curie–Weiss law with a Curie constant of C = 9.006(3) cm3 K mol−1 and the Weiss constant θ = −28.383(4) K (Fig. 3a). The C value gives rise to an average g value of 2.53, being normal for an octahedral Co2+ ion. The large negative value of the Weiss constant indicates the AF interaction between Co2+ ions. The magnetization of 1 at 2 K increases slowly and linearly with the applied field. The field-dependent magnetization at the highest field 5 T is 2.38Nβ (Fig. 3b), far below the saturation value of 9Nβ expected for three spin-only Co2+ ions, but close to the value of one spin-only Co2+ ion, revealing the presence of antiferromagnetic interactions in the material which may be associated with spin frustrated Co3 triangles.21 Nevertheless, we cannot establish an appropriate magnetic model to determine the magnetic coupling constant of metal ions, as 1 is a complicated metal crown type 3 D framework.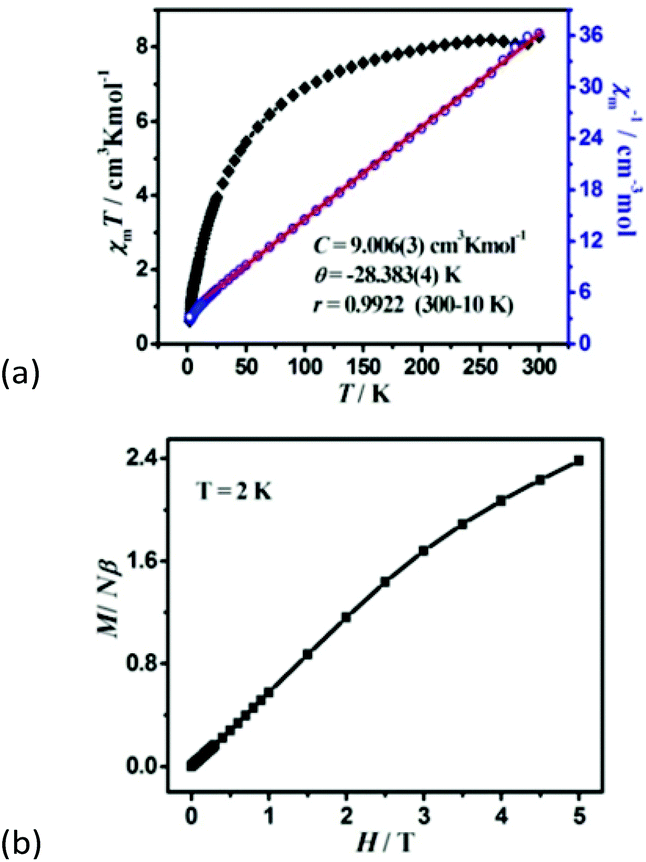 | ||
| Fig. 3 (a) The χmT versus T plots and the Curie–Weiss fitting of χm−1versus T curves for 1 under an applied dc 1000 Oe field per Co3 unit, (b) field dependence of magnetization at 2 K. | ||
For 2, the χmT value per Co4 unit is 11.69 cm3 K mol−1 at room temperature, which is much higher than the expected value of 7.5 cm3 K mol−1 for four magnetically isolated high-spin Co2+ ions with S = 3/2 and g = 2.00, owing to the significant orbital contribution of Co2+ ions in an octahedral environment. The χmT value at room temperature corresponds to effective magnetic moments 4.84μB per Co, which is in accordance with the documented orbital contribution of the high-spin Co2+ ion. As the temperature decreases, the χmT value first decreases smoothly to a minimum of 2.08 cm3 K mol−1 at about 6.29 K, which is caused by spin–orbit coupling or antiferromagnetic interactions. Upon further lowering the temperature, the χmT increases abruptly to a maximum value of 7.13 cm3 K mol−1 at 3.49 K, and finally drops to 4.31 cm3 K mol−1 at 2 K (Fig. 4a). This is a characteristic of the spin-canted antiferromagnetic behaviour, and further confirmed by the field dependence of magnetic susceptibility at 2 K, being larger at a smaller field (Fig. S8†). Between the temperature range of 300 to 9.0 K, the inverse molar susceptibility vs. T plot is linear and obeys the Curie–Weiss law with the Curie constant C = 13.03(4) cm3 K mol−1 and Weiss constant θ = −38.67(5) K. The C value gives rise to an average g value of 2.64, being normal for an octahedral Co2+ ion. The negative Weiss constant θ of 2 indicates the AF interaction between Co2+ ions.
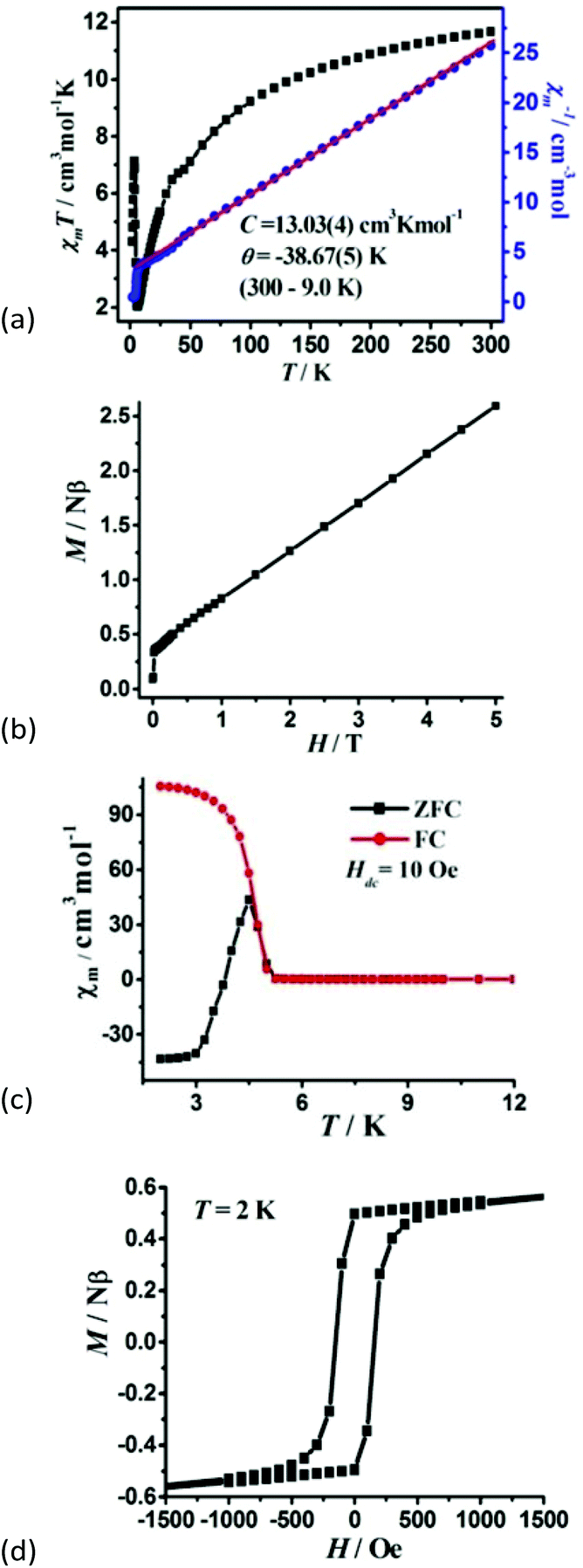 | ||
| Fig. 4 (a)The χmT versus T plots and the Curie–Weiss fitting of χm−1versus T curves under an applied dc 1000 Oe field per Co4 unit, (b) field dependence of magnetization at 2 K, (c) FC and ZFC magnetization at 10 Oe, and (d) the hysteresis loop at 2 K for 2. | ||
The M vs. H curve at 2 K displays the magnetization increase rapidly to 0.34Nβ at a low field of 365 Oe and further reaches linearly 2.28Nβ per Co4 unit at 5 T (Fig. 4b), far below the saturation value of 12Nβ for four spin-only Co(II) ions, suggesting a canted antiferromagnetic behaviour. Field-cooled (FC) and zero-field-cooled (ZFC) magnetizations are performed under an applied field of 10 Oe, and the bifurcation at 5.27 K indicates the occurrence of long-range magnetic ordering below this temperature (Fig. 4c). A narrow hysteresis loop is observed at 2 K with the remnant magnetization MR of 0.524Nβ and the coercive field of 204 Oe for 2 (Fig. 4d). The canting angle can be roughly estimated through the equation sin(γ) = MR/MS to be 3.49°, assuming MS = 8.60Nβ for four octahedral Co2+ ions (at low temperatures, the effect spin S = 1/2 with an average g of 4.3 for octahedral Co2+ ions). It should be pointed out that weak ferromagnets with large canting angles are relatively rare.22 Generally, spin-canting arises due to two factors: (i) the single-ion magnetic anisotropy and (ii) anti-symmetric exchange interaction, namely the Dzyaloshinskii–Moriya interaction (DMI). The study of the structure of 2 reveals that the large canting angle originates from the anti-symmetric exchange in the zigzag Co4 cluster.
Neglecting single-ion anisotropy or spin–orbit coupling of metal ions, simulation of the temperature dependence of χmT values for 2 has been attempted with the Heisenberg Hamiltonian equation (1):
| Ĥex = −2J1(S1S2 + S3S4) − 2J2S2S3 | (1) |
Two exchange coupling constants were adopted for the zigzag Co4 cluster based complex 2: J1 stands for the exchange interaction of Co(1)⋯Co(2) ions, while J2 stands for that of two Co2 ions with S1 = S2 = S3 = 3/2. The best parameters fitted in the range 10–300 K are J1 = −4.3 cm−1, J2 = −13.5 cm−1, and g = 2.65 for 2 (Fig. 5). The negative J value indicates antiferromagnetic interactions between Co2+ ions within the Co4O6 cluster, which can be understood based on structural features. There are three magnetic delivered pathways between Co⋯Co ions: two μ2-O bridges with the Co–O–Co angle of 103.32°, 102.52° and 100.82°, and one syn–syn carboxylate bridge. Generally, it transfer AF interactions that the angle of Co–O–Co greater than 100° and carboxylate syn–syn mode.23 Therefore, it is reasonable to assume that intra-cluster Co4 shows AF coupling.
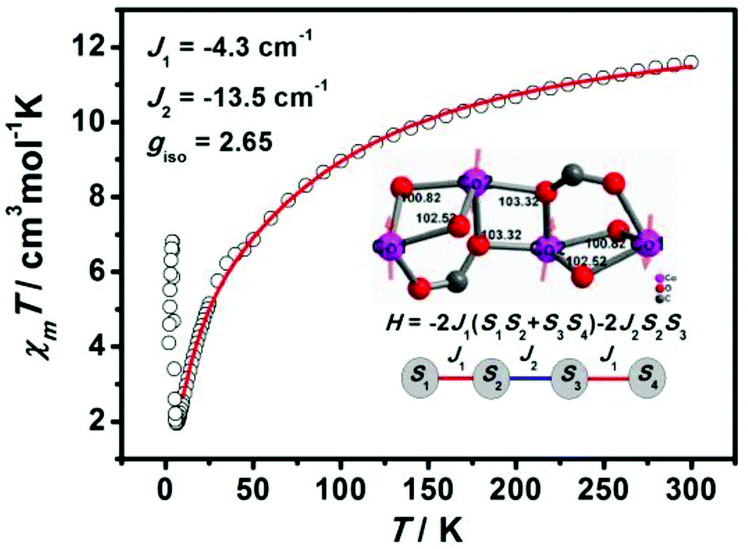 | ||
| Fig. 5 The best fitting of experimental data using the Heisenberg Hamiltonian model for 2. | ||
3.4 Allylic oxidations of cyclohexene with 1 as the catalyst
For compound 1, there are two terminal water molecules for each Co(II) center in the asymmetric unit, which could be lost and provide coordinative unsaturated metal center as Lewis acid catalytic sites. The oxidation reaction of cyclohexene with t-BuOOH oxidant was used as a catalytic test reaction. To select the optimal experimental conditions, the effects of particle size, the amount of catalyst and temperature were investigated, respectively, when fixed other reaction conditions. In the presence of catalyst 1, the conversion increased with the reaction temperature from 50 to 70 °C, but decreased beyond 70 °C (Table S1,† entries 4–8), which may be due to the decomposition of t-BuOOH without oxidizing cyclohexene. The higher conversion achieved with a smaller amount of 1 (Table S1,† entries 1–4). Thus, the optimal conditions are loading of 0.10 mmol activated 1 at 70 °C for 20 hours under solvent-free conditions (shown in Fig. 6). The TON was as high as about 148 (Table S1,† entry 6). Table S2† shows the comparison of the catalytic properties of activated 1 with some representative catalysts for the allylic oxidation of cyclohexene. Transition metal cobalt salts showed the highest conversion and α,β-unsaturated ketone selectivity,24 but they are homogeneous catalysts, which are difficult to separate and reuse. Compound 1 is a heterogeneous catalyst, which offers advantages over homogeneous catalysts, such as easy separation, efficient recycling, improved handling and process control. It is to be noted that heterogeneous catalysts FeIII/SiO2 and Co-MOF provide higher conversion but need the organic solvent CH3CN.25,26 However, 1 is a green catalyst, and shows conversion close to the highest catalyst Cr-MIL-101 to date.27 Although 1 has a hydrophobic pore (12.7% of the total volume) calculated by PLATON, flexible propyl groups might prevent reactants from entering into the pore, which is confirmed by the negligible N2 adsorption at 77 K with a Langmuir surface area of 41.10 m2 g−1 (Fig. S9†), suggesting that the catalytic reaction primarily occurred on the catalyst surface. As a comparative experiment, 1 (without activated) and 2 were used as catalysts under the same conditions, about 3.8% or 3.3% conversion was obtained after 20 hours, which is close to that without the catalyst, probably due to the saturated octahedral geometry of all Co2+ ions for 1 and 2. It also indicated that coordinately unsaturated Co2+ sites in activated 1 played a significant role in the heterogeneous catalytic process.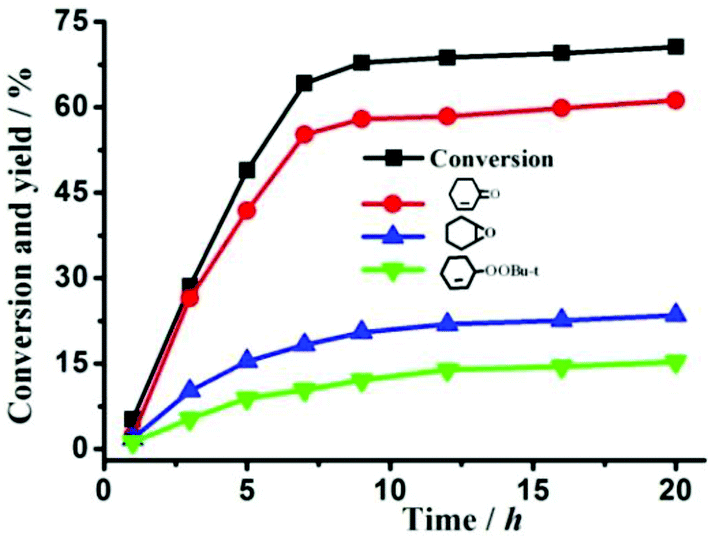 | ||
| Fig. 6 Conversion and product yields for the allylic oxidation of cyclohexene with ground activated 1 as the catalyst at the optimization of reaction conditions. | ||
On the basis of our results and previous oxidation of cyclohexene,27,28 we propose a possible radical mechanism to elucidate the selectivity (Scheme 1). According to the theory of soft and hard acids and bases, in the first step, the Lewis acid Co2+ ions of 1 are preferentially coordinated by an oxygen atom of t-BuOOH. Electrons flow from unsaturated Co2+ centers to the peroxy bond of t-BuOOH, and form tert-butoxy radicals (t-BuO˙). The second step: the t-BuO˙ radical quickly abstracts a hydrogen atom from another t-BuOOH, and creates the t-BuOO˙ radical, which directly reacts with cyclohexene to yield the 3-cyclohexenyl radical. Then, the t-BuOO˙ radical combines with a 3-cyclohexenyl radical to result in t-Bu-cyclohexenyl-1-peroxide, of which the O–O bond thermally decomposes at 70 °C to give the main product 2-cyclohexen-1-one. Meanwhile, the cobalt hydroxy species via fission of the Co–O bond further recover the initial catalyst 1, and the catalytic cycle starts again.
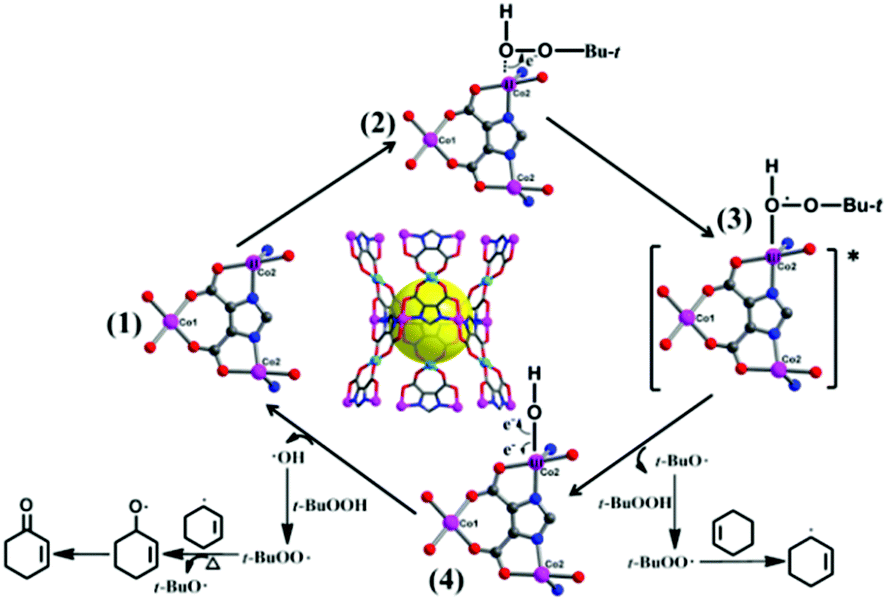 | ||
| Scheme 1 A possible oxidation mechanism of cyclohexene with catalyst 1. | ||
To confirm the heterogeneous nature of the catalyst, activated 1 was removed from the solution by filtration after three hours (approximately 30% conversion). From Fig S10,† we can see that activated 1 is a truly heterogeneous catalyst, no further conversion was being observed. The catalyst was easily separated from the reaction solution by centrifugation and repeatedly washed so that it can be regenerated for catalysis. As shown in Fig. 7, there was no significant decrease in the catalytic efficiency of activated 1 for allylic oxidation of cyclohexene after five recycles under the optimized conditions. The PXRD pattern of the reused catalyst was similar to that of the fresh catalyst 1 (Fig. S7a†), showing that the framework of activated 1 was maintained after the catalytic reaction.
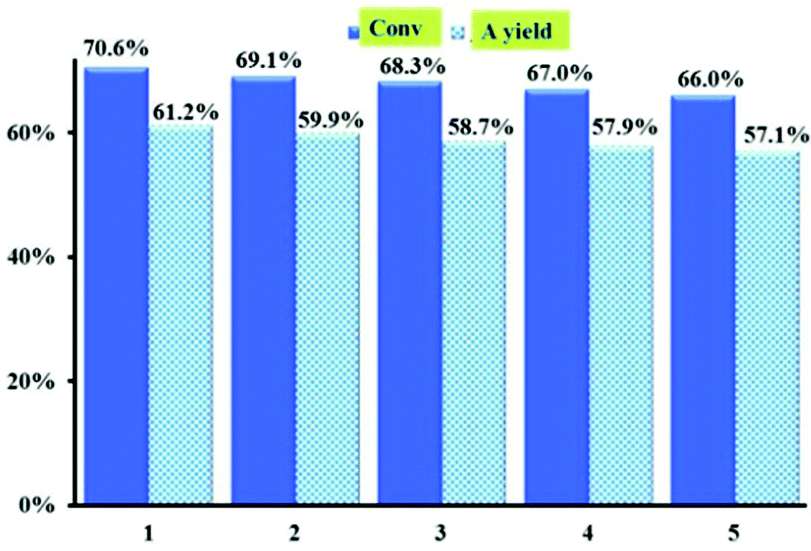 | ||
| Fig. 7 Recycling tests with activated 1 as the catalyst for oxidation reaction of cyclohexene, yielding 2-cyclohexene-1-one. | ||
4. Conclusions
In summary, using 2-position propyl substitutive imidazole-4,5-dicarboxylic acid, two hydrophobic magnetic MOFs with a pcu net respectively constructed by using unique hexagonal Co6 and zigzag Co4 clusters have been successfully synthesized and magnetically characterized. Moreover, activated 1 exhibits highly selective catalytic activity in the allylic oxidation reaction of cyclohexene to form an α,β-unsaturated ketone as the heterogeneous catalyst. This work may provide an effective strategy to construct hydrophobic MOFs through modifying organic ligands and help understand the structure–activity (magnetic and catalytic properties) relationship of Co-MOF materials. In addition, the hydrophobic sites of 1 from the propyl group could be potentially used for separation of alkane molecules.Acknowledgements
Financial support from the 973 Program (2012CB821701), the Ministry of Education of China (Grant IRT1156) and the National Science Fund for Young Scholars (NSFC 20925101 & 21401119) is greatly appreciated. We gratefully acknowledge the assistance in fitting of magnetic properties provided by Jun-Liang Liu.Notes and references
- (a) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2011, 112(2), 675 CrossRef PubMed; (b) N. Stock and S. Biswas, Chem. Rev., 2012, 112(2), 933 CrossRef CAS PubMed.
- (a) J. S. Miller, Chem. Soc. Rev., 2011, 40(6), 3266 RSC; (b) P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40(6), 3249 RSC.
- (a) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44(19), 6804 RSC; (b) A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43(22), 7887 RSC.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34(4), 319 CrossRef CAS PubMed.
- Q.-Q. Wang, V. W. Day and K. Bowman-James, Chem. Commun., 2013, 49(73), 8042 RSC.
- J. Fielden, M. Speldrich, C. Besson and P. Kögerler, Inorg. Chem., 2012, 51(5), 2734 CrossRef CAS PubMed.
- (a) Y. Han, H. Xu, Y. Liu, H. Li, H. Hou, Y. Fan and S. R. Batten, Chem. – Eur. J., 2012, 18(44), 13954 CrossRef CAS PubMed; (b) S. O. H. Gutschke, D. J. Price, A. K. Powell and P. T. Wood, Angew. Chem., Int. Ed., 1999, 38(8), 1088 CrossRef CAS.
- (a) C. Wang, T. Wang, W. Zhang, H. Lu and G. Li, Cryst. Growth Des., 2012, 12(3), 1091 CrossRef CAS; (b) Y. Zhang, X. Luo, Z. Yang and G. Li, CrystEngComm, 2012, 14(21), 7382 RSC.
- (a) I. Serrano, M. I. López, ĺ. Ferrer, A. Poater, T. Parella, X. Fontrodona, M. Solà, A. Llobet, M. Rodrìguez and I. Romero, Inorg. Chem., 2011, 50(13), 6044 CrossRef CAS PubMed; (b) T. Chantarojsiri, Y. Sun, J. R. Long and C. J. Chang, Inorg. Chem., 2015, 54(12), 5879 CrossRef CAS PubMed.
- (a) K. J. Gagnon, C. M. Beavers and A. Clearfield, J. Am. Chem. Soc., 2013, 135, 1252 CrossRef CAS PubMed; (b) H.-Y. Ren, R.-X. Yao and X.-M. Zhang, Inorg. Chem., 2015, 54, 6312 CrossRef CAS PubMed; (c) N. Zhao, F. Sun, H. He, J. Jia and G. S. Zhu, Cryst. Growth Des., 2014, 14, 1738 CrossRef CAS.
- (a) R.-Q. Fang and X.-M. Zhang, Inorg. Chem., 2006, 45(12), 4801 CrossRef CAS PubMed; (b) R.-Q. Fang, X.-H. Zhang and X.-M. Zhang, Cryst. Growth Des., 2006, 6(12), 2637 CrossRef CAS.
- (a) O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjeant, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134(45), 18790 CrossRef CAS PubMed; (b) D. F. Sava, V. C. Kravtsov, J. Eckert, J. F. Eubank, F. Nouar and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131(30), 10394 CrossRef CAS PubMed; (c) Y.-Y. Zhang, X.-Y. Shen, L.-H. Weng and G.-X. Jin, J. Am. Chem. Soc., 2014, 136(44), 15521 CrossRef CAS PubMed.
- (a) R.-R. Zeng, Q.-G. Zhai, S.-N. Li, Y.-C. Jiang and M.-C. Hu, CrystEngComm, 2011, 13(15), 4823 RSC; (b) F. Zhang, Z. Li, T. Ge, H. Yao, G. Li, H. Lu and Y. Zhu, Inorg. Chem., 2010, 49(8), 3776 CrossRef CAS PubMed; (c) J.-H. Deng, D.-C. Zhong, X.-Z. Luo, H.-J. Liu and T.-B. Lu, Cryst. Growth Des., 2012, 12(10), 4861 CrossRef CAS.
- (a) C.-T. He, L. Jiang, Z.-M. Ye, R. Krishna, Z.-S. Zhong, P.-Q. Liao, J. Xu, G. Ouyang, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2015, 137(22), 7217 CrossRef CAS PubMed; (b) C.-T. He, J.-Y. Tian, S.-Y. Liu, G. Ouyang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4(1), 351 RSC.
- CrysAlisPro, Agilent Technologies, Version 1.171.37.35 (release 13-08-2014 CrysAlis171.NET) Search PubMed.
- Sheldrick, SHELX-97: Program for X-ray Crystal Structure Solution and Refinement, Göttingen University, Germany, 1997 Search PubMed.
- (a) S. Bhattacharya, M. Gnanavel, A. J. Bhattacharyya and S. Natarajan, Cryst. Growth Des., 2014, 14(1), 310 CrossRef CAS; (b) J. P. Zhao, W. C. Song, R. Zhao, Q. Yang, B. W. Hu and X. H. Bu, Cryst. Growth Des., 2013, 13, 2858 CrossRef CAS.
- (a) C. G. Perkins, J. E. Warren, A. Fateeva, K. C. Stylianou, A. Mclennan, K. Jelfs, D. Bradshaw and M. J. Rosseinsky, Microporous Mesoporous Mater., 2012, 157, 24 CrossRef CAS; (b) Z.-Y. Liu, E.-C. Yang, L.-L. Li and X.-J. Zhao, Dalton Trans., 2012, 41, 6827 RSC.
- D. Aguila, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromi, Chem. Commun., 2011, 47(2), 707 RSC.
- J.-D. Leng, S.-K. Xing, R. Herchel, J.-L. Liu and M.-L. Tong, Inorg. Chem., 2014, 53(11), 5458 CrossRef CAS PubMed.
- (a) R. X. Yao, Y. L. Qin, F. Ji, Y. F. Zhao and X. M. Zhang, Dalton Trans., 2013, 42(18), 6611 RSC; (b) E.-C. Yang, Z.-Y. Liu, T.-Y. Liu, L.-L. Li and X.-J. Zhao, Dalton Trans., 2011, 40(32), 8132 RSC.
- R.-X. Yao, X. Cui, J. Wang and X.-M. Zhang, Chem. Commun., 2015, 51(24), 5108 RSC.
- P. Díaz-Gallifa, O. Fabelo, J. Pasán, L. Cañadillas-Delgado, J. Rodríguez-Carvajal, F. Lloret, M. Julve and C. Ruiz-Pérez, Inorg. Chem., 2014, 53(11), 5674 CrossRef PubMed.
- E. F. Murphy, T. Mallat and A. Baiker, Catal. Today, 2000, 57, 115 CrossRef CAS.
- J. Mao, X. Hu, H. Li, Y. Sun, C. Wang and Z. Chen, Green Chem., 2008, 10, 827 RSC.
- M. Tonigold, Y. Lu, A. Mavrandonakis, A. Puls, R. Staudt, J. Möllmer, J. Sauer and D. Volkmer, Chem. – Eur. J., 2011, 17, 8671 CrossRef CAS PubMed.
- I. Y. Skobelev, A. B. Sorokin, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2013, 298, 61 CrossRef CAS.
- Y. H. Cao, H. Yu, F. Peng and H. J. Wang, ACS Catal., 2014, 4, 1617 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1409462–1409463. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00159e |
| This journal is © the Partner Organisations 2016 |