
Synthesis and photophysical properties of multi-Ru2+ terpyridine complexes: from di-nuclear linear to star-shaped hexa-nuclear architectures†
Jie
Yuan‡
,
Zhilong
Jiang‡
,
Die
Liu
,
Yiming
Li
and
Pingshan
Wang
*
College of Chemistry and Chemical Engineering, Central South University, Changsha-410083, Hunan, China. E-mail: chemwps@csu.edu.cn; Fax: +0086-731-88830651
First published on 2nd December 2015
Abstract
A series of terpyridine based Ru2+ complexes G1, G2, G3, and G4 were designed and synthesized, the solubility of complexes was enhanced by introducing a multiple tailed long aliphatic chain; full characterization has been performed with 1H NMR, 13C NMR, ESI-MS and UV-vis measurements. Electrochemical behaviours of these complexes have been investigated.
Comprehensive work related to metallo–organic self-assembly had been exploited1–4 and it has a great influence in the field of supramolecular chemistry. Recently, particular attention has been focused on pyridine and polypyridine based metallo-supramolecules, which are assembled from the interaction of metal ions with predesigned multifunctional ligands.5 Many structure-to-property related architectures were developed by the use of the geometries and donor-types of transition-metal ions.6,7 Among these investigations, the terpyridine based ruthenium complex has found extensive applications, such as in water oxidation catalysis,8 cancer chemotherapy,9 photochemistry9 and so on. In this paper, a series of multi-nuclear Ru2+ complexes have been designed and synthesized, and their photophysical and electrochemical properties have been studied.
The strategy for realizing multi-Ru2+ complexation is initially focused on the ligands possessing multiple terpyridine motifs. The para-positioned bis-terpyridines of g1, g2 and g3 were obtained from published procedures with an effective modification; hexa-terpyridine functionalized benzene was also synthesized from a known method.9g1–g4 reacted with the mono-adduct TpyRuCl3 [Ru(T)Cl3] to afford the dinuclear linear metallo-complexes when using bis-terpyridine, and a hexanuclear complex when using hexaterpyridine.10 The multi-Ru2+ complexes G1–G4 are estimated to result in better photophysical and stable properties compared to the mono-Ru2+ complex.10 In order to increase the solubility of the multi-Ru2+ complexes, an aliphatic chain with twelve carbons was introduced into the tails of the metallo-molecules G1–G4; the following experiments proved that the complexes have good solubility in most organic solvents or mixed solvents, such as CHCl3/MeOH, CH2Cl2/MeOH, CH3CN, CHCl3/CH3CN, DMF and so on, which could benefit the characterization and further intermolecular assembly.10
Results and discussion
Ligand g1 (Scheme 1 and ESI†) was prepared by condensing 2-acetylpyridine with terephthalaldehyde to generate 1,4-bis-terpyridinyl benzene g1 under solvent free basic conditions. Ligand g2 was prepared from 4-([2,2′:6′,2′′-terpyridine]4′-yl)-phenyl boronic acid and 4-bromophenyl-terpyridine utilizing a Suzuki coupling with Pd(PPh3)4 as the catalyst. Ligand g3 was prepared from 1,4-dibromobenzene and 4-([2,2′:6′,2′′-terpyridine]4′-yl)-phenyl boronic acid by a two-fold Suzuki coupling reaction. And the ligand g4 was obtained from hexabromobenzene and 4-([2,2′:6′,2′′-terpyridine]4′-yl)-phenyl boronic acid by a six-fold Suzuki coupling reaction according to a literature method.9 The paramagnetic, RuIII mono-capping adduct [Ru(T)Cl3] (Scheme 1) was then obtained by refluxing 4-(4-(dodecyloxy)phenyl)-terpyridine (T) with RuCl3 according to a published method.10 The dinuclear complexes G1–G3 were obtained similarly by reacting g1–g3 with 2.1 equiv. of the mono-adduct [Ru(T)Cl3] under the heat conditions, and the resulting crude product was purified by aluminium column chromatography to give the desired G1–G3, as red solids. As shown in Fig. 1, the 1H NMR spectrum of G1 exhibits two characteristic tpyH3′,5′ resonances, indicating the formation of the dual metallo-core composite. The chemical shifts of tpyH3′,5′ were shifted downfield to 9.51 and 9.28 ppm compared with the peaks at 8.82 and 8.73 ppm in g1 and T, respectively. Moreover, a significant upfield shift for the tpyH6,6′′ signal occurred upon complexation due to the shielding effect of the perpendicular terpyridine counterpart with the metal center.10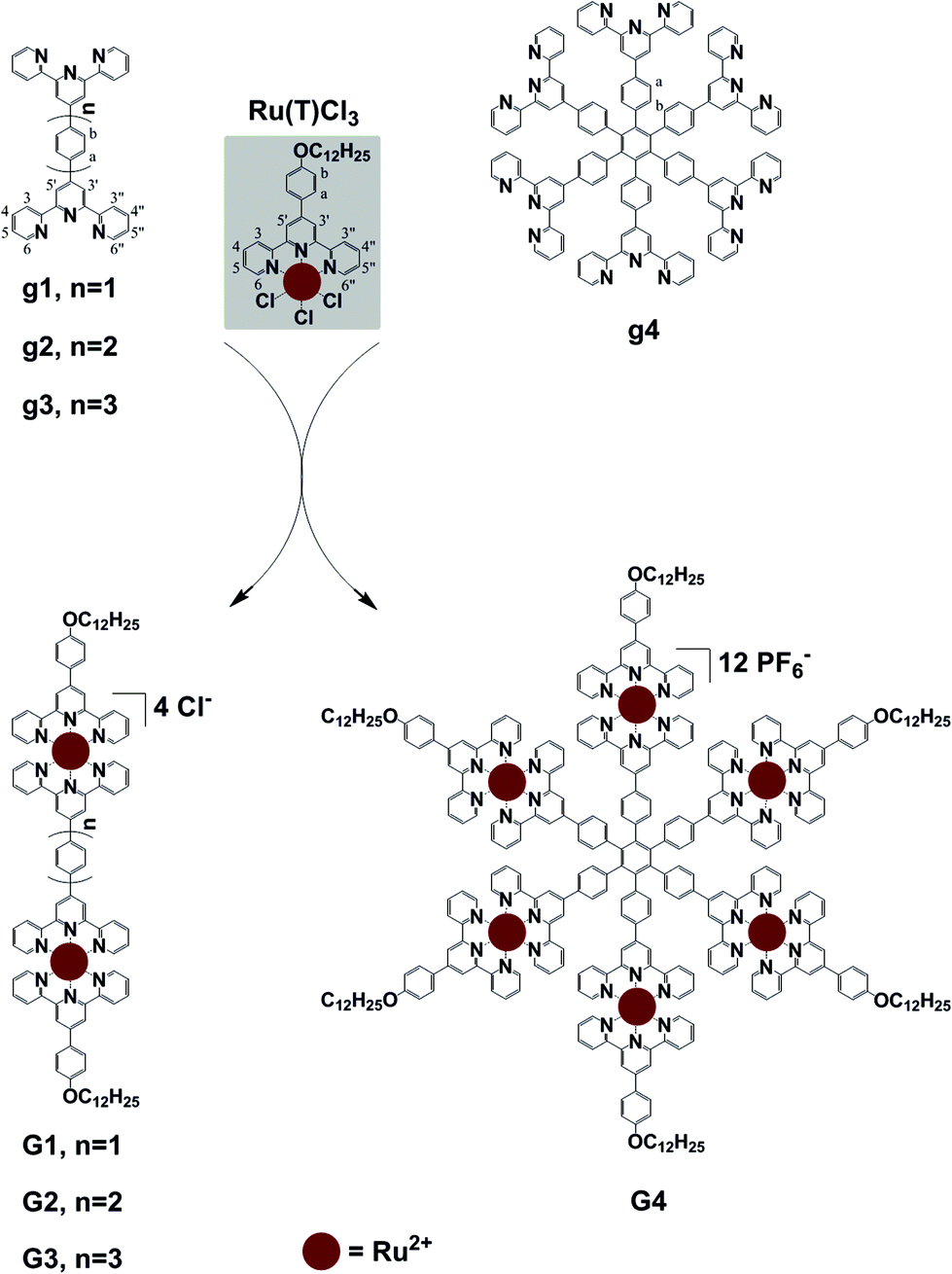 | ||
| Scheme 1 Preparation of the self-assembled multi-Ru2+ complexes G1, G2, G3, and G4. | ||
 | ||
| Fig. 1 1H NMR spectra of ligand g1 and T in CDCl3 and complex G1 in CD3OD. | ||
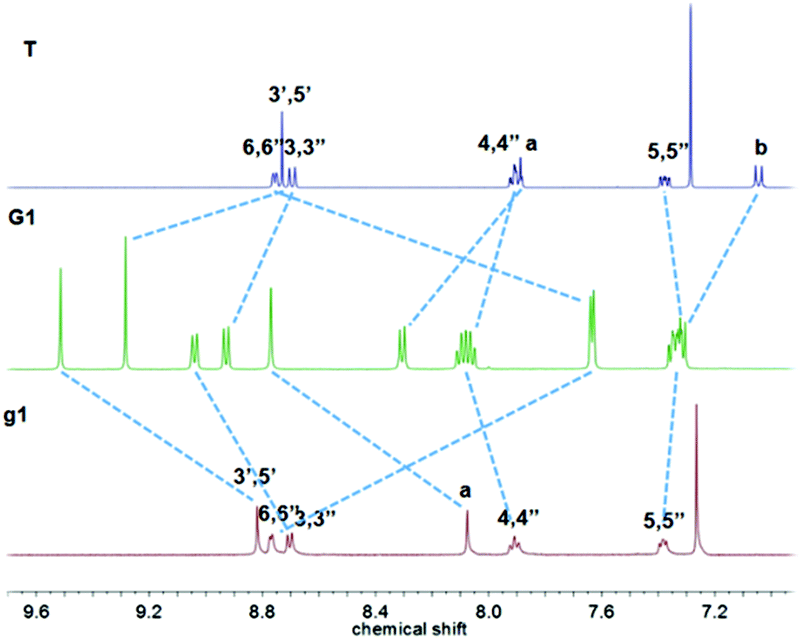
As shown in Fig. 2, complex G2 reflected two units of tpyH3′,5′ resonances at 9.43 and 9.28 ppm compared with the peaks at 8.81 and 8.73 ppm in g2 and T; the considerable upfield shift for the tpyH6,6′′ proton also occurred upon complexation, which was consistent with the formation of the dual-core complex.
 | ||
| Fig. 2 1H NMR spectra of ligand g2 and T in CDCl3 and complex G2 in CD3OD. | ||
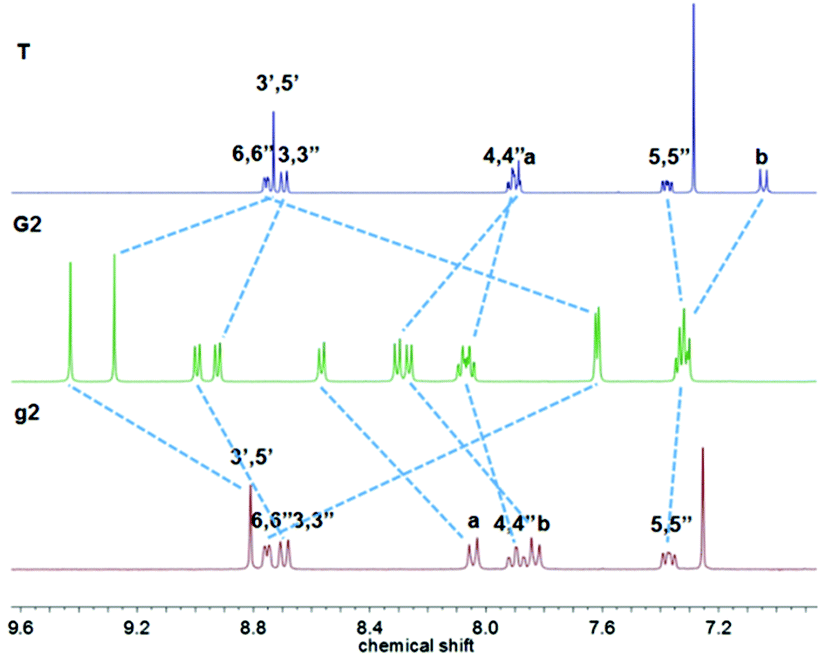
The red solid complex G3's (Fig. 3) two tpyH3′,5′ peaks were shifted downfield to 9.40 and 9.27 ppm compared with the peaks at 8.83 and 8.73 ppm for g3 and T, respectively.
 | ||
| Fig. 3 1H NMR spectra of ligand g3 and T in CDCl3 and complex G3 in CD3OD. | ||
Hexanuclear complex G4 had also been prepared similarly to G1 by reacting g4 with 6.5 equivalents of [Ru(T)Cl3], and the product was finally obtained after anion exchange with NH4PF6 as a deep-red powder. The 1H NMR spectra (Fig. 4) presented two attributed peaks of the downfield tpyH3′,5′ at 8.96 and 8.95 ppm compared with the peaks at 8.59 and 8.73 in g4 and T, respectively.
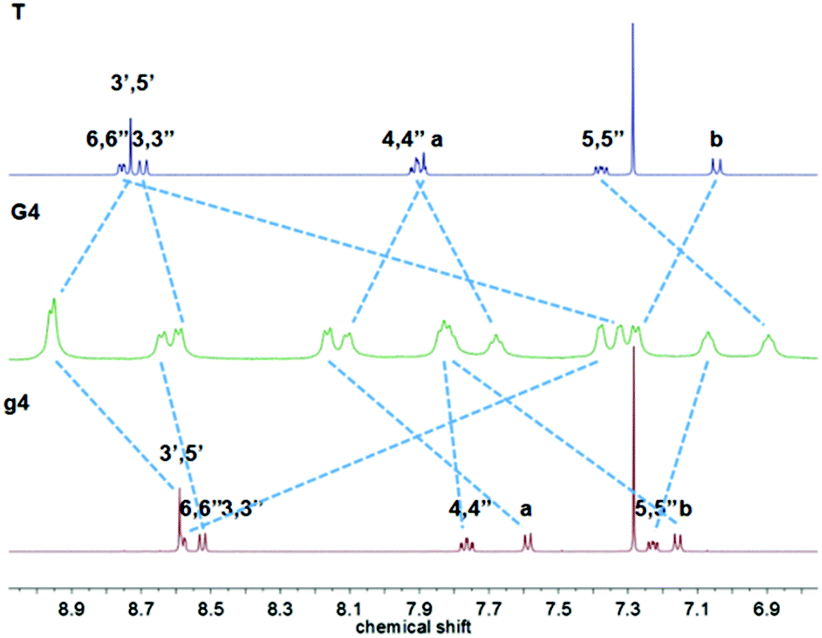 | ||
| Fig. 4 1H NMR spectra of ligand g4 and T in CDCl3 and complex G4 in CD3CN. | ||
Complexes G1–G4 have been characterized additionally by mass spectrometry as shown in Fig. 5, and the results have been recorded in the positive ion reflection mode to verify the complex structure by comparing with the corresponding theoretical distributions. For G1, G2 and G3, the corresponding 2+ to 4+ charge states can be assigned to [G-nCl−]n+ (n = 2, 3, 4) ions. Similarly, the charge states of G4 ranging from 4+ to 9+ can be found at m/z 1662.2, 1300.8, 1060.3, 887.8, 759.0 and 658.2, respectively.
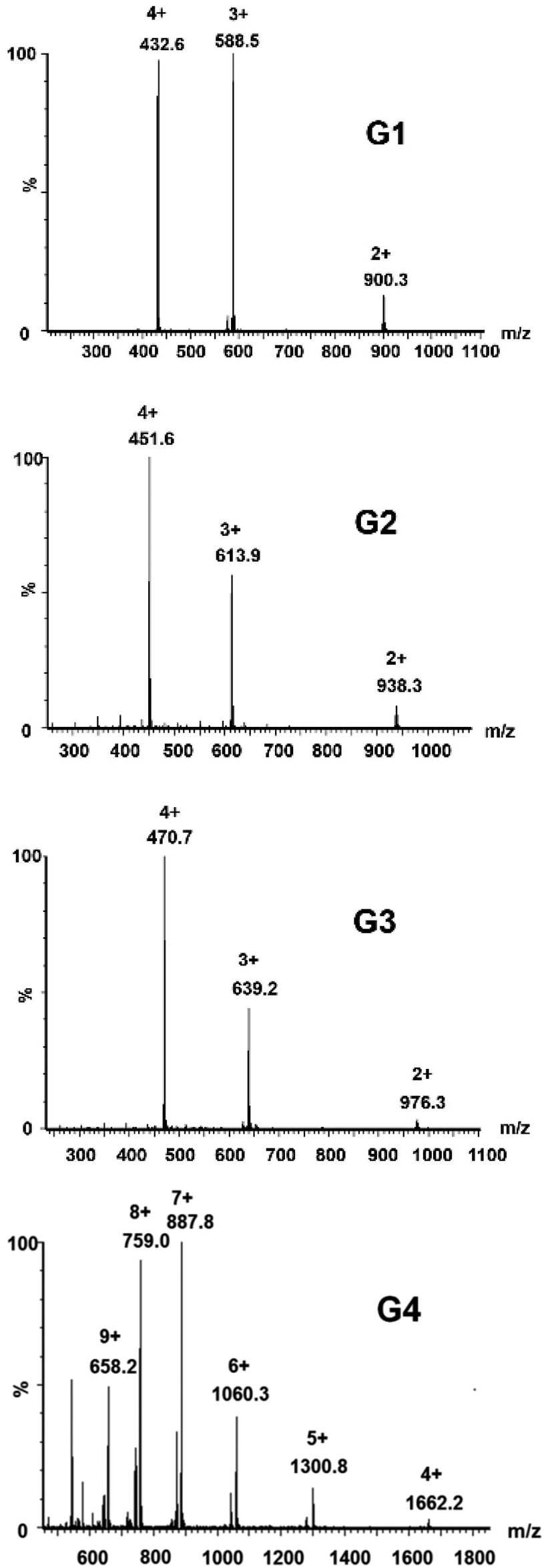 | ||
| Fig. 5 Q-TOF-MS spectra of complexes G1–G4. | ||
Absorption and emission spectroscopy
The UV-vis spectrum (Fig. 6) was examined in MeOH (G1–G3) and CH3CN (G4), respectively. The characteristic absorption band around 500 nm (resultant absorptions of G1–G4 were at 502, 498, 495 and 493 nm, correspondingly) for all complexes was assigned to metal-to-ligand-charge-transfer (MLCT) transitions, corresponding to the electronic transition from the d-orbital of multi-ruthenium atoms to the π*-orbital of the ligands. Slightly hypochromic shifts of the absorption bands were observed along with the increase of the benzene ring of the bis-terpyridine bridge linkages, which may be due to the energy level of the π*-orbital or the electron withdrawing effects ascending according to the increase of the conjugated effect from G1 to G4, which led to the larger band gap between the π*-orbital and the d-orbital of Ru2+. The emissions of complexes G1–G4 had been detected at 77 K (Fig. 6B), which agree with the literature reports.7,11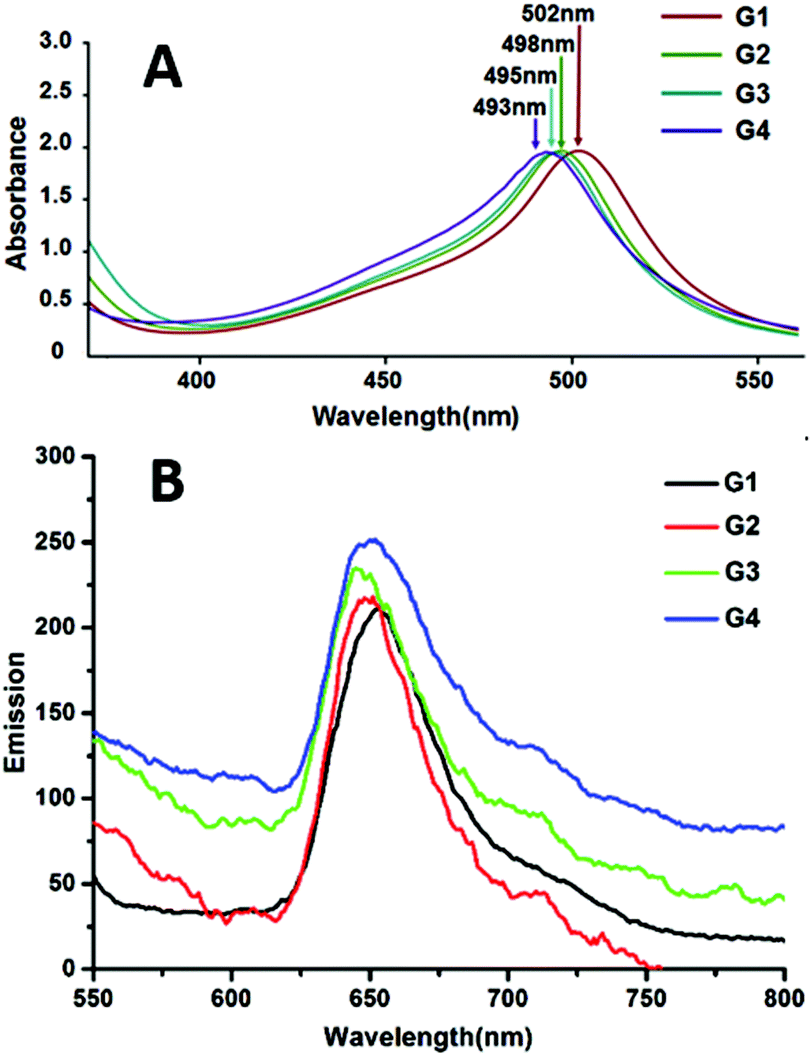 | ||
| Fig. 6 (A) Absorption spectra of the dinuclear complexes G1, G2, G3 (7.5 × 10−6 M, RT) and the hexanuclear complex G4 (2.3 × 10−6 M, RT) in CH3CN. (B) Emission spectra of the dinuclear complexes G1, G2, G3 (7.5 × 10−7 M, 77 K) and the hexanuclear complex G4 (5.8 × 10−7 M, 77 K) in CH3CN. | ||
Electrochemical properties
The electrochemical properties of complexes G1–G4 were investigated by cyclic voltammetry. As shown in Fig. 7, all complexes illustrated one reversible oxidation couple of Ru2+/Ru3+ and the moderate onset oxidation potentials, G1 to G4 were determined to be 1.345, 1.320, 1.305, and 1.298 V vs. Hg/Hg2Cl2, respectively. Obviously, the oxidation potential increased along with the expansion of the benzene ring of terpyridine bridges from G1 to G4. The enhanced conjugation of the terpyridine bridge linkages may be the reason for the decrease of oxidation potentials.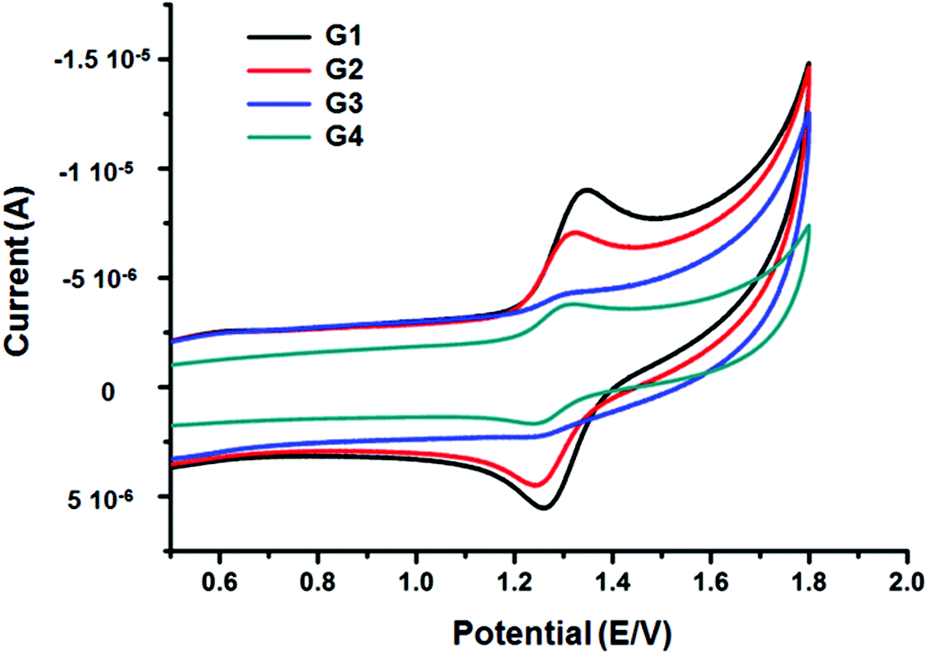 | ||
| Fig. 7 Cyclic voltammograms of the complexes G1, G2, G3 and G4 (in 0.1 M Bu4NPF6, CH3CN solution). | ||
Experimental section
Material
All materials were purchased through J & K Chemical Technology and used without further purification. 4-Dodecyloxy-benzaldehyde, 2-acetylpyridine, bromobenzaldehyde, 1,4-dibromobenzene, hexabromobenzene and RuCl3·3H2O were also purchased through J & K Chemical Technology.Synthesis of 4′-(4-(dodecyloxy)phenyl)-2,2′:6′,2′′-terpyridine (T)
To a solution of 4-dodecyloxy-benzaldehyde (2.70 g, 9.3 mmol) and 2-acetylpyridine (2.25 g, 18.6 mmol) in 100 ml EtOH, NaOH powder (1.12 g, 27.9 mmol) was added. After stirring at 25 °C for 10 h, aqueous NH3·H2O (30 mL) was added and the mixture was refluxed overnight. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was purified by column chromatography (Al2O3) (CH2Cl2/PE = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and a white powder was obtained (2.15 g, 48.5%). 1H NMR (400 MHz, CDCl3): δ 8.76–8.75 (d, J = 4 Hz, 2H), 8.73 (s, 2H), 8.70–8.68 (d, J = 8 Hz, 2H), 7.93–7.90 (m, 2H), 7.89 (s, 2H), 7.39–7.36 (m, 2H), 7.06–7.03 (d, J = 12 Hz, 2H), 4.07–4.04 (m, 2H), 1.88–1.81 (m, 2H), 1.55–1.47 (m, 2H), 1.41–1.30 (m, 16H), 0.92–0.89 (m, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 160.14, 156.44, 155.83, 149.84, 149.11, 136.85, 130.47, 128.50, 123.75, 121.37, 118.25, 114.86, 68.16, 31.94, 29.69, 29.66, 29.62, 29.60, 29.44, 29.37, 29.29, 26.07, 22.71, 14.14. Q-TOF-MS (m/z): Calcd for 493.3; found for 494.3 (M + 1).
:1) and a white powder was obtained (2.15 g, 48.5%). 1H NMR (400 MHz, CDCl3): δ 8.76–8.75 (d, J = 4 Hz, 2H), 8.73 (s, 2H), 8.70–8.68 (d, J = 8 Hz, 2H), 7.93–7.90 (m, 2H), 7.89 (s, 2H), 7.39–7.36 (m, 2H), 7.06–7.03 (d, J = 12 Hz, 2H), 4.07–4.04 (m, 2H), 1.88–1.81 (m, 2H), 1.55–1.47 (m, 2H), 1.41–1.30 (m, 16H), 0.92–0.89 (m, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 160.14, 156.44, 155.83, 149.84, 149.11, 136.85, 130.47, 128.50, 123.75, 121.37, 118.25, 114.86, 68.16, 31.94, 29.69, 29.66, 29.62, 29.60, 29.44, 29.37, 29.29, 26.07, 22.71, 14.14. Q-TOF-MS (m/z): Calcd for 493.3; found for 494.3 (M + 1).
Synthesis of [Ru(T)Cl3]
To a solution of RuCl3 (288 mg, 1.1 mmol) in ethanol (60 ml), 4′-(4-(dodecyloxy)phenyl)-2,2′:6′,2′′-terpyridine (493.68 mg, 1 mmol) was added, and then the suspension was refluxed for 12 h. After the mixture had been cooled, the obtained dark brown solid was filtered and washed with MeOH, and subsequently dried in vacuo to yield a dark brown solid (526 mg, 75%) which was used directly for next steps.1,4-Bis([2,2′:6′,2′′-terpyridin]-4′-yl)benzene ( g1 ) was synthesized according to a literature procedure starting with 2-acetylpyridine and benzene-1,4-dialdehyde, and the obtained product was recrystallized from glacial acetic and washed with ethanol, and the solid was dried in vacuo.12–141H NMR (500 MHz, CDCl3) δ: 8.82 (s, 2H), 8.77–8.76 (d, J = 5 Hz, 2H), 8.71–8.70 (d, J = 5 Hz, 2H), 8.08 (s, 2H), 7.92–7.89 (m, 2H), 7.40–7.37 (m, 2H).
4′-(4-Bromophenyl)-2,2′:6′2′′-terpyridine was synthesised according to a published method.15
Synthesis of 4,4′-di([2,2′:6′,2′′-terpyridin]-4′-yl)-1,1′-biphenyl (g2)16
NaOH (0.16 g, 4.0 mmol) was added to a solution of 4′-(4-bromophenyl)-2,2′:6′2′′-terpyridine (0.58 g, 1.5 mmol) and 4-([2,2′:6′,2′′-terpyridine]4′-yl)-phenyl boronic acid (0.71 g, 2.0 mmol), following the addition of the catalyst tetrakistriphenylphosphine palladium (0.2 g). The mixture was then refluxed at 90 °C for 18 h under N2. After cooling to room temperature, the solvent was evaporated under reducing pressure and the residue was purified by flash column chromatography (Al2O3) (CH2Cl2/PE = 2:1) to give g2 as a white solid (0.65 g, 71%). 1H NMR (301 MHz, CDCl3) δ: 8.81 (s, 2H), 8.76–8.74 (d, J = 6.02 Hz, 2H), 8.71–8.68 (d, J = 9.03 Hz, 2H), 8.06–8.03 (d, J = 9.03 Hz, 2H), 7.91–7.87 (m, 2H), 7.84–7.82 (d, J = 9.03 Hz, 2H), 7.39–7.35 (m, 2H).
4,4′′-Di([2,2′:6′,2′′-terpyridin]-4′-yl)-1,1′:4′,1′′-terphenyl ( g3 ) 17 was obtained as a white solid (0.446 g, 65%) according to a similar procedure of g2. 1H NMR (500 MHz, CDCl3) δ: 8.83 (s, 2H), 8.77–8.76 (d, J = 5 Hz, 2H), 8.71–8.70 (d, J = 5 Hz, 2H), 8.06–8.04 (d, J = 10 Hz, 4H), 7.92–7.89 (m, 2H), 7.84–7.82 (d, J = 10 Hz, 4H), 7.81 (s, 4H), 7.39–7.37 (m, 2H).
Ligand g4 was prepared from hexabromobenzene and 4-([2,2′:6′,2′′-terpyridine]4′-yl)-phenyl boronic acid according to a literature procedure.91H NMR (500 MHz, CDCl3) δ: 8.59 (s, 6H), 8.58–8.57 (d, J = 5 Hz, 12H), 8.53–8.52 (d, J = 5 Hz, 12H), 7.78–7.75 (m, 12H), 7.60–7.58 (d, J = 10 Hz, 12H), 7.24–7.22 (m, 12H), 7.17–7.15 (d, J = 10 Hz, 12H).
Synthesis of G1
In a suspension of T (35 mg, 0.05 mmol) and g1 (13.5 mg, 0.025 mmol) in ethylene glycol (8 ml), N-ethylmorpholine (4 drops) was mixed; the mixture was heated for 16 h. The resulting red solution was allowed to cool down, and then adding diethyl ether, a red solid was precipitated. After purification on a column of aluminium oxide (CH2Cl2/CH3OH = 100/2), G1 was obtained as a red solid (23 mg, 46%). 1H NMR (500 MHz, CD3OD) δ: 9.51 (s, 4H, g1Tpy-H3′,5′), 9.28 (s, 4H, TTpy-H3′,5′), 9.05–9.03 (d, J = 10 Hz, 4H, g1Tpy-H3,3′′), 8.94–8.92 (d, J = 10 Hz, 4H, TTpy-H3,3′′), 8.77 (s, 4H, g1Ph-Ha), 8.31–8.30 (d, J = 10 Hz, 4H, TPh-Ha), 8.11–8.05 (m, 8H, g1,TTpy-H4,4′′), 7.64–7.63 (d, J = 5 Hz, 8H, g1,TTpy-H6,6′′), 7.36–7.30 (m, 12H, g1,TTpy-H5,5′′, TPh-Hb), 4.22–4.19 (m, 4H), 1.95–1.89 (m, 4H), 1.63–1.57 (m, 4H), 1.50–1.31 (m, 32H), 0.95–0.92 (m, 6H). 13C NMR (126 MHz, CD3OD) δ: 161.53, 158.50, 158.42, 155.89, 155.30, 152.01, 151.89, 148.90, 147.37, 138.50, 138.05, 128.94, 128.82, 128.51, 127.63, 127.49, 124.79, 124.54, 121.43, 120.58, 115.28, 68.06, 62.91, 53.37, 31.67, 29.38, 29.36, 29.33, 29.13, 29.07, 28.97, 25.79, 22.33, 13.03. Q-TOF-MS (m/z): Calcd for [G1-2Cl]2+, [G1-3Cl]3+ and [G1-4Cl]4+: 900.3, 588.5, 432.6; Found: 900.3, 588.5, 432.6. Elemental Analysis: C102H102N12O2Ru2·2PF6·2Cl·CH2Cl2; Calcd for C, 56.86; H, 4.82; N, 7.72; Found: C, 60.08; H, 4.87; N, 7.54.Synthesis of G2
A similar procedure was used for the preparation of G1 as a red solid (29 mg, 60%). 1H NMR (500 MHz, CD3OD) δ: 9.43 (s, 4H, g2Tpy-H3′,5′), 9.28 (s, 4H, TTpy-H3′,5′), 9.00–8.98 (d, J = 10 Hz, 4H, g2Tpy-H3,3′′), 8.93–8.92 (d, J = 5 Hz, 4H, TTpy-H3,3′′), 8.57–8.56 (d, J = 5 Hz, 4H, g2Ph-Ha), 8.31–8.30 (d, J = 5 Hz, 4H, TPh-Ha), 8.27–8.26 (d, J = 5 Hz, 4H, g2Ph-Hb), 8.10–8.04 (m, 8H, g2,TTpy-H4,4′′), 7.62–7.61 (d, J = 5 Hz, 8H, g2,TTpy-H6,6′′), 7.35–7.30 (m, 12H, g2,TTpy-H5,5′′, TPh-Hb), 4.21–4.19 (m, 4H), 1.94–1.89 (m, 4H), 1.63–1.57 (m, 4H), 1.50–1.31 (m, 32H), 0.95–0.92 (m, 6H). 13C NMR (126 MHz, MeOD) δ: 161.54, 158.54, 158.47, 155.76, 155.35, 151.97, 151.93, 148.84, 148.01, 141.78, 138.03, 136.34, 128.93, 128.53, 128.34, 127.90, 127.59, 127.48, 124.65, 124.53, 121.21, 120.58, 115.28, 70.15, 69.13, 68.05, 62.91, 31.68, 29.39, 29.37, 29.34, 29.13, 29.08, 28.97, 25.79, 22.34, 13.03. Q-TOF-MS (m/z): Calcd for [G2-2Cl]2+, [G2-3Cl]3+ and [G2-4Cl]4+: 938.3, 613.9, 451.6; found: 938.3, 613.9, 451.6. Elemental Analysis: C108H106N12O2Ru2·4Cl·6C2H5OH; Calcd for C, 64.79; H, 6.43; N, 7.56. Found: C, 64.80; H, 6.67; N, 6.97.Synthesis of G3
A similar procedure was applied to obtain G1 as a red solid (27 mg, 52%). 1H NMR (500 MHz, CD3OD) δ: 9.40 (s, 4H, g3Tpy-H3′,5′), 9.27 (s, 4H, TTpy-H3′,5′), 8.98–8.96 (d, J = 10 Hz, 4H, g3Tpy-H3,3′′), 8.92–8.91 (d, J = 5 Hz, 4H, TTpy-H3,3′′), 8.51–8.50 (d, J = 5 Hz, 4H, g3Ph-Ha), 8.30–8.29 (d, J = 5 Hz, 4H, TPh-Ha), 8.19–8.18 (d, J = 5 Hz, 4H, g3Ph-Hb), 8.09–8.04 (m, 12H, g3,TTpy-H4,4′′, g3Ph-Hc), 7.62–7.60 (d, J = 10 Hz, 8H, g3,TTpy-H6,6′′), 7.34–7.30 (m, 12H, g3,TTpy-H5,5′′, TPh-Hb), 4.21–4.19 (m, 4H), 1.94–1.89 (m, 4H), 1.62–1.57 (m, 4H), 1.50–1.31 (m, 32H), 0.95–0.92 (m, 6H). 13C NMR (101 MHz, CD3OD) δ: 162.41, 161.54, 158.50, 158.44, 155.70, 155.34, 138.08, 138.07, 138.06, 128.93, 128.16, 127.75, 127.63, 127.52, 127.47, 121.14, 120.58, 115.31, 95.42, 88.77, 78.17, 77.84, 77.51, 69.13, 68.07, 60.90, 31.69, 31.67, 30.47, 29.40, 29.38, 29.37, 29.35, 29.15, 29.08, 29.06, 28.98, 25.81, 22.36, 22.33, 13.10, 13.08. Q-TOF-MS (m/z): Calcd for [G3-2Cl]2+, [G3-3Cl]3+ and [G3-4Cl]4+: 976.3, 639.2, 470.7; found: 976.3, 639.2, 470.7. Elemental Analysis: C114H110N12O2Ru2·4Cl·12C2H5OH; Calcd for C, 64.54; H, 7.01; N, 6.64. Found: C, 64.96; H, 7.02; N, 5.91.Synthesis of G4
To a suspension of T (14.5 mg, 0.021 mmol) and g4 (6 mg, 0.003 mmol) in ethylene glycol (8 ml), N-ethylmorpholine (4 drops) was added; the mixture was heated for 2 days. The resulting red solution was allowed to cool down to room temperature, and then was extracted with diethyl ether to obtain a red solid, followed by purification on a column of aluminium oxide (CH2Cl2/CH3OH = 100:2) to obtain G4 as a red solid (8 mg, 37%). 1H NMR (400 MHz, CD3CN) δ: 8.96 (s, 12H, g4Tpy-H3′,5′), 8.95 (s, 12H, TTpy-H3′,5′), 8.65–8.63 (d, J = 8 Hz, 12H, g4Tpy-H3,3′′), 8.61–8.59 (d, J = 8 Hz, 12H, TTpy-H3,3′′), 8.18–8.16 (d, J = 8 Hz, 12H, g4Ph-Ha), 8.12–8.10 (d, J = 8 Hz, 12H, TPh-Ha), 7.85–7.80 (m, 24H, g4Tpy-H4,4′′, g4Ph-Hb), 7.70–7.66 (m, 12H, TTpy-H4,4′′), 7.39–7.38 (d, J = 4 Hz, 12H, g4Tpy-H6,6′′), 7.33–7.32 (d, J = 4 Hz, 12H, TTpy-H6,6′′), 7.29–7.27 (d, J = 8 Hz, 12H, TPh-Hb), 7.09–7.06 (m, 12H, g4Tpy-H5,5′′), 6.91–6.88 (m, 12H, TTpy-H5,5′′), 4.19–4.16 (m, 12H), 1.89–1.85 (m, 12H), 1.56–1.53 (m, 12H), 1.44–1.33 (m, 96H), 0.92–0.90 (m, 18H). 13C NMR (126 MHz, CD3CN) δ: 161.35, 158.31, 158.13, 155.50, 155.16, 152.38, 152.29, 148.10, 147.50, 142.75, 142.71, 137.89, 137.64, 134.63, 132.78, 129.17, 128.52, 127.45, 127.11, 124.49, 124.46, 123.39, 121.32, 121.31, 120.89, 115.53, 68.34, 31.66, 29.39, 29.38, 29.33, 29.09, 28.92, 25.72, 22.41, 13.40. Q-TOF-MS (m/z): Calcd for [G4-4PF6]4+, [G4-5PF6]5+, [G4-6PF6]6+, [G4-7PF6]7+, [G4-8PF6]8+ and [G4-9PF6]9+: 1662.2, 1300.8, 1060.3, 887.8, 759.0, 658.2; found: 1662.2, 1300.8, 1060.3, 887.8, 759.0, and 658.2, respectively. Elemental Analysis: C331H321N36O6Ru6·4Cl·8PF6·12C2H5OH; Calcd for C, 57.93; H, 5.38; N, 6.85. Found: C, 57.97; H, 5.49; N, 6.69.
Conclusions
In summary, a series of new bimetallic Ru2+ terpyridine complexes G1, G2 and G3 were reported, and the hexametallic Ru2+ terpyridine based G4 was prepared from Ru(T)Cl3 and the corresponding hexa-terpyridine ligands. These complexes’ behaviours were explored by absorption spectra and cyclic voltammograms. Photophysical and electrochemical properties of the complexes had been found to be finely tuned by the differed conjugation of the bis-terpyridine bridges. The maximum absorption wavelength had an obvious hypochromic shift from 502 nm for G1 to 493 nm for G4 when the conjugation of the terpyridine bridge linkage increased. Likewise, the oxidation potential exhibited a slight decrease (47 mv) from G1 to G4. The results described here could facilitate the understanding of the relationship of multi-Ru2+ complexes’ properties and structures, which could benefit the further design and preparation of multi-Ru2+-based terpyridine complexes with desired photophysical properties and metallo–liquid crystal materials.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21274165), and the Distinguished Professor Research Fund from Central South University. The authors acknowledge the NMR measurements from The Modern Analysis and Testing Center of CSU.Notes and references
- (a) Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed; (b) R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed; (c) I. A. Riddell, Y. R. Hristova, J. K. Clegg, C. S. Wood, B. Breiner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 2723 CrossRef CAS PubMed; (d) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063 CrossRef CAS PubMed; (e) P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502 CrossRef CAS; (f) M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456 CrossRef CAS PubMed; (g) P. Mobian, J.-M. Kern and J.-P. Sauvage, J. Am. Chem. Soc., 2003, 125, 2016 CrossRef CAS PubMed; (h) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561 CrossRef CAS; (i) A. E. V. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207 CrossRef CAS PubMed; (j) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed; (k) S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1–9 CrossRef CAS PubMed; (l) M. Barboiu, G. Vaughan, R. Graff and J.-M. Lehn, J. Am. Chem. Soc., 2003, 125, 10257 CrossRef CAS PubMed; (m) M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52 CrossRef CAS.
- T. Schroder, R. Brodbeck, M. C. Letzel, A. Mix, B. Schnatwinkel, M. Tonigold, D. Volkmer and J. Mattay, Tetrahedron Lett., 2009, 50, 5939–5942 Search PubMed.
- Z. Zhao, Y. R. Zheng, M. Wang, J. B. Pollock and P. J. Stang, Inorg. Chem., 2010, 49, 8653–8655 CrossRef CAS PubMed.
- X. Liu, C. L. Stern and C. A. Mirkin, Organometallics, 2002, 21, 1017–1019 CrossRef CAS.
- (a) U. S. Schubert, H. Hofmeier and G. R. Newkome, ModernTerpyridine Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) G. R. Newkome, P. Wang, C. N. Moorefield, T. J. Cho, P. P. Mohopatra, S. Li, S.-H. Hwang, O. Lukoyanova, L. Echegoyen, J. A. Palagallo, V. Lancu and S.-W. Hla, Science, 2006, 312, 1782 CrossRef CAS PubMed.
- (a) E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC; (b) M. W. Cooke and G. S. Hanan, Chem. Soc. Rev., 2007, 36, 1466 RSC; (c) E. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133 RSC; (d) D. G. Kurth and M. Higuchi, Soft Matter, 2006, 2, 915 RSC; (e) G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689 CrossRef CAS PubMed.
- J. Hummel, A. Winter, A. Baumgaertel, N. Risch and U. S. Schubert, Synlett, 2010, 1, 61–66 Search PubMed.
- (a) R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS PubMed; (b) R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802 CrossRef CAS PubMed.
- (a) J. L. Wang, X. P. Li, I. F. Hsieh, Y. Cao, C. N. Moorefield, C. Wesdemiotis, S. Z. D. Cheng and G. R. Newkome, J. Am. Chem. Soc., 2011, 133, 11450–11453 CrossRef CAS PubMed; (b) R. Passalacqua, F. Loiseau, S. Campagna, Y.-Q. Fang and G. S. Hanan, Angew. Chem., Int. Ed., 2003, 42, 1607 CrossRef PubMed; (c) Y. Li, Z. Jiang, J. Yuan, D. Liu, T. Wu, C. N. Moorefield, G. R. Newkome and P. Wang, Chem. Commun., 2015, 51, 5766 RSC; (d) E. Wachter, D. K. Keidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649 RSC.
- (a) E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans., 1994, 1409–1418 RSC; (b) H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373 RSC; (c) C. Tschierske, Angew. Chem., Int. Ed., 2000, 39, 2454 CrossRef CAS.
- A. Gamliel, M. Afri and A. A. Frimer, Free Radicals Biol. Med., 2008, 44, 1394–1405 CrossRef CAS PubMed.
- E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans, 1992, 24, 3467–3475 RSC.
- F. Krohnke, Synthesis, 1976, 1–24 CrossRef.
- T. K. Sievers, A. Vergin, H. Möhwald and D. G. Kurth, Langmuir, 2007, 23, 12179–12184 CrossRef CAS PubMed.
- T. Mutai, J. D. Cheon, S. Arita and K. Araki, J. Chem. Soc., Perkin Trans. 2, 2001, 1045–1050 RSC.
- F. S. Han, M. Higuchi and D. G. Kurth, Org. Lett., 2007, 9, 559–562 CrossRef CAS PubMed.
- R. R. Pal, M. Higuchi, Y. C. Negishi, T. Tsukuda and D. G. Kurth, Polym. J., 2010, 42, 336–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, and NMR and mass spectra. See DOI: 10.1039/c5qi00181a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |